Nat Rev Neurosci
. Author manuscript; available in PMC: 2018 Jun 5.
Published in final edited form as: Nat Rev Neurosci. 2018 Jun;19(6):323–337. doi: 10.1038/s41583-018-0001-8
Intrinsic mechanisms of neuronal axon regeneration
Marcus Mahar 1,iD, Valeria Cavalli 1,*
- Author information
- Copyright and License information
PMCID: PMC5987780 NIHMSID: NIHMS966948 PMID: 29666508
The publisher's version of this article is available at Nat Rev Neurosci
Abstract
Permanent disabilities following CNS injuries result from the failure of injured axons to regenerate and rebuild functional connections with their original targets. By contrast, injury to peripheral nerves is followed by robust regeneration, which can lead to recovery of sensory and motor functions. This regenerative response requires the induction of widespread transcriptional and epigenetic changes in injured neurons. Considerable progress has been made in recent years in understanding how peripheral axon injury elicits these widespread changes through the coordinated actions of transcription factors, epigenetic modifiers and, to a lesser extent, microRNAs. Although many questions remain about the interplay between these mechanisms, these new findings provide important insights into the pivotal role of coordinated gene expression and chromatin remodelling in the neuronal response to injury.
이 논문은
2018년 Nature Reviews Neuroscience에 게재된 리뷰 기사로,
신경 축삭 재생의 내재적 메커니즘을 다룹니다.
저자는 Marcus Mahar와 Valeria Cavalli로,
중추신경계(CNS)와 말초신경계(PNS) 재생의 차이를 강조하며,
상처 후 전사 및 후성유전 변화가 재생을 주도하는 과정을 합성적으로 분석합니다.
주요 초점은
PNS의 강력한 재생 메커니즘을 통해
CNS 재생 장애를 극복할 통찰을 제공하는 것입니다.
초록 (Abstract)
CNS 상처는 축삭 재생 실패로 영구 장애를 초래하나,
PNS 상처는 강력한 재생으로 감각/운동 기능 회복을 이끕니다.
이는 상처된 뉴런에서 광범위한 전사 및 후성유전 변화에 의존하며,
전사인자, 후성유전 수정자, miRNA의 조화된 작용을 통해 일어납니다.
actions of transcription factors,
epigenetic modifiers and, to a lesser extent,
microRNAs
https://pmc.ncbi.nlm.nih.gov/articles/PMC4888057/

최근 연구는 이러한 메커니즘의 상호작용을 밝혔으나,
여전히 많은 질문이 남아 있습니다.
Inhibitory factors present in the CNS tissue environment and the poor intrinsic regenerative capacity of adult CNS neurons are obstacles to axon regeneration and functional recovery1–4. Aberrant synaptogenesis, in which injured CNS axons at the lesion site remain engaged in a synaptic function or maintenance state rather than switching to a regenerative state, has also emerged as a barrier to axonal regrowth4,5. The failure of damaged adult CNS axons to regrow results in permanent disabilities for individuals with spinal cord injury or stroke. As such, control of central axon regeneration is both a major unmet medical need and an unsolved problem in neurobiology. By contrast, functional recovery can often occur following injury in the peripheral nervous system (PNS) because peripheral neurons retain the ability to self-repair and reactivate intrinsic growth programmes after injury. However, severe insults to peripheral nerves can also lead to permanent neurological deficits, including failure of reinnervation and the development of chronic pain. In those cases, recovery is slow and limited even when nerve grafts provide a bridge to promote axon regeneration6,7. To solve these problems and promote functional recovery in the adult mammalian CNS and PNS, a major focus of research has been the identification of the molecular mechanisms that trigger a regenerative response and enable meaningful, long-distance axon regeneration.
Two primary approaches have been used to understand the cell-intrinsic mechanisms underlying axon regeneration: studying the mechanisms governing the developmental decline of axon growth competency in the CNS and studying the injury-induced activation of a pro-regenerative programme in the PNS and other regenerative systems5,8. During neuronal development, axon growth competence decreases as neurons form synapses to transmit information and integrate into functional neuronal circuits. The pro-regenerative cellular changes after axon injury are thought to recapitulate some of the developmental processes that occur before synapse formation, thus switching the neuron from an active, electrically transmitting state back to an electrically silent, growth-competent state4,9. Using peripheral sensory neurons and model organisms, such as Caenorhabditis elegans and zebrafish, many intrinsic signalling pathways that trigger the regenerative response have been identified. An overview of the various model organisms and injury paradigms used to study axon regeneration is given in BOX 1. More recently, studies have implicated the activity of various transcription factors, epigenetic modifiers of DNA methylation and chromatin structure, and microRNAs (miRNAs) that together activate the pro-regenerative transcriptional programme.
Box 1 | Models used to study intrinsic axon regeneration mechanisms.
Mammalian sensory neurons with cell bodies in the dorsal root ganglia share unique features of both CNS and peripheral nervous system neurons because they project both a peripheral axonal branch into peripheral nerves and a central axonal branch through the dorsal root into the spinal cord. Whereas injury to the peripheral axon elicits a regenerative response, injury to the central axon fails to do so51,123. The activation of a pro-regenerative programme following peripheral axon injury is illustrated by the conditioning injury paradigm, in which a sensory neuron exposed to a prior peripheral nerve lesion exhibits a dramatic improvement in peripheral axon regeneration following subsequent injury when compared with that of a naive neuron104,213–214. This paradigm has also been used to reveal that activation of a pro-regenerative programme by a prior peripheral nerve lesion can partially overcome the inhibitory environment of the CNS to improve regeneration of the central axonal branch after injury215.
Spinal cord contusion injuries or partial spinal cord transections have also been used to study ascending sensory axon regeneration in the CNS31,166. Additionally, the regeneration of descending fibres can be examined in these models, and studies have shown that different neuronal subtypes possess different regenerative capacities216.
Another widely used model of CNS injury is a crush injury to the optic nerve, the axons of which project from retinal ganglion cells (RGCs) in the retina and synapse in the superior colliculus. Injury to the optic nerve initiates both regenerative and apoptotic responses, with apoptosis as the ultimately dominant response66. Genetic manipulation of RGCs has been shown to be capable of enhancing both survival and regeneration93.
Axon injury responses have also been investigated in non-mammalian vertebrates and invertebrates. The ability of vertebrate zebrafish to fully recover following complete spinal cord transection has provided unique insights into the role of non-neuronal cells in the regenerative response17. The nematode Caenorhabditis elegans provides several unique advantages, including optical transparency, the possibility of genetic manipulation and the availability of methods to axotomize single axons with a laser14. A number of different injury paradigms have also been used in the larval and adult nervous system of the fruitfly Drosophila melanogaster, which also enables the power of live imaging to be combined with genetic manipulation16. Other animals used to study injury responses include Aplysia, which has been instrumental in demonstrating the existence of positive injury signals217, lamprey, which possess both regenerating and non-regenerating CNS neurons17, and the axolotl, which can also regenerate its spinal cord following complete transection218. More details for each experimental model can be found in REF8.
In this Review, we describe the most recent studies that have focused on understanding how injured neurons revert to a growth-competent state. We review what is currently known about the early injury signals elicited at the site of injury in regeneration-competent axons and then focus on the intracellular communication mechanisms required to change the genetic and epigenetic programme of injured neurons in order to allow axon regeneration to occur. Other aspects of axon regeneration, which include the roles of the cellular environment, axon–soma signalling and growth cone formation and dynamics have been extensively covered in recent reviews4,5,8,10–13 and will not be discussed here. This Review largely focuses on studies on mice, as recent reviews have covered axon regeneration in other model systems including C. elegans14,15, Drosophila melanogaster16 and fish17.
