
Curr Opin Immunol. Author manuscript; available in PMC 2017 Dec 1.
Published in final edited form as:
Curr Opin Immunol. 2016 Dec; 43: 81–88.
Published online 2016 Oct 17. doi: 10.1016/j.coi.2016.09.008
PMCID: PMC5292941
NIHMSID: NIHMS823883
PMID: 27764715
Understanding mechanisms of autoimmunity through translational research in vitiligo
James P Strassner1 and John E Harris1,*
Author information Copyright and License information PMC Disclaimer
The publisher's final edited version of this article is available at Curr Opin Immunol
Abstract
Vitiligo is an autoimmune disease of the skin that leads to life-altering depigmentation and remains difficult to treat. However, clinical observations and translational studies over 30-40 years have led to the development of an insightful working model of disease pathogenesis: Genetic risk spanning both immune and melanocyte functions is pushed over a threshold by known and suspected environmental factors to initiate autoimmune T cell-mediated killing of melanocytes. While under cellular stress, melanocytes appear to signal innate immunity to activate T cells. Once the autoimmune T cell response is established, the IFN-γ-STAT1-CXCL10 signaling axis becomes the primary inflammatory pathway driving both progression and maintenance of vitiligo. This pathway is a tempting target for both existing and developing pharmaceuticals, but further detailing how melanocytes signal their own demise may also lead to new therapeutic targets. Research in vitiligo may be the future key to understand the pathogenesis of organ-specific autoimmunity, as vitiligo is common, reversible, progresses over the life of the individual, has been relatively well-defined, and is quite easy to study using translational and clinical approaches. What is revealed in these studies can lead to innovative treatments and also help elucidate the principles that underlie similar organ-specific autoimmune diseases, especially in cases where the target organ is less accessible.
백반증은
삶을 변화시키는 탈색으로 이어지는
피부의 자가 면역 질환으로 여전히 치료가 어려운 질환입니다.
하지만 30~40년에 걸친 임상 관찰과 중개 연구를 통해 질병 발병에 대한 통찰력 있는 작동 모델이 개발되었습니다:
면역 및 멜라닌 세포 기능 모두를 아우르는 유전적 위험은
알려진 또는 의심되는 환경 요인에 의해 임계치를 넘어
멜라닌 세포의 자가 면역 T 세포 매개 사멸을 유발합니다.
세포 스트레스를 받는 동안
멜라닌 세포는 선천성 면역에 신호를 보내
T 세포를 활성화하는 것으로 보입니다.
자가 면역 T 세포 반응이 확립되면
IFN-γ-STAT1-CXCL10 신호 전달 축이
백반증의 진행과 유지를 주도하는 주요 염증 경로가 됩니다.
이 경로는 기존 의약품과 개발 중인 의약품 모두에게 매력적인 표적이지만, 멜라닌 세포가 스스로 사멸 신호를 보내는 방법을 더 자세히 연구하면 새로운 치료 표적으로 이어질 수도 있습니다. 백반증은 흔하고 가역적이며 평생에 걸쳐 진행되며 비교적 잘 정의되어 있고 중개 및 임상 접근법을 사용하여 연구하기가 쉽기 때문에 백반증 연구는 장기 특이적 자가 면역의 발병 기전을 이해하는 미래의 열쇠가 될 수 있습니다. 이러한 연구에서 밝혀진 내용은 혁신적인 치료법으로 이어질 수 있으며, 특히 표적 장기에 대한 접근성이 떨어지는 경우 유사한 장기 특이적 자가면역 질환의 근간이 되는 원리를 규명하는 데 도움이 될 수 있습니다.
Introduction
Vitiligo is a common, but under-recognized, autoimmune disease of the skin in which melanocytes are specifically targeted and destroyed by skin-infiltrating, autoreactive CD8+ T cells. Patients develop patchy areas of depigmented skin (Figure 1) that become disfiguring, and thus negatively impact their quality of life (1-3). Approximately 1% of the global population is affected and the disease is reversible via melanocyte regeneration; however, the needs of this population remain unmet as current medical therapies are only moderately effective in reversing depigmentation. This is partly due to the fact that current treatments for vitiligo broadly dampen the immune response and do not specifically target the cells or pathways that are principally responsible for melanocyte destruction (2,3).
