beyond reason
동맥경화는 혈관내피세포에서 죽은세포 청소과정(efferocytosis)이 제대로 이루어지지 않은 것이 원인이라는 논문
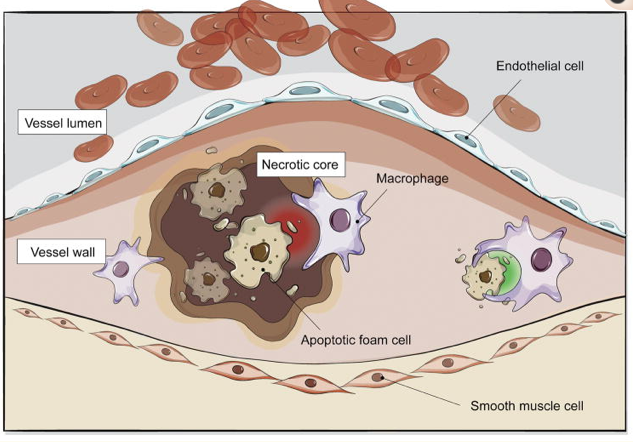
Impaired efferocytosis contributes to atherosclerosis
Diseased and apoptotic cells in the growing atherosclerotic plaque are not recognized for efficient phagocytic clearance by lesional macrophages. While the mechanisms which drive this pathology are still an area of active investigation, emerging data suggests that this defect may be due to impaired ‘eat me’ (green) and ‘don’t eat me’ (red) signaling that renders these cells ‘inedible’. As a result, foam cells accumulate to promote lesion expansion and apoptotic tissue undergoes secondary necrosis to accelerate vascular inflammation and lesion instability.
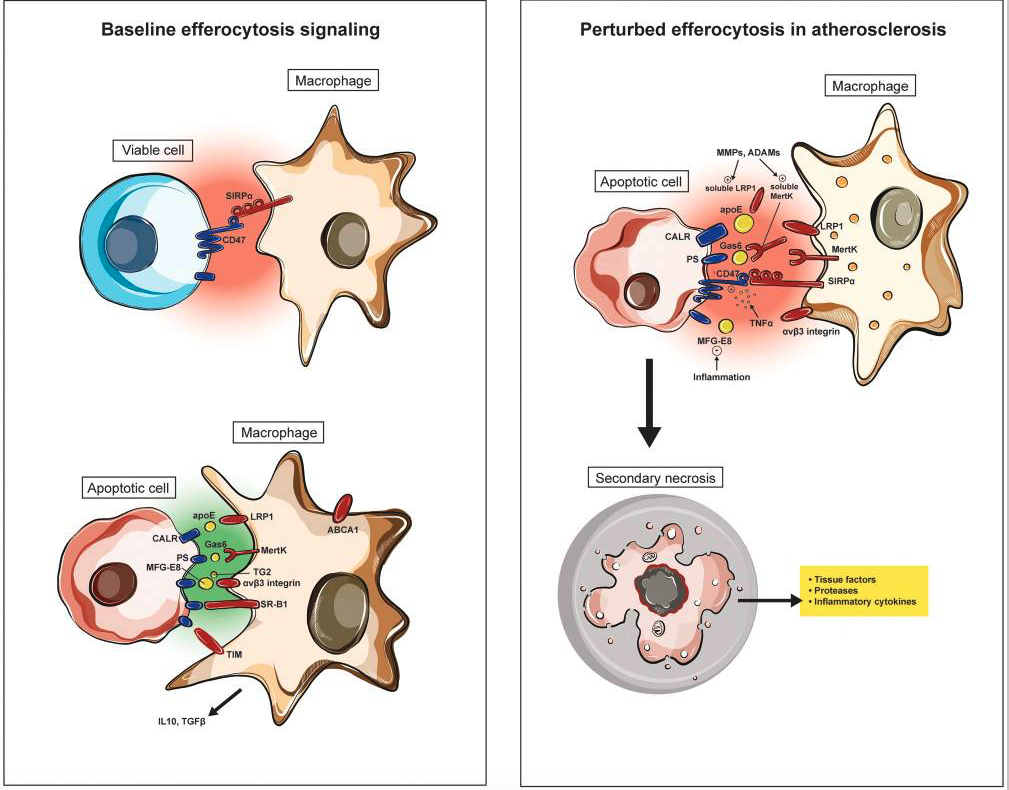
Impaired efferocytosis signaling in vascular disease
Experimental data suggests that pro-phagocytic signals (including calreticulin, Mfg-e8 and Mertk) are reduced in atherosclerosis due to inflammation, post-translational modifications and/or genetic variability. Exacerbating this loss of ‘eat me’ signaling is a concomitant upregulation of the CD47-Sirp-α ‘don’t eat me’ pathway, which furthers decreases the ‘edibility’ of cells within the necrotic core. The end result is that apoptotic cells in the growing plaque becomes poor substrates for phagocytic cells such as macrophages and dendritic cells. Such uncleared cells become secondarily necrotic and release additional proinflammatory stimuli, thus promoting a positive feedback loop.