Injury signalling
In regeneration-competent neurons, injury signalling occurs in two distinct temporal phases: a rapid phase that is dictated by a retrograde calcium wave that propagates into the soma18–21 and a slow phase characterized by the retrograde transport of signalling molecules by motor proteins10,22.
Rapid injury signals
In regeneration-competent neurons, axonal membrane rupture combined with the opening of voltage-dependent sodium channels and the inversion of Na+/Ca2+ exchange pumps promote an influx of calcium ions at the injury site10. A calcium wave is propagated to the soma by the actions of L-type voltage-gated calcium channels (VGCCs) and the release of calcium from the endoplasmic reticulum via ryanodine receptors10,18,20,21,23–25. The intracellular calcium reaches millimolar concentrations in both the axon and soma after injury19 and plays several key roles in the initial stages of regeneration, including roles in membrane resealing26,27, growth cone assembly11, translocation and activation of specific epigenetic modifiers28, local protein synthesis29 and activation of additional signalling molecules and transcription factors10. However, proper regulation of this intracellular calcium is required, as excess concentrations can lead to cell death30 and the activity of specific VGCCs is inhibitory to regeneration24,31. Although the influence of calcium in regenerating neurons is widespread, this Review focuses on its effects on signalling pathways, transcription and epigenetics (FIG. 1).
Fig. 1. Signalling events following peripheral axon injury.
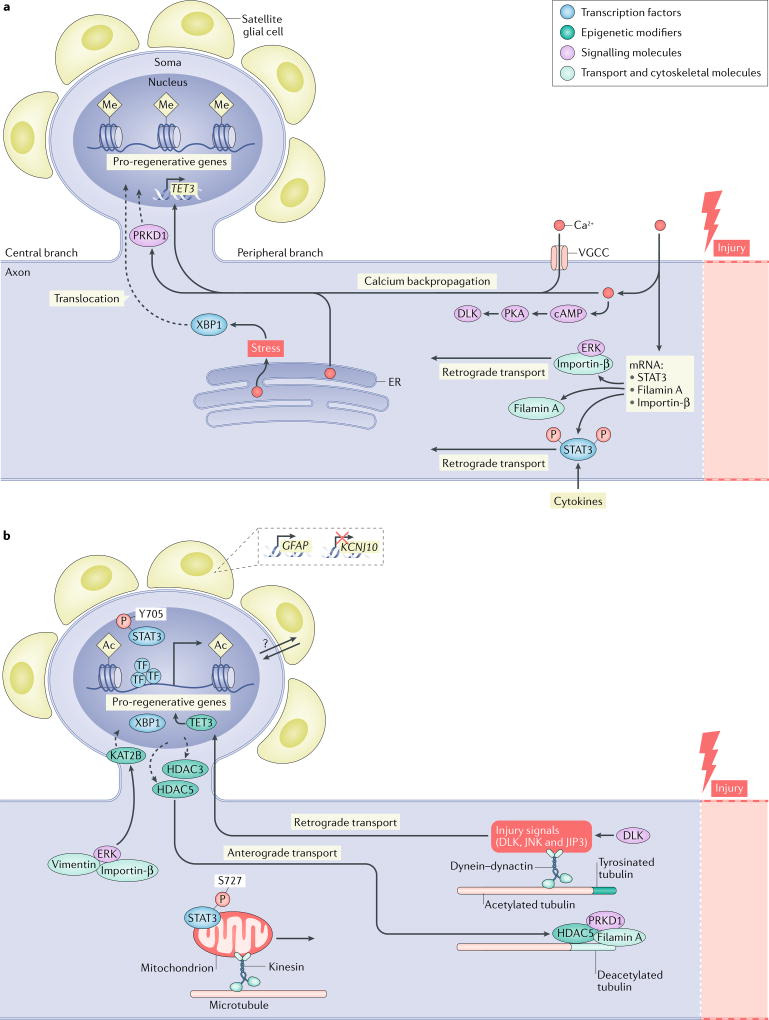
a | Early injury signalling events. Following injury, calcium rapidly flows into the damaged axon and is actively propagated to the cell soma by voltage-gated calcium channels (VGCCs) and the release of calcium by internal stores in the endoplasmic reticulum (ER). The release of stored calcium also triggers cellular stress pathways in the axon, resulting in the splicing of X-box-binding protein 1 (XBP1) and its translocation to the nucleus91. The increase in calcium activates pro-regenerative pathways at the site of injury and in the cell soma. For example, near the injury site, calcium activates cAMP, which in turn activates protein kinase A (PKA). PKA then activates dual leucine zipper-bearing kinase (DLK), a key mediator of later regenerative events34. Similarly, calcium at the injury site facilitates both the local translation of signal transducer and activator of transcription 3 (STAT3)57, importin-β58 and filamin A247, and the association of extracellular-signal-related kinase (ERK) with importin-β, which allows them to be retrogradely transported52. The backpropagation of calcium to the cell soma promotes the translocation of serine/ threonine-protein kinase D1 (PRKD1) to the nucleus, which promotes histone deacetylase 5 (HDAC5) nuclear export20. The calcium backpropagation also facilitates the expression of the methylcytosine dioxygenase TET3, which promotes post-injury demethylation of DNA28. In addition to the actions of calcium, other pathways are triggered by the injury: in the axon, the transcription factor STAT3 is phosphorylated, probably through the activity of cytokines, and is retrogradely transported to the nucleus to activate regenerative programmes57. b | Late injury signalling events. In the axon, DLK enables the retrograde transport of several injury signalling proteins, including JUN N-terminal kinase (JNK), JNK-interacting protein 3 (JIP3) and DLK itself, to the nucleus53,54,63. This process is also facilitated by an increase in tyrosinated tubulin near the injury site64. Phosphorylated STAT3 can localize either to mitochondria (when phosphorylated at S727) to increase ATP synthesis or to the nucleus (when phosphorylated at Y705) to influence transcription of pro-regenerative genes67. ERK, which is protected by vimentin during its retrograde transport, causes the histone acetyltransferase KAT2B to translocate into the nucleus52,166. Conversely, HDAC5 and HDAC3 are exported from the nucleus in response to PRKD1 nuclear translocation20 and HDAC5 is transported to the growth cone, where it interacts with filamin A and PRKD1 to deacetylate microtubules167,247. The changes in subcellular location of these histone modifiers, along with the calcium-dependent increase in TET3 (REF.28), cause a transition from repressive DNA and histone methylation (Me) into permissive histone acetylation (Ac). This transition likely results in a relaxation of the DNA around pro-regenerative genes, allowing binding by pro-regenerative transcription factors and expression of pro-regenerative genes20,28,179. Additional signalling may also be provided by surrounding satellite glial cells, which respond to axon injury by downregulating the ATP-dependent inwardly rectifying potassium channel Kir4.1 (KCNJ10)242 and upregulating glial fibrillary acidic protein (GFAP)231. The molecular mechanisms by which satellite glial cells can sense a distant axon injury remain largely unknown (indicated by the question mark). P, phosphorylation.
One multifaceted signalling molecule downstream of calcium is the second messenger cAMP. In rat dorsal root ganglion (DRG) neurons, cAMP levels correlate with growth capacity32. Generated by the calcium-dependent enzyme adenylyl cyclase after peripheral injury, cAMP affects the activity of several important proteins, including dual leucine zipper-bearing kinase (DLK; also known as MAP3K12), a highly conserved pro-regenerative kinase21,33–35, and cAMP-responsive element-binding protein 1 (CREB1), an important pro-regenerative transcription factor32,36,37. Although cAMP induction is important for the activity of several pro-regenerative pathways, induction of the pro-regenerative programme requires additional factors: cAMP stimulation activates only a fraction of injury-induced genes in peripheral sensory neurons and is not sufficient to promote growth38.
