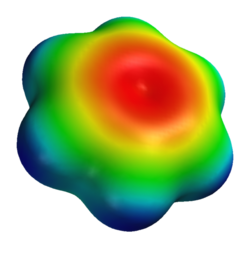
벤젠 분자에 대해 계산된 전위장입니다.
양자화학은 분자 양자역학이라고도 하며, 양자역학을 화학 시스템에 적용하는 데 중점을 둔 물리화학의 한 분야로, 특히 원자 수준에서 분자, 재료, 용액의 물리적·화학적 특성에 대한 전자 기여를 양자역학적으로 계산하는 데 중점을 둡니다. [1]
이 계산들은 계산이 가능하면서도 계산된 파동 함수에 중요한 기여와 구조, 스펙트럼, 열역학적 특성과 같은 관측 가능한 특성에 대한 정보를 최대한 많이 포착하기 위해 체계적으로 적용된 근사를 포함합니다. 양자화학은 분자 역학과 화학 속도론에 미치는 양자 효과 계산에도 관심을 가집니다.
양자 화학 연구는 원자, 분자, 이온의 전자 바닥 상태와 여기 상태에 초점을 맞추었습니다. 이러한 계산은 화학 반응을 경로, 중간체, 전이 상태에 대해 설명할 수 있게 합니다. 분광학적 특성도 예측할 수 있습니다. 일반적으로 이러한 연구는 전자파동 함수가 핵 위치(즉, 본-오펜하이머 근사)에 의해 단열적으로 매개변수화된다고 가정합니다. 반경험적 방법, 밀도 함수 이론, 하트리-폭 계산, 양자 몬테카를로 방법, 결합 클러스터 방법 등 다양한 접근법이 사용됩니다.
슈뢰딩거 방정식의 계산 해를 개발하여 전자 구조와 분자 역학을 이해하는 것은 양자화학의 핵심 목표입니다. 이 분야의 진보는 여러 도전을 극복하는 데 달려 있습니다. 여기에는 소분계의 결과 정확도를 높이는 필요성과, 계산 시간이 원자 수만큼 거듭제곱 증가하는 스케일링 고려에 의해 제한되는 대형 분자의 크기를 더 크게 하는 것 등이 포함됩니다. [2]
역사
일부는 양자화학의 탄생을 슈뢰딩거 방정식의 발견과 수소 원자에 적용한 것으로 본다. 그러나 1927년 발터 하이틀러(1904–1981)와 프리츠 런던의 논문은 양자화학 역사상 첫 번째 이정표로 자주 인정받는다. [3] 이는 양자역학이 이원자 수소 분자에 처음으로 적용되었고, 따라서 화학 결합 현상에 적용된 사례였다. [4] 그러나 그 이전에는 길버트 N. 루이스가 1916년 논문 『원자와 분자』[5]에서 원자가 전자의 첫 작동 모델을 개발한 중요한 개념적 틀이 제시되었다. 요시카츠 스기우라[6][7]와 S.C. 왕도 중요한 기여를 했다. [8] 1930년대 내내 라이너스 폴링이 쓴 일련의 논문들은 하이틀러, 런던, 스기우라, 왕, 루이스, 존 C. 슬레이터의 원가 개념과 그 양자역학적 기초에 관한 연구를 새로운 이론적 틀에 통합했다. [9] 많은 화학자들은 폴링의 1939년 저서 『화학 결합의 본질과 분자 및 결정의 구조: 현대 구조화학 입문』을 통해 양자화학 분야에 입문했다. 이 책에서 그는 이 연구(현재 널리 원가결합 이론으로 불림)를 요약하고 화학자들이 따라갈 수 있도록 양자역학을 설명했다. [10] 이 교재는 곧 많은 대학에서 표준 교재가 되었습니다. [11] 1937년, 한스 헬만은 러시아어와 독일어로 양자화학에 관한 책을 최초로 출판한 것으로 보인다. [13]
이후 몇 년 동안 이 이론적 기초는 서서히 화학 구조, 반응성, 결합에 적용되기 시작했습니다. 앞서 언급한 연구자들 외에도, 어빙 랭뮤어, 로버트 S. 멀리켄, 맥스 본, J. 로버트 오펜하이머, 한스 헬만, 마리아 괴퍼트 마이어, 에리히 휘켈, 더글라스 하트리, 존 레나드-존스, 블라디미르 폭 등이 이 분야의 초기 시기에 중요한 진전과 중요한 기여를 했다.
