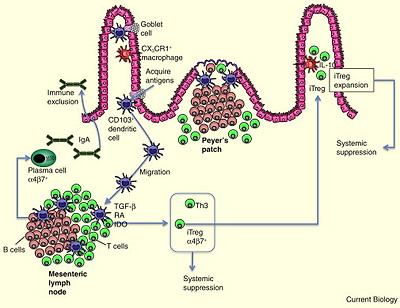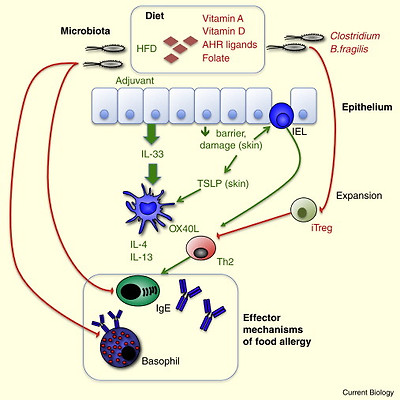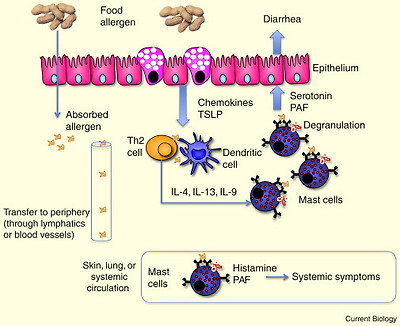