- nature
- npj precision oncology
- review
- article
Enhancing adoptive cell therapy: future strategies for immune cell radioprotection in neuro-oncology
- Review
- Open access
- Published: 29 July 2025
Enhancing adoptive cell therapy: future strategies for immune cell radioprotection in neuro-oncology
npj Precision Oncology volume 9, Article number: 264 (2025) Cite this article
1581 Accesses
Abstract
Adoptive cell therapy (ACT), particularly chimeric antigen receptor T cell (CAR T) therapy, has emerged as a promising approach in cancer treatment, demonstrating efficacy in hematological malignancies but facing challenges in brain tumors. The combination of ACT with radiation therapy (RT) offers a potential strategy to enhance therapeutic outcomes, as RT can stimulate immune responses by promoting antigen presentation and T cell recruitment. However, a major hurdle is the radiosensitivity of immune cells, leading to their rapid depletion within the radiation field, which undermines the benefits of this combination. This review explores strategies to increase the radioresistance of immune cells, highlighting the need for innovative radioprotective approaches. We discuss the potential of extremophile-derived molecules, such as the Damage Suppressor protein from tardigrades, as novel radioprotectants that could be integrated into ACT protocols. Furthermore, we address key considerations for clinical trial design, including the sequencing of RT and ACT, dosing parameters, and safety considerations. By bridging insights from extremophile biology and immuno-oncology, this work aims to optimize the efficacy of ACT in the challenging context of brain tumors, paving the way for enhanced treatment strategies in neuro-oncology.
초록
입양 세포 치료(ACT),
특히 키메라 항원 수용체 T 세포(CAR T) 치료는
혈액암에서 효능을 입증하며 암 치료의 유망한 접근법으로 부상했으나
뇌종양에서는 어려움을 겪고 있다.
방사선 치료(RT)는
항원 제시와 T 세포 유입을 촉진하여 면역 반응을 자극할 수 있으므로,
ACT와 RT의 병용은 치료 결과를 향상시킬 잠재적 전략을 제공한다.
그러나 주요 장애물은
면역 세포의 방사선 민감성으로,
방사선 조사 영역 내에서 면역 세포가 급속히 소실되어 이 병용 요법의 이점을 약화시킨다.
본 리뷰는
면역 세포의 방사선 저항성을 높이는 전략을 탐구하며,
혁신적인 방사선 보호 접근법의 필요성을 강조한다.
우리는 극한생물 유래 분자,
예를 들어 느림보동물(tardigrade)의 손상 억제 단백질(Damage Suppressor protein)과 같은 물질이
ACT 프로토콜에 통합될 수 있는 새로운 방사선 보호제로서의 잠재력을 논의한다.
또한 방사선 치료(RT)와 항암치료(ACT)의 순서,
투여량 매개변수,
안전성 고려사항 등 임상시험 설계의 핵심 고려사항을 다룬다.
극한생물학 및 면역종양학의 통찰력을 연결함으로써,
본 연구는 뇌종양이라는 어려운 환경에서 ACT의 효능을 최적화하고
신경종양학 분야의 향상된 치료 전략을 위한 길을 열기 위해 노력한다.
Similar content being viewed by others
Immunomodulation by radiotherapy in tumour control and normal tissue toxicity
Article 01 July 2021

Article Open access01 July 2025

Article Open access16 October 2022
Introduction
Adoptive cell therapy (ACT) including chimeric antigen receptor (CAR) T-cell therapy has shown promising early signals of activity in treating select patients with brain tumors1. ACT is a type of immunotherapy that uses a patient’s own immune cells to help fight disease, such as cancer. CAR T cells are patient-isolated T cells genetically engineered ex vivo to regain cancer-fighting properties. The efficacy of CAR T for solid tumor types, including brain tumors, remains elusive. However, the approach holds promise as evidenced by (i) responses in small series of brain tumor patients2,3; (ii) success of CAR T in B cell neoplasm settings in which six therapies have been FDA approved4; and (iii) recent FDA approval of a similar cell therapy approach for advanced melanoma5. The autologous in vitro- expanded tumor-infiltrating lymphocyte (TIL) lifileucel was approved by the FDA in 2024 as the first TIL therapy to treat cancer confirming the future use of TILs in mainstream practice. There are also promising TIL early efficacy data in lung cancer6, but these findings are yet to be replicated in immunologically “cold” tumors such as glioma5. Existing ACT modalities have shown limited efficacy against many brain tumors7. This challenge may stem from various mechanistic hurdles, including the scarcity of the brain tumor microenvironment (TME), impaired T cell trafficking to the tumor, downregulated checkpoint molecule expression, tumor heterogeneity, immunosuppressive TME, and lack of tumor antigen presentation4 (Fig. 1a).
서론
키메라 항원 수용체(CAR) T 세포 치료를 포함한
채택 세포 치료(ACT)는
특정 뇌종양 환자 치료에서 유망한 초기 활성 신호를 보여주고 있다1.
chimeric antigen receptor (CAR) T-cell therapy
ACT는
환자의 자체 면역 세포를 활용하여 암과 같은 질병과 싸우는 데 도움을 주는
면역요법의 한 유형이다.
https://www.nature.com/articles/s41408-021-00459-7

CAR T 세포는
환자로부터 분리된 T 세포를
체외에서 유전자 조작하여 암 퇴치 능력을 회복시킨 것이다.
뇌종양을 포함한 고형 종양 유형에 대한
CAR T의 효능은 여전히 불분명하다.
그러나
(ii) FDA 승인을 받은 6가지 치료법이 존재하는 B세포 신생물 환경에서의 CAR T 성공 사례4;
(iii) 진행성 흑색종에 대한 유사한 세포 치료법 접근법의 최근 FDA 승인5을 통해
이 접근법의 가능성은 입증되었다.
자가 체외 확장 종양 침윤 림프구(TIL) 치료제
라이필루셀(lifileucel)은
2024년 FDA 승인을 받아 암 치료용 최초의 TIL 치료제로,
TIL의 주류 임상 적용 가능성을 입증하였다.
폐암에서도 유망한 TIL 초기 효능 데이터가 보고되었으나6,
이러한 결과는 교모세포종과 같은 면역학적으로 ‘차가운’ 종양에서는 아직 재현되지 못하고 있다5.
기존의 세포독성 항암제(ACT) 치료법은
많은 뇌종양에 대해 제한된 효능만을 보여왔다7.
이러한 어려움은
뇌종양 미세환경(TME)의 희소성,
종양으로의 T세포 이동 장애,
체크포인트 분자 발현 저하,
종양 이질성,
면역억제성 TME,
종양 항원 제시 부족 등
다양한 기전적 장벽에서 비롯될 수 있다4(그림 1a).
Fig. 1: Using an ultra-radioresistant extremophile gene to radioprotect T cells and potentiate ACT.
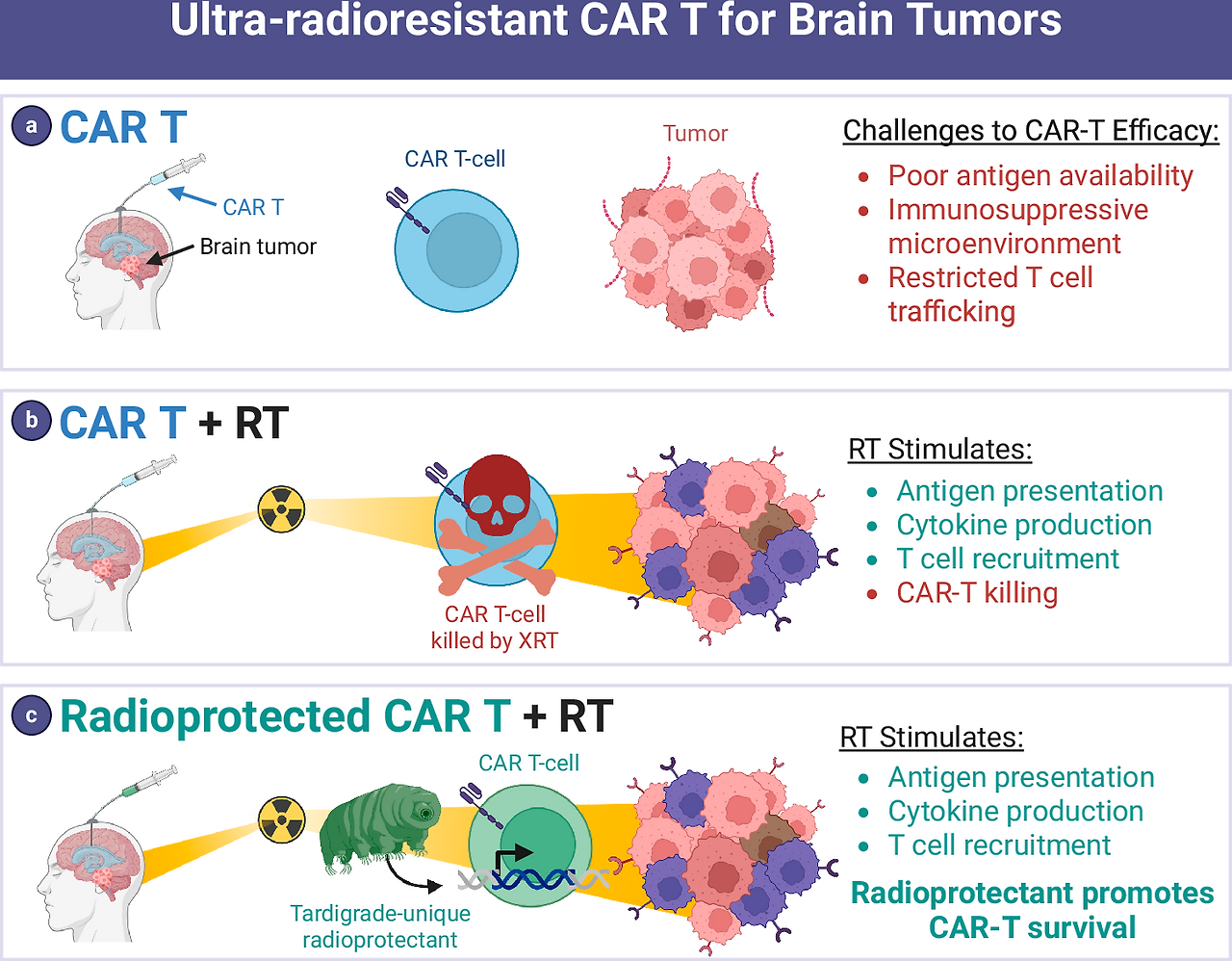


a Poor immunogenicity is a key barrier to efficacy for CAR T cells in brain tumors. b RT added to CAR T therapy has immunostimulatory effects that may potentiate CAR T therapy. But RT also kills T cells that enter the radiation field which has a counterproductive effect. c Radioprotectants such as a gene from the ultra-radioresistant extremophile Tardigrade are expressed in CAR T cells to protect them from RT, allowing immune stimulation from RT to synergize with CAR T.
