Re:The Macrophage ‘Do not eat me’ signal, CD47, is a clinically validated c
작성자문형철작성시간20.09.02조회수347 목록 댓글 0beyond reason
참고) 위키디피아
CD47 (Cluster of Differentiation 47) also known as integrin associated protein (IAP) is a transmembrane protein that in humans is encoded by the CD47 gene. CD47 belongs to the immunoglobulin superfamily[5] and partners with membrane integrins and also binds the ligands thrombospondin-1 (TSP-1) and signal-regulatory protein alpha (SIRPα).[6] CD-47 acts as a don't eat me signal to macrophages of the immune system which has made it a potential therapeutic target in some cancers, and more recently, for the treatment of pulmonary fibrosis.[7] CD47 is involved in a range of cellular processes, including apoptosis, proliferation, adhesion, and migration. Furthermore, it plays a key role in immune and angiogenic responses. CD47 is ubiquitously expressed in human cells and has been found to be overexpressed in many different tumor cells.[6][8] Expression in equine cutaneous tumors has been reported as well.[9]
The Macrophage ‘Do not eat me’ signal, CD47, is a clinically validated cancer immunotherapy target

CD47 is a potent ‘do not eat me’ signal that enables cancer cells to evade detection by the innate immune system, thereby avoiding destruction by first responder cells, such as macrophages. CD47 overexpression is common in solid and hematological tumors including acute leukemia, non-Hodgkin’s lymphoma (NHL), colorectal, and ovarian cancers. In many malignancies, its expression correlates with an aggressive phenotype and an overall poor clinical prognosis. Inhibition of CD47 signaling enhances macrophage phagocytic activity, and in preclinical models, leads to impaired tumor growth, inhibition of metastatic spread, and tumor regression. Enhancing the innate immune system’s antitumor response by removing macrophage inhibitory signals is analogous to how classic immune checkpoint inhibitors augment T-cell-mediated adaptive immunity by targeting CTLA4 and PD-1. Furthermore, CD47 blockade and enhanced phagocytosis by antigen-presenting cells can enhance antigen priming of T cells, thereby providing a bridge between the innate and adaptive arms of the immune system. Thus, the inhibition CD47 signaling is a promising strategy for treating a broad range of cancers. A growing number of CD47-targeting agents have recently entered clinical trials.
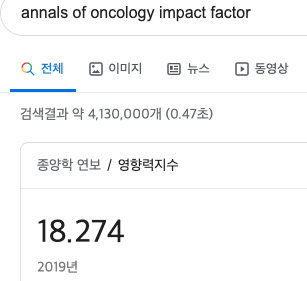
CD47 is a 50 kDa cell surface glycoprotein that is ubiquitously expressed in normal and malignant tissues [1]. It inhibits cellular phagocytosis through its interaction with signal receptor protein-alpha (SIRPα), which is expressed on phagocytic cells, such as macrophages and dendritic cells. However, blocking CD47 alone is insufficient to cause phagocytosis because additional prophagocytic signals, such as calreticulin are also required [2]. Most normal cells, with the notable exception of aging red blood cells (RBCs), lack these prophagocytic signals and remain unaffected by CD47 blockade [2]. In contrast, because cancer cells frequently express prophagocytic signals, the inhibition of CD47 signaling renders them susceptible to phagocytosis (Figure 1) [2, 3].
Normal tissues express low levels of CD47, but overexpression in tumors is common and may predict poor clinical outcomes. High expression of CD47 was first well characterized in human AML blasts [4] and overexpression was subsequently found in a broad range of solid tumors including cancers of the ovary, breast, colon, bladder, prostate, brain, and liver, as well as in other hematological malignancies such as NHL, chronic myeloid leukemia in blast crises, acute lymphoblastic leukemia, multiple myeloma and others . Thus, the overexpression of CD47 on tumors is widespread, and its presence often heralds a worse clinical prognosis.