Another set of genes that are rapidly activated by calcium-dependent cAMP signalling and may contribute immediately following injury are immediate early genes39, which are important for neuronal activity-induced responses and plasticity40. However, because few studies focus on early post-injury time points, the contribution of these genes (with the exception of the transcription factor AP-1 (JUN)) to axon regeneration remains largely unstudied. In fact, many neuronal activity-induced molecules are common to various axon regenerative responses: these responses include the calcium-dependent nuclear export of histone deacetylase 5 (HDAC5)20,41 and the activation of CREB1 (REFS36,42,43), CREB-binding protein (CBP; also known as CREBBP)40,44–46, serum response factor (SRF)47–49 and mitogen-activated protein (MAP) kinases49–51. A better understanding of both the similarities and differences between the neuronal responses to activity and those to injury may provide key insights into the early post-injury events that set the stage for later pro-regenerative transcriptional changes.
Delayed injury signals
The slower phase of injury signalling is mediated by retrograde transport of several injury-responsive signalling proteins, including extracellular-signal-related kinases (ERKs)52, DLK, JUN N-terminal kinase (JNK)53–56 and the transcription factor signal transducer and activator of transcription 3 (STAT3)57. Following axon injury, ERK1 and ERK2 are phosphorylated and associate with both vimentin and locally translated importin-β in a calcium-dependent manner. This association allows for both binding to dynein for retrograde transport and protection from dephosphorylation52,58. Several lines of evidence have demonstrated the importance of ERK1 and/or ERK2 activation in the regenerative response. Studies have shown that pharmacological inhibition of dual specificity MAP kinase 1 (MAPKK1) or MAPKK2 (MAP kinase kinases (MAPKKs) that are upstream of ERK) or the genetic deletion of vimentin significantly reduces peripheral axon regeneration52. Furthermore, constitutive activation of serine/threonine-protein kinase B-raf (BRAF; a MAPKK kinase (MAPKKK) that is upstream of MAPKK1 and MAPKK2) is sufficient to promote robust axon regrowth into the adult spinal cord59.
Interestingly, the DLK–JNK pathway also appears to function in a calcium-dependent manner. Studies in multiple organisms have revealed that DLK is a key sensor of local injury that informs the cell soma about axonal injury35,56,60,61. Pharmacologically induced microtubule instability activates DLK60 as does cAMP (via protein kinase A (PKA)) in both D. melanogaster and mammals34 (FIG. 1). Downstream of DLK signalling, JNK is both activated and retrogradely transported to the nucleus, where it activates the transcription factor JUN53–55,62. DLK activity is also necessary for the immediate retrograde transport of another injury signalling protein, phosphorylated STAT3 (pSTAT3), but is dispensable for the transport of both JNK and STAT3 after the early injury stage63. Local alterations in the microtubule cytoskeleton elicited by injury, particularly an increase in tyrosinated α-tubulin, facilitate the retrograde transport of these injury signals64. These studies demonstrate the connection between the microtubule cytoskeleton in sensing axon injury and the mechanisms by which information about the injury is transmitted to the soma.
Although axon injury activates several pathways in an analogous manner in diverse types of neurons, contextual cues that are specific to those neuronal populations can result in a dramatically different interpretation of the injury signals. Studies in retinal ganglion cells (RGCs) suggest that cofactor interactions are one mechanism of specifying the cellular response following DLK activation65. Whereas DLK signalling in peripheral neurons is necessary for the activation of several pro-regenerative pathways63, in RGCs it promotes cell death66. In the latter context, DLK interacts with leucine zipper-bearing kinase (LZK; also known as MAP3K13), another MAPKKK, to activate a set of transcription factors that leads to RGC death after optic nerve injury65.
In uninjured, mature neurons, STAT3 mRNA is localized to the axon, and the protein remains in an inactive state localized to the cytoplasm57. Upon injury, axonal STAT3 mRNA is locally translated near the injury site. Both Janus kinases (JAKs) and MAPKKs phosphorylate STAT3: JAKs phosphorylate Y705 and MAPKKs phosphorylate S727 (REF.67). Although it is known that JAKs are activated via membrane glycoprotein 130 (gp130; also known as IL6Rβ) signalling68, it is unclear which ligands are responsible for gp130 activation and their source. Several studies have suggested that interleukin 6 (IL-6)69–71 and CC-chemokine ligand 2 (CCL2) produced by macrophages72 are responsible for gp130 activation in DRG neurons, whereas astrocytic expression of ciliary neurotrophic factor (CNTF)73 and leukaemia inhibitory factor (LIF)74 activate gp130 in RGCs. pSTAT3 is retrogradely transported by the molecular motor dynein to the cell soma57. In RGCs, it was recently demonstrated that the phosphorylation site serves as a localization signal for STAT3, with phosphorylation of Y705 directing it to the nucleus (where it influences transcription) and phosphorylation of S727 directing it to the mitochondria (leading to increased ATP synthesis)67. STAT3 phosphorylation at Y705 also plays a role in increasing the interaction of STAT3 with stathmin75, which antagonizes the microtubule destabilizing activity of stathmin76 and rescues axon degeneration in a mouse model of motor neuron disease77.
In summary, axon injury leads to the activation of several signalling pathways and, in many cases, local generation and activation of transcription factors or epigenetic modifiers78 that convey specific post-injury cellular changes from the site of axon injury to the soma (FIG. 1). Injury location along the length of the axon is also a key determinant of the cell body response: proximal, but not distal, axon injury leads to severe loss of rubrospinal neurons and facial motor neurons79,80. Similarly, RGC loss is more severe and more rapid when the optic nerve is injured close to the eye than when it is injured far from the eye81,82. These studies suggest that the availability of trophic support from exogenous sources and, possibly, intracellular mechanisms that can sense length83 contribute to the relationship between the cellular response and the injury location.
Pathways involved in translation
Beyond the classically defined injury signals, which are activated in the axon and travel back to the soma, other signalling pathways have recently been shown to be involved in the control of axon regeneration. Exposure of neurons to growth-limiting molecules, such as myelin-derived Nogo, myelin-associated glycoprotein (MAG) and chondroitin sulfate proteoglycans (CSPGs), activates the enzyme poly(ADP-ribose) polymerase 1 (PARP1), causing the accumulation of poly(ADP-ribose), which limits axon growth84. Pharmacological inhibition or genetic deletion of PARP1 facilitates axon regeneration over non-permissive substrates in cultured cortical and DRG neurons by a mechanism that is independent of histone poly(ADP-ribosylation) (PARylation) or the activation of regeneration-associated genes (RAGs)84. This system is conserved across species, as removal of poly(ADP-ribose) from proteins by poly(ADP-ribose) glycohydrolases (PARGs) enhances axon regeneration in C. elegans neurons85. Additionally, PARP is locally activated in the injured optic nerve84; however, inhibition of PARP is insufficient to enhance axonal regeneration in either the optic nerve crush injury or thoracic spinal cord injury mouse models86. This is consistent with the fact that downstream of Nogo, MAG and CSPGs, other pathways cause growth cone collapse87. Although PARP inhibitors alone may not be sufficient to promote recovery, it will be interesting to see whether these pathways can synergize in combinatorial approaches.