The Role of Efferocytosis in Atherosclerosis
Abstract
The necrotic core has long been a hallmark of the vulnerable atherosclerotic plaque. While apoptotic cells are cleared very quickly in almost all other tissue beds, their removal appears to be significantly impaired in the diseased blood vessel. Emerging evidence indicates that this phenomenon occurs due to a defect in ‘efferocytosis’, the process by which apoptotic tissue is recognized for engulfment by phagocytic cells such as macrophages. Genetic and experimental data suggest that efferocytosis is impaired during atherogenesis due to dysregulation of so-called ‘eat me’ ligands which govern the ‘edibility’ of cells undergoing programmed cell death. Highlighted below is a summary of recent data indicating that efferocytosis is a major unappreciated driver of lesion expansion, but also a reversible defect that can potentially be targeted as a means to prevent plaque progression.
What is efferocytosis?
Even in health, the human body turns over more than one million cells per second through a process known as programmed cell death (PrCD), or apoptosis1, 2. This process occurs for a variety of physiological reasons (e.g. negative T cell selection), but is largely driven by the regular homeostatic turnover of aged and senescent cells3. To remove these superfluous cells, the body engages in an evolutionarily conserved process known as programmed cell removal (PrCR), or efferocytosis4, 5. Efferocytosis is a term derived from the Greek (meaning to carry the dead to the grave), and refers to the phagocytic engulfment of a cellular corpse4, 6, 7. Both professional phagocytes (e.g. macrophages) and non-professional (e.g. neighboring) cells participate in efferocytosis.
Because the body must carefully ensure that only apoptotic cells are removed, and that there is no off-target clearance of healthy tissue, efferocytosis is a highly regulated process. Dozens of molecules have now been implicated in the cross-talk which occurs between a dying cell and a potential phagocyte4, 8, 9. These include: 1. Chemoattractant ‘find me’ ligands which recruit phagocytes to the area of cell death; 2. ‘Bridging molecules’ that play an opsonin-like role while linking phagocytes to their target cells; and 3. Cell-surface ‘eat me’ ligands which physically engage and transactivate engulfment receptors on the phagocyte to initiate PrCR. Importantly, these molecules are counterbalanced by so-called ‘don’t eat me’ ligands that are ubiquitously expressed on viable cells, but are rapidly downregulated during PrCD10. While these signaling pathways remain an area of intense research, it is clear that highly specific regulation of these apparently redundant ‘eat me’ and ‘don’t eat me’ molecules ultimately determines whether a cell is viewed as ‘inedible’, and thus ignored by a phagocytic cell, or if it will be marked for engulfment and clearance from the body1.
Evidence that efferocytosis is impaired in atherosclerosis
An interesting aspect of efferocytosis is the speed and efficiency with which the process occurs9, 11. It has been estimated that under almost all conditions, cells undergoing apoptosis are recognized and targeted for clearance in a matter of minutes. Indeed, these cells are cleared so efficiently that it is nearly impossible to identify TUNEL-positive apoptotic bodies (A.B.’s) in any healthy tissue bed12, including organs with very high rates of cellular turnover such as the thymus, tonsil and bone marrow. In those rare instances when a pathologist is able to document an A.B. in healthy tissue, it is commonly in close proximity to a phagocytic cell. Such co-localization suggests that these apoptotic cells are in the process of being identified for clearance, and likely would not have been observed had the tissue been sampled a few minutes later.
One of the few exceptions to this rule is within the atherosclerotic plaque. Unlike the rest of the body, ‘free’ apoptotic bodies (which are not in close proximity to macrophages or other professional phagocytes) are commonly observed in the growing lesion. Careful quantification of the numbers of ‘free’ vs ‘associated’ A.B.’s has led Martinet and colleagues to conclude that the capacity for efferocytosis is reduced nearly 20-fold in plaque compared to elsewhere in the body13. Atheromas also contain far fewer phagocytes that have ingested multiple - as opposed to single - cellular corpses, again suggesting an impairment in efferocytosis13. While apoptosis is known to be increased during plaque formation, the capacity of the healthy phagocytes is normally so high that Tabas and colleagues have concluded in their review that the problem is not the consequence of too much cell death, but rather too little cell clearance6.
How impaired efferocytosis might promote vascular disease
The accumulation of apoptotic and necrotic debris has long been associated with atherogenesis and plaque vulnerability14–17. At its most simplistic level, expansion of the necrotic core can be viewed as contributing to plaque expansion, which in turn impinges on luminal flow, thus reducing coronary perfusion. However, impaired efferocytosis likely induces a number of other maladaptive changes beyond this mechanical impediment to blood flow. Many of these may be directly causal for atherosclerosis and plaque vulnerability13.