전자 구조
원자 또는 분자의 전자 구조는 그 전자들의 양자 상태이다. [14] 양자 화학 문제를 해결하는 첫 단계는 보통 슈뢰딩거 방정식(상대론적 양자화학에서는 디랙 방정식)을 전자 분자 해밀토니안으로 푸는 것이며, 보통 본-오펜하이머(B–O) 근사를 사용한다. 이를 분자의 전자 구조 결정이라고 한다. [15] 비상대론적 슈뢰딩거 방정식의 정확한 해는 수소 원자에 대해서만 얻을 수 있다(다만 B-O 근사 내에서 수소 분자 이온의 결합 상태 에너지에 대한 정확한 해는 일반화된 램버트 W 함수를 통해 확인되었다). 다른 모든 원자 및 분자 시스템은 세 개 이상의 "입자"의 운동을 포함하기 때문에, 슈뢰딩거 방정식은 해석적으로 풀 수 없으므로 근사 및/또는 계산적 해를 찾아야 한다. 이 문제들에 대한 계산적 해를 찾는 과정은 계산 화학이라는 분야의 일부이다. [2]
원가 결합 이론
주요 문서: 원가 결합 이론
앞서 언급했듯이, 하이틀러와 런던의 방법은 슬레이터와 폴링에 의해 원가결합(VB) 방법으로 확장되었다. 이 방법에서는 주로 원자 간의 쌍별 상호작용에 초점을 맞추며, 따라서 고전 화학자들의 결합 도면과 밀접하게 연관되어 있다. 이 방법은 분자가 형성될 때 원자의 오비탈이 결합하여 개별 화학 결합을 형성하는 방식을 중점으로 하며, 궤도 혼성과 공명이라는 두 가지 핵심 개념을 통합한다. [16]
공유 결합은 두 원자가 반쯤 채워진 원자 오비탈들이 겹쳐져 전자 쌍을 형성할 때 형성됩니다. 시스템의 세기와 에너지는 겹침의 양에 따라 달라집니다. 원자들이 함께 움직이면서 오비탈이 겹치기 시작하고, 전자들은 서로의 핵의 인력을 느끼기 시작합니다. 또한 두 원자가 가까이 있을 때 너무 강해지는 반발력도 발생하기 시작합니다. 두 원자 사이의 이상적이고 가장 안정적인 길이는 결합 거리로, 이는 가장 낮은 에너지 구성을 만드는 반발력과 인력의 결합입니다.
오비탈의 방향은 어떤 결합이 형성되는지에 큰 영향을 미칠 수 있다. 각 원자에서 하나의 원자 궤도가 직접 겹치면 시그마(σ) 결합이 형성된다. 이는 두 개의 s-오비탈, 즉 s-오비탈과 p-오비탈, 또는 두 개의 p-오비탈에서 만들어질 수 있다. 파이(π) 결합은 두 개의 p-오비탈이 좌우로 겹쳐져 형성된다. 파이 결합은 겹치는 p-오비탈의 위상이 같을 때만 형성된다. [17]
분자 궤도 이론
주요 문서: 분자 궤도 이론
원가 결합 이론에 대한 대안적 접근법은 1929년 프리드리히 훈트와 로버트 S. 멀리켄에 의해 개발되었는데, 전자는 분자 전체에 걸쳐 비국재화된 수학적 함수로 기술된다. 헌트-멀리켄 접근법 또는 분자 궤도(MO) 방법은 화학자에게는 직관적이지 않지만 VB 방법보다 분광학적 특성을 더 잘 예측합니다. VB 이론과 달리, MO 이론은 단순히 전자 밀도가 한 영역에서 겹쳐져 결합을 형성하는 데 초점을 맞추지 않고, 전체 분자를 하나의 시스템으로 설명합니다. 이로 인해 시스템에 대한 더 복잡한 이해가 이루어집니다. 이 접근법은 하트리-폭 방법과 이후 하트리-폭 기법의 개념적 기반이다. [2]
MO 계산은 분자의 궤도, 파동함수, 에너지를 산출하며, 이 분자는 서로 다른 원자 오비탈의 전자로 채워질 수 있습니다. 이 원자 오비탈들은 서로 다른 원자에서 나오며, 분자 오비탈은 원자 오비탈의 선형 결합이 됩니다.
밀도 함수 이론
주요 문서: 밀도 함수 이론
토마스-페르미 모델은 1927년에 토마스와 페르미에 의해 독립적으로 개발되었다. 이는 파동함수 대신 전자 밀도를 기반으로 다전자 시스템을 기술하려는 최초의 시도였으나, 전체 분자 처리에는 큰 성공적이지 못했다. 이 방법은 현재 밀도 함수 이론(DFT)으로 알려진 것의 기초를 제공했습니다. 현대의 DFT는 밀도 함수를 네 항으로 나누는 콘-샴 방법을 사용합니다; 코언-샴 운동 에너지, 외부 퍼텐셜, 교환 및 상관 에너지입니다. DFT 개발의 큰 부분은 교환 및 상관관계 용어를 개선하는 데 있습니다. 이 방법은 하트리-폭 이후 방법보다 덜 발전했지만, 계산 요구량이 훨씬 낮아(순수 함수에 대해 n/3 이하로 스케일링하는 경우) 더 큰 다원자 분자와 심지어 거대분자까지 다룰 수 있습니다. 이러한 계산 비용의 저렴함과 MP2 및 CCSD(T)(하트리-폭 이후 방법)와 비교할 만한 정확도 덕분에 계산 화학에서 가장 인기 있는 방법 중 하나가 되었습니다. [18]
화학 역학
또 다른 단계는 분자의 운동을 연구하기 위해 전체 분자 해밀토니안으로 슈뢰딩거 방정식을 푸는 것입니다. 슈뢰딩거 방정식의 직접 해법은 양자 동역학이라 하며, 반고전 근사 내의 해는 반고전 동역학이라고 합니다. 순수하게 고전적인 분자 운동 시뮬레이션은 분자 동역학(MD)이라고 불립니다. 또 다른 동역학 접근법은 혼합 양자-고전 동역학이라는 하이브리드 프레임워크입니다; 또 다른 하이브리드 프레임워크는 파인만 경로 적분 공식화를 사용하여 분자 역학에 양자 보정을 더하는 경로 적분 분자 역학이라고 합니다. 고전 및 양자 몬테카를로 방법과 같은 통계적 접근법도 가능하며, 상태의 평형 분포를 설명하는 데 특히 유용합니다. [2]
단열 화학 역학
주요 문서: 본-오펜하이머 근사
단열 역학에서 원자 간 상호작용은 위치 에너지 표면이라 불리는 단일 스칼라 퍼텐셜로 표현됩니다. 이것이 바로 1927년 본과 오펜하이머가 도입한 본-오펜하이머 근사이다. 이 기술의 선구적 응용은 1927년 라이스와 램스퍼거, 1928년 카셀에 의해 이루어졌으며, 1952년 마커스가 1935년 아이링이 발전시킨 전이 상태 이론을 고려하여 RRKM 이론으로 일반화했다. 이 방법들은 전위 표면의 몇 가지 특성으로부터 단일 분자 반응 속도를 간단히 추정할 수 있게 합니다. [2]