백반증은
흔하지만 잘 알려지지 않은
피부의 자가 면역 질환으로,
멜라닌 세포가 피부 침윤성 자가 반응 CD8+ T 세포에 의해
특이적으로 표적이 되어 파괴되는 질환입니다.
환자는 탈색된 피부(그림 1)의 반점 부위에
흉터가 생겨 삶의 질에 부정적인 영향을 미칩니다(1-3).
전 세계 인구의 약 1%가 이 질환을 앓고 있으며
멜라닌 세포 재생을 통해 질환을 되돌릴 수 있지만,
현재의 의료 요법은 탈색을 되돌릴 수 있는 효과가 중간 정도에 불과하기 때문에
이러한 인구의 요구는 여전히 충족되지 않고 있습니다.
이는 부분적으로는
현재의 백반증 치료법이 면역 반응을 광범위하게 약화시키고
멜라닌 세포 파괴를 주로 담당하는 세포나 경로를 구체적으로 표적으로 삼지 않기 때문입니다(2,3).
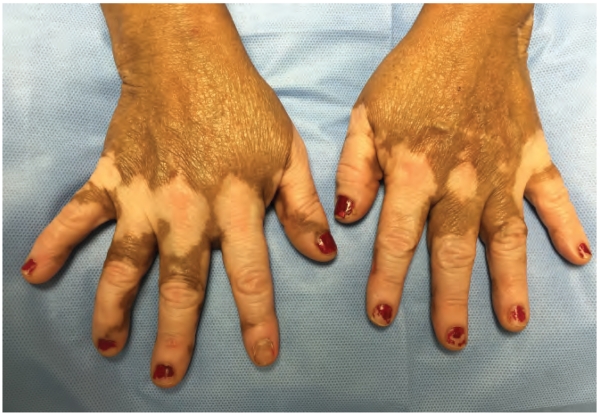
“Disfiguring white patches characteristic of vitiligo.”
To improve the treatment of vitiligo, researchers have studied the disease for over thirty years, advancing our understanding of its pathogenesis. Much of this work has been done directly on patients and their tissues, and thus has led to an advanced understanding of this organ-specific autoimmune disease within the human context. While the mechanism of disease initiation is not yet fully defined, a confluence of genetics and cellular stress most likely provide the fuel and spark, respectively (4). The subsequent CD8+ T cell response is then responsible for the destruction of melanocytes in the skin. Recent insights gleaned from basic and translational studies indicate that vitiligo could be more effectively treated by specifically targeting the pathways that allow CD8+ T cells to initiate and continue their attack on melanocytes (5).
백반증 치료를 개선하기 위해 연구자들은
30년 이상 백반증을 연구하며
그 발병 기전에 대한 이해를 발전시켜 왔습니다.
이러한 연구의 대부분은
환자와 그 조직을 대상으로 직접 수행되었으며,
그 결과 이 장기 특이적 자가면역 질환에 대한 이해가 인간의 맥락에서 발전하게 되었습니다.
질병의 발병 메커니즘은
아직 완전히 밝혀지지 않았지만,
유전학과 세포 스트레스의 결합이
각각 연료와 불꽃을 제공할 가능성이 가장 높습니다(4).
이후 이어지는 CD8+ T 세포 반응은
피부의 멜라닌 세포를 파괴합니다.
최근 기초 및 중개 연구에서 얻은 통찰력에 따르면
CD8+ T 세포가 멜라닌 세포에 대한 공격을 시작하고
계속할 수 있도록 하는 경로를 구체적으로 표적으로 삼아 백반증을 더 효과적으로 치료할 수 있습니다(5).