그림 1: 초방사선 내성 극한생물 유전자를 이용한 T 세포 방사선 보호 및 CAR-T 치료 효과 증강.
a 낮은 면역원성은 뇌종양에서 CAR-T 세포의 효능을 저해하는 주요 장벽이다. b CAR-T 치료에 방사선 치료(RT)를 병용하면 면역 자극 효과를 통해 CAR-T 치료 효과를 증강시킬 수 있다. 그러나 방사선 치료는 방사선 조사 영역에 진입한 T 세포를 사멸시켜 역효과를 유발하기도 한다. c 초고방사선 내성 극한생물인 느림보동물(Tardigrade) 유전자와 같은 방사선 보호제를 CAR T 세포에 발현시켜 방사선 치료로부터 보호함으로써, 방사선 치료의 면역 자극 효과가 CAR T 치료와 시너지 효과를 발휘할 수 있도록 한다.
Several ACT modalities are currently in trial for central nervous system (CNS) tumors, mainly recurrent glioblastoma, with mixed results. Given the heterogeneity of glioblastoma and CNS tumors, most successful trials are targeting multiple tumor antigens with CARs. For example, one clinical study saw no clinical efficacy in recurrent glioblastoma patients cotreated with CAR T-cell therapy targeting epidermal growth factor receptor (EGFR) III and the anti-PD1 antibody pembrolizumab8. However, an early phase I clinical trial for recurrent glioblastoma saw success in CAR T cells engineered to target both EGFR III and wildtype EGFR9. All patients saw tumor regression, though two of the three patients eventually showed reoccurrence9. Similarly, a study using CAR T-cells targeting both EGFR and IL13Rα2 saw regression in 8 of 13 patients with measurable disease at the time of infusion10. One confirmed partial response by Modified Response Assessment in Neuro-Oncology criteria was observed10. However, patients in both studies exhibited grade 3 adverse events, including neurotoxicity, encephalopathy, and fatigue9,10. Further trials of CAR T treatments for brain tumors show limited efficacy and are summarized in review11. These inconsistent results and small sample sizes highlight the need for further investigation in CAR therapies for brain tumors.
Radiation therapy (RT) is the standard treatment of brain tumors, offering an opportunity to combine with ACT. The majority of brain tumor patients are candidates for RT with curative or palliative intent12. However, the effects of RT on the tumor immune microenvironment are complex. Many of these processes occur acutely after each fraction of RT in a time-limited fashion. These include stimulation of antigen presentation, induction of cytokine release by neoplastic cells and resident immune cells, downregulation of checkpoint protein expression on tumor and immune cells, and attraction of lymphocytes to the tumor site13,14,15. On the other hand, a major obstacle is that many immune cells are radiosensitive and undergo cell death upon entering the RT field16,17,18,19, hampering the ability of RT to stimulate engineered immune cells or other adaptive immune responses13,14 (Fig. 1b). It stands to reason that maximal immune stimulation occurs as tumors are first exposed to RT during the initial fractions of definitive, fractionated RT. Yet this is the precise time when RT is likely killing any CAR T cells or lymphocytes that may enter the radiation field to sample antigens and elicit adaptive immune responses.
Approaches to increase radioresistance of immune cells may hold promise for enhancing ACT efficacy. Increasing radioresistance could overcome the major challenge to combining ACT with RT, which is that many immune cells are highly radiosensitive and die upon entering the radiation field. Engineering radioprotected immune cells could address a critical barrier to the effective combination of ACT with RT (Fig. 1c). The concept of “radioprotecting” T cells for cell therapy has seen limited exploration. Notably, a recent study reported the overexpression of superoxide dismutase 2 (SOD2) in CAR T cells, which yielded promising results in a preclinical model involving head and neck cancer20. However, SOD2’s radioprotective effects may be modest21,22. HeLa cells with SOD2 overexpression still showed signs of DNA damage and reactive oxygen species (ROS) buildup after treatment with 5 Gy21, emphasizing the need for a broader investigation into various candidate genes and the potential of potent radioprotectants derived from extremophiles. Thus, there is an unmet need to identify methods to protect immune cells from RT-induced death to optimize the immune-stimulatory benefits of RT. There is also a pressing need for a deeper understanding of the molecular pathways involved in radioresistance, which could significantly enhance the efficacy of ACT when combined with RT.
Extremophiles, organisms that thrive in extreme environments like high radiation, temperature, or desiccation, possess unique molecular mechanisms to safeguard cellular integrity, making their proteins attractive for radioprotective applications in cell therapy. For instance, the tardigrade-derived Damage Suppressor (Dsup) protein binds to nucleosomes, reducing hydroxyl radical-induced DNA damage and double-strand breaks (DSBs), significantly enhancing cell survival under ionizing radiation without impairing normal functions23,24. Notably, local Dsup mRNA nanoparticle delivery has been shown to effectively radioprotect nearby healthy tissue in mice undergoing RT for orthotopic oral cancer25. Other promising proteins include PprI from Deinococcus radiodurans, which activates DNA repair and reduces apoptosis26; TRID1 from tardigrades, which promotes DNA repair through phase separation and repair machinery recruitment27; and SASP from Bacillus subtilis, which binds to DNA to provide a robust protective shield28.
Here we review the literature that could provide insights on approaches to radioprotect immune cells to stimulate ACT for brain tumors. We delineate knowledge gaps and opportunities to advance immune-cell radioprotectors and rationally combine ACT with RT. We highlight extremophile organisms as an intriguing source of radioprotective molecules that could be applied to human immune cells. A variety of approaches to screen for ideal radioprotectors are considered. Safety and ACT manufacturing considerations are evaluated. Finally, we consider different clinical situations in which clinical trials for radioprotected ACT and RT combination treatments could be deployed. Along with this, we review considerations for clinical trial design, ACT administration, rational sequencing of ACT and RT delivery, and selection of RT fractionation schemes.
현재 중추신경계(CNS) 종양, 주로 재발성 교모세포종을 대상으로 여러 ACT 치료법이 임상시험 중이며 결과는 혼재되어 있다. 교모세포종 및 중추신경계 종양의 이질성을 고려할 때, 대부분의 성공적인 임상시험은 CAR을 통해 여러 종양 항원을 표적화하고 있습니다. 예를 들어, 한 임상 연구에서는 표피 성장 인자 수용체(EGFR) III를 표적화하는 CAR T 세포 치료와 항-PD1 항체 펨브롤리주맙8을 병용 치료한 재발성 교모세포종 환자에서 임상적 효능이 나타나지 않았습니다. 그러나 재발성 교모세포종을 대상으로 한 초기 1상 임상시험에서는 EGFR III와 야생형 EGFR을 모두 표적하도록 설계된 CAR T세포가 성공을 거두었다9. 모든 환자에서 종양 퇴행이 관찰되었으나, 3명 중 2명은 결국 재발을 보였다9. 마찬가지로, EGFR과 IL13Rα를 모두 표적하는 CAR T세포를 사용한 연구에서는 투여 시점에 측정 가능한 질환을 가진 13명의 환자 중 8명에서 퇴행이 관찰되었다10. 신경종양학 반응 평가 기준(mRAST)에 따른 부분 반응 1건이 확인되었다10. 그러나 두 연구의 환자 모두 신경독성, 뇌병증, 피로감 등 3등급 이상 부작용을 나타냈다9,10. 뇌종양에 대한 CAR T 치료의 추가 임상시험은 제한된 효능을 보였으며, 이 내용은 리뷰 논문에서 요약되어 있다11. 이러한 일관성 없는 결과와 소규모 표본 크기는 뇌종양에 대한 CAR 치료법의 추가 연구 필요성을 강조한다.
