- nature
- scientific reports
- articles
- article
Development of gut microbiota during the first 2 years of life
- Article
- Open access
- Published: 31 May 2022
Development of gut microbiota during the first 2 years of life
- Mona-Lisa Wernroth,
- Sari Peura,
- Anna M. Hedman,
- Susanne Hetty,
- Silvia Vicenzi,
- Beatrice Kennedy,
- Katja Fall,
- Bodil Svennblad,
- Ellika Andolf,
- Göran Pershagen,
- Jenny Theorell-Haglöw,
- Diem Nguyen,
- Sergi Sayols-Baixeras,
- Koen F. Dekkers,
- Stefan Bertilsson,
- Catarina Almqvist,
- Johan Dicksved &
- Tove Fall
Scientific Reports volume 12, Article number: 9080 (2022) Cite this article
Abstract
Although development of microbiota in childhood has been linked to chronic immune-related conditions, early childhood determinants of microbiota development have not been fully elucidated. We used 16S rRNA sequencing to analyse faecal and saliva samples from 83 children at four time-points during their first 2 years of life and from their mothers. Our findings confirm that gut microbiota in infants have low diversity and highlight that some properties are shared with the oral microbiota, although inter-individual differences are present. A considerable convergence in gut microbiota composition was noted across the first 2 years of life, towards a more diverse adult-like microbiota. Mode of delivery accounted for some of the inter-individual variation in early childhood, but with a pronounced attenuation over time. Our study extends previous research with further characterization of the major shift in gut microbiota composition during the first 2 years of life.
초록
유아기 미생물군집 발달이
만성 면역 관련 질환과 연관되어 있음에도 불구하고,
미생물군집 발달의 초기 결정 요인은 완전히 규명되지 않았다.
우리는 16S rRNA 시퀀싱을 활용하여
생후 2년 동안 4개 시점에서 수집한 83명의 유아와
그들의 어머니의 대변 및 타액 샘플을 분석하였다.
본 연구 결과는
영아의 장내 미생물군집이 낮은 다양성을 보이며,
개인 간 차이는 존재하지만 일부 특성이 구강 미생물군집과 공유됨을 확인하였다.
생후 2년간 장내 미생물군집 구성은
성인형 미생물군집과 유사한 더 다양한 형태로 상당한 수렴 현상을 보였다.
분만 방식은
유아기 초기 개인 간 변이의 일부를 설명했으나,
시간이 지남에 따라 현저히 감소하였다.
본 연구는
생후 첫 2년간 장내 미생물군 구성의 주요 변화를 추가로 규명함으로써
기존 연구를 확장합니다.
Similar content being viewed by others

Changes in the intestinal microbiota of Japanese children during the first 3.5 years of life
Article Open access26 November 2024
Article Open access08 April 2024

Early life microbial succession in the gut follows common patterns in humans across the globe
Article Open access14 January 2025
Introduction
Microbial colonization of the gut is essential for natural maturation of the intestine and immune system in infants, and early life imbalances in the gut microbiota have been suggested to increase the risk of chronic immune-related conditions such as allergy1 and type 1 diabetes2. Like the gut, the oral cavity also harbours a diverse microbial community and the composition of the oral microbiota has been associated with dental health3 and cardiovascular disease4.
Microbial colonization of the gut starts at birth, or even in utero5 and maternal vaginal, gut and skin constitute important bacterial inocula. The gut microbiota undergoes most of its development very early in life and major changes in the composition of the gut microbiota have been observed until the child is 2 to 3 years of age2,6,7. However, an adult level gut microbial diversity might not be reached even at 5 years of age7. The diversity and colonization patterns have been proposed to be influenced by factors such as mode of delivery2, having siblings2, gestational age2, birth weight8, antibiotics use during the first years of life2, presence of furry pets in the household2 and diet2. Mode of delivery is recognised as a major determinant of microbiota composition, and differentially delivered infants have contrasting gut microbiota composition throughout the first years of life2,9,10,11,12. However, longitudinal studies following children from birth through their first 2 years of life investigating the role of mode of delivery are scarce9,12. Further, prenatal factors such as exposure to antibiotics, might be of importance and a recent review by Grech et al.13 highlighted the lack of longitudinal studies investigating the effect of maternal exposures on gut microbiota development in children. Thus, there is a need to better understand the role of mode of delivery and other factors of importance for shaping the gut microbiota composition in a growing child.
A study of the oral and gut microbiota in 3-day-old children suggested that more species and strains are shared between their own oral and gut microbiota in children than in their mothers14. The duration of these similarities is still unclear and warrants further investigation.
The aim of the present study was to investigate environmental exposures of relevance for gut microbiota development in children, following them longitudinally from birth until age 2, and to describe associations between gut and oral microbiota. The study was based on repeated faecal and saliva samplings in a cohort of 83 children and their mothers and leveraged the unique availability of high-quality national registers and questionnaire data to obtain information on maternal and birth characteristics.
서론
장내 미생물 군집화는
영아의 장 및 면역 체계 자연적 성숙에 필수적이며,
생후 초기 장내 미생물 군집 불균형은
알레르기1 및 제1형 당뇨병2과 같은 만성 면역 관련 질환 위험 증가와 연관된 것으로 제안되어 왔다.
장과 마찬가지로 구강 내에도 다양한 미생물 군집이 서식하며,
구강 미생물 군집 구성은 치아 건강3 및 심혈관 질환4과 연관성이 확인되었다.
장내 미생물 군집화는
출생 시, 또는 자궁 내5에서 시작되며
모체의 질, 장 및 피부가 중요한 세균 접종원이 된다.
장내 미생물 군집은 생후 매우 이른 시기에 대부분 발달하며,
2~3세까지 장내 미생물 군집 구성에 큰 변화가 관찰됩니다2,6,7.
그러나
5세에도 성인 수준의 장내 미생물 다양성에 도달하지 못할 수 있습니다7.
다양성과 정착 패턴은 분만 방식2, 형제자매 유무2, 임신
주수2, 출생 체중8, 생후 초기 항생제 사용2, 가정 내 털 있는 반려동물 보유 여부2 및 식이2 등이
영향을 미친다고 제안되어 왔다.
분만 방식은 미생물군 구성의 주요 결정 요인으로 인정되며,
분만 방식이 다른 영아들은 생후 초기 전반에 걸쳐
상반된 장내 미생물군 구성을 보인다2,9,10,11,12.
그러나
출생부터 생후 2년까지의 아동을 추적 관찰하며
분만 방식의 역할을 조사한 종단 연구는 드물다9,12.
또한 항생제 노출과 같은 태아기 요인도 중요할 수 있으며,
Grech 등의 최근 리뷰13은 모체 노출이 아동의 장내 미생물군집 발달에 미치는 영향을 조사한 종
단 연구의 부족을 강조하였다.
따라서
성장기 아동의 장내 미생물군 구성 형성에 중요한 분만 방식 및
기타 요인의 역할을 더 잘 이해할 필요가 있다.
생후 3일 된 영아의 구강 및 장내 미생물군 연구에 따르면,
영아의 구강과 장내 미생물군 간에는 모체보다
더 많은 종과 균주가 공유되는 것으로 나타났다14.
이러한 유사성의 지속 기간은
아직 불분명하며 추가 연구가 필요하다.
본 연구의 목적은
출생부터 2세까지 아동을 종단적으로 추적하여
장내 미생물군집 발달과 관련된 환경적 노출 요인을 조사하고,
장내 및 구강 미생물군집 간의 연관성을 기술하는 것이었다.
본 연구는 83명의 아동과 그 어머니를 대상으로 한 코호트에서
반복적인 분변 및 타액 샘플링을 기반으로 수행되었으며,
모체 및 출생 특성에 대한 정보를 얻기 위해
고품질 국가 등록 자료와 설문조사 데이터의 독보적인 가용성을 활용하였다.
Materials and methods
The Born into Life cohort
This study is based on the prospective longitudinal cohort study Born into Life15. The cohort consists of women who lived in Stockholm County, Sweden, enrolled in the LifeGene study and became pregnant at any time between 2010 and 201216. Children were delivered at the same clinic and were included in the cohort upon delivery. The cohort comprised 107 pregnant women, while the associated child cohort, with consent from the parents to follow the children also after birth, comprised 93 children (Fig. 1).