The Genetics Underlying Vitiligo
It is clear that vitiligo is an autoimmune disease, as many of the risk alleles that are associated with vitiligo are immune-specific genes, and vitiligo appears more frequently in family members of patients with autoimmune diseases, including type 1 diabetes, pernicious anemia, and autoimmune thyroiditis (6-9). Similar to other autoimmune diseases, dysregulation of antigen presentation is implicated as several HLA-haplotypes confer risk to developing disease: SNPs in risk alleles HLA-A*02 (10), HLA-DRB1, and HLA-DQA1 (11) lead to increased expression of MHC, which can allow for enhanced immune activation (10,11). NLRP1 is also identified to play a role, possibly through increased IL-1β processing (12); however, whether this is limited to disease initiation or also affects progression is unclear. Other immune genes involved in T cell signaling include PTPN22, TSLP, CCR6, IL2RA, UBASH3A, FOXP3 and GZMB (7), implicating both innate and adaptive responses, as well as regulatory T cells.
Melanocyte-specific genes are implicated in disease pathogenesis as well. TYR encodes tyrosinase, the enzyme that performs the rate-limiting step in melanogenesis (pigment production), and modifications of this enzyme are also associated with anti-melanoma tumor surveillance (13,14). OCA2, involved in melanosomal transport of tyrosine, as well as MC1R, a hormone receptor that promotes melanogenesis, are also implicated (15). Xbp1 (X-box binding protein 1) also confers risk of vitiligo and is more highly expressed in lesional skin (16), suggesting Xbp1 regulation could be involved in maintaining homeostasis in melanocyte surveillance (4). It is a key mediator of the unfolded protein response (UPR), which responds to endoplasmic reticulum (ER) stress (17), but also has additional roles in antigen loading and regulation of dendritic cell (DC) functions (18). Other autoimmune diseases are associated with hypomorphic variants in XBP1 that contribute risk and lead to ER stress and autoinflammation (19).
백반증과 관련된 많은 위험 대립 유전자가 면역 특이 유전자이며 백반증은 제1형 당뇨병, 악성 빈혈, 자가 면역 갑상선염을 포함한 자가 면역 질환 환자의 가족 구성원에게 더 자주 나타나기 때문에 백반증은 자가 면역 질환이라는 것이 분명합니다(6-9). 다른 자가면역 질환과 마찬가지로, 항원 제시의 조절 장애는 여러 HLA 일배열형이 질병 발병 위험에 영향을 미치기 때문에 관련되어 있습니다: 위험 대립유전자 HLA-A*02(10), HLA-DRB1, HLA-DQA1(11)의 SNP는 MHC의 발현을 증가시켜 면역 활성화가 강화될 수 있습니다(10,11). NLRP1도 IL-1β 처리 증가를 통해 역할을 하는 것으로 확인되었지만(12), 이것이 질병 시작에만 국한되는지 아니면 진행에도 영향을 미치는지는 불분명합니다. T세포 신호 전달에 관여하는 다른 면역 유전자로는 선천성 및 적응성 반응과 조절 T세포를 모두 포함하는 PTPN22, TSLP, CCR6, IL2RA, UBASH3A, FOXP3 및 GZMB(7)가 있습니다.
멜라닌 세포 특이 유전자는 질병 발병에도 관여합니다. TYR은 멜라닌 생성(색소 생성)에서 속도 제한 단계를 수행하는 효소인 티로시나아제를 암호화하며, 이 효소의 변형은 항흑색종 종양 감시와도 관련이 있습니다(13,14). 티로신의 멜라노솜 수송에 관여하는 OCA2와 멜라닌 생성을 촉진하는 호르몬 수용체인 MC1R도 관련되어 있습니다(15). Xbp1(X-박스 결합 단백질 1)도 백반증의 위험을 증가시키며 병변이 있는 피부에서 더 많이 발현되어(16), Xbp1 조절이 멜라닌 세포 감시의 항상성 유지에 관여할 수 있음을 시사합니다(4). 또한 소포체(ER) 스트레스에 반응하는 UPR(언폴딩 단백질 반응)의 핵심 매개체이며(17), 항원 로딩과 수지상 세포(DC) 기능 조절에도 중요한 역할을 합니다(18). 다른 자가 면역 질환은 XBP1의 저형성 변이와 관련이 있으며, 이는 위험에 기여하고 ER 스트레스와 자가 염증을 유발합니다(19).
