Re: Re: Ubiquitin과 단백분해 in neuronal function and dysfunction - nature 리뷰
작성자문형철작성시간20.08.24조회수1,146 목록 댓글 0beyond reason
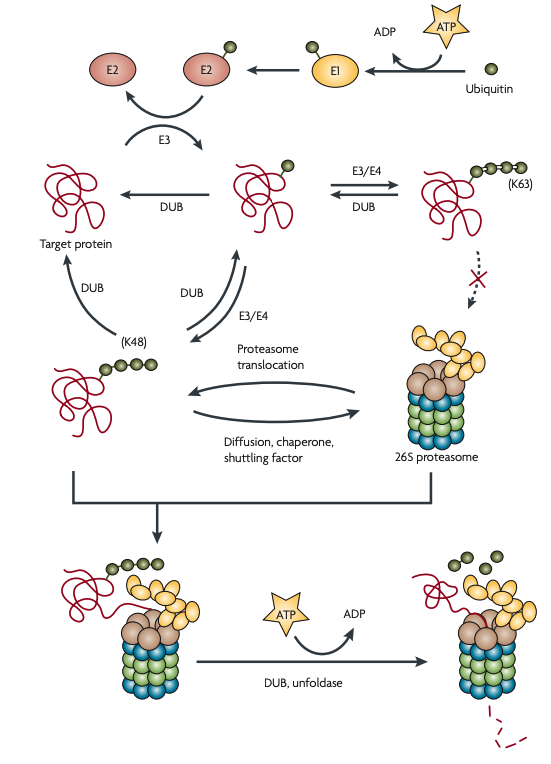
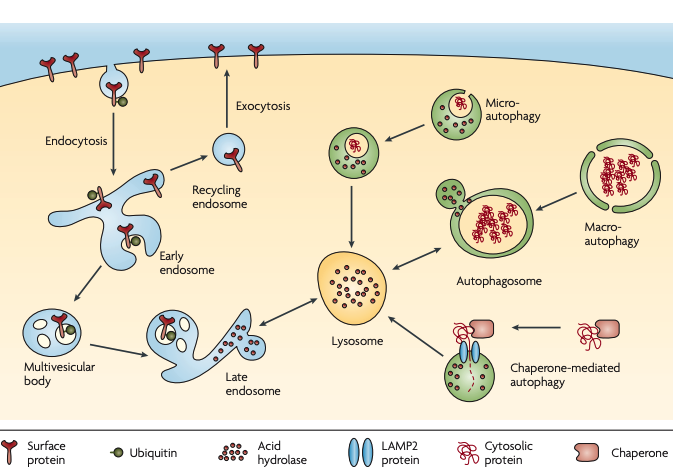
Figure 2 | Lysosomal pathways. Lysosomes are organelles that contain acid hydrolases that break down biopolymers and other biomolecules17. The primary catabolic route of plasma membrane proteins is endocytosis followed by sorting to late endosomes and lysosomes. Many types of signals can regulate endocytosis and sorting, including monoubiquitylation and K63-polyubquitylation (not shown)7,145. Endocytosed membrane proteins first arrive at early endosomes and subsequently get sorted into recycling endosomes (for return to the surface by exocytosis) or multivesicular bodies146 (for transport to late endosomes or lysosomes). The acid hydrolases in the lumen of lysosomes (pH 4–5) and late endosomes (pH 5–6) are highly active in acidic environments but loose their activities in the cytosol (pH ~7.2). The confinement and the pH-dependence of hydrolases provide dual safeguards17,147. Intracellular proteins can enter lysosomes through several autophagic mechanisms18. In macroautophagy, large amounts of cytosolic materials or even organelles are surrounded by a double-membrane structure (autophagosome) that fuses with lysosomes. In microautophagy, a small amount of the cytoplasm is internalized through lysosomal invagination. In chaperone-mediated autophagy, proteins that have been unfolded by the chaperone translocate into the lysosome through interactions with lysosome-associated membrane protein 2 (LAMP2)148.