Figure 1
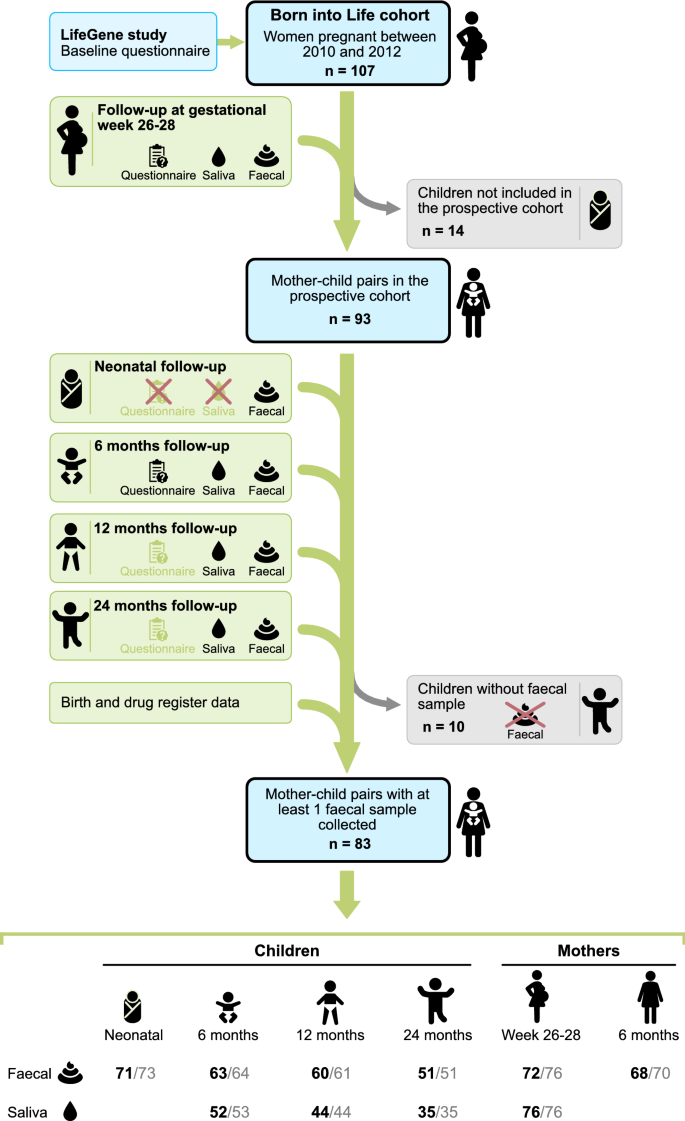
Flow chart of the study population. The cohort consists of pregnant women who lived in Stockholm, Sweden enrolled in the LifeGene study. After obtaining parental consent, the children were included in the prospective cohort (n = 93). A red cross indicates that questionnaires or saliva samples were not administrated. Light green questionnaire symbol indicates that the answers were not used in the current study. Number of samples with at least 12,588 reads reported in black font. Number of samples sequenced are reported in grey font. The figure is made by Andreas Dahlin at Visualize your Science.
Phenotypic information
Repeated questionnaires and linkage to national health registers were used to collect the phenotypic information. Linkage to national health registers was enabled through use of the unique individual Swedish personal identity number.
From the Medical Birth Register, we collected information relating to the birth (sex, date of birth, mode of delivery, gestational age, and birthweight) and the maternal characteristics such as parity, maternal body mass index (BMI), and smoking in early pregnancy. Information on exclusive breastfeeding was provided from the Born into Life questionnaire at 6 months. We obtained information on dispensed systemic antibiotics (ATC: J01) including date of dispense from the Swedish Prescribed Drug Register. Prenatal antibiotic exposure was defined as any maternal dispensed prescription of oral antibiotics during pregnancy. Information on exposure to furry pet, defined as presence of a cat or a dog in the household, was obtained from the questionnaire administrated at gestational week 26–28. We retrieved information on maternal highest education level from the LifeGene questionnaire.
Faecal and saliva samples within the Born into Life cohort
Both faecal and saliva samples were collected following a standardized protocol17. Faecal samples were collected from the children 2–4 days after birth and at 6, 12, and 24 months of age, whereas maternal samples were collected at gestational week 26–28, and 6 months post-partum. The neonatal faecal samples were collected by the midwife at the routine health check-up after birth. The remaining faecal samples were collected by the parents the day before visits to the test centre. Faecal samples were kept frozen at home and transported to the test centre in a cool transport container to prevent thawing. Subsequently, faecal samples were stored at − 80 °C until processing. Saliva samples from children were collected in the evenings at 6, 12, and 24 months of age using cotton swabs placed into Salivette tubes (Salivette–Sarstedt), whereas samples from mothers were collected in Salivette tubes according to a standard protocol one evening during gestational week 26–2818. The saliva samples were stored overnight at standard refrigerator temperature. Ethical approval for the study was granted by the Regional Ethics Review Board in Stockholm, Sweden (reference numbers 2011/192–31/2, 2011/1127–31/3, and 2014/1081–32 with amendments) and complied with the World Medical Association Declaration of Helsinki regarding ethical conduct of research. Informed consent for each participant in the maternal cohort was obtained during enrolment into the study and both parents gave informed consent for their children.
Microbiota analysis
The analysis order was randomly assigned across samples and DNA extraction was performed separately for faecal and saliva samples. DNA from 407 faecal samples (249 samples from children, 158 from mothers) and 241 saliva samples (156 samples from children, 85 samples from mothers) were all extracted using the MO BIO PowerSoil DNA kit (MO BIO, Solana beach, CA, USA) and the MiniG1600 homogeniser (SPEX SamplePrep, Metuchen, NJ, USA) according to the protocol provided by the manufacturer. Five negative controls were included in the saliva DNA extraction procedure. After PCR amplification of the v3-v4 regions of 16S rRNA gene (PCR protocol is available in18), the amplified fragments were purified with AMPure XP beads and DNA concentration was quantified using Qubit (ThermoFisher Scientific) with the dsDNA high-sensitivity assay. All samples and negative controls were subjected to gel electrophoresis to assess presence of gel bands and product size after each PCR amplification and purification step, and selected samples were also analysed using a bioanalyzer (Agilent Technologies, Santa Clara, CA, USA) to ensure high DNA quality and correct band size. Four of the negative controls showed no amplification, while the fifth showed some evidence of contamination. The final amplicon libraries were also verified by bioanalyzer analysis. In total, 407 faecal and 216 saliva samples with individual dual barcodes produced sufficient amount of product. The faecal samples were randomly allocated into three pools and saliva samples into two pools of 120–150 samples, each pooled in equimolar amounts. The pools were sequenced using Illumina MiSeq technology (v3 chemistry, 2*300 bp) at Science for Life Laboratory (Uppsala University, Sweden). A total of nine negative control samples were included in the sequencing, whereof eight had very low number of reads, and the previously suspected contaminated sample had a significant number of reads. The resulting sequence reads were processed using the software Mothur19 following the standard recommended procedure20, except that clustering of operational taxonomic units (OTUs) was done using VSEARCH (abundance-based greedy clustering)21 as implemented in Mothur. No batch effects with regards to sequencing pools were detected by principal coordinate analysis (PCoA). After visual inspection of the distribution of read counts, samples with fewer than 12,588 reads were removed (1 saliva sample and 9 faecal samples) and the remaining samples were rarefied to this sequencing depth using Mothur. A cut-off of 97% sequence identity was used to delineate OTUs. The taxonomic classification of OTUs was done using the SILVA database (version 132, released on Dec 13, 2017)22,23,24. Across more than 34 million pair-end reads, we observed 733,773 OTUs with 709,652 of these with less than 10 reads. These were discarded as recommended in Franzén et al.25 to reduce the number of spurious OTUs. The remaining 24,115 OTUs comprised 85% of the total amount of reads. The removed OTUs were most commonly classified as belonging to the family Lachnospiraceae (25%), unclassified Clostridiales (15%) and unclassified Bacteria (14%). Taxonomic classification based on SILVA was largely confirmed on genus level for the top 20 OTUs by BLAST to the NCBI 16S data base accessed on January 19, 2022.