비단열 화학 역학
주요 문서: 바이브로닉 결합
비단열 역학은 여러 결합된 위치 에너지 표면(분자의 서로 다른 전자 양자 상태에 해당)간의 상호작용을 수행하는 것으로 구성됩니다. 결합 항들은 진동 결합이라고 불립니다. 이 분야의 선구적인 연구는 1930년대 슈투켈베르크, 란다우, 제너가 현재 란다우-제너 전이로 알려진 현상에서 수행했다. 이 공식은 회피한 교차 근처에서 두 단열 퍼텐셜 곡선 간의 전이 확률을 계산할 수 있게 해줍니다. 스핀 금지 반응은 반응물에서 생성물로 진행할 때 스핀 상태가 최소 한 가지 이상 변화하는 비단열 반응의 한 유형입니다. [2]
참고 문헌
- 원자 물리학
- 계산 화학
- 응집물질 물리학
- 카르-파리넬로 분자 역학
- 전자 위치 함수
- 국제 양자 분자 과학 아카데미
- 분자 모델링
- 물리 화학
- 양자 계산 화학
- 양자 화학 및 고체 상태 물리학 소프트웨어 목록
- QMC@Home
- 생명의 양자 측면
- 양자 전기화학
- 상대론적 양자 화학
- 이론 물리학
- 스핀 금지 반응
참고문헌
- 맥쿼리, 도널드 A. (2007). 양자 화학 (2판). 대학교 과학 도서. ISBN 978-1891389504.
- 맥쿼리, 도널드 A.; 사이먼, 존 D. (200). 물리화학: 분자적 접근법. 캘리포니아 소살리토: Univ. Science Books. ISBN 978-0-935702-99-6.
- 하이틀러, W.; 런던, F. (1927). "Wechselwirkung neutraler Atome und homopolare Bindung nach der Quantenmechanik". Zeitschrift für Physik. 44권 (6–7): 455–472. 비브코드: 1927ZPhy... 44..455H. doi:10.1007/BF01397394.
- 코워스, W. (1989). "하이틀러-런던 접근법의 기원, 발전 및 중요성". 양자화학에서의 관점. 국제 양자 분자 과학 아카데미. 6권. 도르드레흐트: 슈프링거. 145–159쪽. doi:10.1007/978-94-009-0949-6_8. ISBN 978-94-010-6917-5.
- 루이스, G.N. (1916). "원자와 분자". 미국 화학회 저널. 38 (4): 762–785. 참고 코드: 1916JAChS.. 38.762L. doi:10.1021/ja02261a002.
- 스기우라, Y. (1927). "Über die Eigenschaften des Wasserstoffmoleküls im Grundzustande". Zeitschrift für Physik. 45권 (7–8): 484–492. 비브코드: 1927ZPhy... 45..484S. doi:10.1007/BF01329207.
- 나카네, 미치요 (2019). "일본 양자 물리학 발전에 대한 스기우라 요시카츠의 기여". Berichte zur Wissenschaftsgeschichte. 42권 4호: 338–356. doi:10.1002/bewi.201900007. PMID 31777981.
- 왕, S. C. (1928-04-01). "새로운 양자역학에서의 정상 수소 분자 문제". 피지컬 리뷰. 31 (4): 579–586. 참고 코드: 1928PhRv... 31..579W. doi:10.1103/PhysRev.31.579.
- 폴링, 라이너스 (1931년 4월 6일). "화학 결합의 본질. 양자역학과 분자 구조에 대한 패러자성 감수성 이론에서 얻은 결과를 적용하는 것". 미국 화학회 저널. 53 (4): 1367–1400. 참고 코드: 1931JAChS.. 53.1367P. doi:10.1021/ja01355a027 – 오리건 주립대학교 도서관 경유.
- 폴링, 라이너스 (1939). 화학 결합의 본질과 분자 및 결정의 구조: 현대 구조화학 입문 (1판). 코넬 대학교 출판부.
- 노먼, 제레미. "폴링, "화학 결합의 본질"을 출판하다. 정보의 역사. 2023년 7월 11일에 확인함.
- Хельман, Г. (1937). Квантовая химия. Главная редакция технико-теоретической литературы, 모스크바와 레닌그라드.
- 헬만, 한스 (1937). Einführung in die Quantenchemie. 듀티케, 라이프치히, 빈.
- 사이먼스, 잭 (2003). "6장. 전자 구조". 이론화학 입문 (PDF). 영국 케임브리지: 케임브리지 대학교 출판부. ISBN 0521823609.
- 마틴, 리처드 M. (2008-10-27). 전자 구조: 기본 이론과 실용 방법. 케임브리지: 케임브리지 대학교 출판부. ISBN 978-0-521-53440-6.
- 샤이크, S.S.; 하이버티, P.C. (2007). 원자가 결합 이론에 관한 화학자 가이드. 와일리-인터사이언스. ISBN 978-0470037355.
- "5.4 원가결합 이론 | 일반 대학 화학 I. courses.lumenlearning.com. 2025-12-01에 확인함.
- 해리슨, N. M. "밀도 함수 이론 입문". 제국 과학기술의학대학.
출처
- 앳킨스, P.W. (2002). 물리화학. 옥스퍼드 대학교 출판부. ISBN 0-19-879285-9.