방사선 치료(RT)는 뇌종양의 표준 치료법으로, ACT와의 병용 기회를 제공한다. 대부분의 뇌종양 환자는 완치 또는 완화 목적을 위한 RT 대상자이다12. 그러나 RT가 종양 면역 미세환경에 미치는 영향은 복잡하다. 이러한 과정의 상당수는 각 방사선 치료 분획 후 시간 제한적으로 급성적으로 발생합니다. 여기에는 항원 제시 자극, 종양 세포 및 상주 면역 세포에 의한 사이토카인 분비 유도, 종양 및 면역 세포의 체크포인트 단백질 발현 하향 조절, 종양 부위로의 림프구 유인 등이 포함됩니다13,14,15. 반면, 주요 장애물은 많은 면역 세포가 방사선 민감성을 지녀 방사선 치료 영역에 진입하면 세포 사멸을 겪는다는 점이다16,17,18,19. 이는 방사선 치료가 공학적으로 조작된 면역 세포나 기타 적응성 면역 반응을 자극하는 능력을 저해한다13,14 (그림 1b). 종양이 확정적 분할 방사선 치료의 초기 분획 동안 처음으로 방사선에 노출될 때 최대 면역 자극이 발생한다는 것은 당연한 이치이다. 그러나 바로 이때 방사선 치료가 항원을 탐색하고 적응성 면역 반응을 유발하기 위해 방사선 조사 영역에 진입할 수 있는 CAR T 세포나 림프구를 사멸시킬 가능성이 높다.
면역 세포의 방사선 저항성을 높이는 접근법은 ACT 효능 향상에 유망할 수 있다. 방사선 저항성 증가는 많은 면역 세포가 고도로 방사선 민감성을 보이며 조사 영역 진입 시 사멸한다는 점, 즉 ACT와 방사선 치료 병용의 주요 난제를 극복할 수 있다. 방사선 보호 기능을 가진 면역 세포를 공학적으로 설계하는 것은 ACT와 방사선 치료의 효과적 병용에 대한 핵심 장벽을 해결할 수 있다(그림 1c). 세포 치료를 위한 T 세포의 “방사선 보호” 개념은 제한적으로만 탐구되었다. 특히 최근 연구에서는 CAR T 세포에서 슈퍼옥사이드 디스뮤타제 2(SOD2)의 과발현이 두경부암을 포함한 전임상 모델에서 유망한 결과를 보였다고 보고되었다20. 그러나 SOD2의 방사선 보호 효과는 미미할 수 있다21,22. SOD2 과발현 HeLa 세포는 5 Gy 방사선 치료 후에도 DNA 손상과 활성산소종(ROS) 축적 징후를 보였으며21, 이는 다양한 후보 유전자에 대한 광범위한 연구와 극한성 미생물 유래 강력한 방사선 보호제의 잠재력 탐구가 필요함을 강조한다. 따라서 방사선 치료(RT)의 면역 자극 효과를 극대화하기 위해 면역 세포를 RT로 인한 사멸로부터 보호하는 방법을 규명할 미충족 요구가 존재한다. 또한 방사선 저항성에 관여하는 분자 경로를 심층적으로 이해할 시급한 필요성이 있으며, 이는 방사선 치료와 병용 시 세포 면역 치료(ACT)의 효능을 크게 향상시킬 수 있다.
극한 환경(고방사선, 고온, 건조 등)에서 생존하는 극한생물은 세포 무결성을 보호하는 독특한 분자적 메커니즘을 지니고 있어, 세포 치료에서의 방사선 보호 응용을 위한 단백질 후보로 주목받고 있다. 예를 들어, 느림보동물(tardigrade) 유래 손상 억제자(Dsup) 단백질은 뉴클레오솜에 결합하여 하이드록실 라디칼에 의한 DNA 손상과 이중 가닥 절단(DSBs)을 감소시켜, 정상 기능을 저해하지 않으면서 이온화 방사선 하에서 세포 생존율을 현저히 향상시킵니다23,24. 특히, 국소적 Dsup mRNA 나노입자 전달은 생체 내 구강암에 대한 방사선 치료를 받는 생쥐에서 인근 건강한 조직을 효과적으로 방사선 보호하는 것으로 나타났습니다25. 다른 유망한 단백질로는 DNA 복구를 활성화하고 세포 사멸을 감소시키는 Deinococcus radiodurans의 PprI26; 위상 분리와 복구 기구 동원을 통해 DNA 복구를 촉진하는 느림보동물류의 TRID127; 그리고 DNA에 결합하여 강력한 보호막을 제공하는 Bacillus subtilis의 SASP28 등이 있다.
본고에서는 뇌종양에 대한 ACT를 촉진하기 위해 면역 세포를 방사선으로부터 보호하는 접근법에 대한 통찰력을 제공할 수 있는 문헌을 검토한다. 우리는 면역 세포 방사선 보호제 개발을 진전시키고 ACT와 RT를 합리적으로 결합하기 위한 지식 격차와 기회를 규명한다. 인간 면역 세포에 적용될 수 있는 방사선 보호 분자의 흥미로운 공급원으로 극한생물체를 강조한다. 이상적인 방사선 보호제를 선별하기 위한 다양한 접근법을 고려한다. 안전성 및 ACT 제조 관련 고려사항을 평가한다. 마지막으로 방사선 보호된 ACT와 방사선 치료(RT) 병용 요법에 대한 임상 시험을 적용할 수 있는 다양한 임상 상황을 검토한다. 이와 함께 임상 시험 설계, ACT 투여, ACT와 RT 투여의 합리적 순서, RT 분할 방사선 요법 선택에 대한 고려사항을 검토한다.
Immune-cell types used for ACT and their radiation sensitivity
Several immune-cell types have been explored for ACT, including T cells, natural killer (NK) cells, macrophages, and more recently, unconventional lymphocytes such as γδ T cells and invariant natural killer T (iNKT) cells (Fig. 2)29,30,31. Each immune-cell type offers unique advantages in terms of tumor recognition, cytotoxicity, and persistence. Understanding their role within the tumor microenvironment and their radiosensitivity is crucial for optimizing radioprotected ACT in combination with radiotherapy (RT), a standard cancer treatment for brain tumors.
ACT에 사용되는 면역 세포 유형과 그 방사선 민감도
ACT에 활용 가능한 여러 면역 세포 유형이 탐구되어 왔으며, 여기에는 T 세포, 자연살해(NK) 세포, 대식세포, 그리고 최근에는 γδ T 세포 및 불변 자연살해 T(iNKT) 세포와 같은 비전통적 림프구 등이 포함됩니다(그림 2)29,30,31. 각 면역 세포 유형은 종양 인식, 세포독성, 지속성 측면에서 고유한 장점을 제공합니다. 뇌종양의 표준 치료법인 방사선 치료(RT)와 병용하여 방사선 보호형 ACT를 최적화하기 위해서는 종양 미세환경 내에서의 역할과 방사선 감수성을 이해하는 것이 중요하다.
Fig. 2: Baseline radiosensitivity of cell types used for ACT.
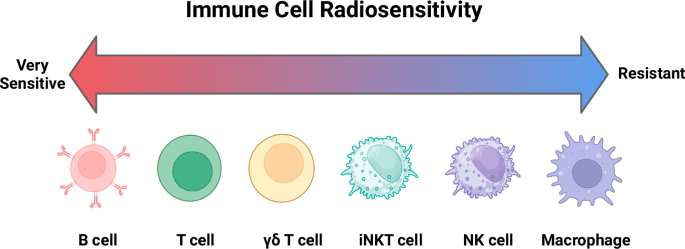
This figure summarizes the inherent radiosensitivity and other effects of irradiation on various immune-cell types commonly used in adoptive cell therapy (ACT). While radiosensitivity is likely highly context-dependent, in general B cells are highly sensitive to radiation and undergo apoptosis at even low doses. T cells are sensitive, with sublethal doses (<2 Gy) causing activation, while higher doses (>2 Gy) induce apoptosis. γδ T cells are relatively resistant to radiation and maintain their cytotoxic functions. Invariant natural killer T cells (iNKT cells) exhibit moderate resistance to radiation. Natural killer (NK) cells and macrophages are relatively radioresistant.
A general theme for all immune-cell types is that RT may stimulate responses at low doses but leads to cell death or decreased responsiveness at higher doses32. Patients undergoing standard-of-care RT for glioblastoma, the most aggressive and malignant primary brain tumor, experience worsening lymphopenia (lymphocyte depletion) during RT17. The risk of lymphopenia is related to the volume of tissue being irradiated18,19. While concurrent chemotherapy is a risk factor, RT alone likely induces lymphopenia via distinct mechanisms18,19. Therefore, methods to modulate the radiosensitivity of lymphocytes may be of interest to decrease the risk of lymphopenia and maximize the efficacy of ACT. Understanding the exact radiosensitivity of lymphocytes, however, is imperfect as many experimental methods and results differ, as shown in review33. Additionally, some research suggests that sex and age can affect the radiosensitivity of lymphocytes33.