We excluded 10 mother–child pairs because no faecal sample was collected for the child and our final study population therefore included 245 faecal samples and 131 saliva samples from 83 children, and 140 faecal samples and 76 saliva samples from 80 mothers (Fig. 1). In total 58 children had faecal samples from at least 3 sampling occasions and 38 from all 4 occasions.
Born into Life 코호트 내 대변 및 타액 샘플
표준화된 프로토콜17에 따라 대변 및 타액 샘플을 모두 수집했습니다. 대변 샘플은 출생 후 2~4일 및 생후 6개월, 12개월, 24개월에 아동으로부터 채취했으며, 모체 샘플은 임신 26~28주 및 분만 후 6개월에 채취했습니다. 신생아 대변 샘플은 출생 후 정기 건강 검진 시 조산사가 채취했습니다. 나머지 대변 샘플은 검사 센터 방문 전날 부모가 채취했습니다. 대변 샘플은 가정에서 냉동 보관 후 해동을 방지하기 위해 냉장 운반 용기에 담아 검사 센터로 운송되었습니다. 이후 대변 샘플은 처리 전까지 −80°C에서 보관되었다. 아동의 타액 샘플은 6개월, 12개월, 24개월 시기에 저녁 시간대에 Salivette 튜브(Salivette–Sarstedt)에 면봉을 넣어 채취하였으며, 모체의 샘플은 임신 26~28주 기간 중 한 저녁 시간대에 표준 프로토콜에 따라 Salivette 튜브에 채취하였다18. 타액 샘플은 표준 냉장고 온도에서 하룻밤 동안 보관되었습니다. 본 연구에 대한 윤리적 승인은 스웨덴 스톡홀름 지역 윤리 심사 위원회(참조 번호 2011/192–31/2, 2011/1127–31/3, 2014/1081–32 및 수정안)로부터 획득되었으며, 연구 윤리 수행에 관한 세계의학협회 헬싱키 선언을 준수하였습니다. 모성 코호트 참여자 각각에 대한 사전 동의는 연구 참여 등록 시 획득되었으며, 자녀의 경우 양 부모 모두 사전 동의를 제공했습니다.
미생물군 분석
분석 순서는 샘플별로 무작위로 할당되었으며, 분변 및 타액 샘플에 대해 별도로 DNA 추출을 수행했습니다. 407개의 대변 샘플(아동 249개, 어머니 158개)과 241개의 타액 샘플(아동 156개, 어머니 85개)에서 추출한 DNA는 모두 제조업체가 제공한 프로토콜에 따라 MO BIO PowerSoil DNA 키트(MO BIO, Solana beach, CA, USA)와 MiniG1600 균질기(SPEX SamplePrep, 미국 뉴저지주 메튜첸 소재)를 사용하여 제조사 제공 프로토콜에 따라 DNA를 추출하였습니다. 타액 DNA 추출 과정에는 5개의 음성 대조군이 포함되었습니다. 16S rRNA 유전자의 v3-v4 영역을 PCR 증폭한 후(PCR 프로토콜은 18 참조), 증폭된 단편은 AMPure XP 비드로 정제되었으며, Qubit(ThermoFisher Scientific)의 dsDNA 고감도 분석법을 사용하여 DNA 농도를 정량화하였습니다. 모든 샘플과 음성 대조군은 각 PCR 증폭 및 정제 단계 후 겔 전기영동을 통해 겔 밴드 존재 여부와 생성물 크기를 평가했으며, 선별된 샘플은 고품질 DNA 및 정확한 밴드 크기를 확인하기 위해 바이오애널라이저(Agilent Technologies, Santa Clara, CA, USA)로도 분석했다. 음성 대조군 4개는 증폭이 전혀 관찰되지 않았으며, 나머지 1개는 일부 오염 증거가 확인되었습니다. 최종 증폭산물 라이브러리 역시 바이오애널라이저 분석을 통해 검증되었습니다. 개별 이중 바코드가 부여된 총 407개의 분변 샘플과 216개의 타액 샘플에서 충분한 양의 증폭산물이 생성되었습니다. 분변 샘플은 무작위로 3개 풀로, 타액 샘플은 120~150개 샘플로 구성된 2개 풀로 각각 동등한 몰농도로 풀링되었다. 풀링된 샘플은 스웨덴 웁살라 대학교 소속 생명과학연구소(Science for Life Laboratory)에서 일루미나 MiSeq 기술(v3 화학, 2*300 bp)을 사용하여 시퀀싱되었다. 총 9개의 음성 대조군 샘플이 시퀀싱에 포함되었으며, 이 중 8개는 매우 낮은 리드 수를 보였고, 이전에 오염이 의심되었던 샘플은 상당한 수의 리드를 나타냈다. 결과 시퀀스 리드는 표준 권장 절차20에 따라 Mothur 소프트웨어19를 사용하여 처리되었으나, 운영 분류군(OTU)의 클러스터링은 Mothur에 구현된 VSEARCH(풍부도 기반 탐욕적 클러스터링)21을 사용하여 수행되었다. 주요 좌표 분석(PCoA)을 통해 시퀀싱 풀과 관련된 배치 효과는 검출되지 않았다. 리드 수 분포를 육안으로 확인한 후, 12,588개 미만의 리드를 가진 샘플(타액 샘플 1개 및 분변 샘플 9개)을 제거하고, 남은 샘플은 Mothur를 사용하여 이 시퀀싱 깊이로 희석 처리했다. 97% 서열 동일성 기준을 사용하여 OTU를 구분했다. OTU의 분류학적 분류는 SILVA 데이터베이스(버전 132, 2017년 12월 13일 공개)를 사용하여 수행되었습니다22,23,24. 3,400만 개 이상의 페어엔드 리드에서 733,773개의 OTU가 관찰되었으며, 이 중 709,652개는 10개 미만의 리드를 가졌습니다. Franzén 등25의 권고에 따라 허위 OTU 수를 줄이기 위해 이들 OTU는 제외되었습니다. 잔여 24,115개 OTU가 전체 리드량의 85%를 차지했습니다. 제거된 OTU는 주로 Lachnospiraceae과(25%), 분류되지 않은 Clostridiales목(15%), 분류되지 않은 박테리아(14%)로 분류되었습니다. SILVA 기반 분류학적 분류는 2022년 1월 19일 접속한 NCBI 16S 데이터베이스에 대한 BLAST를 통해 상위 20개 OTU의 속 수준에서 대체로 확인되었습니다.
우리는 아이의 대변 샘플을 수집하지 못했기 때문에 10개의 모자 쌍을 제외했으며, 따라서 최종 연구 대상에는 83명의 아이로부터 245개의 대변 샘플과 131개의 타액 샘플, 그리고 80명의 어머니로부터 140개의 대변 샘플과 76개의 타액 샘플이 포함되었습니다(그림 1). 총 58명의 아동은 최소 3회 이상의 채취 시점에서 분변 샘플을 확보했으며, 38명은 4회 모두의 샘플을 확보했습니다.
Statistical analysis
The gut microbiota was assessed with faecal samples and the oral microbiota with saliva samples.
Outcomes
We considered five different aspects of gut microbiota composition as outcomes: (1) alpha diversity (taxonomic diversity of microbiota within samples) assessed by the inverse Simpson index26, (2) beta diversity (overall community differences between samples) assessed by Bray–Curtis distance, (3) relative abundance of each bacterial phylum, (4) relative abundance of each bacterial family (only analysed if there was a significant association at the phylum level) and (5) relative abundance of the 20 most common OTUs among the children (Supplementary Table S1).
Alpha diversity and beta diversity were calculated at the OTU level with the R package vegan27. All analyses were performed in R version 4.0.028. Beta diversity was illustrated by PCoA plots and these plots were for descriptive purposes only. The relative abundance of each bacterial phylum and family was calculated for those phyla and families present in at least 75% of the faecal samples. The most common OTUs were selected by rank-transforming OTU abundance within each sample (with absent OTUs given the lowest possible rank), and choosing the 20 OTUs with the highest average rank across all samples. These 20 OTUs represented 51% of the reads in the faecal samples from children.
Distance-based similarity analysis
To understand the microbial communities shared between mothers and their children, as well as between unrelated individuals (non-self comparisons), and between faecal and saliva samples, we calculated Bray–Curtis and Jaccard distances between the samples. For non-self comparisons, we first calculated the distance for all possible relevant comparisons, and thereafter calculated the mean distance per individual. Lastly, we calculated the mean value with 95% confidence interval (CI) and performed t-tests for all possible comparisons. Results with a two-sided p < 0.05 were considered to be statistically significant and no adjustments for multiple comparisons were performed.