- 앳킨스, P.W.; 프리드먼, R. (2005). 분자 양자역학 (4판). 옥스퍼드 대학교 출판부. ISBN 978-0-19-927498-7.
- 앳킨스, P.W.; 프리드먼, R. (2008). 양자, 물질, 변화: 물리적 변화에 대한 분자 접근법. 맥밀란. ISBN 978-0-7167-6117-4.
- 베이더, 리처드 (1994). 분자 속의 원자: 양자 이론. 옥스퍼드 대학교 출판부. ISBN 978-0-19-855865-1.
- 크레이머, 크리스토퍼 J (2004). 계산 화학의 핵심: 이론과 모델(2판). 와일리. ISBN 9780470091821. OCLC 55887497.
- 드랄, 파블로 O. (2023). 드랄, 파블로 O. (편집). 기계 학습 시대의 양자화학. 네덜란드 암스테르담: 엘스비어. ISBN 9780323900492. LCCN 2024443666. OCLC 1294286017.
- 가브로글루, 코스타스; 시모이스, 아나 (2011). 물리학도 화학도 아니다: 양자화학의 역사. MIT 출판부. ISBN 978-0-262-01618-6.
- 카플러스, M.; 포터, R. N. (1971). 원자와 분자: 물리화학 학생을 위한 입문서. 벤자민-커밍스 출판사. ISBN 978-0-8053-5218-4.
- 랜다우, L.D.; 리프시츠, E.M. (1977). 양자역학: 비상대론 이론. 이론물리학 과정. 3권. 퍼가몬 프레스. ISBN 0-08-019012-X.
- 레빈, I. (2008). 물리화학 (6판). 맥그로우-힐 사이언스. ISBN 978-0-07-253862-5.
- 컬슨, 찰스 알프레드 (1991) [1979]. 로이 맥위니 (편집). 콜슨의 발언스 (3판). 옥스퍼드 대학교 출판부. ISBN 9780198551454. OCLC 468330825.
- 폴링, L. (1954). 일반 화학. 도버 출판사. ISBN 0-486-65622-5.
- 폴링, L.; 윌슨, E. B. (1963) [1935]. 양자역학 입문과 화학에 대한 응용. 도버 출판사. ISBN 0-486-64871-0.
- 버나드 풀먼; 풀먼, 알베르테 (1963). 양자 생화학. 뉴욕 및 런던: 아카데믹 프레스. ISBN 90-277-1830-X.
- 스케리, 에릭 R. (2006). 주기율표: 그 이야기와 중요성. 옥스퍼드 대학교 출판부. ISBN 0-19-530573-6. 화학, 특히 주기계가 양자역학으로 환원된 정도를 고려한다.
- 사이먼, Z. (1976). 양자 생화학과 특이 상호작용. 테일러 & 프랜시스. ISBN 978-0-85626-087-2.
- 사보, 아틸라; 오스트룬드, 닐 S. (1996). 현대 양자 화학: 고급 전자 구조 이론 입문. 도버. ISBN 0-486-69186-1.
외부 링크
화학의 분야분석적이론적신체무기물유기농생물학적학제간 연구참고
카테고리:
- 이 페이지는 2026년 1월 6일 13:54 (UTC)에 마지막으로 편집되었습니다.
화학적 동역학
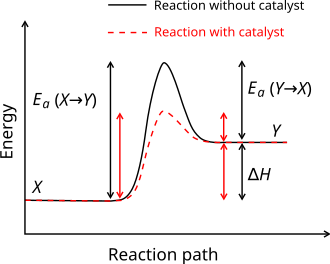
촉매가 발열 반응에서 활성화 에너지를 어떻게 낮추는지 보여주는 반응 좌표 다이어그램.
화학 반응 속도론은 반응 속도론이라고도 하며, 화학 반응의 속도를 이해하는 데 중점을 두는 물리화학의 한 분야입니다. 이는 반응이 일어나는 방향을 다루지만 그 자체로는 반응 속도에 대해 아무것도 알려주지 않는 화학 열역학과는 다릅니다. 화학 반응 속도론은 실험 조건이 화학 반응 속도에 어떻게 영향을 미치는지, 반응의 기전 및 전이 상태에 관한 정보를 제공하는 연구를 포함하며, 화학 반응의 특성을 설명할 수 있는 수학적 모델을 구축하는 것도 포함합니다.
역사
화학 반응학의 선구적인 연구는 1850년 독일 화학자 루트비히 빌헬미가 수행했다. [1] 그는 자당의 역전 속도를 실험적으로 연구했으며, 이 반응의 반응 속도 속도를 결정하기 위해 통합 속도 법칙을 사용했다. 그의 작품은 34년 후 빌헬름 오스트발트의 주목을 받았다. 1864년, 피터 와게와 카토 굴드베리는 질량 작용 법칙을 발표했는데, 이는 화학 반응의 속도가 반응 물질의 양에 비례한다는 것이다. [2][3][4]
반 't 호프는 화학 동역학을 공부했으며, 1884년에 유명한 "Études de dynamique chimique"를 출판했다. [5] 1901년 그는 "용액 내 화학 역학 법칙과 삼투압 법칙을 발견함으로써 기여한 탁월한 공로"로 최초의 노벨 화학상을 수상했다. [6] 반 트 호프 이후, 화학 동역학은 반응 속도의 실험적 결정에 관여했으며, 그로부터 속도 법칙과 속도 상수를 도출하는 연구가 되었다. 반응 속도가 농도와 무관한 0차 반응, 1차 반응, 2차 반응에 대해 비교적 단순한 속도 법칙이 존재하며, 다른 반응들에 대해서도 도출할 수 있습니다. 기본 반응은 질량 작용 법칙을 따르지만, 단계별 반응의 속도 법칙은 여러 기본 단계의 속도 법칙을 결합하여 도출되어야 하며, 상당히 복잡해질 수 있습니다. 연속적인 반응에서는 속도 결정 단계가 종종 동역학을 결정합니다. 연속적인 1차 반응에서는 정상 상태 근사가 속도 법칙을 단순화할 수 있다. 반응의 활성화 에너지는 아레니우스 방정식과 아이링 방정식을 통해 실험적으로 결정됩니다. 반응 속도에 영향을 미치는 주요 요인으로는 반응물의 물리적 상태, 반응물의 농도, 반응이 일어나는 온도, 그리고 반응 내에 촉매가 존재하는지 여부가 포함됩니다.