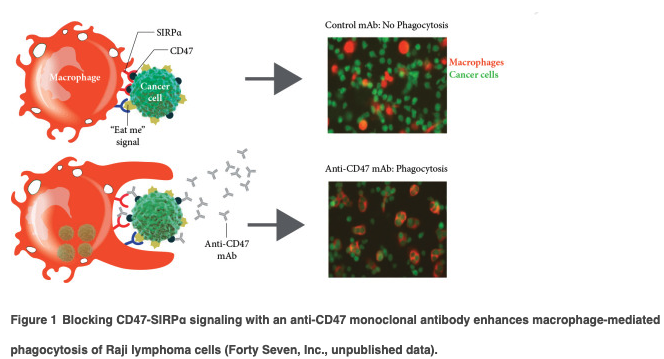
Inhibitors of CD47 are broadly active in preclinical antitumor models (see supplementary Appendix, available at Annals of Oncology online) and this activity is enhanced when combined with cancer targeting antibodies that provide exogenous prophagocytic signals. For example, synergistic suppression of tumor growth is observed when CD47-targeting agents are combined with monoclonal antibodies, such as rituximab, cetuximab, or trastuzumab (Figure 2). These antibodies all have active Fc domains that bind to Fc-gamma receptors on macrophages resulting in a stimulation of phagocytosis. This mechanism provides a rationale for exploring combinations of antitumor antibodies with CD47 targeting agents in clinical trials.
Finally, CD47 blockade can also enhance antitumor T-lymphocyte-mediated adaptive immune responses. The phagocytosis of tumor cells facilitates tumor antigen processing and presentation to T cells leading to enhanced priming of cytotoxic T lymphocytes. Thus, CD47-mediated antitumor effects may directly involve both the innate and adaptive immune systems. This provides a strong scientific rationale for evaluating CD47 targeting agents in combination with checkpoint inhibitors in clinical trials.
Despite widespread expression of CD47 in normal tissues, non-human primates tolerated treatment with IgG4-based CD47-blocking agents in preclinical models, even at high exposures []. This is due to the absence of prophagocytic signals on most normal tissues, which renders them unaffected by CD47 blockade. However, aging RBCs are a notable exception. Older RBCs gradually lose CD47 expression and accumulate prophagocytic signals, eventually undergoing phagocytic removal by splenic red pulp and sinusoidal macrophages in liver and bone marrow, respectively. Thus, agents that unequivocally block CD47 signaling can cause treatment-related anemia. However, younger RBCs are not susceptible to CD47-mediated phagocytosis. In preclinical toxicology studies with the anti-CD47 antibody, Hu5F9-G4 (5F9), the use of an initial, low priming dose followed by higher maintenance doses is effective in mitigating the anemia associated with CD47 blockade and reduces the expression of CD47 on circulating erythrocytes []. In non-human primate studies, 5F9 was not associated with cytopenias other than anemia []; however, other anti-CD47 targeting agents, such as the SIRPα-IgG1 Fc fusion protein, TTI-621, can cause reversible thrombocytopenia, lymphopenia, neutropenia, and monocytopenia in addition to anemia [].
There are two broad classes of CD47-targeting agents currently in clinical development: IgG4 monoclonal antibodies and SIRPα-Fc fusion proteins (Table 1) (supplementary Appendix and Figure S1, available at Annals of Oncology online). Several of these agents are showing favorable clinical toxicity profiles and promising antitumor activity has been reported in patients with NHL, cutaneous T-cell lymphoma (CTCL), and ovarian cancer. Activity has been seen both as monotherapy and in combination with agents such as rituximab, which can enhance prophagocytic signals on CD20-expressing cancer cells. Additional agents that directly target SIRPα on immune cells are also in development, but none are yet in the clinic.
Hu5F9-G4 (5F9) is a humanized IgG4 monoclonal antibody (mAb) that binds to CD47 and enables the phagocytosis of human cancer cells [
]. 5F9 was engineered with a human IgG4 isotype to minimize potential off target effects on normal tissues. 5F9 binds to CD47 on RBCs and tumor tissues and its antitumor effects are primarily dependent on inhibition of CD47 signaling, although for optimal activity, an IgG4 Fc domain is required. In preclinical testing, 5F9 is broadly active in a wide range of hematologic malignancies including AML, NHL, CTCL, acute lymphoblastic anemia, and multiple myeloma (MM) [
], and in solid tumors such as breast, ovarian, colon, liver, brain, and other cancers [
,
,
].