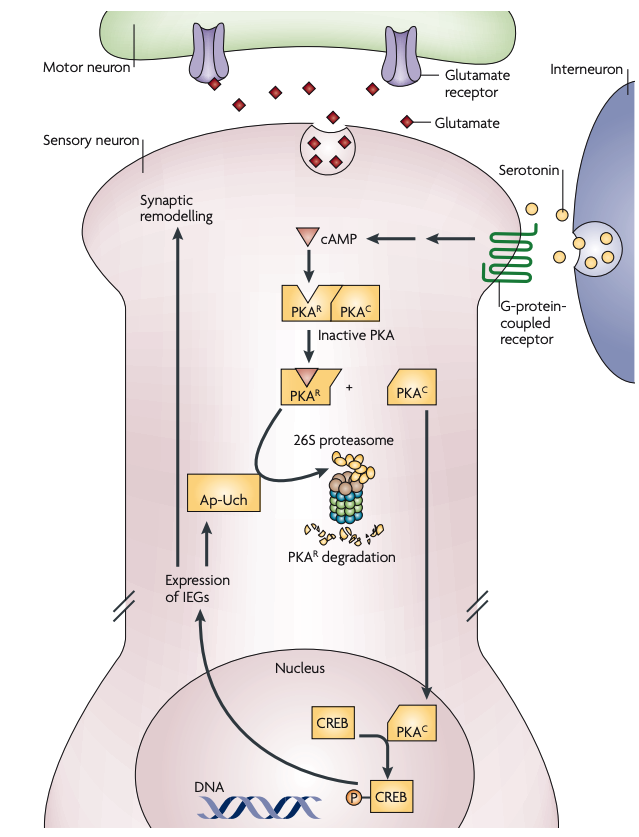
Figure 3 | Aplysia LTF: regulation of PKA by the UPS. In baseline conditions, the catalytic subunit of protein kinase A (PKAC) is bound to the regulatory subunit of PKA (PKAR) and remains inhibited. When serotonin is released, adenylyl cyclase is activated through G-protein-coupled receptors, leading to increased cyclic AMP levels in the postsynaptic cell. The competitive binding of cAMP to PKAR releases PKAC, which in turn translocates into the nucleus and phosphorylates cAMP-response-element-binding protein (CREB). Phosphorylated CREB promotes the transcription of immediate-early genes (IEGs), initiating a cascade of cellular processes that leads to synaptic remodelling. One of these IEGs is Aplysia ubiquitin C-terminal hydrolase (Ap-Uch). Ap-Uch is a deubiquitylating enzyme that binds to the proteasome and enhances the degradation of ubiquitylated substrates, including PKAR. This positive-feedback control can lead to long-lasting PKA activities that underlie long-term facilitation (LTF)23,25–27. UPS, ubiquitin–proteasome system.
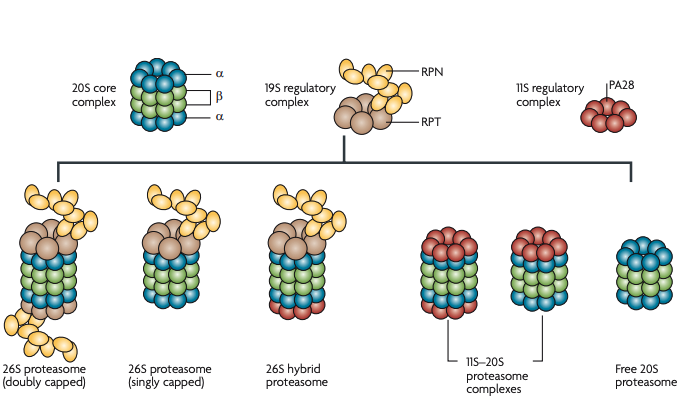
Figure 4 | Proteasome structure and heterogeneity. The proteasome is a large, multisubunit protease complex114. The 20S core complex is composed of four heptameric rings of α1–7 and β1–7 subunits. The proteolytic activities are conferred by three of the β subunits in the interior of the barrel. The 19S regulatory complex, or PA700, contains a hexameric ring of AAA-ATPase subunits (RPT1–6) and ~12 non-ATPase (RPN) subunits. The 19S complex receives the K48-polyubiquitylated substrate, removes the ubiquitin and, using ATPase activity, unfolds the protein for translocation into the 20S chamber114. Another complex that regulates the 20S complex, called 11S, REG or PA28, is a heptameric ring of PA28 subunits (α, β and γ) and is highly abundant in mammalian cells. This complex is thought to activate the 20S complex by widening the pore115. The mix and match of 20S complexes, 19S complexes and 11S complexes gives rise to a variety of proteasomes116. Owing to experimental difficulties in characterizing proteasome heterogeneity, the nomenclature of proteasome subtypes is not yet standardized in the literature. Different proteasome subtypes and the names that we have used for them in this article are shown. In this article we use ‘26S proteasome’ to denote singly and doubly capped species as well as hybrid proteasomes, because all three subtypes can degrade K48-polyubiquitylated proteins.