First, it is important to note that every time a phagocyte engulfs a dying cell it effectively doubles its intracellular content18. Because mammalian cells have difficulty metabolizing cholesterol, efferocytes must initiate reverse cholesterol transport machinery to avoid the intracellular accumulation of the membrane-derived lipids they have just ingested. Normally, externalized phosphatidylserine (PS) present on the surface of apoptotic cells upregulates ABCA1 in macrophages, which promotes the efflux of cholesterol to ApoA118, 19. Interestingly, necrotic cells (which also express PS) do not elicit a similar response18, and defective efferocytosis signaling has now been shown to suppress critical reverse cholesterol transport pathways in vascular cells19. The end result is the formation of foam cells, which are widely recognized as key drivers of atherosclerosis.
In addition to promoting salutary survival mechanisms that prevent the accumulation of potentially toxic cellular contents20, 21, physiologic efferocytosis is also known to directly suppress inflammation. When a macrophage has successfully cleared a dying cell, beneficial factors such as IL-10 and TGF-β are released, presumably to signal that no additional inflammatory cells need to be recruited to the site of injury22. The opposite occurs when a phagocyte is unable to clear an A.B., however, and nonresolving inflammation or autoimmunity may ensue19, 23, 24. Thus, failed efferocytosis appears to trigger a molecular switch in phagocytic cells, converting them from champions of inflammation resolution to drivers of vascular inflammation.
A final point to consider is that the reason apoptosis is classically considered an ‘immunologically silent’ form of cell death may be because dying cells are normally cleared before they have had the opportunity to undergo secondary necrosis4, 22. When efferocytosis is impaired, however, apoptotic bodies very rapidly experience breakdown of their cell membranes, which leads to the release of previously-sequestered intracellular contents into the interstitium16. These factors - which include plaque destabilizing proteases16, cytokines which promote plaque angiogenesis, and thrombogenic tissue factors25 – each could accelerate atherosclerosis and promote lesion vulnerability.
Taken together, defects in efferocytosis could theoretically effect much more than the physical growth of the interlesional necrotic core. Rather, they may also stimulate plaque inflammation (via release of chemokinetic cytokines), foam cell accumulation (via impaired reverse cholesterol transport) and plaque vulnerability (via atherothrombotic changes within the ECM). What has classically been viewed as a simple ‘waste management’ issue may actually play a larger role in the vascular biology of atherosclerosis (Figure 1).

Diseased and apoptotic cells in the growing atherosclerotic plaque are not recognized for efficient phagocytic clearance by lesional macrophages. While the mechanisms which drive this pathology are still an area of active investigation, emerging data suggests that this defect may be due to impaired ‘eat me’ (green) and ‘don’t eat me’ (red) signaling that renders these cells ‘inedible’. As a result, foam cells accumulate to promote lesion expansion and apoptotic tissue undergoes secondary necrosis to accelerate vascular inflammation and lesion instability.
Why is efferocytosis impaired in atherosclerosis?
It is not yet understood why efferocytosis is dysfunctional within the atherosclerotic plaque, when it appears to be so highly preserved elsewhere throughout the healthy human body13. Conceptually, the accumulation of apoptotic debris within the plaque must be due to at least one of the following problems, as previously outlined by Thorp and Tabas6: 1. Overwhelming apoptosis within the plaque; 2. An impairment in the phagocytic capacity of lesional macrophages; and/or 3. Reduced ‘edibility’ of apoptotic vascular cells.
While programmed cell death is certainly known to be increased during atherosclerosis26, experimental data suggest that the efferocytic system should have sufficient capacity to deal with this increase in apoptosis, at least initially2, 8. For example, studies in which macrophage apoptosis is induced in the beginning stages of lesion formation have found that triggered cell death actually prevents plaque expansion and promotes lesion stabilization27–29. Similarly, forced SMC apoptosis (accomplished by Bennett and colleagues via cell-specific expression of the human diphtheria toxin receptor), has no deleterious effect in normal vessels, and only becomes harmful when induced in the late stages of plaque development30. Thus, investigators have concluded that overwhelming apoptosis is not the reason uncleared cells accumulate in the diseased vessel wall, at least in the early stages of plaque development.