Intrinsic Stress in Vitiligo
There is a growing body of literature that suggests a role for cellular stress in the development of autoimmunity, including vitiligo (4,20). Vitiligo patients exhibit signs of stress in the skin. Reactive oxygen species (ROS), principally H202, are elevated (21), and when compared to primary melanocytes generated from an unaffected individual, melanocytes from a vitiligo patient exhibit slower growth, demanding catalase supplementation as well as a host of growth factors (22-25). Melanocytes from vitiligo patients have a dilated ER (22), a tell-tale sign of ER stress, and intracellular staining of equally aged cell lines suggests there is dysregulation of protein expression (26), a precipitating factor that can initiate ER stress.
We have previously discussed the hypothesis that melanocytes could be uniquely prone to stress because of their functional and environmental niche in the skin (4,20). Melanogenesis requires the coordination of several energy-intensive processes within the melanocyte that result in generation of ROS. This includes the production of large amounts of protein components of the melanin pathway, melanin synthesis, and orchestrating organelle movement in the cell for pigment distribution. In addition, the redox reactions involved in chemical transformation of tyrosine to melanin directly yield potentially harmful byproducts, including H202. Moreover, the melanocyte’s physical position in the skin allows it to be subjected to UV light, another source that can generate ROS (20,27).
백반증을 포함한 자가 면역의 발달에서 세포 스트레스의 역할을 시사하는 문헌이 점점 더 많아지고 있습니다(4,20). 백반증 환자는 피부에 스트레스 징후가 나타납니다. 주로 H202와 같은 활성 산소 종(ROS)이 증가하며(21), 백반증 환자의 멜라닌 세포는 영향을 받지 않은 사람의 멜라닌 세포와 비교할 때 성장 속도가 느려서 카탈라아제 보충과 다양한 성장 인자가 필요합니다(22-25). 백반증 환자의 멜라닌 세포는 ER 스트레스의 징후인 확장된 ER을 가지고 있으며(22), 동일하게 숙성된 세포주의 세포 내 염색은 ER 스트레스를 유발할 수 있는 단백질 발현의 조절 장애가 있음을 시사합니다(26).
우리는 이전에 멜라닌 세포가 피부의 기능적 및 환경적 틈새 때문에 스트레스를 받기 쉽다는 가설에 대해 논의한 적이 있습니다(4,20). 멜라닌 생성은 멜라닌 세포 내에서 에너지 집약적인 여러 과정의 조정을 거쳐야 하며, 그 결과 ROS가 생성됩니다. 여기에는 멜라닌 경로의 다량의 단백질 성분 생산, 멜라닌 합성, 색소 분포를 위한 세포 내 소기관 이동 조율 등이 포함됩니다. 또한 티로신이 멜라닌으로 화학적으로 변환되는 산화 환원 반응은 H202와 같은 잠재적으로 유해한 부산물을 직접 생성합니다. 또한 피부에서 멜라닌 세포의 물리적 위치
Extrinsic Insults Damage Melanocytes and Can Initiate Vitiligo
Additional in vitro experiments in which the melanocytes were exogenously stressed using reagents that generate ROS resulted in the death of melanocytes from vitiligo patients at far lower exposure levels compared to primary cells derived from healthy donors (26,28). This would suggest that melanocytes from vitiligo patients possess a specific defect that increases their susceptibility to stress. These data have also been paired with relevant clinical observations that vitiligo has a strong environmental component. While there is increased risk to develop vitiligo in families with vitiligo or other organ-specific autoimmune diseases, studies in monozygotic twins reveal only 23% concordance of disease between them (8), suggesting that while genes confer a significant risk for the development of vitiligo, a large proportion is independent of genes, and thus likely incorporates environmental factors as well (20). Importantly, many of these factors and their mechanisms of action in vitiligo have been described. The chemical monobenzyl ether of hydroquinone (MBEH) was first shown to cause vitiligo in tannery workers who developed depigmentation after wearing rubber gloves containing the phenol, and a prescription cream containing the phenol can now be used to electively exacerbate disease in patients aiming for an even skin tone without pigmentation (29,30).