RNA processing has also been shown to control axon regenerative capacity. The RNA processing enzymes RNA 3´-terminal phosphate cyclases (RTCA and RTCB) act to inhibit axon regeneration after nerve injury in C. elegans and D. melanogaster88,89. By contrast, protein archease, an RNA ligase cofactor that functions downstream of RTCA, serves as a pro-regeneration factor89. These RNA processing enzymes are involved in the unconventional mRNA splicing of X-box-binding protein 1 (XBP1), a stress sensor involved in the unfolded protein response. XBP1 is required for regeneration and appears to have a role that is conserved in C. elegans, D. melanogaster and mice89–91. Another type of stress response, involving glucocorticoid release and increased expression of glucocorticoid receptors in sensory neurons, is also integral to the regenerative response after nerve injury71.
Mammalian target of rapamycin (mTOR) is a key molecule involved in another very well-established pathway that can be manipulated to promote regeneration92. mTOR is activated following peripheral but not central axon injury, and genetic activation of mTOR through the deletion of upstream negative regulators, including phosphatase and tensin homologue (PTEN), hamartin (TSC1) and tuberin (TSC2), increases axon regeneration in both the PNS and CNS93–96. However, both the upstream and downstream mechanisms involved in mTOR function in axon regeneration are not well understood. Crosstalk between mTOR and STAT3 signalling pathways97, as well as negative feedback by the mTOR downstream target ribosomal protein S6 kinase-β1 (S6K1)98, suggest that a deeper understanding of this master regulator of axon growth is needed.
The relevance of cell-specific injury responses is emerging with the observation that in RGCs, PTEN deletion increases regeneration almost exclusively in the α-RGC subtype99. Whether this is a result of an epigenetic barrier (see below and BOX 2) that is present in other RGC subtypes remains to be determined. Cell-type-specific responses may also result from other mechanisms, such as post-transcriptional modification of RNA, that affect gene expression (and are referred to as epitranscriptomic mechanisms)100. Because RNA modifications were proposed to affect activity-induced RNA editing and localization in the brain101 and are known to affect cortical development102, it is likely that RNA modifications have an important role in axon regeneration. Indeed, nerve injury was recently shown to induce an increase in N6-methyladenosine, the most abundant modification of mRNA, in transcripts encoding many RAGs, and deletion of the enzymes responsible for this epitranscriptomic mark was shown to reduce axon regeneration in the PNS103.
Box 2 | Epigenetic modifiers and transcription factors in the regulation of gene expression.
Gene expression is regulated by the coordinated action of epigenetic modifiers and transcription factors219. Epigenetic mechanisms are defined as covalent chemical modifications of DNA and histones, which play an important role in defining gene expression through the modulation of chromatin structure and function. DNA can be modified by methylation (resulting in 5-methylcytosine marks) and demethylation pathways, which may produce intermediate products (such as 5-hydroxymethylcytosine)220. Whereas DNA methylation had been thought to be static in cells after mitosis, it is now clear that this is a very dynamic modification of the DNA in both peripheral nervous system and CNS neurons221. DNA is also packaged around histone octamers, which have tails that can be post-translationally modified in various ways, including through acetylation, methylation and phosphorylation165. These modifications are performed by enzymes called ‘writers’, are removed by another class of enzymes called ‘erasers’ and serve as recognition sites for proteins called ‘readers’. The genomic specificity of writers and erasers is largely dictated by the proteins with which they complex222. Readers and transcription factors recognize post-translational motifs or sequence motifs, respectively, and these motifs serve as binding sites for the complex. In the case of transcription factors, the presence of DNA methylation can alter their cognate motif and therefore affect where they bind222–223. Cumulatively, these modifications affect the overall compaction and packaging of DNA around histones, which alter the ability of transcriptional machinery to bind DNA224. Additional layers of gene regulation are provided by the packaging of chromatin, which is regulated by histone occupancy and can either facilitate or block transcription factor binding and the initiation of transcription, and the association of DNA into topologically associated domains in which regions of DNA are tethered together spatially224,225.
Transcriptional changes
In the two decades following the discovery that peripheral axon regeneration is transcription-dependent104, an increasing focus has been put on uncovering and understanding the roles of axon injury-induced genes105. More recently, advances in next-generation sequencing have enabled many studies to characterize the post-injury transcriptional changes. These studies have identified more than 1,000 differentially expressed genes and uncovered many novel regulators of axon regeneration106. One group of proteins that have received particular attention in these studies is transcription factors. With their ability to bind regulatory DNA elements, such as promoters and enhancers, transcription factors can influence the expression of large sets of genes in a cell-type-specific or context-specific manner. Additionally, exogenous expression of certain sets of transcription factors can instruct cells to undergo complex cellular changes, such as reprogramming from one cell type to another107. As such, understanding which transcription factors are responsible for inducing the post-injury transcriptional changes that promote regeneration in peripheral neurons may enable CNS regeneration to be boosted through combinatorial manipulation.
Transcription factors activated by injury signaling
As previously discussed, many transcription factors are activated directly downstream of injury signalling pathways. For example, JNK signalling leads to the activation of JUN and increased expression of the cAMP-dependent transcription factor ATF3 (REFS55,108,109). In DRG neurons, JUN activation occurs in response to peripheral, but not central, axon injury51. Interestingly, JUN and ATF3 are sufficient to promote regeneration only under certain cellular conditions. Although overexpression of ATF3 can promote peripheral nerve regeneration109, it fails to do so in several models of CNS injury110,111. Conversely, JUN overexpression can promote regeneration in cultured cortical110,112,113 and DRG neurons114; however, as described above, its activation downstream of DLK also promotes cell death in RGCs66. Whereas neuronal JUN is clearly necessary for peripheral axon regeneration115, JNK-mediated phosphorylation of JUN appears to not be required for its full function in facial nerve regeneration116. Importantly, co-expression of ATF3 and JUN can cooperate to improve regeneration in DRG neurons to a greater extent than can be achieved by the expression of either transcription factor alone114. However, this cooperative effect does not occur in cortical neurons110. These results make it clear that the extent to which genetic manipulation can impact regeneration is strongly influenced by cellular context and the competency of the neurons to respond (BOX 3).
Box 3 | Cellular context and neuronal competency for axon regeneration.
Genetic manipulations that have focused on overexpression of regeneration-associated genes or transcription factors have yielded limited success114,143,146,148. This lack of success could be because more than one factor is necessary to fully stimulate the growth process; however, another explanation is that the cellular context dictates the competency of the neurons to respond to a given genetic manipulation. Indeed, multiple factors influence the dynamics of chromatin interactions during development201,226, and each cell type is likely to have a specific, unique chromatin organization. In the context of cellular reprogramming, the accessibility of the chromatin to a given transcription factor dictates the effectiveness of the conversion227. Differences in chromatin accessibility may also lead to different outcomes, as genomic regions that are differentially bound by given transcription factors or differences in signalling pathways may lead to different cellular responses, such as the cell death and regeneration that are promoted by transcription factor SOX11 (REF143) and dual leucine zipper bearing kinase (DLK)65, respectively. As such, therapies that address both cellular competency and the manipulation of gene expression may be necessary.
Several members of the SMAD family of transcription factors are also involved in peripheral axon regeneration. Activated downstream of the injury-induced phosphatidylinositol 3-kinase (PI3K)–glycogen synthase kinase 3 (GSK3) signalling pathway, SMAD family member 1 (SMAD1) is both phosphorylated and upregulated after peripheral injury, leading to its accumulation in the nucleus117,118. Once in the nucleus, phosphorylated SMAD1 (pSMAD1) interacts with the histone acetyltransferase p300 (p300 HAT) to promote the expression of several pro-regenerative target genes119. Indeed, transcriptional regulation of these genes by pSMAD1 is necessary for sciatic nerve regeneration119, and pSMAD1 activation by bone morphogenetic protein 2 (BMP2) and/or BMP4 is sufficient to promote axon growth in DRG neurons in vitro117. One interesting recent study also demonstrated that activation of SMAD2 and/or SMAD3 by activin signalling after axon injury in a wild-derived inbred mouse strain (CAST/Ei mice)120 confers a greater regenerative potential after peripheral and central injury when compared with other previously derived mouse strains121. Accordingly, exogenous activin is sufficient to increase peripheral regeneration in poorly regenerating C57Bl/6 mice and was shown to modestly increase optic nerve regeneration121.