Conversely, several lines of evidence suggest that phagocytes may become less effective during atherogenesis. For example, macrophage ‘skewing’ toward the proinflammatory ‘M1’ phenotype in the plaque may reduce the number of beneficial ‘M2’ macrophages, thus inducing a relative deficit of those macrophages known to have a high phagocytic capacity31, 32. Additionally, reactive oxygen species such as peroxynitrite present in the plaque may impair the ability of macrophages to phagocytose apoptotic bodies13 and dendritic cells are known to also have reduced efferocytic capacity as they mature33, 34. While lipid-laden foam cells have been shown to retain their phagocytic ability at least to some extent13, 35, 36, vascular SMCs, which are known to possess potent ‘non-professional’ efferocytic capabilities, lose this functionality when exposed to oxidized lipids37. Thus, it is highly likely that the number or ratio of ‘competent’ efferocytes is reduced in or near the necrotic core, either through direct loss (e.g. apoptosis) or via the expansion of less effective phagocyte subpopulations (e.g. M1 macrophages and foamy SMCs).
Perhaps most importantly, diseased and dying vascular cells appear to become ‘poor substrates’ which are less ‘appealing’ to phagocytic cells than apoptotic tissue elsewhere in the body. This likely occurs for a number of reasons, including: 1. Genetic changes in subjects predisposed to atherosclerotic disease; 2. Inflammation-dependent modifications to efferocytosis signaling molecules within the plaque; and 3. The downstream sequelae of intralesional oxidized LDL.
First, studies investigating the heritable component of coronary artery disease (CAD) have found that carriers of the top genome-wide association study (GWAS) risk allele38–40 have reduced intraplaque expression of a key ‘eat me’ ligand known as Calreticulin19. As a result, these subjects develop larger lesions replete with diseased vascular cells that are presumably resistant to phagocytic clearance. The Calreticulin axis is discussed in further detail below, but these data suggest that impaired efferocytosis may be the mechanism underlying the most important commonly inherited locus for atherosclerosis (the 9p21 locus), and may explain how these variants promote risk independently of all known traditional risk factors (e.g. hypertension, dyslipidemia, diabetes and smoking)41.
Second, emerging evidence has now linked inflammatory signaling directly to intra-arterial defects in efferocytosis-related ligand expression (Figure 2). For example, TNF-α, a cytokine known to be causal for- and upregulated in- atherosclerosis, triggers expression of a key ‘don’t eat me’ molecule known as CD47 on the surface of apoptotic vascular SMCs42. Similarly, inflammation related to toll-like receptor signaling may suppress the expression of pro-efferocytic ligands such as MFGE843, and inactivating post-translational modifications to other key ‘eat me’ ligands such as MerTK and LRP1 are induced under inflammatory conditions6, 8, 44, 45, as discussed below. Because failed efferocytosis induces the secretion of TNF-α16, and TNF-α renders cells even more resistant to clearance, it is highly likely that an endless loop is active in the plaque, and that this may explain why efferocytosis is so impaired during atherogenesis. These data directly support the ‘inflammatory hypothesis’ of atherosclerosis46.

Experimental data suggests that pro-phagocytic signals (including calreticulin, Mfg-e8 and Mertk) are reduced in atherosclerosis due to inflammation, post-translational modifications and/or genetic variability. Exacerbating this loss of ‘eat me’ signaling is a concomitant upregulation of the CD47-Sirp-α ‘don’t eat me’ pathway, which furthers decreases the ‘edibility’ of cells within the necrotic core. The end result is that apoptotic cells in the growing plaque becomes poor substrates for phagocytic cells such as macrophages and dendritic cells. Such uncleared cells become secondarily necrotic and release additional proinflammatory stimuli, thus promoting a positive feedback loop.
Finally, the oxidized LDL present within the atherosclerotic plaque may also directly render apoptotic cells inedible13. OxLDL induces the generation of autoantibodies which decorate and presumably mask oxidized ‘eat me’ ligands on the surface of dying cells in the lesion47, 48. OxLDL is also known to directly compete with apoptotic bodies for scavenger receptors, making them less likely to be cleared through competitive inhibition23.
Taken together, we are still learning why PrCR becomes defective during atherogenesis13 and with ageing49. However, the problem appears to involve multiple pathways, including processes that impair the capacity of the phagocytes present in the plaque and factors that render diseased vascular cells less likely to be cleared. As with most other aspects of vascular biology, genetic and inflammatory factors appear to play central roles in the pathobiology of lesional efferocytosis, and will likely emerge as areas of focus for future translational studies.
Pathways already specifically linked to efferocytosis in vascular disease
Several factors involved in PrCR have now been experimentally linked to apoptotic debris retention or necrotic core expansion in the growing atherosclerotic plaque (Table). These include the bridging molecule, complement C1q62, the phagocyte receptor, transglutaminase 2 (TG2)63, and potentially other ‘eat me’ ligands such as lysoPC and Fas65, 67. However, it is likely that some phagocytosis-related molecules have greater relevance to vascular disease than others, and emerging studies have identified a handful of efferocytosis pathways that appear to have a disproportionate impact on atherogenesis.