고르반과 야블론스키는 화학 역학의 역사를 세 시대로 나눌 수 있다고 제안했다. [7] 첫 번째는 반 't 호프 파동으로, 화학 반응의 일반 법칙을 찾고 운동론을 열역학과 연관시키는 것입니다. 두 번째는 특히 연쇄 반응에 중점을 둔 반응 메커니즘에 중점을 둔 세메노프-힌셸우드 파동이라고 부를 수 있습니다. 세 번째는 아리스와 화학 반응 네트워크의 상세한 수학적 설명과 관련이 있습니다.
반응 속도에 영향을 미치는 요인
반응물의 특성
반응 속도는 반응하는 물질에 따라 달라집니다. 산/염기 반응, 염분 형성, 이온 교환은 보통 빠른 반응입니다. 분자 간 공유 결합 형성과 큰 분자가 형성될 때는 반응이 느려지는 경향이 있습니다.
반응물 분자 내 결합의 성질과 강도는 생성물로 변환되는 속도에 큰 영향을 미칩니다.
물리적 상태
반응물의 물리적 상태(고체, 액체 또는 기체)도 변화율의 중요한 요소입니다. 반응물들이 수성 용액과 같이 같은 상일 때, 열운동에 의해 접촉하게 됩니다. 하지만 이들이 서로 다른 상에 있을 때는 반응물이 반응물들 간의 경계면에 한정됩니다. 반응은 접촉 부위에서만 일어날 수 있으며; 액체와 기체의 경우, 액체 표면에 위치해 있습니다. 반응을 완성하려면 격렬한 흔들기와 저어주기도 필요할 수 있습니다. 즉, 고체 또는 액체 반응물이 더 미세하게 분열될수록 단위 부피당 표면적이 커지고, 다른 반응물과의 접촉도 많아져 반응이 더 빨라집니다. 예를 들어, 불을 피울 때 나무 조각과 작은 가지를 사용하는데, 큰 통나무로 바로 시작하는 것은 아닙니다. 유기화학에서 물 반응은 균질 반응이 이질적 반응(용질과 용매가 제대로 혼합되지 않은 반응)보다 더 빠르게 일어난다는 규칙의 예외입니다.
고체 상태의 표면적
고체에서는 표면에 있는 입자들만이 반응에 참여할 수 있습니다. 고체를 더 작은 조각으로 으깨면 표면에 더 많은 입자가 존재하고, 이들과 반응물 입자 간의 충돌 빈도가 증가하여 반응이 더 빠르게 일어납니다. 예를 들어, 셔벗(분말)은 매우 미세한 말산(약한 유기산)과 탄산수소나트륨의 혼합물입니다. 입 안의 침과 접촉하면 이 화학물질들은 빠르게 녹아 반응하여 이산화탄소를 방출하고 탄산지 같은 느낌을 제공합니다. 또한, 불꽃놀이 제조업체들은 고체 반응물의 표면적을 조절하여 불꽃놀이 연료의 산화 속도를 조절하여 다양한 효과를 만들어냅니다. 예를 들어, 미세하게 분리된 알루미늄이 포탄 안에 갇혀 있으면 격렬하게 폭발합니다. 더 큰 알루미늄 조각을 사용하면 반응이 느려지고, 타는 금속 조각이 튀어나오면서 불꽃이 발생합니다.
집중
주요 문서: 요금 방정식
이 반응은 반응 물질 간의 충돌에 의해 발생합니다. 분자나 이온이 충돌하는 빈도는 농도에 따라 달라집니다. 분자가 더 빽빽할수록 충돌하고 서로 반응할 가능성이 높아집니다. 따라서 반응물 농도가 증가하면 반응 속도도 증가하는 반면, 농도가 낮아지면 보통 반대 효과가 나타납니다. 예를 들어, 연소는 공기(산소 21%)보다 순수 산소에서 더 빠르게 일어납니다.
속도 방정식은 반응 속도가 반응물 및 존재하는 다른 종의 농도에 대한 상세한 의존성을 보여줍니다. 수학적 형태는 반응 메커니즘에 따라 달라집니다. 주어진 반응의 실제 속도 방정식은 실험적으로 결정되며 반응 메커니즘에 대한 정보를 제공합니다. 속도 방정식의 수학적 표현은 종종 다음과 같이 주어집니다.
v=dcdt=k∏나는c나는m나는

여기 k

는 반응 속도 상수입니다. c나는

는 반응물 I의 몰 농도이며, m나는

이 반응물의 부분 반응 순서입니다. 반응물의 부분 순서는 실험적으로만 결정할 수 있으며, 종종 화학양론계수로는 알 수 없습니다.