Exposures
We investigated the following potential determinants of child gut microbiota composition as exposures: age, maternal gut microbiota composition at gestational week 26–28, mode of delivery (vaginally and by caesarean section), parity, gestational age, birth weight, antibiotic treatment (both prenatal and postnatal), and the presence of furry pet in the household.
Potential confounders of each association were selected for adjustment using the d-separation criteria applied on Directed Acyclic Graphs, DAGs29,30, taking into account prior knowledge regarding their effect on the exposure and gut microbiota (Supplementary Fig. S1). We identified the potential confounders for each model separately.
Factors associating with the oral microbiota in this cohort have been previously analysed in18 and will thus not be presented here.
Gut microbiota—models
The outcomes alpha diversity and relative abundances were analysed using ordered logistic regression models (cumulative logit)31. To take the repeated measurements design into account, clustered robust standard errors with participant identity as cluster were used. The outcome beta diversity was analysed using the permutational multivariate ANOVA (PERMANOVA) method32, as implemented in the R package vegan. To calculate the type II sum of squares, the wrapper function adonis_II from the R package RVaideMemoire33 was used. The PERMANOVA analyses were performed for each sampling time-point separately. The homogeneity of within-group variation was checked using PERMDISP.
The gut microbiota development over time in children was analysed using statistical models with age of the child as the only independent variable. Age was modelled using restricted cubic splines with 4 knots at the 5th, 35th, 65th, and 95th percentiles to allow for nonlinear associations.
To investigate the effect of the exposures on gut microbiota development in children, we fitted models including the exposure, age, the interaction between exposure and age, and potential confounders (Table 1).
Table 1 Outcomes, exposure, and confounders included in the statistical models.
Continuous exposures were included as restricted cubic splines, with 3 knots at the 10th, 50th, and 90th percentiles. The interaction was based only on the linear part of the spline for age of the child, gestational age, birth weight, and maternal gut microbiota composition. When the p for the interaction was ≥ 0.05 (no multiple testing correction) the model was refitted without the interaction. Ordinal regression model-based predictions of mean relative abundance of phyla, families and OTUs were calculated and plotted for models including a continuous exposure and/or an interaction, otherwise only the odds ratio (OR) was reported.
The number of children exposed to antibiotics during the first year of life was low (n = 7) in our study. We therefore restricted our analysis on any association between antibiotics and the gut microbiota composition sampled at 24 months.
To evaluate the maternal gut microbiota composition as an exposure, alpha diversity, relative abundance of phyla, families and relative abundance of the 20 most common OTUs in gut microbiota in children were compared in turn as outcomes (e.g. maternal alpha diversity was explored in association with child alpha diversity).
The results from the gut microbiota models (Table 1) including four different aspects of gut microbiota composition as outcomes [(1) alpha diversity, (2) beta diversity (3) phylum, and (4) OTU], were then evaluated jointly for statistical significance using a false discovery rate (FDR) of 10%, where FDR was calculated using the Benjamini–Hochberg method34. The FDR cut-off corresponded to a nominal p of 0.0213. Findings driven by a single influential observation were not considered or discussed further but are reported in Supplementary Table S2. Family level analyses were only performed if there was a significant association at the phylum level.
Results
Of the 83 children (50 boys and 33 girls), 15 children were delivered by caesarean section (10 emergency, 4 elective, 1 missing information on type) (Table 2). The median birth weight was 3,530 g, and the median gestational age was 40 weeks. Overall, 18 children were exposed to prenatal antibiotic treatment and among them 8 children were exposed ≤ 90 days before birth. At the 12 months sampling occasion, 7 children had been prescribed antibiotics, and 15 children at the 24 months sampling occasion. In the latter group 6 children were exposed within 90 days before the sampling. Children delivered by caesarean section were not overrepresented in the group of children treated with antibiotics, only 1 child delivered by caesarean section had antibiotic treatment prescribed before the 12 months sampling occasion. Overall, we detected 17,757 and 5,326 OTUs (OTUs with at least 10 reads) in faecal and saliva samples of the children, respectively.
Table 2 Descriptive characteristics of the 83 mother–child pairs included in the study.
Gut microbiota became more homogeneous with age of the child
We observed an increase in alpha diversity of the gut microbiota with increasing age of the children (p < 0.001, Supplementary Fig. S2, Supplementary Table S2).
Figure 2 illustrates the children and their mother’s overall composition of the gut microbiota at different time points. The Bray–Curtis distance-based similarity analysis indicated that the largest changes in microbiota composition occurred during the first year of life (Fig. 3a, Supplementary Table S3). For example, the self-comparison between neonatal and 12 months showed a larger difference in composition than the comparison between 12 and 24 months (p < 0.001, Fig. 3a, Supplementary Table S3 and S5), despite the same 12-month timespan between samplings. However, all of the self-comparisons across time in children were found to be less similar than self-comparison in mothers (p < 0.001, Fig. 3a, Supplementary Table S3 and S5), indicating a more stable microbiota in mothers, despite the pregnancy. Furthermore, the non-self comparison presented in Fig. 3a indicated that the Bray–Curtis distance between faecal samples from different children also became smaller with age, and the similarity at 24 months was for example higher than at 12 months (p < 0.001, Fig. 3a, Supplementary Table S3 and S5). The similarity to the mothers’ samples also increased with age (Fig. 3b, Supplementary Table S4) and the similarity at 24 months was for example higher than the similarity at 12 months (p < 0.001, Supplementary Table S6) In summary, these results indicate that the gut microbiota in children matured with age and became less variable between different children as compared to the earlier stages of their lives.
Figure 2
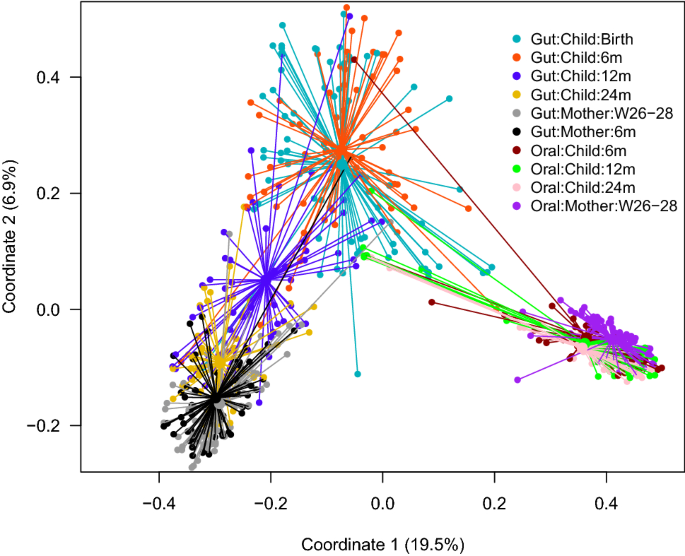
Principal coordinate analysis plot (Bray–Curtis distances) illustrating the children and their mother’s overall composition of the gut and oral microbiota at different time points, from birth (neonatal sample) of the child to 24 months and from mothers at 26–28 weeks of gestation and 6 months after delivery. W gestational week and m month.
Figure 3
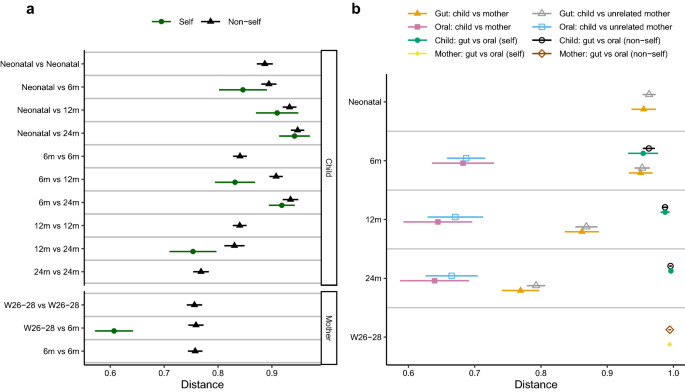
Comparison of microbiota composition based on Bray–Curtis distances. The shorter the distance between any pair of comparisons indicate greater similarity between microbial communities collected at those times. W gestational week and m month. Symbols represent means and error bars 95% confidence intervals. (a) Comparisons of gut microbiota at different sampling times, where distances between different sampling points within the same child/mother (self), and distances between unrelated children (non-self) and between unrelated mothers (non-self) are denoted. (b) Comparisons between gut and oral microbiota, and between children and their mothers and unrelated mothers. The distance between child and mother is based on the maternal sample at gestational week 26–28.