Although these studies clearly demonstrate the importance of SMADs in the pro-regenerative response, little is known about either the genes downstream of SMAD transcriptional regulation or the partners with which the SMAD transcription factors bind. Because SMADs have a low binding affinity for DNA, binding generally requires the recruitment of additional transcription factors or epigenetic modifiers122. Several binding partners have been previously identified for SMAD2 and SMAD3, including pro-regenerative transcription factors and epigenetic modifiers such as JUN, ATF3, cellular tumour antigen p53 (TP53), p300 HAT and the histone acetyltransferase KAT2B122. Whether these interactions occur in neurons after injury and affect transcriptional regulation is yet to be determined.
As with the previously discussed transcription factors, STAT3 is specifically activated in response to peripheral axon injury but not spinal cord or dorsal root injury123. This injury-induced activation is necessary for the initiation of growth but not for axonal elongation: mice with genetic deletion of STAT3 exhibit a delayed regenerative response in peripheral nerves124. The role of STAT3 in initiating axon regeneration was further demonstrated in studies in which overexpression of STAT3 increased outgrowth and collateral sprouting of central DRG branches after a dorsal column lesion124. STAT3 overexpression also enhances remodelling of lesioned corticospinal tract (CST) fibres and induces de novo formation of collaterals from unlesioned CST fibres, which promotes functional recovery after spinal cord injury125. Indeed, overexpression of a hyperactivated form of STAT3 can also modestly promote optic nerve regeneration67,126. Accordingly, inhibition of JAK2, an upstream activator of STAT3, reduces regenerative growth both in vitro and in vivo127, whereas activation of the JAK–STAT pathway using cytokines such as CNTF can increase RGC survival and modestly promote regeneration128,129. The limited extent to which these cytokines can increase regeneration is thought to be due to the injury-induced increase in suppressor of cytokine signalling 3 (SOCS3) expression, a potent JAK–STAT inhibitor. Indeed, genetic deletion of SOCS3 significantly increased regeneration when combined with expression of CNTF and knockout of the tumour suppressor PTEN129,130.
Developmentally regulated transcription factors
Although many of the previously discussed transcription factors have been discovered in the context of adult peripheral axon injury, the decline in axon growth competency in the CNS following synapse formation has also proved useful in discovering regeneration-associated transcription factors. In particular, many of these studies have focused on the Krueppel-like factor (KLF) family of transcription factors, which have been shown to regulate the growth competency of RGCs and other CNS neurons131–133. Interestingly, examination of sequence similarity among KLF family members showed that those that share a high degree of sequence similarity also have similar effects on regeneration131. For example, overexpression of KLF4, the expression of which increases during development in RGCs, potently inhibits axon regeneration in RGCs, whereas its deletion promotes axon regeneration in vivo after optic nerve injury131. Similarly, deletion of KLF9, the expression of which also increases during development, promotes long-distance optic nerve regeneration in a JNK3-dependent manner134. Conversely, both KLF6 (REFS131,135) and KLF7 (REFS131,132), which are developmentally downregulated, promote growth when overexpressed in CNS neurons in both mammals and zebrafish136 (TABLE 1).
Table 1.
Pro-regenerative transcription factors and their effects on axon regeneration
Transcription factorNecessary forregeneration in PNS?Sufficient forregeneration in the PNS?Sufficient forregeneration in the CNS?
| ATF3 | Yes108 | Yes109,114,148 | No109,110,148 |
| CREB1 | Unknown | Yes36 | Yes36,113 |
| HIF1α | Yes138 | Yes138 | Unknown |
| JUN | Yes115,116 | Yes114 | Yes110,112,113 |
| KLF6 | Unknown | Unknown | Yes131,135 |
| KLF7 | Unknown | Unknown | Yes131,132 |
| MYC | Unknown | Unknown | Yes137 |
| SMAD1 | Yes117,118,249 | No118 | Unknown |
| SMAD2 | Unknown | Unknown | Unknown |
| SMAD3 | Unknown | Unknown | Unknown |
| SOX11 | Yes140 | Yes141 | Yes142,143 |
| SRF | Yes48 | Yes48 | Yes145 |
| STAT3 | Yes124 | Unknown | Yes67,125,126,250 |
| TP53 | Unknown | Yes144 | Yes46,144 |
| XBP1 | Yes91 | Yes91 | Unknown |
Several studies have manipulated transcription factors to alter the regenerative potential of neurons both in vitro and in vivo. Experiments in which the transcription factor has been knocked down via short hairpin RNA or small interfering RNA or genetically deleted have shown that several are necessary for PNS regeneration. Conversely, overexpression of various transcription factors has demonstrated their ability to boost regeneration in either the peripheral nervous system (PNS) or CNS. ATF3, cAMP-dependent transcription factor ATF3; CREB1, cAMP-responsive element-binding protein 1; HIF1α, hypoxia-inducible factor 1α; JUN, transcription factor AP-1; KLF, Krueppel-like factor; MYC, Myc proto-oncogene protein; SMAD, SMAD family member; SOX11, transcription factor SOX11; SRF, serum response factor; STAT3 signal transducer and activator of transcription 3; TP53, cellular tumour antigen p53; XBP1, X-box-binding protein 1.
Identifying combinations of pro-regenerative transcription factors
In addition to the previously highlighted transcription factors, several others — namely Myc proto-oncogene protein (MYC)137, hypoxia-inducible factor 1α (HIF1α)138, CREB1 (REFS36,113), SOX11 (REFS139–143), TP53 (REFS46,144), SRF48,145 and XBP1 (REFS90,91) — have been identified to have roles in axon regeneration (see TABLE 1 and REFS146,147). As little is known about the mechanisms by which these transcription factors are activated after injury, their regulation during development or their downstream targets, they will not be further discussed in detail here. However, as this list continues to grow, it has become increasingly clear that new methods are needed to identify those factors that work cooperatively to establish the pro-regenerative gene regulatory network. Although interactions between the known pro-regenerative transcription factors have been extensively studied in other cell types, it is critical to understand their interactions in the context of axon injury. This need is further highlighted by recent studies in which combinatorial co-expression of a few of these transcription factors had limited to no ability to boost regeneration above single-factor overexpression in poorly regenerating neurons110,148.