Investigation into the mechanism of action of MBEH has revealed that it works directly upon tyrosinase-positive, pigment-producing cells initiating several events: melanogenesis is inhibited, ROS are produced, the melanocyte undergoes ER stress, the UPR is activated, autophagy pathways are initiated, and exosomes are released (31). Others argue that vitiligo-inducing phenols act via tyrosinase related-protein 1 (TRP1), rather than directly through TYR, although both implicate melanocyte-specific function in toxicity (32). In these experiments, phenols such as 4-tertiary butyl phenol (4-TBP) and MBEH were used in vitro to initiate these cellular responses, and revealed that activation of stress could elicit an immune response, as the stressed melanocytes produced inflammatory cytokines IL-6 and IL-8 (33). Phenol-induced stress of melanocytes leads to the activation of DC and enhanced activation of T cells in co-culture experiments, possibly through the release of HSP70i, a pro-inflammatory signal (31,34,35).
More recently, van den Boorn and colleagues discovered that MBEH-induced haptenization of melanocyte antigens and phenol-induced stress creates an inflammatory environment within the skin that leads to the recruitment of natural killer (NK) cells and the formation of NK memory, which is then capable of continuing autoimmune attack on even remote, unexposed skin (36). Stressed melanocytes may release cell-specific antigens via exosome secretion (31) combined with NK mediated killing of melanocytes to promote DC activation and antigen presentation, which contributes to priming of a T cell response. While some details are still missing, the potential for stressed melanocytes to initiate the autoimmune attack in vitiligo is clear.
ROS를 생성하는 시약을 사용하여 멜라닌 세포에 외인성 스트레스를 가한 추가 시험관 실험에서는 건강한 기증자에서 추출한 일차 세포에 비해 훨씬 낮은 노출 수준에서 백반증 환자의 멜라닌 세포가 사멸하는 것으로 나타났습니다(26,28). 이는 백반증 환자의 멜라닌 세포가 스트레스에 대한 취약성을 증가시키는 특정 결함을 가지고 있음을 시사합니다. 이러한 데이터는 백반증이 강력한 환경적 요소를 가지고 있다는 관련 임상 관찰 결과와도 일치합니다. 백반증 또는 기타 장기 특이적 자가 면역 질환이 있는 가족에서 백반증 발병 위험이 증가하지만, 일란성 쌍둥이를 대상으로 한 연구에 따르면 이들 사이의 질병 일치율은 23%에 불과하여(8) 유전자가 백반증 발병에 상당한 위험을 부여하지만 상당 부분은 유전자와 무관하며 따라서 환경 요인도 포함될 가능성이 높다는 것을 시사합니다(20). 중요한 것은 이러한 요인 중 많은 부분과 백반증의 작용 메커니즘이 설명되었다는 점입니다. 화학 물질인 모노벤질 에테르 오브 하이드로퀴논(MBEH)은 페놀이 함유된 고무 장갑을 착용한 후 탈색이 발생한 제혁소 근로자에게 백반증을 유발하는 것으로 처음 밝혀졌으며, 이제 이 페놀이 포함된 처방 크림은 색소 침착 없이 고른 피부 톤을 목표로 하는 환자에서 선택적으로 질병을 악화시키는 데 사용될 수 있습니다(29,30).