The results based on the Jaccard distance are presented in Supplementary Fig. S3, Table S3, S4, S7 and S8. The mean Jaccard distance was in general lager than the Bray–Curtis distance, but the same pattern for comparisons between groups was observed independent of the metric.
At the phylum level we noted an association of age with an increase in relative abundance of Firmicutes (p < 0.001), a decrease of Proteobacteria (p < 0.001), an inverse U-shaped association with highest prevalence at 6 months for Actinobacteria (p = 0.005) and a non-linear association between Bacteroidetes and age (p = 0.015) (Fig. 4a,b). At 24 months, the phylum distribution in children was similar to the mothers. The analysis at family level indicated that the observed association with age was mainly driven by Bifidobacteriaceae for the phylum Actinobacteria and by Enterobacteriaceae for the phylum Proteobacteria (Supplementary Fig. S4). For Firmicutes no single major contributor at the family level was observed (Supplementary Fig. S4).
Figure 4
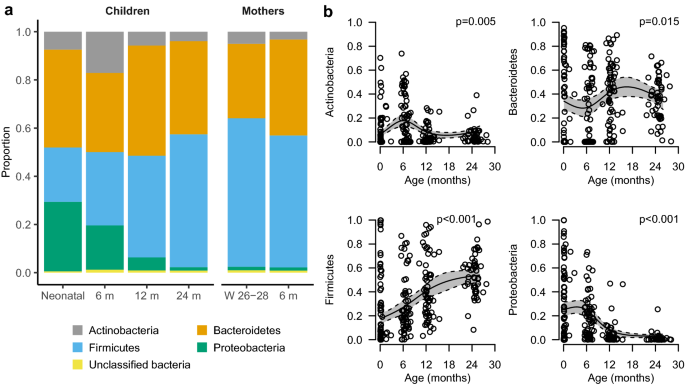
Bacterial phyla present in at least 75% of the faecal samples. a Stacked bar charts of the relative distribution of phyla detected in the gut at different time points. (b) Model-based predictions of relative abundance of phyla in the gut of children plotted against age. Lines indicate mean values (solid) and 95% confidence intervals (dashed). The y-axis shows the relative abundance in proportion. The predictions are shown from the 10th to the 90th percentile of age. W gestational week and m month.
Age was associated with 18 of the 20 most common OTUs (Supplementary Fig. S5 and S6, Supplementary Table S2). Faecalibacterium (OTU8), Blautia (OTU22), Agathobacter (OTU13), and Anaerostipes (OTU31) were initially rare, but appeared at 12 and 24 months; these OTUs were present also in mothers (Supplementary Fig. S5).
Similarity between gut and oral microbiota
All of the 20 most abundant OTUs detected in faecal samples from the children were also present in at least one of the saliva samples from children. Streptococcus (OTU3, OTU17), Veillonella (OTU5) and Haemophilus (OTU6) were present in at least 93% of the saliva samples, while the other 16 OTUs were present in 2% to 34% of the samples. The median number of detected OTUs in samples from children (age 6 to 24 months) was 590 and 488 in faecal and saliva samples, respectively.
We compared the gut microbiota composition and oral microbiota composition at 6, 12 and 24 months and the distance-based similarity analysis showed a small increase in Bray–Curtis distance with age (24 months vs 6 months p < 0.001 and 24 months vs 12 months p = 0.013, Fig. 3b, Supplementary Table S4 and S6). However, already at 6 months of age, the mean Bray–Curtis distance was as large as 0.95 (95% CI: 0.93–0.98) increasing to 1.00 (0.99–1.00) at 24 months (Supplementary Table S4). The same pattern was observed for the Jaccard distance (Supplementary Table S4 and S8). The median number of shared OTUs between faecal and saliva samples was 13.5, 9.0, and 8.0 at 6, 12, and 24 months in children, respectively, and 6.0 shared OTUs in mothers. Veillonella (OTU5) was the most frequently shared OTU between faecal and saliva samples at 6 months of age. It was detected in both the faecal and saliva sample in 92% (46/50), 82% (36/44) and 43% (15/35) of the children at 6, 12, and 24 months (Supplementary Table S9), respectively, and in 39% (26/67) of the mothers (Supplementary Table S10). The median abundance of shared OTUs in faecal samples decreased from 9.5% at 6 months to 2.6% at 24 months and was then similar in abundance in mothers (median: 2.2%). This indicates that the gut and oral microbiota share some characteristics in infants, but that with time these communities become more distinct. We observed no differences between self-comparisons and non-self comparisons, of oral and gut microbiota, at any of the sampling occasions (Children: 6 months p = 0.97, 12 months p = 0.95, 24 months p = 0.62; Mothers p = 0.91, Fig. 3b, Supplementary Tables S4 and S6).
Mode of birth differentiated gut microbiota in children
The composition of gut microbiota of a child was almost as similar to the microbiota of an unrelated mother as to their own mother (Fig. 3b). We observed an inverse association between the mother’s and child’s relative abundance of the phylum Bacteroidetes (Supplementary Fig. S7, Supplementary Table S2) and a positive association for an OTU belonging to the genus Veillonella (OTU5) (Supplementary Fig. S8, Supplementary Table S2). The overall gut microbiota composition differed between children delivered vaginally and by caesarean section at birth (PERMANOVA p = 0.007, PERMDISP = 0.46, Supplementary Fig. S9), but not at later sampling occasions. Compared with vaginally delivered children, children delivered by caesarean section had a lower relative abundance of Bacteroidetes and the strength of the associations decreased with age (Fig. 5). The same type of age-dependent association was also observed for two OTUs belonging to the Bacteroides genus (OTU1 and OTU66) (Fig. 5). Children delivered by caesarean section also had a lower abundance of an additional OTU belonging to the Bacteroides genus (OTU29 (OR = 0.32, p = 0.009)), but no significant interaction between mode of delivery and age was observed. The family level analysis suggests Bacteroidaceae to be a major contributor of the difference observed for Bacteroidetes (Supplementary Fig. S10). Delivery by caesarean section was positively associated with the relative abundance of Firmicutes (Fig. 5), Veillonella (OTU5, OR = 2.19, p = 0.009), Faecalibacterium (OTU8, OR = 2.15, p = 0.003) and Streptococcus (OTU3 (Fig. 5) and OTU17 (OR = 2.28, p = 0.016)). However, as Veillonella (OTU5) and Faecalibacterium (OTU8) represented a minor fraction of the abundance in neonatal samples, it is likely that other OTUs within the Firmicutes phylum are the main contributors to the differences observed in the Firmicutes. Either could any family be identified a main contributor even though we observed an association with Streptococcaceae (Supplementary Fig. S10). Furthermore, delivery by caesarean section was negatively associated with Escherichia-Shigella (OTU2) (Fig. 5).
Figure 5
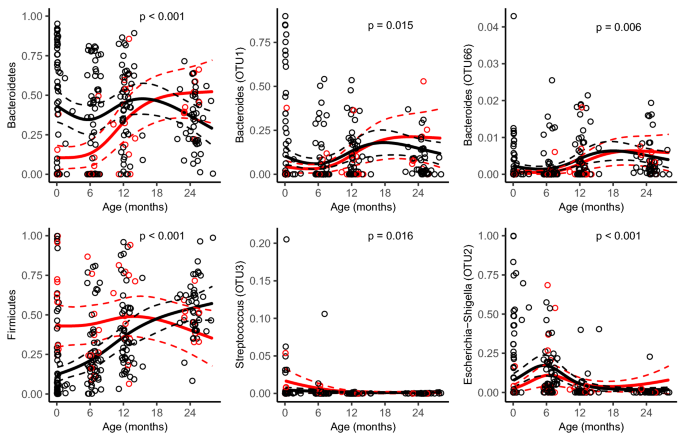
Model-based predictions of relative abundance in bacterial phyla and specific operational taxonomic units (OTUs) in the gut microbiota by mode of delivery plotted against age. Lines indicate mean values (solid) and 95% confidence intervals (dashed), adjusted for potential confounders (see Table 1). P reflect for the interaction between mode of delivery and child age. All continuous adjustment variables are fixed at their median value and all categorical adjustment variables are fixed at their most common category.