미세몰 수준 이하 농도와같이 매우 희석된 용액에서는 분자 충돌이 주로 확산에 의해 지배됩니다. 이러한 조건에서는 반응 분자들이 서로 만나기 전에 더 긴 거리를 이동하는 데 추가 시간이 필요하기 때문에 겉보기 반응 순서가 화학양론적 기대치와 다르게 나타납니다. 이 행동은 피크의 확산 법칙으로 설명할 수 있으며, 분수 반응 차수를 산출하는 프랙탈 반응 속도론과도 일치합니다.
온도
주요 문서: 아레니우스 방정식
온도는 보통 화학 반응 속도에 큰 영향을 미칩니다. 더 높은 온도의 분자는 더 많은 열 에너지를 가집니다. 충돌 빈도가 높은 온도에서 더 높지만, 이 정도만으로는 반응률 증가에 아주 작은 비율만을 차지합니다. 훨씬 더 중요한 점은 반응할 에너지가 충분한(활성화 에너지보다 큰 에너지: E > Ea) 비율이 훨씬 높으며, 이는 분자 에너지의 맥스웰-볼츠만 분포로 자세히 설명된다.
온도가 반응 속도 상수에 미치는 영향은 보통 아레니우스 방정식을 따릅니다. k=Ae−Ea/(RT)

여기서 A는 지수 전 인자 또는 A-인자, Ea는 활성화 에너지, R은 몰기 가스 상수, T는 절대 온도입니다. [8]
주어진 온도에서 반응의 화학적 속도는 A인자 값, 활성화 에너지 크기, 반응물의 농도에 따라 달라집니다. 보통 빠른 반응은 상대적으로 작은 활성화 에너지를 필요로 합니다.
화학 반응 속도가 온도 상승 10°C마다 두 배가 된다는 '경험 법칙'은 흔한 오해입니다. 이는 α(온도 계수)가 종종 1.5에서 2.5 사이인 생물학적 시스템의 특수한 경우에서 일반화된 것일 수 있습니다.
급속 반응의 속도론은 온도 점프 방법으로 연구할 수 있습니다. 이는 온도가 급격히 상승하는 것을 이용하고 평형 회복 시 이완 시간을 관찰하는 것을 포함합니다. 특히 유용한 온도 점프 장치의 형태는 충격관으로, 이는 기체의 온도를 1000도 이상 빠르게 올릴 수 있습니다.
촉매제
주요 문서: 촉매
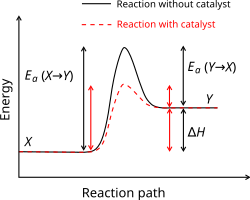
가상의 흡열 화학 반응에서 촉매의 효과를 보여주는 일반 퍼텐셜 에너지 다이어그램. 촉매의 존재는 활성화 에너지가 낮은 새로운 반응 경로(빨간색으로 표시됨)를 열어줍니다. 최종 결과와 전체 열역학은 동일합니다.
촉매는 화학 반응 속도를 변화시키지만 이후에도 화학적으로 변하지 않는 물질입니다. 촉매는 낮은 활성화 에너지에서 새로운 반응 기전을 제공하여 반응 속도를 높입니다. 자가촉매에서는 반응 생성물 자체가 그 반응의 촉매 역할을 하여 양의 피드백을 유도합니다. 생화학적 반응에서 촉매 역할을 하는 단백질을 효소라고 부릅니다. Michaelis–Menten 반응 속도론은 효소 매개 반응의 속도를 설명합니다. 촉매는 평형 위치에 영향을 주지 않으며, 촉매는 역방향과 순방향 반응을 똑같이 빠르게 만듭니다.
특정 유기 분자에서는 특정 치환기가 인접한 그룹 참여 시 반응 속도에 영향을 미칠 수 있습니다. [출처 필요]
압력
기체 반응에서 압력을 높이면 반응물 간 충돌 횟수가 증가하여 반응 속도가 증가합니다. 이는 기체의 활동이 기체의 분압에 정비례하기 때문입니다. 이는 용액 농도를 높이는 효과와 유사합니다.
이 단순한 질량 작용 효과 외에도, 압력에 의해 속도 계수 자체가 변할 수 있습니다. 많은 고온 기상 반응의 속도 계수와 생성물은 불활성 가스가 혼합물에 첨가되면 변합니다; 이 효과의 변형을 감쇠(fall-off)와 화학적 활성화(chemical activation)라고 합니다. 이러한 현상은 발열 또는 흡열 반응이 열 전달보다 빠르게 일어나기 때문이며, 이로 인해 반응하는 분자들은 비열적 에너지 분포(비-볼츠만 분포)를 갖게 됩니다. 압력을 높이면 반응하는 분자들과 시스템 전체의 열 전달 속도가 증가하여 이 효과가 줄어듭니다.
응축상 속도 계수도 압력에 영향을 받을 수 있으나, 이온과 분자는 압축성이 낮아 측정 가능한 효과를 위해서는 꽤 높은 압력이 필요합니다. 이 효과는 종종 다이아몬드 모루를 사용해 연구됩니다.
반응의 동역학은 압력 점프 접근법으로도 연구할 수 있습니다. 이는 압력을 빠르게 변화시키고 평형 회복 시 이완 시간을 관찰하는 것을 포함합니다.
빛의 흡수
화학 반응의 활성화 에너지는 한 반응물 분자가 적절한 파장의 빛을 흡수하여 여기 상태로 전환될 때 제공될 수 있습니다. 빛에 의해 시작된 반응을 연구하는 분야는 광화학이며, 대표적인 예가 광합성입니다.
실험 방법

스핀코 디비전 모델 260 반응 동역학 시스템은 분자 반응의 정확한 속도 상수를 측정했습니다.