T lymphocytes
T cells, particularly T cell receptor (TCR)-transgenic T cells and chimeric antigen receptor (CAR) T cells, have been at the forefront of ACT for cancer. CAR T cells, genetically engineered to express receptors targeting tumor antigens, have shown remarkable efficacy in hematologic malignancies34. Essentially all FDA approvals for ACT anticancer therapies utilized T cells, including six approvals for CAR T cell infusions and an approval for tumor-infiltrating lymphocytes for advanced melanoma patients5,34.
T cells are sensitive to ionizing radiation, which can impair their proliferation and function at high doses. In particular, the D10 (dose required to reduce surviving fraction to 10%) for CD4+ and CD8+T cells is ~3 Gray (Gy)16, 20-fold lower than a standard RT dose of 60 Gy. T cells are killed by direct and indirect DNA damage in radiation fields16. Fluorescence tracking demonstrates T cell recirculation is transiently impaired by radiation therapy to the tumor35. Prolonged or high-dose radiation exposure can induce T cell exhaustion, reduce cytokine production (e.g., interferon-gamma), and increase the expression of inhibitory receptors such as PD-136,37,38.
Of note, sub-lethal doses of radiation (typically <2 Gy) can enhance T cell activation and recruitment to tumors39. Radiation induces the release of neoantigens and pro-inflammatory cytokines, improving T cell recognition of tumor cells40,41. Additionally, RT can upregulate major histocompatibility complex (MHC) molecules and death receptors on cancer cells, enhancing T cell-mediated cytotoxicity42. Some studies also suggest that low-dose RT enhances the trafficking of T cells into the tumor microenvironment (TME) by modulating the expression of chemokines like CXCL9 and CXCL1043.
Natural killer (NK) cells
NK cells are innate immune effectors that recognize and kill tumor cells independently of antigen presentation, through mechanisms such as recognition of downregulated MHC class I molecules or through activating receptors like NKG2D. NK cells offer an advantage in brain tumors that evade T cell immunity by downregulating MHC molecules. NK cells do not induce graft versus host disease (GvHD) and can therefore be collected from allogenic donor sources or cell lines, positing the potential development of universal, radioprotected CAR-NK cells targeting common tumor antigens44.
NK cells are generally more resistant to radiation than T cells45,46. Studies show that NK cell cytotoxicity remains functional at moderate doses of radiation, making them ideal candidates for combination with RT45. Some research suggests that fractionation, in comparison to single, large-dose RT, may improve the cytotoxicity and expansion of NK cells47. In cancers such as prostate cancer48, non-small cell lung cancer49, and hepatocellular carcinoma50, increased levels of NK cells in the blood have been observed following RT.
Radiation can induce the upregulation of stress ligands (e.g., MICA/B, ULBP1-6) on tumor cells, enhancing NK cell recognition and killing51. However, high doses of radiation (>8 Gy) can impair NK cell proliferation and effector functions, including degranulation and cytokine production46. Additionally, radiation can alter the expression of NK cell ligands on tumor cells, either enhancing or diminishing NK cell cytotoxicity depending on the radiation dose and tumor type51. Interestingly, pre-treatment of the tumor site with low-dose radiation has been shown to prime the tumor for NK cell-mediated lysis, suggesting a synergistic effect when combined with NK-based ACT52. This makes NK cells a promising candidate for combinatory therapies involving radiation.
Macrophages
Macrophages play a dual role in cancer, either promoting tumor progression (M2-like macrophages) or mediating tumor destruction (M1-like macrophages). Adoptive transfer of macrophages reprogrammed toward an M1 phenotype is an emerging strategy in ACT53. These macrophages can be engineered to enhance their phagocytic activity against cancer cells or to produce pro-inflammatory cytokines within the TME.
Macrophages are relatively radioresistant compared to T and NK cells54. Low to moderate doses of radiation (≤2 Gy) can induce polarization of macrophages toward an M1 phenotype, promoting anti-tumor activity55,56. Radiation enhances macrophage-mediated phagocytosis by upregulating “eat me” signals (e.g., calreticulin) on tumor cells and increasing the production of inflammatory cytokines such as TNF-α and IL-1257,58. However, macrophages exposed to high-dose radiation (>2 Gy) may undergo apoptosis or shift toward an M2-like immunosuppressive phenotype, supporting tumor growth and immune evasion58. Radiation also affects the recruitment of macrophages into the TME by inducing the expression of macrophage-attracting chemokines, such as CCL259. This can lead to the infiltration of both pro-tumorigenic and anti-tumor macrophages59, highlighting the complexity of their role in radiation-enhanced immune responses.
γδ T cells
γδ T cells represent a small subset of T cells that recognize non-peptide antigens and exhibit MHC-independent tumor recognition60. They have garnered interest for ACT due to their broad tumor specificity and cytotoxic potential30. γδ T cells may be useful for pediatric brain tumors, which have lower mutational loads61.
γδ T cells are relatively radioresistant and maintain their cytotoxic functions even after moderate doses of radiation62,63. Radiation-induced stress ligands on tumor cells, such as NKG2D ligands, enhance the recognition and killing of tumor cells by γδ T cells51. Moreover, γδ T cells can proliferate and produce cytokines such as IFN-γ in irradiated tumors, further promoting anti-tumor immunity64. The combination of γδ T cell-based ACT with low-dose RT has shown promise in preclinical studies, as radiation not only primes tumors for γδ T cell recognition but also enhances the local recruitment of these cells51,59,65.
Invariant natural killer T (iNKT) cells
Invariant natural killer T (iNKT) cells are a subset of T cells that bridge innate and adaptive immunity by recognizing glycolipid antigens presented by CD1d molecules66. Their ability to produce large amounts of cytokines, such as IFN-γ and IL-4, makes them potent activators of anti-tumor immune responses66.
iNKT cells show moderate sensitivity to radiation67. Relatively little is known about the impact of radiation therapy on iNKT activity with inconsistent findings in the literature, with some proposing that radiation decreases iNKT anti-tumor activity67,68. However, iNKT remain an intriguing target for ACT because they do not induce GvHD, similar to NK cells69. iNKT cells also have two target ligands: the natural CD1d ligand and CAR-targeted antigen69. CD1d is expressed in several brain tumors, including glioblastoma70 and medulloblastoma71. Radiation also upregulates the expression of intercellular adhesion molecule-1 (ICAM-1), which is expressed in some gliomas and binds to LFA-1 on the surface of iNKT cells72,73,74. However, like other immune cells, high-dose radiation can reduce iNKT cell viability and function67, emphasizing the need for dose optimization when combining iNKT cell ACT with RT.
Each immune-cell type utilized in ACT for cancer presents distinct advantages and limitations regarding radiation sensitivity. T cells, NK cells, macrophages, γδ T cells, and iNKT cells all exhibit varied responses to radiation, which can be leveraged to enhance their anti-tumor efficacy in combination with radiotherapy. Figure 3 shows each unique CAR cell type and how radiation may increase the anti-tumor response. A strategic combination of ACT with radiotherapy holds significant promise for improving clinical outcomes in cancer treatment, but careful consideration of radiation dosing is critical to maximize synergistic effects while minimizing damage to immune effector cells.
Fig. 3: Anti-tumor effects of radiation on CAR immune cells.
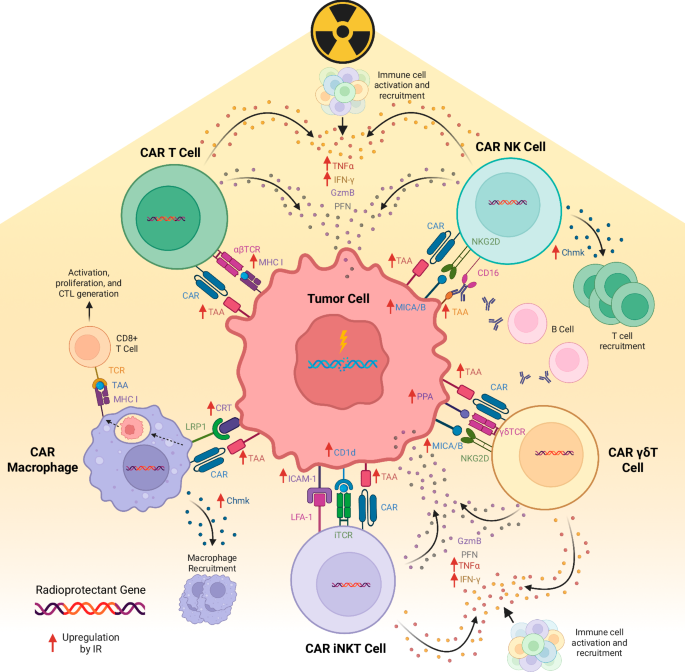
This figure summarizes the anti-tumor impacts of radiation on CAR T, NK, γδ, iNKT, and macrophage cells. Radiation may illicit an immune response via a variety of mechanisms, including upregulation of tumor antigen presentation and increased cytokine release. TAA: tumor associated antigen; CAR: chimeric antigen receptor; GzmB: granzyme B; PFN: perforin; PPA: phosphoantigen; LRP1: low-density lipoprotein receptor 1; MHC I: major histone compatibility complex I; IFNγ: interferon gamma; TNFα: tumor necrosis factor alpha; Chmk: chemokines; CRT: calreticulin; CTL: cytotoxicity T lymphocyte; CD: cluster of differentiation; MICA/B: MHC class I chain-related protein A and B; TCR: T cell receptor; iTCR: invariant T cell receptor.