Clinical trials with 5F9 began in 2014. In the phase I, first-in-human, trial, weekly i.v. infusions were well tolerated, and the expected on-target anemia was successfully mitigated by employing a priming and maintenance dosing schedule (NCT02216409) [
]. Maintenance doses as high as 45 mg/kg weekly were administered without reaching a maximum tolerated dose (MTD). Common treatment related adverse events included anemia, fatigue, fever, chills, headache, hemagglutination, and infusion-related reactions. Rare patients experienced back pain or chest pain during the infusion but these resolved when the infusion was slowed down or stopped temporarily. Hemagglutination on the peripheral blood smear was observed after the first or second dose of 5F9 but was not associated with clinically significant sequelae, despite rigorous monitoring. As expected, 5F9 pharmacokinetics were non-dose proportional; however, kinetics became linear at weekly doses above 10 mg/kg. Evidence of single-agent 5F9 antitumor activity was documented in two heavily pretreated patients with clear cell ovarian and fallopian tube cancers.
In a combination phase Ib/II study, relapsed/refractory NHL patients were treated with escalating doses of 5F9 and rituximab without encountering an MTD (NCT02953509) [
]. Twenty-two heavily pretreated phase Ib NHL patients had a median of four lines of prior systemic treatment (range 2–10). Objective responses were observed in 11/22 (50%) patients, with 8/22 (36%) achieving complete responses. In 15 diffuse large B cell lymphoma (DLBCL) patients, the objective response rate (ORR) was 6/15 (40%) with 5/15 (33%) complete responses (CR), and in 7 indolent lymphoma patients with Follicular Lymphoma (FL), the ORR was 5/7 (71%) with complete responses observed in 3/7 (43%). The median duration of response was not achieved in either group. Over 90% of patients met the formal definition of being rituximab refractory. The magnitude of objective responses seen with this combination in rituximab-refractory patients exceeds that expected from rituximab monotherapy alone. Phase II studies in NHL are ongoing. These observations in rituximab refractory patients provide clinical validation for the hypothesis that CD47 blockade can synergize with antitumor antibodies that add additional prophagocytic signals. Data from additional ongoing 5F9 clinical studies are provided in the supplementary Appendix, available at Annals of Oncology online.
CC-90002 is a humanized IgG4 antibody that entered clinical testing in 2015. CC-9002 was selected to avoid hemagglutination and it can facilitate the in vitro phagocytosis of both hematological malignancies and solid tumors [
]. In xenograft studies, CC-90002 is active against several tumors including lenalidomide-resistant MM, triple-negative breast cancer, and AML. CC-90002 is currently being evaluated in two phase I clinical trials. The first is a single-agent dose escalation study in patients with advanced solid tumors and hematological malignancies that will expand to treat CD20-positive NHL patients in combination with rituximab (NCT02367196). The second is a dose escalation and expansion study in patients with AML and high-risk myelodysplastic syndrome (NCT02641002). As of October 2018, the solid tumor and NHL combination study is ongoing, while the AML monotherapy trial has closed. No clinical results have been formally reported.
Two additional anti-CD47 IgG4 monoclonal antibodies recently entered clinical development. These include TI-061 [
] and SRF231 (NCT03512340) [
]. Both are treating patients with advanced solid and hematologic cancers, but no clinical results are available.
TTI-621 is a fusion protein composed of wild-type human SIRPα fused to an active IgG1 Fc domain. TTI-621 a decoy receptor that binds to human CD47 with micromolar affinity, while the IgG1 domain provides a potent prophagocytic signal that can engage with Fc-gamma receptors on effector cells [
]. In preclinical studies, TTI-621 controlled the growth of aggressive AML and lymphoma xenografts [
]. TTI-621 binds minimally to human RBCs, but it does bind to human leukocytes and platelets [
].