Table
Table summary of putative ‘find me’, ‘eat me’ and ‘don’t eat me’ molecules that have been shown to alter atherosclerosis or features of plaque vulnerability, with notes about the animal models used and an interpretation of the authors’ findings.
| Gene | Type of molecule | Mouse Model | In vivo findings | In vitro findings and additional notes | Expression | Ref |
|---|---|---|---|---|---|---|
| Mertk | Phagocytic receptor | Mertkkd;Apoe−/− | No change in plaque size (after 10 or 16 weeks of HFD) Increased:
| Reported to bind the bridging molecule, Gas6 | Present on macrophages | 50 |
| Mertk | Phagocytic receptor | Ldlr−/−transplanted with Mertk−/− bone marrow | Increased:
|
| Present on MΦ. Reportedly not present on vascular SMCs | 51 |
| Lrp1 | Phagocytic receptor | SMC Lrp−/−;Ldlr−/− | Increased:
| Activation of PDGF-receptor signaling | Expressed on SMCs | 52 |
| Lrp1 | Phagocytic receptor | Ldlr−/−transplanted with MΦLrp1−/− bone marrow | Increased:
|
| 53 | |
| Lrp1 | Phagocytic receptor | Ldlr−/−transplanted with MΦLrp1−/− bone marrow | Increased:
|
| 54 | |
| Lrp1 | Phagocytic receptor | MΦ Lrp1−/−;Apoe−/− Ldlr−/−transplanted with MΦLrp1−/−;Apoe−/− bone marrow | Increased:
|
| 55 | |
| Lrp1 | Phagocytic receptor | Ldlr−/−transplanted with MΦLrp1−/−, MΦLrp1−/−;Apoe−/− bone marrow Treated with TNF-α inhibitor | Increased:
|
| 56 | |
| SR-B1 | Phagocytic receptor | Apoe−/−transplanted with SR-B1−/−; Apoe −/− bone marrow Ldlr−/−transplanted with SR-B1−/−, SR-B1−/−; Apoe −/−bone marrow | Increased:
| Reduced phagocytic capacity and anti-inflammatory cytokine production, more inflammatory cytokine production in SR-B1−/−macrophages | 57 | |
| Tim-1/Tim-4 | Phagocytic receptor | Ldlr−/− treated with Tim-1 or Tim-4 blocking antibodies | Increased:
| Reduced phagocytic capacity. Increased IL6 and MCP1 in Tim4 Ab treated macrophages | Present on T cells (Tim-1), macrophages and dendritic cells (Tim-4) | 58 |
| G2A | Possible phagocytic receptor | G2A−/−;Apoe−/ − Apoe−/−transplanted with G2A−/−; Apoe −/− bone marrow Ldlr−/−transplanted with G2A−/−;Ldlr−/−bone marrow | Increased:
|
| 59 | |
| CX3CL1 | Find me ligand | CX3CL1−/−;Apoe−/ − CX3CL1−/−;Ldlr−/ − | Decreased:
| Effects possibly due to reduced chemoattractant and adhesive properties | 60 | |
| Mfge8 | Bridging molecule | Ldlr−/−transplanted with Mfge8−/− bone marrow | Increased:
|
| Present on ECs, SMCs and MΦ. Downregualted on MΦs during atherogenesis | 61 |
| C1q | Bridging molecule | C1q−/−;Ldlr−/−with standard diet | Increased:
|
| 62 | |
| TG2 | Bridging molecule | Ldlr−/−transplanted with TG2−/− bone marrow | Increased:
|
| Expressed in plaque intimal cells | 63 |
| Gas-6 | Bridging molecule | Gas6−/−;Apoe−/ − with standard diet | No change in overall plaque size Increased:
| Expressed by ECs, SMCs and MΦ in plaque. High in ruptured plaque, TCFA | 64 | |
| Fas | May stimulate Find me signaling | Faslpr/lpr;Apoe−/− | Increased:
| Enlarged spleen, thymus, lymph node Renal damage Reduced circulating T cells, cholesterol, oxidized phosphor lipid on apoB-100 containing lipoprotein, bone mineral density | 65 | |
| Fas ligand | May stimulate Find me signaling | Gld (fasL mutation);Apoe−/− | Increased:
|
| 66 | |
| Calreticulin | Eat me ligand | Cdkn2b−/−;Apoe−/ −(animals have reduced Calr, indirectly suggesting a role for this gene which was confirmed by in vitro assays) | Increased:
|
| 19 | |
| CD47 | Don’t eat-me ligand | Apoe −/− treated with CD47 blocking antibody | Decreased:
|
| Upregulated in advanced atherosclerosis and in the necrotic core | 42 |
Mfge8
First among these is the secreted glycoprotein, milk fat globule-EGF factor 8 (Mfge8, also known as lactadherin)61. Mfge8 functions as a bridging molecule that tethers the phagocyte to its target, by simultaneously binding both αvβ3 integrin on the macrophage and externalized phosphatidylserine (PS) on the apoptotic body68. Mallat and colleagues reported that Mfge8 is expressed in human vascular tissue, but appears to be reduced in advanced plaque, particularly in lesions with a high burden of TUNEL-positive apoptotic cells. In animal models, atheroprone LDL receptor knockout mice (ldlr−/−) transplanted with Mfge8−/− bone marrow developed systemic inflammation and advanced atherosclerotic plaques which had larger necrotic cores than control animals, even though loss of Mfge8 did not directly increase the susceptibility of macrophages to apoptosis in vitro. Of note, Mfge8 may also bind directly to TG269, another efferocytosis molecule previously linked to reverse cholesterol transport and plaque development63. Taken together, these data suggest that Mfge8 produced by bone marrow-derived macrophages is necessary to maintain efferocytosis and prevent inflammation, and that loss of this factor may promote atherosclerosis.