ACT and chemotherapy
While we propose radioprotected ACT in combination with standard of care RT, many oncologic treatment plans also involve chemotherapies. Radioprotecting gene candidates that modulate DNA damage responses and cell cycle arrest may also decrease the chemosensitivity of the immune-cell75. However, chemotherapeutics that preferentially target immune-cell subtypes may negatively affect CAR T efficacy75. If ACT must be administered with chemotherapy, researchers may consider engineered expression of chemo-resistance proteins75. Modifying cell cycle and DNA damage proteins should be viewed with caution and suicide genes should be added in case of unexpected proliferation (See “Radioprotector safety considerations”)76. Of note, some chemotherapeutics may have an immuo-stimulatory effect on the TME that may improve CAR T cell trafficking to the tumor. For example, CAR T cell trafficking to the TME improved in mice preconditioned with temozolomide, a standard chemotherapeutic for glioblastoma, which was linked to decreased regulatory T cell populations in the TME77.
Mechanisms of radioprotection
Figure 4 outlines various mechanisms of radioprotection, each with unique implications for efficacy and safety. One mechanism, physically protecting DNA from direct damage by radiation, is likely to be safe, as this simply preserves the integrity of the genetic material without altering cellular processes. Similarly, the reduction of indirect DNA damage caused by reactive oxygen species (ROS) appears to be a generally safe approach. ROS scavenging neutralizes harmful free radicals without disrupting normal cellular functions and may be more universally applicable across species due to its fundamental nature in biology. These strategies aim to protect cells from radiation-induced damage without interfering with critical regulatory processes, making them promising candidates for clinical application.
Fig. 4: Mechanisms of radioprotection.
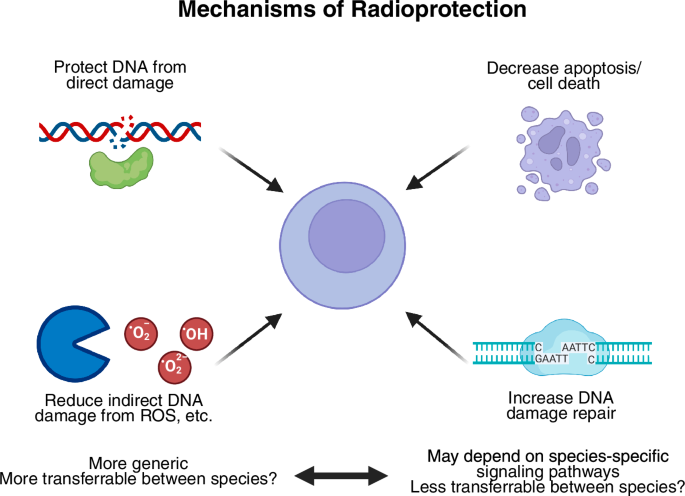
Radioprotective agents mitigate radiation-induced damage through multiple mechanisms. These include protection of DNA from direct damage, reduction of indirect DNA damage by scavenging reactive oxygen species (ROS), and promotion of DNA damage repair. Additionally, radioprotective strategies may involve decreasing apoptosis and cell death. While reducing ROS is a broadly applicable and potentially safer strategy, inhibiting apoptosis could carry risks, such as increasing the potential for carcinogenesis. Mechanisms related to ROS scavenging are likely to be more transferable between species, while apoptosis modulation may depend on species-specific pathways, raising concerns about their safety and efficacy across different models.
However, other mechanisms may raise safety concerns. For example, reducing apoptosis could have unintended consequences, such as increasing the risk of carcinogenesis. Apoptosis is a natural defense mechanism that eliminates damaged or potentially cancerous cells. Dampening this process might allow cells with genomic damage to survive and proliferate, potentially leading to tumorigenesis. Similarly, interventions that enhance DNA repair might inadvertently preserve cells with incomplete or improper repair of damaged DNA, increasing the potential for mutations that could lead to cancer. These mechanisms would require careful evaluation in clinical trials to assess the risks and benefits before being considered for non-oncologic applications, such as protecting stem cell allografts during transplantation. It is crucial that any potential radioprotective strategy be scrutinized not only for efficacy but also for long-term safety to prevent unintended consequences like carcinogenesis.
Extremophiles as a source of radioprotectors
Bridging extremophile biology and immuno-oncology may provide an avenue to radioprotect immune cells for ACT. Extremophiles are organisms that are ultra-tolerant to temperature, pressure, and irradiation extremes. Indeed, a number of bacteria, archaea, fungi, and animal extremophiles exist that can tolerate enormous doses of irradiation (>1000 Gy or 1000 times the human lethal dose, Fig. 5). The genetic basis for this protection is being uncovered by genomic and functional studies, providing a unique opportunity for radioprotection applications. The genetic basis for extremophile radioresistance involves molecules that (i) physically protect DNA from direct damage; (ii) mitigate indirect DNA damage via metabolic activities such as scavenging of reactive oxygen species; (iii) provide improved or redundant DNA damage repair machinery; (iv) alter apoptosis pathways; or (v) modulate cell signal transduction23,24,28,78,79,80. This raises the opportunity to apply emerging extremotolerant technology to T cells to enable therapeutic approaches.
Fig. 5: Radioresistant extremophile organisms.
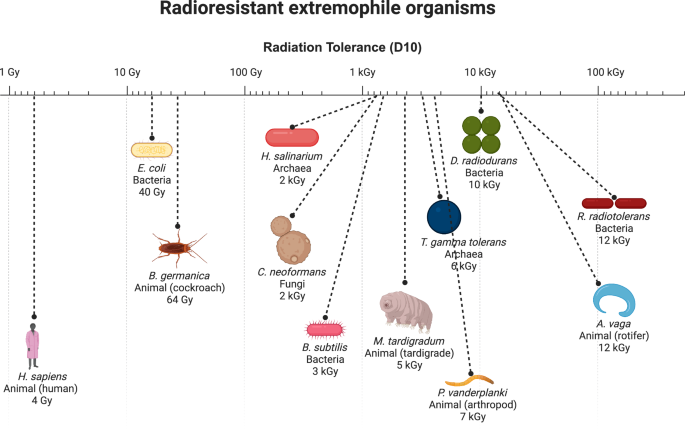
Select radioresistant extremophile organisms are shown, compared to humans. Radiation dose in Gray (Gy) for D10 (dose required to kill 90% of organism sample) when available or LD50 (dose required to kill 50% of organism sample) is shown. Note log scale indicating >1000X radioresistance compared to humans for many organisms.
Among the candidates, the Damage Suppressor (Dsup) protein from tardigrades stands out. Tardigrades are water-dwelling, eight-legged micro-animals that can be found everywhere from high-altitude mountaintops to the deep sea. Tardigrades have an extraordinary ability to tolerate immense doses of radiation (>5000 Gy) that would be lethal to most other life forms23,24. In the most stress-tolerant Tardigrade species, Ramazzottius varieornatus, the Tardigrade-unique damage suppressor (Dsup) protein colocalizes with DNA (nucleosomes in particular) and protects from hydroxyl radicals, protecting cells from radiation-induced DNA damage and cell death23,24. Transfection of Dsup into immortalized human cells enabled expression of the Dsup protein with no reduction in cell proliferation23. In addition to Dsup, additional radiosensitizers in tardigrades have been explored, including the DOPA (dihydroxyphenylalanine) dioxygenase gene (DODA1), tardigrade-specific radiation-induced disordered protein (TRID1), ubiquinol–cytochrome c reductase (bc1) synthesis protein (BCS1), and NADH dehydrogenase (ubiquinone) 1 beta subcomplex subunit 8 protein (NDUFB8)27. DODA1 leads to the production of betalins, a plant pigment with radical-scavenging properties27. TRID1 assists with liquid-liquid phase separation and enhances the recruitment of DNA repair protein to the double strand break (DSB) sites27. NDUFB8 and BCS1 are non-tardigrade specific proteins part of the mitochondrial respiratory chain complex assembly that are upregulated in tardigrades27. These proteins accelerate NAD+ regeneration for PARP1-mediated DNA repair27.
Other candidate radioprotectors include D. radiodurans PprI, which stimulates DNA repair and radioprotects human cells and mice26 and B. subtilis small acid soluble protein (SASP) which binds and potently protects DNA28. Sulfiredoxin from C. neoformans is strongly induced post-irradiation and radioprotects fungi80. Also, specific heat shock proteins have been linked to maximal irradiation survival response in the rotifer R. vega81. Molecules that mitigate direct or indirect DNA damage may be most likely to function in human cells, while molecules that have more complicated functions (such as in damage repair complexes and signaling pathways) may be less likely to do so.
Expression of foreign proteins, or “xenoproteins,” may present its own challenges in the pathway to engineering radioprotected immune cells. Extremophile-derived proteins may cause unexpected effects when expressed in human T cells. Future work could engineer these proteins to mitigate these issues by rationally combining key domains from these proteins with structurally similar human proteins.