반응 속도의 실험적 측정은 반응물이나 생성물의 농도가 시간에 따라 어떻게 변하는지 측정하는 것을 포함합니다. 예를 들어, 시스템 내 다른 반응물이나 생성물이 빛을 흡수하지 않는 파장에서 반응물의 농도를 분광광도측정법으로 측정할 수 있습니다.
최소 몇 분이 소요되는 반응의 경우, 반응물이 관심 온도에서 혼합된 후 관측을 시작할 수 있습니다.
빠른 반응
더 빠른 반응을 위해서는 반응물을 혼합하고 지정된 온도에 도달하는 데 걸리는 시간이 반감기와 비슷하거나 더 길 수 있습니다. [9] 느린 혼합 단계 없이 빠른 반응을 시작하는 특별한 방법들은 다음과 같습니다.
- 혼합 시간을 밀리초 단위로 줄일 수 있는 스톱드 플로우 방법[9][10][11]은 제한이 있습니다. 예를 들어, 가스나 용액을 혼합하는 데 걸리는 시간을 고려해야 하며, 반감기가 약 0.1초 미만일 경우 적합하지 않습니다.
- 온도 도약법과 압력 점프와 같은 화학적 이완 기법으로, 처음에는 평형 상태에 있던 예혼합 시스템이 급격한 가열 또는 감압으로 교란되어 더 이상 평형 상태가 아니게 만들고, 다시 평형 상태로 이완하는 과정을 관찰합니다. [9][12][13][14] 예를 들어, 이 방법은 일반 조건에서 반감기가 1 μs 이하인 H 3O+ + OH−를 연구하는 데 사용되었습니다. [9][14]
- 플래시 광분해는 레이저 펄스가 자유 라디칼과 같은 고흥분된 종을 생성하고, 그 반응을 연구하는 과정입니다. [11][15][16][17]
균형
화학 반응 속도론이 화학 반응의 속도에 관한 반면, 열역학은 반응이 일어나는 정도를 결정합니다. 가역적 반응에서는 순방향 및 역방향 반응의 속도가 같아질 때(동적 평형 원리) 화학 평형에 도달하고, 반응물과 생성물의 농도가 더 이상 변하지 않을 때입니다. 예를 들어, 질소와 수소를 결합해 암모니아를 생성하는 하버-보쉬 공정이 이를 증명합니다. 벨로우소프–자보틴스키 반응과 같은 화학 시계 반응은 성분 농도가 오랜 시간 진동할 수 있음을 보여주며, 결국 평형에 도달할 수 있음을 보여준다.
자유 에너지
일반적으로 반응의 자유 에너지 변화(ΔG)는 화학 변화가 일어날지 여부를 결정하지만, 동역학은 반응의 속도를 설명합니다. 반응은 매우 발열되어 매우 긍정적인 엔트로피 변화를 가질 수 있지만, 반응이 너무 느리면 실제로는 일어나지 않습니다. 반응물이 두 가지 생성물을 생성할 수 있다면, 일반적으로 열역학적으로 가장 안정적인 생성물이 형성되지만, 반응이 운동 반응 제어 하에 있다고 할 때를 제외하고는 그렇지 않습니다. 커틴-해밋 원리는 두 반응물이 빠르게 상호변환하여 각각 다른 생성물로 전환될 때 생성물 비율을 결정할 때 적용됩니다. 자유 에너지 관계에서 반응 속도 상수를 예측할 수 있습니다.
동위원소 효과는 반응물 중 하나의 원자가 그 동위원소로 대체될 때 화학 반응 속도의 차이를 말합니다.
화학 반응 속도론은 화학공학에서 화학 반응기 내 체류 시간과 열 전달, 그리고 고분자 화학에서 몰 질량 분포에 관한 정보를 제공합니다. 또한 부식 공학에 관한 정보도 제공합니다.
응용 및 모델
화학 반응 반응 속도를 설명하는 수학적 모델은 화학자와 화학공학자들에게 식품 분해, 미생물 성장, 성층권 오존 분해, 생물학적 시스템의 화학과 같은 화학 과정을 더 잘 이해하고 설명할 수 있는 도구를 제공합니다. 이 모델들은 화학 반응기 설계 또는 개조에도 활용되어 제품 수율 최적화, 제품 분리 효율 향상, 환경 해로운 부산물 제거 등이 가능합니다. 예를 들어, 중탄화수소를 가솔린 및 경성기체로 촉매 해킹할 때, 동역학 모델을 사용하여 중화수소가 휘발유로 가장 높은 수율을 내는 온도와 압력을 구할 수 있습니다.
화학 동역학은 상미분방정식 해법(ODE-solving)과 곡선 적합의 함수로서 전문 패키지에서 모델링을 통해 자주 검증되고 탐구됩니다. [18]
수치 방법
경우에 따라 방정식은 해석적으로 풀 수 없지만, 데이터 값이 주어지면 수치적 방법으로 풀 수 있습니다. 이를 구현하는 방법은 소프트웨어 프로그램을 사용하는 것과 오일러 방법과 같은 수학적 방법을 사용하는 두 가지가 있습니다. 화학 반응 속도론용 소프트웨어의 예로는 i) 화학 반응을 수치적으로 시뮬레이션하고 실제 데이터와 비교할 수 있는 Java 앱인 Tenua, ii) 계산 및 추정을 위한 Python 코딩, iii) 반응을 모델링, 회귀, 적합 및 최적화하는 Kintecus 소프트웨어 컴파일러가 있습니다.
- 수치 적분: 1차 반응 A→ B에 대해
반응물 A의 미분 방정식은 다음과 같습니다:d[A]dt=−k[A]

또한 다음과 같이 표현할 수 있습니다.d[A]dt=f(t,[A])

이는 다음과 같습니다. y′=f(x,y)

오일러와 룬게-쿠타 방법으로 미분방정식을 풀려면 초기 값이 필요합니다.