Mertk
The second efferocytic factor clearly linked to atherosclerosis is Mer receptor tyrosine kinase (mertk). This receptor is present on the surface of phagocytic cells and mediates engulfment of apoptotic bodies via the critical bridging molecule, Gas670. Mallat and colleagues previously reported that MERTK is present on macrophages (but not SMCs) in human atherosclerotic plaque, and that atheroprone ldlr−/− mice transplanted with mertk−/− bone marrow develop significantly larger lesions that have larger necrotic cores, more uncleared apoptotic cells and increased inflammation, relative to control animals51. These findings were extended by Tabas and colleagues, who found that mice carrying a kinase-defective form of Mertk (MertkKD) generated lesions with more TUNEL-positive cells and more plaque necrosis than control animals on the apoe−/− background50. Mertk is also required for the clearance of apoptotic cardiomyoctyes after myocardial infarction, and its deficiency has been shown to promote cardiomyopathy in mouse models, extending its relevance to other cardiovascular disorders71. Of interest, the mertk receptor can be cleaved by metalloproteinases into an inactive soluble form (solMER), and this process leads to competitive inhibition of efferocytosis by providing a decoy receptor for Gas644. Because solMer shedding is enhanced by pro-inflammatory stimuli commonly observed in vascular disease44, 72, it is possible that this post-translational modification may play a role in suppressing efferocytosis during atherogenesis.
Calr/LRP1 and CD47
The final and perhaps most exciting PrCR pathway that has been linked to atherosclerosis is the one involving the pro-phagocytic Calr/LRP1 axis, and its counterbalancing ‘don’t eat me’ molecule, CD47. Calreticulin is a highly conserved chaperone protein that is now known to be upregulated and redistributed on the surface of cells undergoing programmed cell death10. After physically associating with phosphatidylserine (the other key ‘eat me’ ligand found on apoptotic cells), Calr transactivates LRP1 on the surface of adjacent phagocytic cells and induces engulfment. Emerging evidence suggests that Calr and LRP1 are critical mediators of efferocytosis, as supported by the fact that global knockout of either factor is embryonically lethal73, 74.
Tissue-specific modulation of LRP1 has confirmed its central role in atherosclerosis. For example, ldlr−/−mice lacking LRP1 in the SMC (SM22Cre+/LRPflox/flox) develop dramatic, near-occlusive atherosclerotic lesions and aortic aneurysms52. Similarly, a series of studies by Fazio and colleagues have confirmed that loss of LRP1 in bone marrow derived macrophages impairs efferocytosis and promotes vascular inflammation, necrotic core accumulation and lesion growth, without having any impact on systemic lipid levels53, 54. The fact that loss of this efferocytosis receptor on either professional (e.g. macrophages) or non-professional (e.g. vascular SMCs) phagocytes was sufficient to significantly increase atherosclerosis highlights the importance of this pathway in vascular disease.
While no studies have specifically investigated conditional or cell-specific knockout of Calr in murine atherosclerosis models, other evidence has confirmed a role for LRP1’s pro-efferocytic ligand in the prevention of atherosclerosis. For example, carriers of the risk allele at the chromosome 9p21 GWAS locus have now been shown to have reduced intraplaque expression of Calr due to an inherited defect in TGFB signaling19, 39. Mice deficient in one of the top 9p21 candidate genes (Cdkn2b) have reduced Calr expression and develop markedly larger atherosclerotic plaques that have several features of lesion instability including larger necrotic cores19. In vitro, apoptotic vascular SMCs deficient in Calr not only resist clearance by neighboring cells, but also promote juxtacrine changes in co-cultured macrophages, including a propensity to adopt a foam-cell phenotype, suppress reverse cholesterol transport, and secrete pro-atherosclerotic cytokines. Interestingly, these in vitro defects can be reversed with exogenous Calr peptide, suggesting that targeted reactivation of efferocytosis could prevent macrophage inflammation in atherosclerosis.