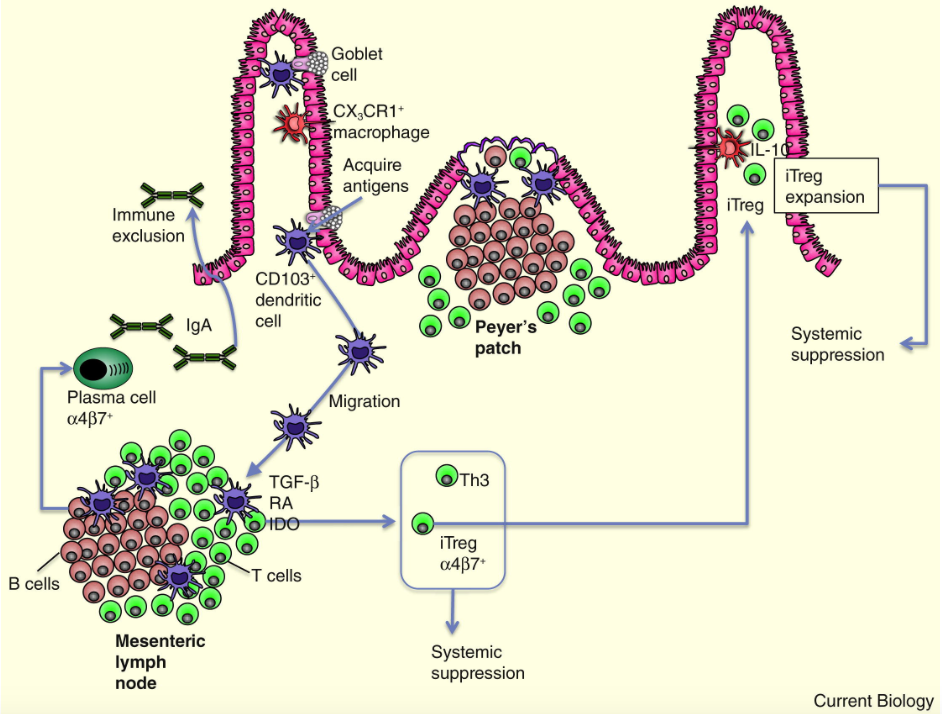
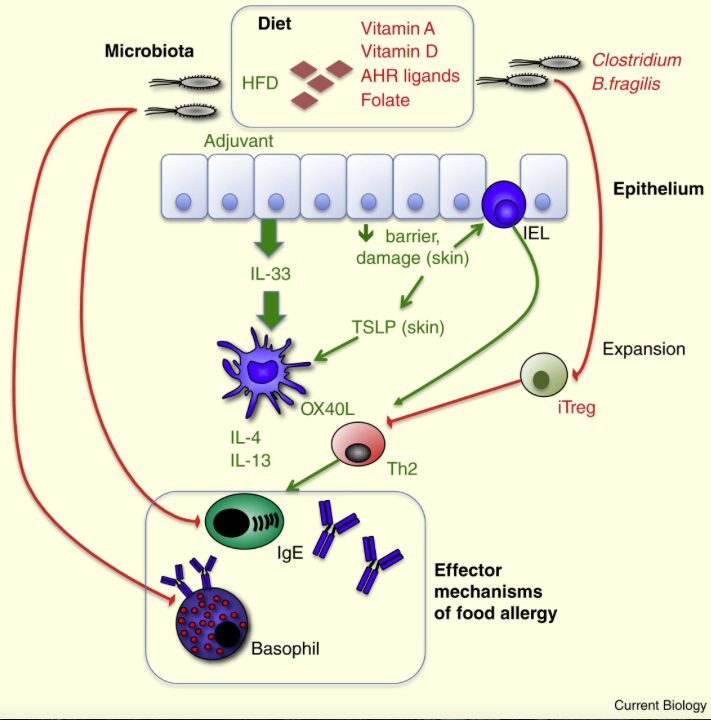
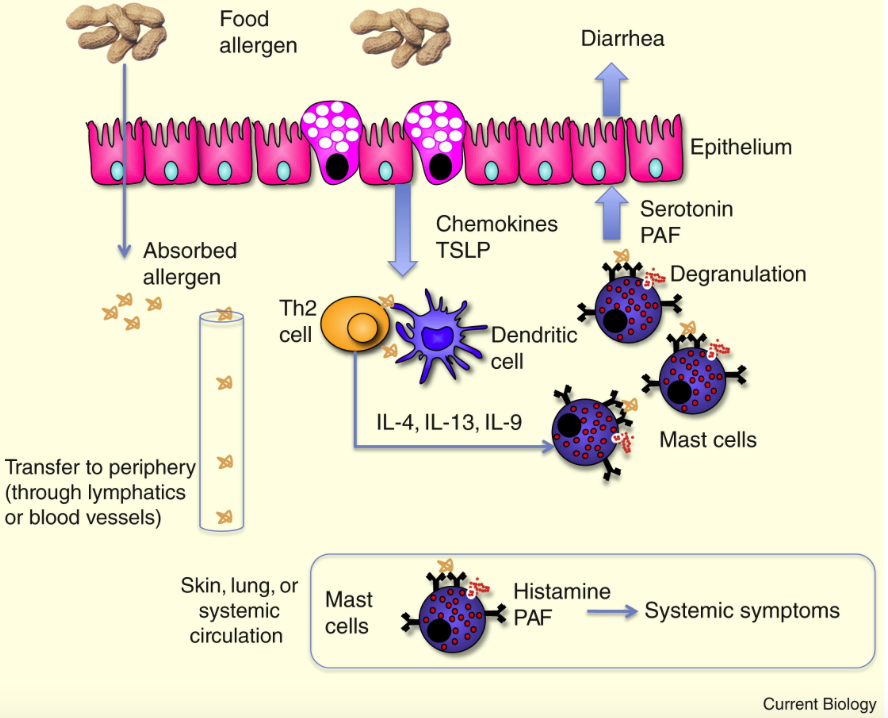
Glossary of immunology terminology.