Diminished association of some factors with gut microbiota with child age
First-born children had a higher relative abundance of Haemophilus (OTU6, OR = 2.11, p = 0.005) than later-born children. Gestational age was negatively associated with Firmicutes (age pinteraction = 0.016) and Streptococcus (OTU3, age pinteraction = 0.021) and the strength of the associations decreased with age (Supplementary Fig. S11). Birth weight was associated with the overall gut microbiota composition of the child at the age 1 year (PERMANOVA p = 0.020) and two OTUs belonging to the genus Bacteroides (OTU1: p = 0.007, OTU29: p = 0.013, Supplementary Fig. S12).
Prenatal exposure to antibiotics was associated with a lower abundance of Blautia (OTU22, OR = 0.47, p = 0.016). We did not find any association between antibiotic treatment during the first 2 years of life and gut microbiota of the child at the age 2 years.
The presence of a furry pet in the household was associated with a lower relative abundance of the phylum Actinobacteria and an OTU belonging to the genus Bifidobacterium (OTU21) in the child (Supplementary Fig. S13). However, these associations diminished with age as there was an interaction between pet exposure and age of the child (Actinobacteria: pinteraction = 0.012, OTU21: pinteraction < 0.001). Furthermore, the family level analysis suggests Bifidobacteriaceae as the major contributor of the difference observed for Actinobacteria (Supplementary Fig. S14) between pet exposed and non-exposed children.
Discussion
In this Swedish cohort of 83 children, we observed an age-dependent development of the gut microbiota, with an adult-like microbial composition attained around the age of 2 years. Our distance-based similarity analyses indicated that the similarity between the gut and oral microbiota decreased after 6 months of age, although already at this age, the microbiota were highly dissimilar, and the two communities were clearly different at 2 years. Mode of delivery differentiated the overall gut microbiota composition. Caesarean section was associated with a lower relative abundance of the phylum Bacteriodetes, with the family Bacteroidaceae being the major contributor and a higher relative abundance of the phylum Firmicutes. As expected, these associations were strongest during the first 6 months of life and diminished thereafter.
A major part of the maturation of the microbiota composition seems to occur in children at an early age. However, a recent study showed that several bacterial taxa are acquired late in childhood and that differences in microbiome structure and diversity still may exist even at 5 years of age7. In concordance with previous studies, we observed a gradual shift in the gut microbiota composition from birth to age 2, with an increased alpha diversity, increased relative abundance of the phylum Firmicutes and decreased relative abundance of the phylum Proteobacteria and Actinobacteria2,9,12. We found the most marked changes to occur during the first year of life. Even though the alpha diversity increased with age, the beta diversity was reduced and more similar to a characteristically adult community composition. Our results also suggest that the gut microbiota composition is rather stable in mothers compared to children, even when comparing samples during and after pregnancy. This finding aligns with a longitudinal study (n = 128 mother–infant pairs) that compared faecal samples from the same individual across time and found samples from mothers 2 months before and after delivery to be more similar than samples from children (2 months vs 6 months)35.
A study, by Ferretti et al.14, of 25 mother–child pairs reported that the number of shared oral—gut species decreased from an average of 9.8 at day 1 to 7.2 species already at 3 days of life, which can be compared with 6 shared oral—gut species in mothers. In addition, faecal samples were reported to have a lower abundance of oral-gut microbiota shared species in mothers than in infants. Our distance-based similarity analyses extends this finding and indicates that the similarity between oral and gut microbiota continue to decrease also after 6 months of age. Ferretti et al.suggested that oral bacterial transition to gut is more relevant in infants than in adults and speculated that this could be a consequence of reduced acid production in children at birth14. However, this could also be due to niche adaptations of the bacterial communities both in the oral cavity and the gut, as these communities develop early in life6,36. One example is Bifidobacterium dentum which have shown adaptation to the intestine via its acid tolerance, mucus adhering properties and metabolic plasticity towards dietary nutrient sources commonly found in the intestine37. In the present study, we lack saliva samples from children before 6 months of age and future work focusing on this period would likely advance the understanding of mechanisms of early microbiota acquisition and development. This could also enable identification of specific environmental sources of microbial inoculation.
Gut microbiota composition appears to be shaped by different factors and is modulated by age over time. In line with previous work, our study identified an association between caesarean section delivery and the overall gut microbiota composition7,9,10. However, no association with alpha diversity was observed. The negative association of delivery by caesarean section with abundance of Bacteroides has been previously observed2,9,12, but while this persisted throughout the first 6 months of life in the present study, the effects then diminished with age. B. dentium species are important regulators of the intestinal immunity38,39 and low abundances have been proposed to partially explain the associations between birth by caesarean section and adverse health effects such as asthma, allergies and atopy40. Our results also add to earlier findings showing that delivery by caesarean section is positively associated with bacteria affiliated with the phylum Firmicutes9, including the genera Veillonella9,11 and Streptococcus11. However, two large longitudinal studies2,41, did not show an association with either of these genera. Veillonella is commonly present in the gastrointestinal tract, mouth, vagina, and breast milk acting as an important link to the immune system42. While the overall patterns suggest that caesarean section has some effect on the development of the gut microbiota composition, presence of different species may be differentially affected.
Mother-to-child transmission of bacteria has been thought to be an important source of bacterial inoculum in children, and earlier studies have reported such transmission of bacterial strains11,43. We did not however observe strong evidence for higher bacterial community similarity within related mother–child pairs compared to unrelated pairs, neither in the gut nor in the oral microbial communities. Other studies performing community similarity analysis based on OTU level have reported comparable results9,44,45,46,47. However, to accurately quantify microbial transmission, strain level profiling is necessary, and we might have underestimated the difference between related and unrelated pairs as individuals frequently share common OTUs. Moreover, it should be noted that the confidence intervals were wide and the power low for detecting moderate similarities.
It is plausible that the impact of mode of delivery on infant gut microbiota development may be at least in part due to the lack of contact with maternal vaginal and intestinal microbiota for children delivered by caesarean section. In addition, differential occurrence of foetal exposure to maternal peripartal antibiotics prophylaxis, with potential effects on postpartum infant microbial colonization, could also mediate the association. Peripartal antibiotics prophylaxis was recommended for all mothers undergoing emergency caesarean section (n = 10 mothers in our study), whilst only on medical indication in elective caesarean section (n = 4) and vaginal deliveries (n = 68). We cannot ascertain use or timing of peripartal antibiotic prophylaxis in our maternal cohort, but a randomized control trial including 20 children in each arm with up to 3 years of follow-up failed to show any effect of different timing of antibiotic treatment, although both groups differed compared with a third group of vaginally delivered children. These findings indicate that there are other more important factors that explain the association of caesarean section with the child’s gut microbiota48.
Discrepancies between studies might be explained by different sampling schemes, sample sizes, hospital routines and methodologically also to the specific diagnostic regions of 16S rRNA gene that are being used. In addition, we performed the analysis of relative abundance at OTU level, while many of the other studies only compared responses at the broader genus level2,11,12,41.
We noted that prenatal exposure to antibiotics was associated with a lower relative abundance of an OTU belonging to the genus Blautia, which has been associated with potential probiotic functions49. These findings are supported by a cohort study on intrapartum antibiotics, including faecal samples collected at 6 weeks and 1 year of age from 266 children50. One explanation for the observed associations, in the present study, with prenatal antibiotics exposure could be changes in the gut and/or vaginal microbiota in the mothers that affect the child at birth or later46. Furthermore, the microbial colonization process could be started already in utero5, a process that may be affected by antibiotics administered during pregnancy.
In line with a large longitudinal study2, we observed that having a furry pet in the household was associated with a lower relative abundance of an OTU within the genus Bifidobacterium. However, our result should be interpreted with caution as information on furry pet was missing for 25 children which could bias the result.