Approaches to identify radioprotectors
Identifying effective radioprotectors requires comprehensive screening strategies that take into account the unique characteristics of human immune cells. One approach is to utilize in vitro genetic screens, which can provide insights into how various candidate genes may confer radioprotection. Both pooled and arrayed screens could be adapted from existing methodologies used to identify costimulatory molecules for CAR T cells82, thus prioritizing radioprotective genes for further study. Executing in vivo screens in animal models may be able to capture aspects of therapeutic efficacy in a more complex biological environment. Computational modeling and in silico analyses can also aid in predicting interactions and outcomes, streamlining the discovery process. Considerations for screens are summarized in Fig. 6.
Fig. 6: Approaches to screen for immune-cell specific radioprotectors.
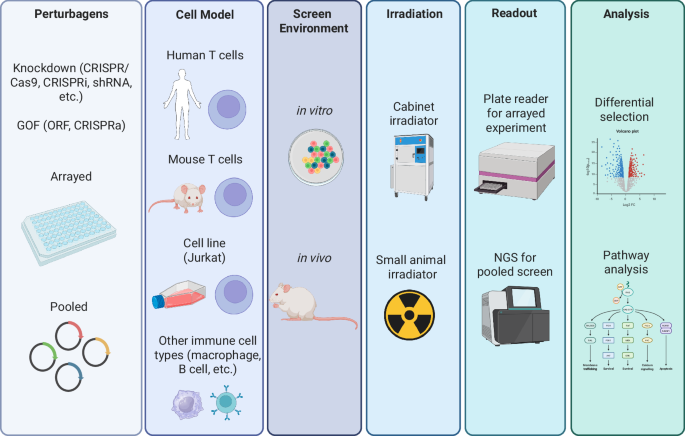
Various strategies can be employed to identify radioprotectors that specifically target immune cells. High-throughput screening of chemical libraries, genetic screens, and functional assays offer ways to discover agents that selectively shield immune cells from radiation damage. Depending on the type of ACT being studied, screens can be conducted in a range of immune-cell types, from easily manipulated cell lines to more clinically relevant, human-derived cells. Screening can be performed in vitro, which allows for more practical, controlled experiments, or in vivo, where complex biological interactions are better represented. Radiation is applied as needed, using tools like cabinet irradiators for in vitro cultures or small animal irradiators for in vivo studies. Readouts can vary depending on the specific goals of the screen, ranging from simple measurements of cell survival or function post-irradiation to more sophisticated next-generation sequencing (NGS) approaches that assess the selection of different perturbations after exposure. Analytical methods can involve straightforward ranking of top perturbations that improve immune-cell survival or function, as well as deeper molecular pathway analyses to gain mechanistic insight. Radioprotectors are also assessed for their ability to maintain immune-cell functionality, prevent apoptosis, or preserve immune-cell subsets during or after radiation. Validation of hits is critical to confirm radioprotective efficacy and ensure that identified candidates not only protect immune cells but also preserve their therapeutic potential. These methods hold promise for identifying radioprotectors that maintain immune competence in ACT, while ensuring therapeutic safety and efficacy.
Logistics of incorporating radioprotectors in cell therapy manufacture
Integrating radioprotectors into ACT manufacturing processes presents logistical challenges and opportunities. Strategies may involve utilizing the same viral vector for gene delivery or opting for separate vectors (Fig. 7), thereby allowing for a modular design that enables the co-expression of radioprotectors alongside therapeutic genes. This flexibility in vector design will facilitate the tailoring of cell therapies to maximize both therapeutic efficacy and safety.
Fig. 7: Formulation of radioresistant CAR T cells.
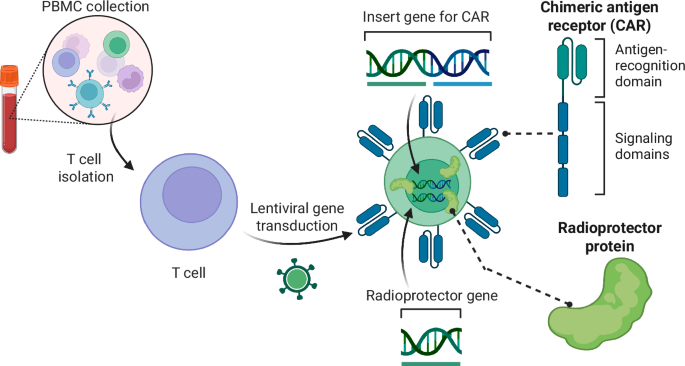
CAR T cells are developed from peripheral blood mononuclear cells (PBMCs) which are harvested, expanded, sorted into T cells, and then tranduced with lentiviral vectors to deliver the CAR gene playload. Our proposal will identify the most potent radioprotector genes to deliver in a similar fashion during the CAR T manufacturing process, producing radioresistant CAR T cells.
RT dose, fractionation, volume considerations, and administration method for combination with ACT
Combining ACT with radiotherapy offers a potent approach to cancer treatment, but the success of this combination may rely heavily on the careful selection of radiation dose, fractionation, treatment volumes, and administration method to ensure synergy while minimizing damage to immune cells, particularly the infused radioprotected ACT cells. Here, we discuss how different RT regimens and anatomic considerations influence ACT-radiotherapy combinations, and the potential role of radioprotection strategies to preserve ACT cells (Fig. 8).
Fig. 8: Considerations for RT delivery in combination with ACT.
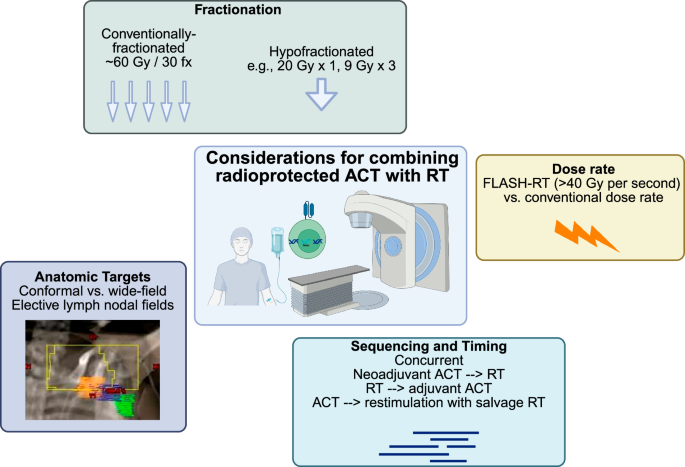
Key factors in optimizing the combination of radiation therapy (RT) with adoptive cell therapy (ACT) are presented. Fractionation options include conventionally-fractionated RT (e.g., 60 Gy in 30 fractions) and hypofractionated RT (e.g., 20 Gy in 1 fraction or 9 Gy in 3 fractions). Dose-rate considerations compare the use of conventional dose rates with FLASH-RT, where doses greater than 40 Gy per second are delivered. Anatomic targets are categorized into conformal vs. wide-field approaches, focusing on elective lymph node fields. Sequencing and timing of treatment strategies include concurrent, neoadjuvant ACT followed by RT, RT followed by adjuvant ACT, and using ACT with salvage RT for restimulation of immune responses.
Fractionated RT
Fractionated RT, where a total radiation dose is divided into smaller daily doses, presents an attractive opportunity to combine with radioprotected ACT. Standard fractionation schemes often use daily doses of 1.8–2.0 Gy, which fall near the lethal dose for 50% of T cells (LC50) at ~3 Gy16. Fractionated delivery allows immune cells time to recover between doses, reducing the risk of overwhelming damage to ACT cells.
This approach may progressively enhance the recruitment of ACT cells to the irradiated tumor over time83. Repeated low-dose exposure can upregulate the expression of stress ligands and increase antigen presentation by tumor cells, thus improving ACT cell recognition and tumor infiltration84. Additionally, fractionated RT spares normal tissues, reducing systemic toxicities that could otherwise impair immune responses.
From a logistical perspective, fractionated RT is commonly used in clinical settings for treating brain tumors. Therefore, its widespread clinical use facilitates the integration of ACT without substantial alterations to current RT protocols. Optimizing the timing and sequencing of ACT infusion within a fractionated RT regimen could maximize the beneficial effects of radiation while minimizing immune-cell depletion.
Hypofractionated RT
Hypofractionated RT, which delivers higher doses of radiation per fraction (e.g., >8 Gy), poses a different set of challenges and opportunities for ACT combinations. High-dose treatments, such as 20 Gy delivered in a single fraction in stereotactic radiosurgery (SRS) for brain metastases, may result in greater tumor cell death and release of tumor antigens85,86. However, the steep increase in radiation dose may overwhelm even radioprotected ACT cells, particularly those sensitive to radiation, such as T cells.
To circumvent the potential for immune-cell depletion, hypofractionated RT may be more effective when administered immediately before ACT infusion rather than concurrently. Pre-irradiation could prime the tumor for ACT by increasing tumor antigenicity and altering the tumor microenvironment to favor immune infiltration without subjecting the infused cells to excessive radiation85,86. Careful scheduling of hypofractionated RT prior to ACT infusion may amplify anti-tumor immune responses by using radiation as a priming tool.