- 오일러 방법은 단순하지만 부정확→.
언제든 y′=f(x,y)
는 다음과 같습니다.y′=dydx
이산이 증가함에 따라 미분을 근사할 수 있습니다:y′=dydx≈ΔyΔx=y(x+Δx)−y(x)Δx
방정식의 미지의 부분은 y(x+Δx)이며, 초기 값 데이터가 있으면 이를 찾을 수 있습니다. - 룬게-쿠타 방법은 오일러 방법보다 더 정확→. 이 방법에서는 초기 조건이 필요합니다: x = x0에서 y = y 0. 문제는 x = x0 + h일 때 y의 값을 찾는 것으로, 여기서 h는 주어진 상수이다.
분석적으로 그 순간 곡선의 지순은 (x0, y0)에 의해 3차 룽게-쿠타 공식으로 주어진다.
1차 일반 방정식에서 룬게-쿠타 방법은 온도와 반응 속도 간의 관계를 나타내는 수학적 모델을 사용합니다. 서로 다른 온도에서 서로 다른 농도에서 반응 속도를 계산하는 것이 가치가 있습니다. 얻어진 방정식은 다음과 같습니다: dr/dt=R/T+rΔH∘/RT2 - 확률적 방법은 미분 속도 법칙과 운동 상수의 확률을 →합니다. 직항률과 역속도 상수를 가진 균형 반응에서는 B에서 A로 변환하는 것보다 A에서 B로 변환하는 것이 더 쉽다. 확률 계산에 관해서는, 매번 무작위 수를 선택해 임계값과 비교하여 반응이 A에서 B로 이어지는지 또는 그 반대로 진행되는지 알 수 있습니다.




참고
- 자가촉매 반응과 질서 생성
- 부식 공학
- 폭발
- 전기화학 속도론
- 화염 속도
- 이종 촉매
- 내재적 저차원 다양체
- MLAB 화학 동역학 모델링 패키지
- 비열 표면 반응
- 포터스휠 실험 데이터에 화학 성분 상수를 맞추기 위한 Matlab 도구 상자
- 반응 진행 속도 역학 분석
참고문헌
- L. 빌헬미, "Ann. Phys. Chem. (포겐도르프)" 제81권, (1850) 413
- C.M. 굴드베리와 P. 와게, "친화에 관한 연구" 포핸들링거 i 비덴스카브스-셀스카벳 이 크리스티아니아 (1864), 35
- P. Waage, "친화 법칙 결정 실험", Forhandlinger i Videnskabs-Selskabet i Christiania, (1864) 92.
- C.M. 굴드베리, "화학적 친화성 법칙에 관하여", 포핸드글링거 및 비덴스카브스-셀스카베트 이 크리스티아니아 (1864) 111쪽
- 호프, J. H. 반트 (야코부스 헨리쿠스 반트); 코헨, 에른스트; 토마스 유언 (1896-01-01). 화학 역학 연구. 암스테르담: F. 뮐러; 런던: 윌리엄스 앤 노게이트.
- 1901년 노벨 화학상, 노벨상 및 수상자, 공식 웹사이트.
- A.N. 고르반, G.S. 야블론스키, 『화학 역학의 세 파동, 자연현상의 수학적 모델링』 10(5) (2015), 1–5쪽.
- 라이들러, K. J. Chemical Kinetics (3판, Harper and Row 1987) p.42 ISBN 0-06-043862-2
- 라이들러, K. J. Chemical Kinetics (3판, Harper and Row 1987) 33-39쪽 ISBN 0-06-043862-2
- Espenson, J.H. 『화학 반응 반응 메커니즘』(2판, McGraw-Hill 2002), p.254-256 ISBN 0-07-288362-6
- Atkins P.와 de Paula J., 『물리화학』(8판, W.H. Freeman 2006), 793쪽, ISBN 0-7167-8759-8
- Espenson, J.H. 『화학 반응 속도론과 반응 기전』 (2판, McGraw-Hill 2002), 256-8쪽 ISBN 0-07-288362-6
- 스타인펠드 J.I., 프란시스코 J.S., 하세 W.L. 『화학 운동학과 동역학』 (2판, 프렌티스-홀 1999) 140-3쪽 ISBN 0-13-737123-3
- Atkins P.와 de Paula J., 『물리화학』(8판, W.H. Freeman 2006) 805-807쪽 ISBN 0-7167-8759-8
- Laidler, K.J. 『화학 반응학』(3판, Harper and Row 1987) 359-360쪽 ISBN 0-06-043862-2
- Espenson, J.H. 『화학 반응 반응 메커니즘』 (2판, McGraw-Hill 2002), p.264-6 ISBN 0-07-288362-6
- 스타인펠드 J.I., 프란시스코 J.S., 하세 W.L. 『화학 운동학과 동역학』 (2판, 프렌티스-홀 1999) 94-97쪽 ISBN 0-13-737123-3
- "화학 동역학: 단순 결합: F + G ⇋ B" (PDF). 문명화된 소프트웨어 주식회사 2015-09-01에 확인함.
외부 링크
화학 동역학에 관한
도서관 자료
- 화학 애플릿 (2009-06-04에 웨이백 머신에 보관됨)
- 워털루 대학교
- 기체 상 반응의 화학 동역학
- 킨피: 운동 방정식 푸는 파이썬 코드 생성기
- 반응 속도 법칙과 반응 프로파일 - 온도, 농도, 용매 및 촉매 - 반응이 얼마나 빨리 진행될지 (SciFox 영상, TIB AV-Portal)