It is important to note, however, that Calr is also expressed on some non-apoptotic cells, suggesting the existence of a counterbalancing mechanism which prevents the off-target clearance of healthy tissue10. Oldenborg and colleagues have now shown that the key ‘don’t eat me’ molecule, CD47, fulfills this role by triggering anti-efferocytic signaling cascades downstream of the SIRPα receptor on phagocytic cells1, 75. During PrCD, CD47 is rapidly downregulated and redistributed away from Calr, thus allowing unopposed LRP1 activation and successful engulfment10.
Paradoxically, CD47 is upregulated in atherosclerosis42. This surprising observation results from a TNF-α-dependent signaling cascade through NFKB, which blunts the fall in CD47 expression normally expected to occur during apoptosis. As a result, these apoptotic vascular cells are rendered inedible and presumably contribute to the growth of the necrotic core. Confirming the critical role of this pathway in atherogenesis, CD47 blocking antibodies were found to have a profound antiatherosclerotic effect in several mouse models, including the ability to prevent progression of established lesions, protect against plaque rupture and induce regression of the necrotic core42. Taken together, the studies provide compelling evidence that perturbations in either pro-efferocytic Calr-LRP1 signaling or anti-phagocytic CD47-SIRP signaling is sufficient to cause atherosclerotic disease, and prioritize these axes for additional investigation.
Opportunities for translation
Efferocytosis has tremendous potential as both a diagnostic and therapeutic target. For example, CD47 is not only upregulated in patients with plaque compared to no plaque, but also in subjects with symptomatic carotid disease (e.g. those with TIA or stroke) compared to those with an asymptomatic stenosis42. Given the data that genetic variants may correlate with alterations in efferocytosis gene expression19, it may be that a biomarker panel that combines genomic risk variants and expression profiling76 could have utility in diagnosing individuals at risk for clinical events due to necrotic core expansion and plaque vulnerability.
Additionally, therapies that appear to specifically reactivate efferocytosis are already being tested in humans. In the oncology field, Weissman and colleagues have recently found that a major mechanism by which cancer cells evade the tumoricidal macrophage is by upregulating ‘don’t eat me’ molecules on their surface77, 78. Accordingly, humanized antibodies and decoy molecules have been developed that interrupt these pathways and restore the phagocytosis of malignant cells79. Preliminary studies suggest that these therapies may have an acceptable toxicity profile in non-human primates80 and phase I cancer studies are already underway (ClinicalTrials.gov: NCT02678338). Assuming that these treatments are found to be safe and effective, there will be a very unusual opportunity to leverage the experience of the immuno-oncology field and rapidly translate these treatments into the cardiovascular realm.
Further, diseases associated with impaired efferocytosis seem particularly susceptible to combination therapies. In cancer models, anti-tumor antibodies such as rituximab synergize with pro-efferocytic therapies to dramatically accelerate tumor clearance79, 81. There is a clear mechanistic justification to consider a similar approach in cardiovascular disease, particularly given the link between vascular inflammation and the imbalance of efferocytosis ligand expression. As described above, levels of functional Mertk72, LRP145 and Mfge843 all appear to be reduced under pro-inflammatory conditions. The hypothesis that anti-inflammatory therapies may restore pro-efferocytic signaling has specifically been tested in the case of CD47, which is now known to be directly downstream of TNF-α. Humans treated with the anti-TNF-α antibodies Infliximab or Etanercept have reduced in vivo expression of CD47, and mice treated with combination anti-TNF-α/anti-CD47 therapy display a modest incremental reduction in atherosclerosis over anti-CD47 alone42. Given that patients treated with anti-TNF-α therapy for rheumatological conditions appear to be protected from myocardial infarction and other adverse cardiovascular outcomes82, there is a strong rationale for combining directed anti-inflammatory and pro-efferocytic therapies in the treatment of atherosclerosis.
A final consideration is that pro-efferocytic therapies may provide an actionable opportunity for the oft-mentioned concept of precision cardiovascular medicine83. Genetic studies pursuing the heritable component of cardiovascular disease have shown that the majority of genome-wide significant loci confer risk for myocardial infarction independently of all classical risk pathways, suggesting a novel mechanism of action which is not being addressed by lipid lowering or antihypertensive therapies38, 84, 85. Given that the top GWAS locus is now known to be associated with a reduction in intravascular ‘eat me’ ligand expression19, it is highly likely that these individuals will particularly benefit from genotype-driven, pro-efferocytic therapy, similar to oncology patients who receive tailored chemotherapy directed against their personal cancer mutation. While it is expected that all subjects with atherosclerosis will benefit from pro-efferocytic therapies, pharmacogenomic-based approaches could lead to outsized effects and even induce plaque regression.