Anatomic RT field considerations
One critical consideration when combining ACT with RT is the anatomic location and size of the radiation field. While modern RT technologies such as intensity-modulated radiation therapy (IMRT) and stereotactic body radiation therapy (SBRT) allow for precise, conformal delivery of radiation, larger or less targeted radiation fields can pose a risk to immune cells within the lymphatic system and circulating immune cells.
Radiation fields that include lymph node basins or areas of high immune-cell trafficking may reduce the availability of functional immune cells for ACT. Lymph node irradiation may deplete T cells and other immune effectors required for ACT efficacy, which could hinder overall treatment outcomes87. Therefore, minimizing the irradiation of at-risk lymphoid structures or employing radioprotective strategies, such as shielding88, may help preserve immune functionality in combination therapy.
FLASH-RT
An emerging technique in the RT field, FLASH radiotherapy (FLASH-RT), involves ultra-high dose-rate radiation delivery, typically delivered at greater than 40 Gy/s89. Studies suggest that FLASH-RT offers the potential to spare normal tissues from the toxicities associated with conventional radiation, while maintaining potent anti-tumor effects90,91. The unique biological mechanisms behind FLASH-RT, such as differential oxygen depletion and modulation of the tumor microenvironment, could also offer a new paradigm for combination with ACT91. Some research suggests that FLASH-RT can overcome hypoxia-mediated tumor resistance, a hallmark of many brain cancers92,93. In glioblastoma, FLASH-RT has also been demonstrated to spare the normal brain from radiation-induced toxicities94.
Although still in experimental stages, FLASH-RT may stimulate CAR T cells and other ACT-based therapies in ways that conventional RT cannot. Furthermore, FLASH-RT may alter the TME in a manner that enhances CAR T cell trafficking and persistence, creating a synergistic effect that could amplify anti-tumor efficacy. In murine models of diffuse midline glioma, FLASH-RT led to the upregulation CD4+ T cells and genes involved in T-cell activation and trafficking on day 10 following treatment in comparison to conventional radiotherapy95. This warrants further investigation into the specific interactions between FLASH-RT and ACT, with an emphasis on understanding the immunological mechanisms involved.
Administration
The blood brain barrier and TME pose crucial considerations for the location of ACT administrations. Though ACTs can reach the brain via intravenous injection, other administration methods may increase ACT population within the CNS, possibly decreasing side effects96. Intrathecal delivery involves direct injection into the cerebral spinal fluid (CSF), bypassing the blood-brain barrier96. Injection can occur into the spine or the ventricles of the brain. While spinal intrathecal delivery, or lumbar punctures, may be useful for single-dose ACT, intraventricular injections, commonly via Ommaya catheters, can be programmed with a pump to deliver repeated doses96. Implantable devices may also offer an easier way to obtain CSF samples to confirm immune activation96. Intra-tumoral delivery of ACT is also possible via convection enhanced delivery (CED) systems. CED systems form a pressure gradient via a microinjection pump, possibly reaching a larger brain volume97. However, the pressure gradient may cause worsening neurotoxicity symptoms, a common side effect in CAR T patients96. Further research on the optimal administration method may maximize the response of radioprotected ACT.
Sequencing of radioprotected ACT and RT
Optimal sequencing of radioprotected ACT and RT is essential to fully leverage the therapeutic potential of these combined modalities. While both ACT and RT are potent treatments on their own, their integration requires careful planning to avoid negative interactions such as radiation-induced immune-cell damage. Incorporating radioprotection strategies into ACT opens new possibilities for safely combining these treatments, potentially allowing for higher radiation doses or more aggressive fractionation schedules. The number of doses should also be considered, as radioprotection may impact the longevity of modified immune cells. Preclinical models and clinical trials are necessary to establish the most effective sequencing protocols.
Preclinical studies to inform sequencing
Preclinical studies are particularly crucial for evaluating how the addition of radioprotective strategies influences the interaction between ACT and RT. In these models, the timing of ACT administration relative to RT can be explored, particularly in the context of radioprotection. For instance, studies can assess whether radioprotected ACT cells retain functionality and viability when delivered before, during, or after RT. Additionally, preclinical research can help define the thresholds of RT dose and fractionation at which radioprotected ACT cells are most effective, without suffering significant damage from radiation. By incorporating radioprotective agents into ACT protocols, it may be possible to use higher radiation doses that would otherwise impair T-cell function. This opens new avenues for combination therapies, but the precise sequencing of these treatments will need to be fine-tuned through preclinical work before transitioning into clinical practice.
Integrating radioprotected ACT into upfront, fractionated RT for curative-intent treatment
For brain cancers where upfront fractionated RT is the standard of care, such as glioblastoma, integrating radioprotected ACT into curative-intent treatment regimens offers an exciting therapeutic strategy. Fractionated RT typically involves daily radiation doses of around 1.8–2.0 Gy delivered over several weeks, creating a potentially favorable environment for ACT. Radioprotected ACT could be administered early in the course of fractionated RT, allowing T cells to persist and accumulate within the tumor over time.
Because radioprotective strategies could enhance the resilience of ACT cells to low daily doses of radiation, this approach may increase the likelihood of immune cells infiltrating the tumor microenvironment and maintaining functionality throughout the RT course. This might improve the overall therapeutic outcome, especially in brain tumors that are traditionally difficult to treat. In this context, radioprotected ACT cells could be infused early in the RT course and allowed to interact with radiation-induced tumor stress signals and antigen presentation over time. Fractionated RT may also create opportunities for selection of radioprotected ACT cells. While normal immune cells will die throughout fractionated RT, radioprotected ACT cells may not, providing a sustained anti-tumor immune response and reducing the risk of T-cell depletion.
Combining salvage RT with radioprotected ACT
For patients undergoing salvage RT, particularly those with recurrent or metastatic cancer, combining radioprotected ACT with higher RT doses or hypofractionated schedules may improve outcomes. In salvage settings, higher RT doses (e.g., 8 Gy per fraction or more) are often required to address treatment-resistant tumors, which can compromise immune-cell viability. The inclusion of radioprotection in ACT protocols allows the possibility of combining these more aggressive RT regimens without overwhelming the immune system.
In these cases, radioprotected ACT could be administered either before or after salvage RT, depending on the patient’s condition and treatment goals. Post-RT administration of radioprotected ACT could allow the immune system to “clean up” any residual tumor cells that survive the high-dose radiation. Alternatively with pre-RT administration of radioprotected ACT, RT could be used to debulk the tumor and prime the tumor microenvironment for subsequent immune attack by radioprotected cells. This combination might offer significant benefits, but the optimal sequencing needs to be explored through preclinical and clinical trials.
Re-stimulation of radioprotected ACT with RT
Radiation has the potential to re-stimulate adoptive immune cells that may have become less effective over time, and radioprotected ACT cells could be especially well-suited for this approach. As tumors progress, ACT cells may experience functional exhaustion, particularly in cases of CAR T-cell therapy. RT can induce tumor cell death85,86 releasing antigens that could “re-prime” the radioprotected ACT cells, thereby restoring or boosting their anti-tumor activity. In this scenario, radioprotected ACT cells would be better able to withstand the re-stimulation process, maintaining their functionality and viability despite the immunosuppressive effects of radiation. This strategy could be particularly useful in treating brain tumors that exhibit immunoediting, where the immune system drives the selection of tumor cells that evade immune detection98. Immunoediting is especially relevant in glioblastoma, which shows poor response to immunotherapies due to tumor herterogeneity99. By re-exposing the tumor to immune surveillance following RT, the radioprotected ACT cells could help target these previously elusive tumor cell populations.
Non-oncologic opportunities
Radioprotective ACT holds promising potential for non-oncologic purposes, such as protecting stem cell allografts during transplantation. One key application could be to reduce the duration and severity of the post-transplant nadir, a period of vulnerability when the patient’s immune system is severely compromised. Typically, patients undergoing stem cell transplantation will receive total body irradiation (TBI) to prep the immune system prior to the transplant100. Patients generally receive 12–15 Gy over 3–4 days100. TBI acts both cytotoxic and immunosuppressive, creating space in the marrow for new cells and decreasing the likelihood of stem cell rejection100. Administering radioprotected ACT prior to radiation could enhance the success and recovery of hematopoietic stem cell transplants, potentially leading to faster immune reconstitution and fewer complications.
Additionally, radioprotected ACT could be explored for specific non-cancerous indications where radiation is necessary but poses risks to healthy cells, such as in certain autoimmune disorders or organ transplants. However, the use of radioprotected ACT for non-oncologic applications would require careful study, as clinical experience builds in oncology. With more research and validation, these innovative approaches could eventually make their way into clinical practice, expanding the therapeutic benefits of radioprotective strategies beyond cancer treatment.
Alternative radioprotection approaches
Other radioprotective strategies include endogenous pathway modulation, pharmacological agents, physical shields, and cellular engineering, each with distinct limitations. Overexpressing human antioxidants like SOD2 reduces oxidative stress but offers limited protection against high radiation doses, while modulating the p53 pathway can prevent apoptosis but risks preserving genetically unstable cells. Pharmacological options like FDA-approved amifostine provide nonspecific ROS scavenging unsuitable for immune cells, and synthetic molecules such as Mn porphyrins lack DNA repair capabilities. Physical approaches, such as nanoparticle shields, can protect cells but add manufacturing complexity and may hinder cell trafficking. Cellular engineering through CRISPR offers precise genetic modifications for radiation resistance, though extensive screening is needed to avoid adverse effects, while leveraging radiation-resistant stem cell-derived immune cells faces scalability challenges.