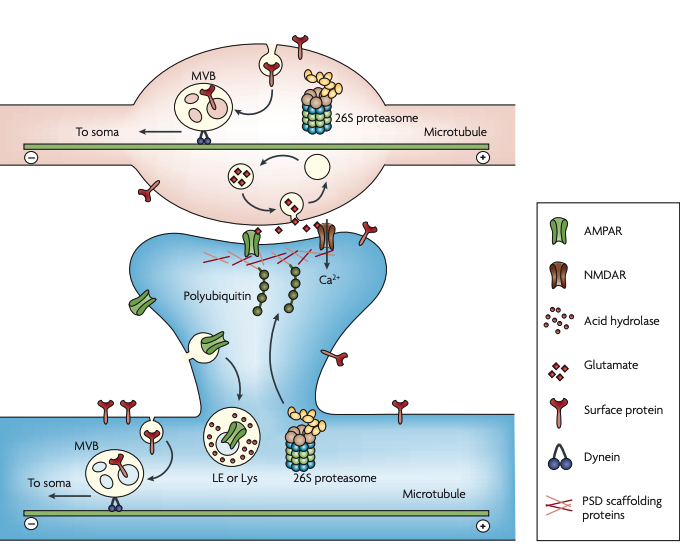
Figure 5 | Degradation of synaptic proteins. A schematic representation of multiple synaptic degradative events mentioned in the text. The ubiquitin–proteasome system (UPS) regulates the recycling and the release of synaptic vesicles at the presynaptic terminal (axonal bouton, top). Released glutamate activates AMPARs (α-amino-3-hydroxy5-methyl-4-isoxazole propionic acid receptors) and NMDARs (N-methyl-d-aspartate receptors) at the postsynaptic terminal (dendritic spine, bottom). This can trigger the polyubiquitylation of postsynaptic density (PSD) scaffolding proteins and the translocation of 26S proteasomes into spines. The degradation of PSD scaffolding proteins leads to the de-anchoring of AMPARs, which are endocytosed at the lateral zone of the spine. Endocytosed AMPARs that have been marked for degradation are sorted into late endosomes (LEs) or lysosomes (Lys), which contain acid hydrolases. In mature hippocampal neurons, endocytosis occurs along the entire dendrite and at the presynaptic terminal but not in the shaft of axons. In both cases of endocytosis, many internalized proteins are transported in multivesicular bodies (MVBs) to the soma for breakdown. Retrograde vesicular traffic requires dynein motors that travel towards the minus end of the microtubule.

Ubiquitin, the proteasome and protein degradation in neuronal function and dysfunction
- Hwan-Ching Tai &
- Erin M. Schuman
Nature Reviews Neuroscience volume 9, pages826–838(2008) Cite this article
1274 Accesses
268 Citations
13 Altmetric
Key Points
In neurons as well as other eukaryotic cells, intracellular proteins are primarily degraded by the ubiquitin–proteasome system (UPS), and membrane proteins by the lysosome system. Ubiquitylation tags proteins for proteasomal degradation and vesicular trafficking.
Protein degradation is important for synaptic plasticity and self-renewal of neurons. The UPS can regulate presynaptic processes such as vesicle release and postsynaptic processes such as glutamate receptor turnover and postsynaptic density (PSD) reorganization.
Many neurodegenerative disorders, such as Alzheimer's disease and Huntington's disease, are characterized by aggregates in the brain of ubiquitin-positive proteins that failed to be degraded. PARK2 (which encodes the E3 ligase parkin) mutations lead to familial Parkinson's disease, suggesting that degradative dysfunction can trigger neurodegeneration. Mutations in many UPS and lysosomal genes are now linked to neurological disorders.
The morphology of neurons creates special demands for protein degradation with respect to membrane-protein turnover and substrate delivery to proteolytic machineries. This may explain why neurons are more susceptible to protein aggregation when protein degradation is impaired.
Protein degradation in axons and dendrites exhibits several unique features, including long-range retrograde transport of endocytosed proteins and the translocation of proteasomes into dendritic spines.
Various UPS components, including E3 ligases, deubiquitylating enzymes, chaperones, shuttling factors and different subtypes of proteasomes, form complex interaction networks in neurons. We know that individual components can have important functions, but detailed mechanisms remain elusive.
Enhancing protein degradation in the brain is a potential therapeutic strategy for aggregate-related neurodegenerative disorders. Another possibility is to enhance molecular chaperones that prevent protein misfolding.
Abstract
Eukaryotic protein degradation by the proteasome and the lysosome is a dynamic and complex process in which ubiquitin has a key regulatory role. The distinctive morphology of the postmitotic neuron creates unique challenges for protein degradation systems with respect to cell-surface protein turnover and substrate delivery to proteolytic machineries that are required for both synaptic plasticity and self-renewal. Moreover, the discovery of ubiquitin-positive protein aggregates in a wide spectrum of neurodegenerative diseases underlines the importance and vulnerability of the degradative system in neurons. In this article, we discuss the molecular mechanism of protein degradation in the neuron with respect to both its function and its dysfunction.