The primary strengths of our study are the combination of oral and gut microbiota, its longitudinal design, following children from birth until age 2, and the use of data from high-quality registers with objectively and prospectively recorded information about maternal and birth characteristics. Some limitations apply. Our platform for 16S rRNA amplicon sequencing did not allow identification of the gut and oral bacteria to the species level and we were not able to characterize the functional properties of microbial changes associated with different exposures. Moreover, we did not investigate the effect of diet on gut microbiota or its possible role as a mediator as the dietary questionnaire information had several limitations. These limitations were (1) 32 mothers did not answer the question at age 6 months regarding diet (2) the pre-determined wide age-categories in the questionnaire (3) only four children in our study were exclusively breastfed for < 3 months, (4) no information of whether breast milk was substituted with solid foods or formula was available and (5) children who were exclusively breast-fed for ≥ 5 months may have been introduced to foods at the 6 months sampling occasion. The inability to include this information in the analysis prevented assessment as to whether diet could mediate the association of mode of delivery with bacterial composition. Although we adjusted the analyses for maternal and birth characteristics, residual confounding may still be present as we did not account for lifestyle factors and our sample size did not allow us to investigate any interactions between exposures. Furthermore, our study only included five children born pre-term (< 37 weeks) and one child with low birth weight (< 2500 g), limiting our power to investigate associations between gestational age and birth weight with gut microbiota variation in children. Additionally, previously decided sampling times did not take the time of antibiotic exposure into account and the low number of children exposed to antibiotics after birth limited our power to detect an association between antibiotics and gut microbiota variation in children. Furthermore, we could not compare different caesarean section types as only four children were delivered by elective caesarean section in our study. For the saliva samples, the overnight refrigerator storage of the samples may have caused some changes in composition. An earlier pilot study showed that room temperature storage of salivary samples for 6 h did not cause any major change in composition, while they noted some changes in specific bacterial taxa levels51. Such changes may have affected our ability to detect similarities between samples in terms of OTU levels. Finally, our study population represents a well-educated population from a predominantly urban area. This may restrict the generalizability of our findings as earlier studies have indicated that differences in geographical6 and socio-economic status52 might influence gut microbiota.
The results from our study, including both repeated saliva and faecal samples from children up to age 2 and from their mothers suggest that (1) the gut microbiota in infants are low in diversity with differences across individuals with regards to composition. (2) During the first 2 years of life, there is notable convergence in community composition towards a more diverse adult-like composition. (3) Oral and gut microbiota communities are distinct in composition in 2-year-old children, but share some similarities at age 6 and 12 months. (4) In infants, perinatal factors, such as mode of delivery, account for some of the observed differences between individuals but the association is attenuated with age.
토론
본 스웨덴 코호트 연구(83명의 아동 대상)에서 우리는 장내 미생물 군집이 연령에 따라 발달하며, 약 2세 경에 성인형 미생물 구성을 획득함을 관찰했다. 거리 기반 유사도 분석 결과, 장내 미생물 군집과 구강 미생물 군집 간의 유사도는 생후 6개월 이후 감소했으나, 이미 이 시점에서 두 군집은 매우 상이했으며, 2세 시점에서는 두 군집이 뚜렷이 달랐다. 분만 방식은 전체 장내 미생물군 구성에 차이를 보였다.
제왕절개는
박테리오데테스문(Bacteriodetes)의 상대적 풍부도가 낮게 나타났으며,
이는 박테리오데테스과(Bacteroidaceae)가 주요 기여자였고,
펌리쿠테스문(Firmicutes)의 상대적 풍부도는 높게 나타났다.
예상대로 이러한 연관성은
생후 첫 6개월 동안 가장 강했으며 이후 감소했다.
미생물군 구성의 성숙 과정 상당 부분은
어린 나이에 발생하는 것으로 보인다.
그러나 최근 연구에 따르면 여러 세균 분류군이 아동기 후반에 획득되며, 5세에도 미생물군집 구조와 다양성의 차이가 여전히 존재할 수 있다7. 기존 연구와 일치하게, 우리는 출생부터 2세까지 장내 미생물군집 구성이 점진적으로 변화하는 것을 관찰했으며, 알파 다양성 증가, Firmicutes 문 상대적 풍부도 증가, Proteobacteria 및 Actinobacteria 문 상대적 풍부도 감소를 확인했다2,9,12. 가장 두드러진 변화는 생후 첫해에 발생하는 것으로 확인되었습니다. 알파 다양성은 연령 증가에 따라 증가했지만, 베타 다양성은 감소하여 성인 특유의 군집 구성과 더 유사해졌습니다. 또한 본 연구 결과는 임신 중 및 이후 시기의 샘플을 비교하더라도, 어머니의 장내 미생물 군집 구성은 아이들에 비해 상대적으로 안정적임을 시사합니다. 이 결과는 동일한 개인의 배변 시기를 비교한 종단 연구(n=128 모자 쌍)와 일치한다. 해당 연구에서는 출산 2개월 전후의 모체 샘플이 아동 샘플(2개월 대 6개월)보다 더 유사한 것으로 나타났다35.
Ferretti 등14의 25쌍 모자 연구에 따르면, 공유 구강-장내 미생물 종의 수는 생후 1일 평균 9.8종에서 생후 3일 만에 이미 7.2종으로 감소했으며, 이는 모체의 6종과 비교할 수 있습니다. 또한 모체의 대변 샘플은 영아에 비해 구강-장내 미생물 공유 종의 풍부도가 더 낮은 것으로 보고되었습니다. 우리의 거리 기반 유사도 분석은 이 결과를 확장하여 구강과 장내 미생물군집 간의 유사도가 생후 6개월 이후에도 계속 감소함을 시사한다. Ferretti 등은 구강 세균의 장 이식이 성인보다 영아에서 더 중요하며, 이는 출생 시 어린이의 산 생성 감소 결과일 수 있다고 제안했습니다14. 그러나 이는 구강 및 장 내 세균 군집이 생애 초기에 발달함에 따라 발생하는 서식지 적응 때문일 수도 있습니다6,36. 한 예로 Bifidobacterium dentum은 산 내성, 점액 부착 특성, 장내에서 흔히 발견되는 식이 영양원에 대한 대사적 가소성을 통해 장에 적응하는 것으로 나타났습니다37. 본 연구에서는 생후 6개월 미만 아동의 타액 샘플이 부족하며, 이 시기에 초점을 맞춘 향후 연구는 초기 미생물군 획득 및 발달 메커니즘에 대한 이해를 진전시킬 가능성이 있습니다. 이는 또한 미생물 접종의 특정 환경적 원천을 식별하는 데 기여할 수 있습니다.
장내 미생물 군집 구성은 다양한 요인에 의해 형성되며, 시간에 따라 연령에 의해 조절되는 것으로 보인다. 기존 연구와 일치하게, 본 연구에서도 제왕절개 분만과 전체 장내 미생물군 구성 간 연관성을 확인하였다7,9,10. 그러나 알파 다양성과의 연관성은 관찰되지 않았다. 제왕절개 분만이 Bacteroides 군집 풍부도와 음의 상관관계를 보인다는 점은 이전 연구에서도 보고된 바 있다2,9,12. 다만 본 연구에서는 생후 6개월 동안 이 상관관계가 지속되었으나, 이후 연령 증가에 따라 그 효과가 감소하였다. B. dentium 종은 장 면역의 중요한 조절자이며38,39, 그 낮은 풍부도는 제왕절개 분만과 천식, 알레르기, 아토피 같은 건강 악화 효과 간의 연관성을 부분적으로 설명하는 요인으로 제시된 바 있다40. 본 연구 결과는 또한 제왕절개 분만이 Veillonella9,11 및 Streptococcus11 속을 포함한 Firmicutes 문에 속하는 박테리아와 양의 상관관계가 있다는 기존 연구 결과를 뒷받침한다. 그러나 두 개의 대규모 종단 연구2,41에서는 이 두 속과 연관성이 관찰되지 않았다. Veillonella는 위장관, 구강, 질, 모유에 흔히 존재하며 면역 체계와의 중요한 연결고리 역할을 한다42. 전반적인 패턴은 제왕절개가 장내 미생물군 구성 발달에 어느 정도 영향을 미친다는 것을 시사하지만, 종별 존재 여부는 차등적으로 영향을 받을 수 있다.