| Innate immunity | An immediate response to infection or damage that is mediated through recognition of conserved patterns. Recognition occurs via pattern recognition receptors, such as Toll-like receptors, NOD-like receptor proteins, and endocytic/phagocytic pattern recognition receptors, such as mannose receptor or DC-SIGN. Effector mechanisms of innate immunity are diverse and include the release of anti-microbial peptides, killing through release of cytotoxic molecules, phagocytosis and intracellular killing of microbes, complement activation, release of innate cytokines (IL-1β, IL-6, and tumor necrosis factor-α, TNF-α) and release of acute-phase proteins from the liver that assist in pathogen clearance. |
| Adaptive immunity | Adaptive immunity is mediated by T and B lymphocytes, which recognize specific antigens through their T-cell receptor (TCR) and B-cell receptor (BCR), respectively. The adaptive immune responses are slower than the innate immune response, but have the property of memory, such that subsequent responses to the same antigen are more rapid. T cells can be divided into cytotoxic (CD8+) and helper (CD4+) T cells. CD4+ T cells can be further subdivided based on the cytokines they secrete. In general, Th1 cells express the transcription factor T-bet, secrete IFN-γ and assist in clearing intracellular pathogens by activating macrophages; Th2 cells express the transcription factor GATA-3, secrete IL-4, IL-5 and IL-13, and provide help to B cells for antibody production and promote allergic responses; Th17 cells express the transcription factor RORγt, secrete IL-17, and play an important role in microbial clearance and a destructive role in autoimmunity. Regulatory T cells suppress these effector T-cell responses, and regulatory T cells come in a variety of different phenotypes. B cells contribute to adaptive immunity by secreting antibodies that can neutralize pathogens, activate complement, and activate antibody-dependent cell-mediated cytotoxicity pathways. Antibodies of the IgE isotype trigger allergic reactions. |
| Dendritic cells | Dendritic cells are resident cells of tissues, acquire antigen and process it for presentation on major histomcompatibility complex (MHC) molecules, and migrate to lymph nodes for presentation of antigen to T lymphocytes. Dendritic cells are the most potent antigen-presenting cells capable of priming naïve T cells. |
| Cytokines | A cytokine is a small protein made by a cell that affects the behavior of other cells. Cytokines can be grouped into categories based on their function. Innate cytokines (IL-1β, IL-6, TNFα) are released by a variety of stromal cells and macrophages early in infection and play a role in pathogen clearance. Th1, Th2, and Th17 cytokines are outlined under ‘adaptive immunity’. Cytokines released from innate cells and antigen-presenting cells can act on naïve T cells to influence their developmental fate. IL-12 promotes the development of Th1 cells, IL-4 promotes the development of Th2 cells, and IL-6 and TGF-β promote the development of Th17 cells. |
| Chemokines and homing molecules | Chemokines are chemotactic cytokines that act on G-protein-coupled surface receptors. Cells respond to chemokines through migration along a chemokine gradient. Tissue-specific homing is mediated through chemokine–chemokine receptor interaction as well as integrin–ligand interactions. Gut homing is mediated by the chemokine receptor CCR9 and its ligand CCL25 (expressed in the small intestine), as well as the integrin α4β7 and its ligand MAdCAM, which is also differentially expressed in the intestine. Skin homing is mediated by the chemokine receptors CCR4 and CCR10 in response to the ligands CCL17, CCL22, and CCL27, which are expressed in the skin. In addition, the integrin cutaneous lymphocyte antigen (CLA) on lymphocytes binds to E-selectin. |
| Regulatory T cells | Regulatory T cells can be CD4+ or CD8+, although the most commonly recognized regulatory T cell is the CD4+ Foxp3+ form. Foxp3+ regulatory T cells can be generated in the thymus (natural regulatory T cells ) or generated from naïve T cells in the periphery (induced regulatory T cells). Regulatory T cells use a variety of suppressive mechanisms, including cytokines (TGF-β and IL-10), cell–cell interaction (for example, involving the surface molecule CTLA-4), and can also suppress by cytolytic activity. |
| Co-stimulatory molecules | These molecules are expressed on the surface of antigen-presenting cells and provide additional signals to shape the nature of the T-cell response. Co-stimulatory molecules can provide an activating signal, such as when B7.1 and B7.2 bind to CD28 on the T-cell surface. Alternatively, inhibitory signals can be provided by co-stimulatory molecules, such as when B7.1 and B7.2 bind to CTLA-4 instead of CD28 and provide an ‘off’ switch to T-cell activation. Expression of other co-stimulatory molecules, such as OX40L, or Notch ligands on antigen-presenting cells can influence the differentiation of naïve T cells through binding to surface receptors on T cells. |
| Innate lymphoid cells | Innate lymphoid cells are innate cells that share characteristics with CD4+T cells, but do not recognize specific antigen within the context of MHC molecules. These cells represent a distinct category of different innate immune cells, but features in common include localization at mucosal sites and production of cytokines that play a role in mucosal homeostasis and regulation of the intestinal microbiota as well as in pathogenesis of intestinal and allergic disease. |