In vivo delivery of CARs and radioprotector genes
The workflow for manufacturing CAR T cells ex vivo for specific patients is long and costly. In vivo delivery proposes the administration of CAR gene or protein payloads enveloped in viral vectors or nanoparticles101. Relevant immune-cell-targeting ligands are fused to the viral envelope protein to ensure the proper cells receive the CAR101. Lentivirus, retrovirus, and adeno-associated virus have been used in mice to deliver CAR payloads with equivalent efficacy in controlling tumor growth to ex vivo CAR delivery101. Nanocarriers delivering mRNA, plasmid, and protein have also been used101. In vivo delivery methods may also be helpful for radioprotector gene candidates. For example, delivery of viral vectors encoding a CAR and radioprotector gene could eliminate the need for ex vivo processing. In vivo delivery is still developing and requires careful control to limit off-target effects while maximizing immune-cell transduction efficiency101.
Future clinical trial designs
Clinical trial design for combining radiation therapy (RT) and radioprotected ACT must thoughtfully consider the optimal sequencing of these treatments to maximize therapeutic efficacy. This involves determining the timing of RT relative to ACT, as well as establishing parameters such as the appropriate ACT dosage, RT fractionation schedules, and dosing to ensure synergistic effects while minimizing toxicity. Additionally, the use of steroids in conjunction with these therapies should be evaluated, as they can impact immune function and treatment outcomes102. Incorporating correlative studies into trial designs will enable researchers to explore the underlying biological mechanisms at play, facilitating a better understanding of how these therapies interact. Moreover, identifying and validating biomarkers of response could provide critical insights, allowing for early outcome readouts that guide subsequent treatment decisions and adjustments, ultimately enhancing the overall effectiveness of the combined approach. A schematic for a potential clinical trial of radioprotected CAR T in combination with conventionally-fractionated RT for newly-diagnosed glioblastoma brain tumor patients is outlined in Fig. 9.
Fig. 9: Concept for clinical trial of radioprotected CAR T and RT.
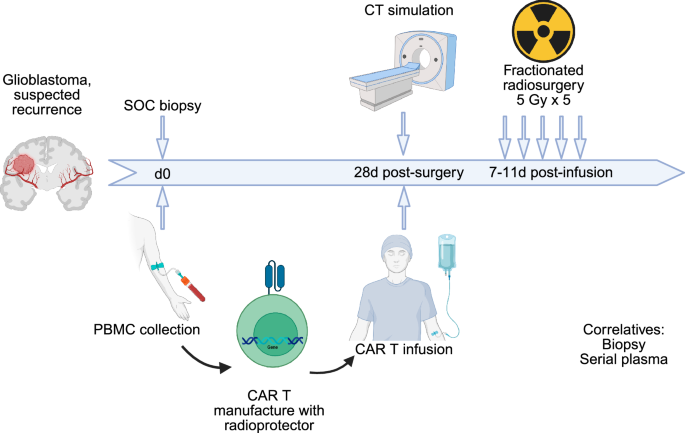
Patients with suspected glioma are recruited to the trial. Patients undergo standard-of-care surgical resection followed by radiation therapy. PBMCs are collected and manufactured into CAR T cells including the radioprotector(s) identified here. CAR T cells are re-infused by day 56, providing time for CAR manufacturing but delivering CAR T early in the RT course when immunogenicity may be highest. Concurrent standard-of-care temozolomide may be given or omitted depending on rapid tumor molecular analysis.
Radioprotector safety considerations
Safety is a paramount consideration when integrating radioprotectors into ACT. First and foremost, the potential for radioprotectors to influence apoptosis or cell cycle pathways raises concerns regarding carcinogenesis. Alterations in these critical regulatory mechanisms may inadvertently promote tumorigenesis, underscoring the necessity for thorough preclinical evaluation and long-term monitoring of patients receiving such therapies. Also, any approach involving the expression of foreign radioprotective proteins must be scrutinized for unforeseen immunogenic effects. The introduction of xenoproteins may elicit immune responses that could compromise the safety and effectiveness of the therapy. Another concern is that CAR T cells expressing genes that modulate the cell cycle or cell senescence could become immortal, potentially leading to rapid cell division, autoimmunity, and/or T cell malignancies. For this reason, researchers should consider integrating suicide genes, such as caspase 9103. Suicide genes may also be helpful in case of severe side-effects following CAR T cell infusions, such as cytokine release syndrome103. Ideal suicide genes could be activated by a biologically inert, bioavailable antibody103.
CAR T trials should also be planned with especially close monitoring and planning for patient safety. Moreover, CAR T cell therapy is known to elicit severe immune reactions, the most notable being neurotoxicity, which can necessitate hospitalization. The introduction of RT may exacerbate these adverse effects, complicating the clinical picture and heightening the need for vigilant monitoring and trial designs that prioritize patient safety. Establishing clear protocols for assessing and managing toxicities will be essential to mitigate these risks effectively. Additionally, the concurrent use of steroids and IL-1R agonists, such as anakinra, should be carefully planned104. While these agents can modulate inflammatory responses, their immunosuppressive effects could potentially undermine the therapeutic efficacy of CAR T cells. Thus, balancing the need for symptom management with the preservation of immune functionality will be crucial. Comprehensive safety evaluations and preclinical studies are necessary to identify and mitigate these risks before advancing to clinical trials. In summary, while the potential benefits of radioprotectors in enhancing ACT are compelling, the associated safety concerns must be addressed through rigorous research, careful clinical trial design, and ongoing patient monitoring to ensure a favorable risk-benefit profile.
Conclusion
The potential to enhance ACT for brain tumors through the radioprotection of immune cells represents a groundbreaking frontier in cancer treatment. Immune-cell types used in CAR therapies exhibit distinct anti-tumor properties that must be carefully leveraged to maximize therapeutic impact. Insights from extremophile biology, coupled with advancements in radioprotective technologies and robust screening methodologies, offer a unique opportunity to overcome the challenges posed by the immunosuppressive tumor microenvironment in brain cancers. However, introducing novel proteins carries inherent risks, underscoring the necessity for extensive preclinical research to optimize the sequencing of ACT and RT for improved efficacy. Collaborative research efforts will be pivotal in translating these innovations into clinical practice, ultimately advancing outcomes for brain cancer patients. Future work should prioritize high-throughput screening to identify additional extremophile-derived proteins with complementary radioprotective functions, alongside the development of sophisticated delivery systems that ensure stable, efficient expression of these genes in immune cells without impairing function. Finally, rigorous safety and immunogenicity testing, including long-term preclinical evaluation of these proteins in CAR T cells, will be essential to validate therapeutic efficacy while minimizing potential adverse effects.
Data availability
No datasets were generated or analysed during the current study.
References
Gallus, M. et al. Chimeric antigen receptor T-cell therapy in patients with malignant glioma—from neuroimmunology to clinical trial design considerations. Neuro Oncol. https://doi.org/10.1093/neuonc/noae203 (2024).
Brown, C. E. et al. Regression of glioblastoma after chimeric antigen receptor T-cell therapy. N. Engl. J. Med. 375, 2561–2569 (2016).
Majzner, R. G. et al. GD2-CAR T cell therapy for H3K27M-mutated diffuse midline gliomas. Nature 603, 934–941 (2022).
Albelda, S. M. CAR T cell therapy for patients with solid tumours: key lessons to learn and unlearn. Nat. Rev. Clin. Oncol. 21, 47–66 (2024).
Mullard, A. FDA approves first tumour-infiltrating lymphocyte (TIL) therapy, bolstering hopes for cell therapies in solid cancers. Nat. Rev. Drug Discov. 23, 238 (2024).
Creelan, B. C. et al. Tumor-infiltrating lymphocyte treatment for anti-PD-1-resistant metastatic lung cancer: a phase 1 trial. Nat. Med. 27, 1410–1418 (2021).
Akhavan, D. et al. CAR T cells for brain tumors: lessons learned and road ahead. Immunol. Rev. 290, 60–84 (2019).
Bagley, S. J. et al. Repeated peripheral infusions of anti-EGFRvIII CAR T cells in combination with pembrolizumab show no efficacy in glioblastoma: a phase 1 trial. Nat. Cancer 5, 517–531 (2024).
Choi, B. D. et al. Intraventricular CARv3-TEAM-E T cells in recurrent glioblastoma. N. Engl. J. Med. 390, 1290–1298 (2024).
Bagley, S. J. et al. Intracerebroventricular bivalent CAR T cells targeting EGFR and IL-13Ralpha2 in recurrent glioblastoma: a phase 1 trial. Nat. Med. https://doi.org/10.1038/s41591-025-03745-0 (2025).
Lin, Y. J., Mashouf, L. A. & Lim, M. CAR T cell therapy in primary brain tumors: current investigations and the future. Front. Immunol. 13, 817296 (2022).
Grunert, M. et al. Radiation and brain tumors: an overview. Crit. Rev. Oncog. 23, 119–138 (2018).
Hovhannisyan, L., Riether, C., Aebersold, D. M., Medova, M. & Zim