So how do we identify combinations of transcription factors that are capable of reprogramming CNS neurons to a regenerative state without altering cell identity? This is a question that is quickly becoming central to ongoing attempts to design therapies for CNS injury. Several lessons can be learned from protocols developed to convert cells into different types and to engineer cell identity. First, these studies have shown that core transcription factors important for the establishment of a particular cellular identity, such as the famous OSKM (OCT4, SOX2, KLF4 and MYC) factors in induced pluripotent stem cells (iPSCs)149,150, cooperatively bind enhancers and promoters to regulate the transcription of downstream gene targets151,152. In the context of regenerating neurons, core pro-regenerative transcription factors would be likely to both repress genes associated with mature neurons engaged in synaptic transmission and activate those associated with growth and survival. Importantly, studies have also shown that each of the core transcription factors regulates the expression of the others, reinforcing cell fate152. Whereas one recent study identified a core network of hub transcription factors involved in peripheral regeneration114, their downstream targets and whether they regulate the expression of the other transcription factors in the context of regeneration are still unclear. Similarly, it is unlikely that the transcription factors that drive the pro-regenerative network are activated simultaneously. As such, examining how the pro-regenerative gene regulatory network develops temporally will be critical for future efforts to recapitulate this process. Finally, reprogramming studies have shown that a successful transition between cellular states requires epigenetic reorganization153. In the context of cellular reprogramming, pioneer transcription factors, which can bind regions of compacted chromatin and recruit epigenetic modifiers, play a substantial role in inducing these changes and are necessary for successful conversion153–155. It will be interesting to see whether one or more pioneer factors play a similar role in post-injury epigenetic changes and whether they are required for inducing long-distance regeneration in CNS neurons. However, it is also possible that even the addition of pioneer transcription factors is insufficient to reprogramme CNS neurons to grow owing to the presence of a growth-repressive chromatin landscape in these cells153,156. Indeed, a decrease in promoter accessibility during cortical development has been observed110, revealing a potential intrinsic constraint to axon growth and regeneration. This constraint may cause a failure reminiscent of the low efficiency observed in cell reprogramming, in which repressive chromatin remains a barrier to deterministic reprogramming153. Detailed analysis of the chromatin accessibility in neurons with differential regenerative capacities will therefore be instrumental in engineering and developing new strategies to direct axon regeneration. Indeed, recent technical developments now enable researchers to study transcription and chromatin accessibility in a cell-type-specific manner157 and have been applied to show that neuronal activity modifies the chromatin accessibility landscape in the adult brain158.
Another limitation of therapeutic strategies in which the overexpression of transcription factors is used to potentiate regeneration is that the response to a given transcription factor may vary according to the cell type in which it is overexpressed. For example, it was recently shown that SOX11, a known pro-regenerative transcription factor in the PNS139, can reprogramme adult non-α-RGCs to a growth-competent state. By contrast, SOX11 expression kills α-RGCs, the cellular subtype that preferentially regenerates following treatments such as PTEN deletion143. SOX11 expression also promotes CST regeneration after spinal cord injury but interferes with functional recovery142. Careful evaluation of the resulting phenotype following transcription factor manipulation will therefore be necessary to evaluate any potential therapies as they should promote both survival and regeneration while maintaining the competency to respond and innervate appropriate targets.
In DRG neurons, differences in the regenerative response have even been observed between subtypes of neurons. Single-cell RNA sequencing (RNAseq) analysis of injured DRG neurons revealed heterogeneous transcriptomic responses159, which suggest that various subtypes of DRG neurons utilize different gene regulatory networks to control regeneration, cell death and neuropathic pain. These studies emphasize the need to investigate molecular mechanisms of axonal injury and regeneration in single cells. With the advent of various single-cell sequencing techniques, including RNAseq and assay for transposase-accessible chromatin using sequencing (ATACseq), these studies are now possible. Although single-cell analysis is not suitable for all studies, future transcriptomic and epigenetic analyses should utilize other available methods such as fluorescence activated cell sorting (FACS)160, laser capture microdissection (LCM)161, translating ribosome affinity purification (TRAP)162 and isolation of nuclei tagged in specific cell types (INTACT)157 to analyse defined populations of neurons.
Epigenetic integration in the nucleus
The term ‘epigenetics’ broadly encompasses the chromatin modifications and changes in DNA structure that affect gene expression but are not directly encoded in the DNA sequence. Epigenetic mechanisms that can induce changes in gene expression include the covalent modification of histones and DNA, which can influence chromatin 3D structure and function. An overview of different types of epigenetic modifiers and how they cooperate with each other and with transcription factors is given in BOX 2. Because epigenetic regulators can affect transcription on a genome-wide scale, yet do so in a cell-type-specific and context-specific manner, they represent interesting targets to promote neural repair. Whereas the role of epigenetics in regulating CNS gene expression during development163 or neural activity is being actively investigated164, its role in axon regeneration is only emerging.
Histone modifications
Although there are well over 200 identified histone modifications165, some of the best characterized involve acetylation of lysine residues in the aminoterminal tail of histones. The addition of these modifications, which are dynamically added by histone acetyltransferases (HATs) and removed by histone deacetylases (HDACs), helps to establish a more relaxed chromatin conformation that is readily bound by transcriptional machinery. Following axon injury, there is a global accumulation of both H3K9 and H3K14 acetylation20,166, and H4 acetylation119 in peripheral neurons, which may, in part, be caused by two recently discovered mechanisms. First, downstream of the injury-induced calcium influx, serine/threonine-protein kinase D1 (PRKD1, also known as PKCµ) is phosphorylated, causing it to translocate to the nucleus20. The increase in nuclear PRKD1 then causes HDAC5 to be exported from the nucleus20. In the axon tip, HDAC5 deacetylates microtubules, thereby increasing growth cone dynamics and promoting axon growth20,167. Similarly, there is evidence that HDAC3 is also exported from the nucleus to some extent20. Activation of ERK signalling also causes epigenetic changes in sensory neurons by stimulating the accumulation of KAT2B in the nucleus, in which it binds and acetylates histones at several pro-regenerative genes166. However, in contrast to peripheral injury, CNS injury fails to stimulate both the nuclear export of HDAC5 and the nuclear import of KAT2B, which may explain the lack of an observable increase in histone acetylation or transcriptional changes following these injuries119,166. Interestingly, in injured RGCs, HDAC3 is imported into the nucleus, where it functions to repress gene expression and promote apoptosis168,169. The lack of a pro-regenerative epigenetic response following CNS injury may explain the failure of overexpressed pro-regenerative transcription factors to produce long-distance CNS regeneration. Notably, both exogenous expression of KAT2B166 and the use of HDAC inhibitors170 are sufficient to modestly promote regeneration after spinal cord injury, and overexpression of p300 HAT can promote some regeneration following optic nerve crush45. However, whether the manipulation of these epigenetic pathways can act synergistically with the overexpression of pro-regenerative transcription factors to promote CNS regeneration is yet to be determined.
Calcium influx, protein kinase C (PKC) signalling and ERK signalling have been linked to changes in histone acetylation in response to injury20,166; however, other signalling pathways also affect chromatin modifications171. Histone modifiers have been identified as downstream effectors of signal transduction pathways in other systems, some of which are known to be activated by axon injury. For example, whereas the phosphorylation of H3S28 and H3S10 occurs downstream of MAPK, PI3K, JAK–STAT and cAMP–CREB signalling, H3K18 acetylation functions downstream of PI3K signalling172. Similarly, changes in cellular metabolism can influence epigenetics and thus transcription173. As the transition to a pro-regenerative state is likely to be metabolically demanding, the differential ability of cell types to cope with this stress may contribute to their ability to successfully regenerate.
DNA modifications
One of the best-studied and most stable epigenetic DNA modifications is the generation of 5-methylcytosine (5mC), an epigenetic mark produced when a methyl group is covalently added to cytosine nucleotides within the DNA. This modification, which is primarily deposited at CpG dinucleotides, is dynamically regulated during neuronal development and in response to various stimuli163,164. Three DNA (cytosine-5)-methyltransferase (DNMT) enzymes — DNMT1, DNMT3A and DNMT3B — are responsible for catalysing this reaction. All three isoforms are expressed in DRG sensory neurons; however, DNMT3A may also be expressed in satellite glial cells in the DRG174,175. Generally associated with transcriptional repression, DNA methylation can be removed by methylcytosine dioxygenases (TET proteins) to activate transcription. TET proteins cannot directly remove methyl groups from DNA but instead sequentially oxidize 5mC to 5-hydroxymethylcytosine (5hmC), 5-formylcytosine (5fC) and 5-carboxylcytosine (5caC). Both 5fC and 5caC can then be targeted by thymine DNA glycosylase-mediated base excision repair, leaving behind an unmethylated cytosine176. Although originally viewed as an intermediate in this demethylation process, 5hmC is now known to be a stable DNA modification in the nervous system177.