어머니로부터 아이로의 세균 전파는 아동의 세균 접종원으로서 중요한 원천으로 여겨져 왔으며, 이전 연구들에서 이러한 세균 균주의 전파가 보고된 바 있다11,43. 그러나 우리는 장내 미생물 군집이나 구강 미생물 군집 모두에서, 관련이 없는 모자 쌍에 비해 관련이 있는 모자 쌍 내에서 세균 군집 유사성이 더 높다는 강력한 증거를 관찰하지 못했다. OTU 수준을 기반으로 군집 유사성 분석을 수행한 다른 연구들9,44,45,46,47에서도 유사한 결과가 보고되었다. 그러나 미생물 전파를 정확히 정량화하려면 균주 수준 프로파일링이 필요하며, 개인 간 공통 OTU가 빈번히 존재하기 때문에 우리는 친족 대조군과 비친족 대조군 간의 차이를 과소평가했을 수 있다. 또한 신뢰 구간이 넓고 중간 정도의 유사성을 검출하는 검정력이 낮았다는 점에 유의해야 한다.
분만 방식이 영아 장내 미생물군 발달에 미치는 영향은 적어도 부분적으로 제왕절개로 분만된 영아가 모체의 질 및 장내 미생물군과 접촉하지 못하기 때문일 수 있다. 또한, 분만 전후 모체 항생제 예방적 투여에 대한 태아의 노출 차이는 분만 후 영아 미생물 군집화에 잠재적 영향을 미쳐 이 연관성을 매개할 수도 있다. 분만 전후 항생제 예방적 투여는 응급 제왕절개술을 시행한 모든 산모(본 연구에서 n=10명)에게 권장된 반면, 선택적 제왕절개술(n=4명) 및 질 분만(n=68명)에서는 의학적 적응증이 있을 때만 시행되었다. 본 연구 모체 코호트에서 분만 전후 항생제 예방적 투여의 사용 여부나 시점을 확인할 수 없었으나, 각 군별 20명의 아동을 대상으로 최대 3년간 추적 관찰한 무작위 대조 시험에서는 항생제 치료 시점의 차이에 따른 효과가 나타나지 않았습니다. 다만 두 군 모두 제3군인 자연분만 출생 아동군과는 차이가 있었습니다. 이러한 결과는 제왕절개술과 아동 장내 미생물군집 간의 연관성을 설명하는 다른 더 중요한 요인들이 존재함을 시사한다48.
연구 간 불일치는 서로 다른 샘플링 계획, 표본 크기, 병원 내 관행, 그리고 방법론적으로도 사용 중인 16S rRNA 유전자의 특정 진단 영역에 기인할 수 있다. 또한, 본 연구에서는 OTU 수준에서 상대적 풍부도 분석을 수행한 반면, 다른 많은 연구들은 더 넓은 속(genus) 수준에서만 반응을 비교하였다2,11,12,41.
본 연구에서는 태내 항생제 노출이 잠재적 프로바이오틱 기능과 연관된 것으로 알려진 Blautia 속의 한 OTU의 상대적 풍부도 감소와 관련이 있음을 확인하였다49. 이러한 결과는 266명의 아동을 대상으로 출생 시 항생제 투여를 포함한 코호트 연구에서 6주 및 1세 시점의 분변 샘플을 수집한 연구50에 의해 뒷받침된다. 본 연구에서 관찰된 태내 항생제 노출과의 연관성에 대한 한 가지 설명은 출생 시 또는 이후에 아이에게 영향을 미치는 모체의 장 및/또는 질 미생물군집 변화일 수 있다46. 또한 미생물 군집화 과정은 이미 자궁 내에서 시작될 수 있으며5, 이 과정은 임신 중 투여된 항생제의 영향을 받을 수 있다.
대규모 종단 연구2와 일치하게, 우리는 가정 내 털이 있는 반려동물의 존재가 Bifidobacterium 속 내 특정 OTU의 상대적 풍부도 감소와 연관되어 있음을 관찰했다. 그러나 털이 있는 반려동물 정보가 25명의 아동에게서 누락되어 결과를 왜곡할 수 있으므로 본 결과는 신중하게 해석되어야 한다.
본 연구의 주요 강점은 구강 및 장내 미생물군집의 병행 분석, 출생부터 2세까지 아동을 추적한 종단적 설계, 그리고 모체 및 출생 특성에 대한 객관적·전향적 기록이 포함된 고품질 등록 데이터의 활용이다. 일부 한계점도 존재한다. 16S rRNA 증폭자 서열 분석 플랫폼은 장내 및 구강 세균을 종 수준까지 식별할 수 없었으며, 다양한 노출과 연관된 미생물 변화의 기능적 특성을 규명하지 못했습니다. 또한 식이 설문지 정보에 몇 가지 한계가 있어 식이가 장내 미생물군집에 미치는 영향이나 매개체로서의 가능성을 조사하지 못했습니다. 이러한 한계는 (1) 32명의 어머니가 생후 6개월 시점 식이 관련 질문에 답변하지 않았고 (2) 설문지의 사전 설정된 광범위한 연령 범주 (3) 연구 대상 아동 중 생후 3개월 미만 완전 모유 수유를 한 아동은 단 4명에 불과했으며, (4) 모유를 고형식이나 분유로 대체했는지 여부에 대한 정보가 없었으며, (5) 5개월 이상 완전 모유 수유를 받은 아동들은 6개월 시료 채취 시점에 이미 고형식을 섭취했을 가능성이 있다는 점이다. 이러한 정보를 분석에 포함하지 못했기 때문에, 분만 방식과 세균 구성 간의 연관성을 식이 습관이 매개할 수 있는지 여부를 평가할 수 없었다. 모체 및 출생 특성에 대해 분석을 조정했음에도, 생활습관 요인을 고려하지 않았고 표본 크기가 노출 간 상호작용을 조사하기에 부족했기 때문에 잔여 혼란 요인이 여전히 존재할 수 있습니다. 또한 본 연구에는 조산아(37주 미만) 5명과 저체중아(2500g 미만) 1명만 포함되어 있어, 임신 주수와 출생 체중이 아동의 장내 미생물군 변이와 연관성을 조사하는 데 한계가 있었습니다. 또한 사전에 결정된 채취 시점은 항생제 노출 시기를 고려하지 않았으며, 출생 후 항생제에 노출된 아동 수가 적어 항생제와 아동 장내 미생물군 변동 간의 연관성 검출력이 제한되었습니다. 게다가 선택적 제왕절개로 분만된 아동이 4명에 불과해 제왕절개 유형 간 비교가 불가능했습니다. 타액 샘플의 경우, 냉장고에서 하룻밤 보관하는 과정에서 일부 구성이 변할 수 있습니다. 선행 예비 연구에서는 타액 샘플을 실온에서 6시간 보관해도 구성에 큰 변화가 없었으나, 특정 세균 분류군 수준에서는 일부 변화가 관찰되었다고 보고했다51. 이러한 변화는 OTU 수준에서 샘플 간 유사성을 검출하는 능력에 영향을 미쳤을 수 있다. 마지막으로, 본 연구 대상 집단은 주로 도시 지역 출신으로 교육 수준이 높은 인구층을 대표한다. 이는 지리적6 및 사회경제적 지위52 차이가 장내 미생물군집에 영향을 미칠 수 있다는 기존 연구 결과에 비추어 볼 때, 본 연구 결과의 일반화 가능성을 제한할 수 있습니다.
본 연구 결과(만 2세 이하 아동 및 그 어머니로부터 반복적으로 채취한 타액 및 분변 샘플 포함)는 다음과 같이 시사합니다: (1) 영아의 장내 미생물군집은 다양성이 낮으며 구성 측면에서 개인 간 차이가 존재합니다. (2) 생후 첫 2년 동안 군집 구성이 성인과 유사한 더 다양한 구성으로 현저히 수렴한다. (3) 2세 아동의 구강 및 장내 미생물 군집 구성은 서로 다르지만, 생후 6개월 및 12개월에는 일부 유사성을 공유한다. (4) 영아에서 분만 방식과 같은 주산기 요인은 개인 간 관찰된 차이의 일부를 설명하지만, 이 연관성은 연령이 증가함에 따라 약화된다.
Data availability
Restrictions apply to the availability of individual level health data, which were used under license and ethical approval for the current study and are not publicly available. Individual level data are however available from the authors upon reasonable request and with permission of the Swedish Ethical Review Authority. Bacterial sequencing data from saliva samples is available at the Sequence Read Archive (SRA) open repository under reference number PRJNA575550.
References