Areas for future study
Although much has been learned about the role of efferocytosis in atherosclerosis over the last decade, several aspects of this process are yet to be explored. A better understanding of the nuances surrounding these pathways in health and disease should enhance our fundamental understanding of vascular biology and facilitate the translation of these findings from bench to bedside.
The first is the potential that pro-efferocytic therapies may have unanticipated drawbacks. For example, CD47 blocking antibodies are generally well tolerated, but do induce erythrophagocytosis and anemia under some conditions42, 77. While this toxicity can be ameliorated with dose escalation or reduced-dose combination therapy approaches42, 80, other pro-efferocytic therapies may not be as specific for diseased tissue. Thus, the spatial and temporal changes in ‘eat me’ and ‘don’t eat me’ ligands that must occur in order to render a cell susceptible to phagocytic clearance need to be defined. Likely, the ratio and physical co-localization of these molecules provides an integrated ‘signature’ that determines a cell’s edibility10. Such information will explain how the body protects against off-target clearance of healthy tissue and should inform the development of pro-efferocytic therapies that do not induce removal of viable cells. Along these lines, the local delivery of pro-efferocytic therapies (e.g. with drug eluting stent86 or targeted nanoparticle technology87) may prove highly effective, and could have a superior safety profile.
Second, the link between cancer and cardiovascular disease requires further investigation19, 42. The recent finding that a common pathway may underlie growth of both tumors and atheromas raises questions about whether the clonal hypothesis of atherosclerosis needs to be revisited88. Cancer stem cells are now known to express high levels of CD47 and they may use this factor to evade immune surveillance89, 90. Whether a similar process occurs on a pre-atherosclerotic clone could be investigated with advanced lineage tracing modalities. Recent studies have suggested there is far more plasticity of vessel wall cells during atherogenesis than previously appreciated91, 92, and it may be that efferocytosis pathways are involved in such cell-fate decision making and expansion of a diseased subset of cells.
Third, the role of the vascular SMC in efferocytosis must be defined. While many of the studies described above focus on macrophage phagocytosis, it is clear that other vascular cells also participate in efferocytosis during plaque development. Elegant in vivo lineage tracing studies have now unequivocally confirmed that SMCs ‘de-differentiate’ and assume a ‘macrophage-like’ phenotype during atherogenesis, based on their ability to upregulate markers previously thought to be ‘macrophage-specific’, such as MAC-291, 92. While the physiologic properties of these ‘phenotypically-modulated’ cells remains an area of intense investigation, in vitro studies suggest that these cells have reduced phagocytic capacity compared to bona fide bone marrow-derived macrophages93. Because careful quantification of advanced lesions from lineage-traced mice has shown that a significant percentage of lesional ‘macrophages’ are actually of smooth muscle and not myeloid origin92, we must determine if their efferocytic program is governed by pathways different from those reported in classical macrophages. If so, different therapies may be required to target specific components of the evolving lesion.
Finally, the intersection with related physiologic pathways and vascular disorders should be pursued. For example, several classical cardiovascular risk factors, including dyslipidemia, smoking and shear stress have been linked to efferocytosis94–96. Statins and PPAR-γ agonists have been shown to enhance efferocytosis, and experimental data supports the concept that enhancing blood flow or targeting of the angiotensin pathway could ameliorate defects in efferocytic signaling31, 95–97. Exciting new work exploring novel processes related to atherosclerosis including necroptosis (programmed cell necrosis)98and autophagy99 has shown that these mechanisms may also regulate efferocytosis, and could become viable theranostic targets, as well. While efferocytosis has been studied in post-infarct cardiomyopathy71, we do not yet know its role in other vascular diseases such as restenosis, pathologic angiogenesis or aneurysm disease. Given the simultaneous link to atherosclerosis, acute inflammation and clearance of apoptotic cardiomyocytes after myocardial infarction, pro-efferocytic therapies should be considered as a potential therapy for the early window after an acute coronary syndrome.
Conclusions
While the preceding 50 years have been marked by major improvements in survival from cardiovascular disease, many of these trends have begun to plateau or even worsen in recent years100. Some of these changes may be due to increases in obesity-related conditions such as diabetes, but others are likely due to the diminishing returns being achieved with the addition of new drugs that target risk factors for which effective therapies already exist. The recent discovery that efferocytosis is impaired in vascular disease represents a major advance in our understanding of the root cause of atherosclerosis and why necrotic debris accumulates over time. The advent of pro-efferocytic therapies that allow for the removal of diseased and apoptotic cells could allow for an entirely new platform of treatment that specifically targets the necrotic core, should these agents prove to be specific and safe in ongoing oncology trials.
Acknowledgments
Sources of Funding: This manuscript was supported by the National Institutes of Health (R01HL12522401 and R01HL12337001 to NJL).