Conflicting results have arisen regarding the importance and dynamics of DNA methylation after peripheral axon injury. In one study, the methylation of CpG islands, the regions of CG-rich DNA typically located near transcription start sites, was assayed following either sciatic or dorsal column injury166. Few regions were found to be differentially methylated following sciatic injury, and comparisons between sciatic and dorsal column injuries also revealed few differences in methylation166. However, in another study, the administration of folate, which increased DNA methylation globally in the spinal cord, increased regeneration of spinal neurons following combined spinal cord and peripheral nerve injury, and folate receptor-α (FRα), was shown to be necessary for peripheral regeneration178. Whether these effects of increased DNA methylation are neuron-specific remains to be determined. Another study reported widespread changes in 5hmC after both sciatic nerve crush and dorsal column transection in mice179. Several pro-regenerative genes, including Atf3, Smad1 and Bdnf, acquired 5hmC marks after peripheral injury, but not after central injury179. This finding is consistent with the finding that TET3 is upregulated28,179 in response to peripheral injury and is necessary for proper regeneration and behavioural recovery28. Although these studies focused on 5hmC, the dynamic regulation of this mark strongly suggests that 5mC is also being dynamically regulated either through demethylation or through conversion into stable 5hmC. Future studies using methods with greater genome coverage and cell-type specificity will be able to more accurately identify regions of differential methylation after injury (FIG. 2).
Fig. 2. Model of epigenetic activation of pro-regenerative genes in response to axon injury.
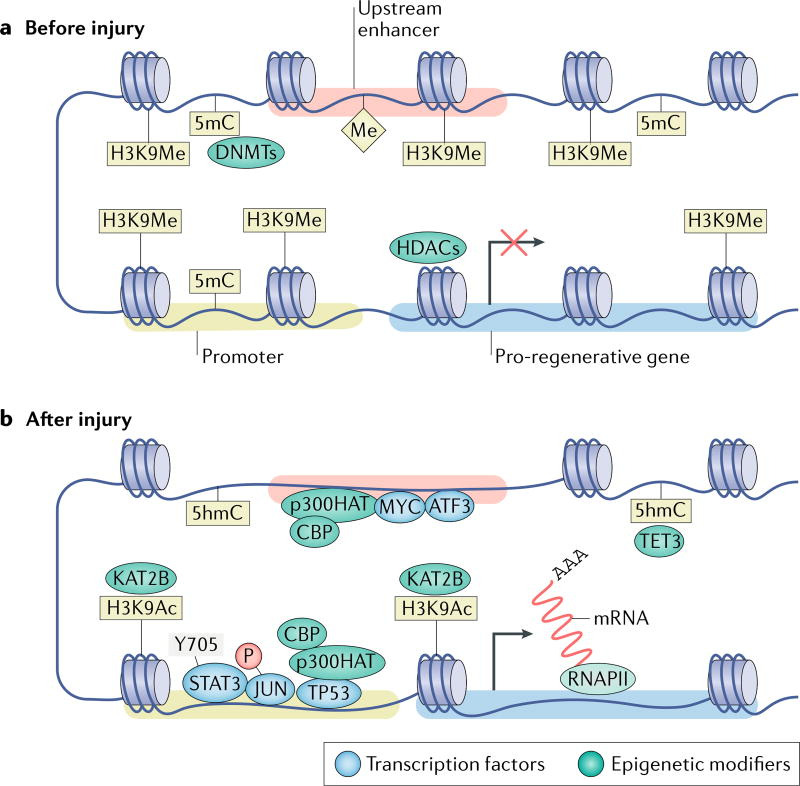
a | When dorsal root ganglion neurons have formed synapses and are electrically active, pro-regenerative gene bodies, promoters and enhancers are maintained in a transcriptionally repressed state through DNA methylation by DNA (cytosine-5)-methyltransferases (DNMTs), histone methylation by yet unknown histone methyltransferases and deacetylation by histone deacetylases (HDACs)20,28,179. b | Following injury, the nuclear export of HDACs20 and import of the histone acetyltransferase (HAT) KAT2B166 causes an increase in acetylation of histone H3K9 (H3K9Ac), whereas the upregulation of the methylcytosine dioxygenase TET3 causes the conversion of repressive 5-methylcytosine (5mC) into 5-hydroxymethylcytosine (5hmC)28. Similarly, CREB-binding protein (CBP) and p300 HAT, HATs that frequently bind to both enhancers and promoters, associate with cellular tumour antigen p53 (TP53) to increase H3K9Ac in pro-regenerative gene promoters144,248. The turnover of transcriptionally repressive marks into activating marks likely causes a relaxation in the chromatin, which should allow pro-regenerative transcription factors — such as Myc proto-oncogene protein (MYC), cAMP-dependent transcription factor ATF3, transcription factor AP-1 (JUN) and signal transducer and activator of transcription 3 (STAT3) — to bind regulatory elements and influence the transcription of downstream pro-regenerative genes. H3K9Me, methylation of histone HeK9; P, phosphorylation; RNAPII, RNA polymerase II.
Role of microRNAs in injured neurons
Despite their ability to regulate more than 60% of the human transcriptome, the importance of miRNAs in axon regeneration is only just beginning to be understood180. For these small (21–23 nucleotide) non-coding RNAs to become active, they must first undergo a series of processing steps181. At least one step of this biogenesis pathway has been identified as being necessary for peripheral nerve regeneration as mice lacking one of the key synthesis enzymes, Dicer, exhibit both reduced axon growth in vitro and reduced functional recovery in vivo182,183. However, whether other proteins involved in the miRNA biogenesis process are also necessary for regeneration is yet to be determined. A potential area of interest will be to determine whether the endoplasmic reticulum stress pathway and the miRNA biogenesis pathway, which are known to regulate each other, do so in the context of axon regeneration184,185. This is possible given that the endoplasmic reticulum stress response promotes regeneration of sensory neurons91,186 and survival of RGCs187.
Similar to other genes and non-coding RNAs, miRNAs are expressed in a tissue-dependent and context-dependent manner. For example, several miRNAs have key roles in neural development and are expressed in a temporally and spatially specific manner that corresponds to these roles188. As such, recent studies have sought to determine the roles of pro-regenerative miRNAs189–193 (reviewed in REF.176). Together, these studies have identified a variety of miRNAs that exhibit upregulated expression in the DRG following sciatic injury in mice and are capable of increasing regeneration in vitro, including miR-431, miR-222 and miR-21, and have implicated several downstream genes as negative regulators of regeneration (including Kremen1 and Spry2 (REFS190,192,193)). Notably, the expression of miR-222 promotes neurite outgrowth in cultured DRG neurons, in part by silencing PTEN192, consistent with the observed decrease in PTEN levels in DRGs following peripheral nerve injury96. One recent study in CAST/Ei mice, which have an increased neuronal regenerative capacity, demonstrated the importance of miRNAs that are downregulated following peripheral injury in this regeneration194. miRNA sequencing was used to identify miR-7048-3p as an miRNA that is downregulated after injury. This downregulation causes an increase in expression of its target achaetescute homologue 1 (ASCL1), a transcription factor that is capable of modestly promoting growth194.