In a phase I trial, of weekly i.v. TTI-621 (NCT02663518) [
] mild to moderate infusion related reactions were reported but clinically significant anemia was not observed. However, at 0.3 mg/kg, thrombocytopenia and hepatic transaminase elevation were dose limiting [
]. Interestingly, even at these low doses, TTI-621 generated objective responses both as monotherapy and in combination with rituximab in 8/32 (25%) patients with refractory DLBCL [
]. Objective responses were observed in 2/8 (25%) DLBCL patients treated with TTI-621 alone and in 6/24 (25%) DLBCL patients treated in combination with rituximab. Two patients experienced complete responses [
]. These observations provide additional clinical validation for the activity of CD47 inhibition in the presence of strong prophagocytic signals.
A second phase I trial is directly injecting TTI-621 into tumors in patients with accessible solid tumors or CTCL/mycosis fungoides (NCT02890368) [
]. Intratumoral injections of TTI-621 were well tolerated with no dose-limiting toxicities observed. In 15 evaluable patients with CTCL (mycosis fungoides/Sezary syndrome), measurable improvements in skin lesions were seen in 13/15 (87%) patients [
], including some with lesions quite distant from the site of injection consistent with a systemic effect. Further investigation of TTI-621 in combination with PD-1 checkpoint inhibitors, oncolytic viruses, IFN-alpha, and radiation therapy is planned (NCT02890368).
A closely related IgG4 SIRPα-Fc fusion protein, TTI-622, also recently entered clinical testing and is anticipated to be less toxic. A phase I dose escalation trial of TTI-622 is underway in patients with advanced relapsed or refractory lymphoma or myeloma with plans to expand into combination treatments with rituximab, anti-PD-1 antibodies, and proteasome inhibitors (NCT03530683).
ALX148 is a fusion protein consisting of a SIRPα N-terminal D1 domain engineered to have over 7000-fold higher affinity for human CD47 than wild-type SIRPα [
]. It is fused to an inactive IgG1 Fc domain that lacks any Fc-mediated effector functions [
]. ALX148 alone does not trigger macrophage-mediated phagocytosis; therefore, its antitumor activity depends on the presence of additional, prophagocytic signals. Consequently, its optimal clinical use will likely require combining with other antitumor antibodies. ALX148 does not cause hemagglutination of human RBCs in vitro, and in preclinical studies, it is broadly active in xenograft models [
].
Clinical trials with ALX148 are ongoing in advanced solid tumor and lymphoma patients (NCT03013218) [
]. In an early phase I study, patients were treated with ALX148 at doses up 30 mg/kg without the use of a priming dose and without reaching an MTD. Two patients experienced dose limiting toxicities (DLTs) consisting of neutropenia with infection and thrombocytopenia with significant bleeding. No monotherapy objective responses were reported although one non-small cell lung cancer patient had a 15% tumor reduction. In a recent update on the clinical experience with ALX148 in combination with pembrolizumab, trastuzumab, and rituximab, no MTD was defined for any combination [
]. In the 39 patients treated with ALX148 and pembrolizumab, 2 partial responses (PRs) were noted in non-small-cell lung and head and neck cancer patients. In the combination with trastuzumab, in 18 patients, a PR was achieved in a HER2+ gastric cancer patient. Enrollment to these combination regimens is ongoing [
].
Targeting CD47, a dominant “don’t eat me” signal for macrophages, represents a novel therapeutic strategy for enhancing antitumor responses mediated by the innate immune system. Clinical validation of this unique mechanism of action is evidenced by the emergence of strong signals of activity in patients with refractory NHL and CTCL. Responses have been observed both as monotherapy and in combinations with agents such as rituximab that can augment prophagocytic signals on cancer cells. This approach holds great promise for the treatment of patients with hematological diseases such as B-cell malignancies, acute leukemia, and CTCL and in patients with solid tumors, such as ovarian and colorectal cancer. Harnessing the power of the innate immune system and first responder cells such as macrophages has the potential to extend the impact of cancer immunotherapies to benefit a broad range of cancer patients.