Despite a recent focus on utilizing miRNAs as therapeutic targets, they have yet to be manipulated to improve axon regeneration in vivo. This is largely due to general issues surrounding the in vivo manipulation of miRNAs195. Another difficulty is the lack of consensus as to which miRNAs are important for regeneration. Indeed, published lists of differentially expressed miRNAs tend to have little overlap with one another, even in studies with identical post-injury time points190,192,193. A few experimental reasons may explain this heterogeneity, including the use of different species, different injury paradigms and the limitations of microarrays. The use of the whole DRG, a tissue with a notoriously heterogeneous population of cells, as an input for miRNA profiling could also have a profound impact on these studies: in addition to neurons and satellite glial cells (BOX 4), macrophages are known to migrate into the DRG after injury196,197. Furthermore, the heterogeneity of injury responses in different sensory neuron subtypes159 is also likely to have influenced these miRNA profiles. It will be important to continue to examine how different neuronal subtypes respond to injury and how the regulation of translation in different populations of sensory neurons relates to their functional recovery.
Box 4 | Contribution of glial cells to peripheral nerve regeneration.
In the mouse peripheral nervous system, the intrinsic mechanisms regulating axon regeneration have traditionally been assumed to involve changes that occur within the neuron. However, satellite glial cells (SGCs), the main type of glial cells in sensory ganglia, ensheathe sensory neuron soma (FIG. 1). Each sensory neuron and its ensheathing SGCs thus form a distinct functional and morphological unit228–230. SGCs are similar to astrocytes in the CNS as they have the capacity to buffer the extracellular environment via potassium and calcium channels and express the intermediate filament glial fibrillary acidic protein (GFAP). Recent studies have provided evidence that distant nerve injury induces morphological and molecular changes in SGCs, suggesting that SGCs can recognize and respond to this stimulus. These changes include an increase in GFAP expression231–236, extracellular-signal-related kinase (ERK) phosphorylation237,238, an increase in SGC coupling via gap junctions239–241, downregulation of the ATP-dependent inwardly rectifying potassium channel Kir4.1 (REF.242) and proliferation231’243,244. Although little is known about how SGCs respond to axon injury, early spontaneous neuronal activity233 and direct communication between the neuronal soma and SGCs231 were proposed to underlie SGC activation. Indeed, robust communication between neurons and SGCs is critical for neuronal excitability and nociception245, and future studies will reveal whether SGCs, similarly to the repair Schwann cells246, participate in the neuronal response to peripheral nerve lesion and control regenerative outcomes.
Conclusion and future directions
Although considerable progress has been made in understanding the mechanisms underlying peripheral nerve regeneration and how these can be manipulated to promote regeneration in the CNS, there are many challenges to overcome in order to develop therapies that achieve complete axon regrowth and functional recovery. Bioinformatics approaches to integrate discoveries related to regenerative capacity across multiple laboratories could help to drive drug discovery, but such studies remain difficult to interpret because of the lack of cell-type-specific data. Although a recent study identified pathways and small molecules that can enhance growth of injured neurons114, the effects observed were small, and future multilevel bioinformatics analyses will benefit from novel neuron-specific data sets. Furthermore, how injured neurons can reprogramme themselves to regrow their axons while maintaining their competency to form synapses and how they can stop growth once regeneration is complete is still not understood. Indeed, studies focused on the temporal regulation of regenerating neurons will help us to understand how this process is initiated after injury, maintained during elongation and successfully shut down upon target reinnervation. Going beyond transcriptomics to understand the use of regulatory elements, such as enhancers and promoters, and the influence of genome topology in the context of axon regeneration will also be important. The cell-type-specific use of enhancer elements and the organization of topologically associated domains are likely to be instrumental in determining the competency of cells to respond properly to axon injury198–202.
Whereas the majority of the pathways discussed in this Review are highly conserved, others exist only in higher mammals or humans. For example, ARMCX1 is a mammalian-specific gene that controls mitochondrial transport during neuronal repair203. Increased mitochondrial transport to injured axons is required for regeneration203,204, and CSPGs can prevent mitochondria from localizing to the growth cone205. There are also human-specific mRNA splice variants and human-specific genes in the nervous system that may play a role in the regenerative response206–208. To determine whether findings in animal model systems are predictive of the injury responses of human neurons, it will be important to determine whether the epigenomic and transcriptional pathways identified in animal models also function in human neurons. For example, in the developing retina, most epigenetic changes are conserved from mice to humans209; however, whether the epigenetic injury responses are conserved in humans is unknown.
Finally, age is another key consideration that is highly relevant to the development of therapeutic strategies. As axon injury in humans can result from a variety of insults at any age, it is important to note that regeneration tends to be increasingly limited with age210,211. Indeed, epigenetic profiling revealed that promoter accessibility decreases with cortical maturation110, and methylation of histone H3K27, a mark that correlates with closed chromatin, increases with age in the brain in medium spiny neurons212. Thus, manipulating epigenetic machinery will likely be critical in efforts to improve regenerative outcomes and may need to be tailored according to age.
Acknowledgments
The authors’ research on these topics has been generously supported by the US National Institute of Health grants NS096034, NS082446 and NS099603, the University of Missouri Spinal Cord Injury Research Program and a Philip and Sima K. Needleman Doctoral Fellowship. The authors thank H. Gabel for helpful comments and critical reading of the manuscript. The authors thank the Cavalli laboratory members for their helpful comments on the manuscript. The authors apologize to those whose studies could not be cited owing to space limitation.
AbbreviationsTranscription factors
Proteins that activate or repress the expression of genes by binding to DNA sequence motifs proximal to a gene’s transcription start site or interacting enhancer regions
Epigenetic modifiers
Proteins that post-translationally modify either DNA or histones, which affects DNA compaction and accessibility for protein binding
MicroRNAs
(miRNAs). Single-stranded RNA molecules of 20–23 nucleotides in length, generated endogenously from a single-stranded hairpin precursor, which act as post-transcriptional inhibitors in association with the RNA-induced silencing complex (RISC)
Motor proteins
Proteins, such as kinesin, dynein and myosin, that use either the microtubule or the actin cytoskeleton for movement by converting chemical energy into mechanical force
Cofactor
Protein that binds with other proteins to form heteromeric complexes that alter or enhance the function of its binding partners
Poly(ADP-ribose)
A negatively charged polymer of ADP-ribose that can be added to proteins. Poly(ADP-ribose) represents a unique post-translational modification that regulates protein function
RNA processing
The process by which an RNA molecule translated from DNA undergoes modifications, including 5′ capping, 3′ polyadenylation, splicing and methylation, before the RNA is translated into a protein
Unfolded protein response
A cellular stress response that is triggered by an excess of unfolded or misfolded proteins in the endoplasmic reticulum
Epitranscriptomic mechanisms
Post-transcriptional RNA modifications that regulate mRNA half-life or translation or otherwise alter biological processes
Next-generation sequencing
High-throughput parallel sequencing of either DNA or RNA
Cytokines
Originally defined as immune system proteins, these proteins are now known to be released by most cells and are important in regulating intercellular communication, cell function and cell survival
Induced pluripotent stem cells
(iPSCs). Cells created from differentiated cell types (for example, fibroblasts) that are reprogrammed by a cocktail of transcription factors (or other approaches) back to a pluripotent state and are capable of differentiating into all three germ layers
Genome topology
The 3D DNA structure that dictates its accessibility to binding by proteins such as transcription factors and epigenetic modifiers
Footnotes
Author contributions
M.M. and V.C. researched data for the article, made substantial contributions to discussions of the content, wrote the article and reviewed and/or edited the manuscript before submission.
Competing interests
The authors declare no competing interests.
References
- 1.Liu K, Tedeschi A, Park KK, He Z. Neuronal intrinsic mechanisms of axon regeneration. Annu. Rev. Neurosci. 2011;34:131–152. doi: 10.1146/annurev-neuro-061010-113723. [DOI] [PubMed] [Google Scholar]