MBEH의 작용 메커니즘에 대한 조사에 따르면 티로시나아제 양성, 색소 생성 세포에 직접 작용하여 멜라닌 생성 억제, ROS 생성, 멜라닌 세포의 ER 스트레스, UPR 활성화, 자가포식 경로 개시, 엑소좀 방출 등의 여러 이벤트를 유발하는 것으로 밝혀졌습니다(31). 다른 사람들은 백반증 유발 페놀이 TYR을 직접 통하지 않고 티로시나아제 관련 단백질 1(TRP1)을 통해 작용한다고 주장하지만, 둘 다 독성에서 멜라닌 세포 특이적 기능과 관련이 있습니다(32). 이 실험에서는 이러한 세포 반응을 시작하기 위해 시험관 내에서 4- 3차 부틸 페놀(4-TBP) 및 MBEH와 같은 페놀을 사용했으며, 스트레스를 받은 멜라닌 세포가 염증성 사이토카인 IL-6 및 IL-8을 생성함에 따라 스트레스 활성화가 면역 반응을 유발할 수 있음을 밝혀냈습니다(33). 페놀로 인한 멜라닌 세포의 스트레스는 공동 배양 실험에서 전염증성 신호인 HSP70i의 방출을 통해 DC의 활성화와 T 세포의 활성화를 강화합니다(31,34,35).
최근에 van den Boorn과 동료들은 MBEH에 의한 멜라닌 세포 항원의 합텐화 및 페놀에 의한 스트레스가 피부 내에 염증 환경을 조성하여 자연 살해(NK) 세포를 모집하고 NK 기억을 형성하여 노출되지 않은 먼 피부에서도 자가 면역 공격을 지속할 수 있다는 사실을 발견했습니다(36). 스트레스를 받은 멜라닌 세포는 엑소좀 분비(31)를 통해 세포 특이적 항원을 방출할 수 있으며, 이는 NK 매개 멜라닌 세포 사멸과 결합하여 DC 활성화 및 항원 제시를 촉진하여 T 세포 반응의 프라이밍에 기여합니다. 일부 세부 사항은 아직 밝혀지지 않았지만 스트레스를 받은 멜라닌 세포가 백반증에서 자가 면역 공격을 시작할 수 있는 가능성은 분명합니다.
CD8+ T Cells Play a Central Role in Depigmentation: Killing Melanocytes
Earlier work detailing the events and pathogenesis in vitiligo demonstrate that CD8+ T cells play a critical role in the destruction of melanocytes. First, patchy infiltrates of T cells were found to localize near melanocytes, the cells responsible for skin pigmentation (37,38). Analysis of vitiligo patient blood using melanocyte antigen-specific tetramers revealed that vitiligo patients have higher frequencies of melanocyte-specific CD8+ T cells in the blood compared to healthy controls, and that these frequencies correlate with total skin involvement (39). The reactivity of these T cells was subsequently tested and found to be capable of killing both melanoma as well as melanocytes derived from the same T cell donors. These T cells possessed a skin homing phenotype, including surface expression of cutaneous lymphocyte antigen (39,40).
Autoreactive T cells kill melanocytes in experiments in which T cells isolated from perilesional vitiligo skin are co-incubated with autologous uninvolved skin. Labeled T cells isolated from vitiligo lesions infiltrated the normal skin, migrated to the epidermal-dermal junction, and were found in close association with dying melanocytes. Depletion of CD8+ T cells prevented melanocyte destruction, whereas enrichment for these cells enhanced it (41), supporting the key role of CD8+ T cells in vitiligo. Our laboratory and others have then expanded on this knowledge by testing melanocyte-reactive T cells in mouse models, revealing that IFN-γ plays a central role in disease pathogenesis (42-45).
We have continued to dissect the role of IFN-γ and IFN-γ-dependent genes in our mouse model and in human tissues. We discovered that the IFN-γ-derived chemokine CXCL10 is essential for driving vitiligo pathogenesis through the recruitment of autoreactive CD8+ T cells to the epidermis, and autoreactive T cells depend upon the chemokine receptor of CXCL10, CXCR3, to home to the skin to kill melanocytes. Moreover, we found that melanocyte-specific CD8+ T cells in vitiligo patients express higher levels of CXCR3, paralleling our findings in the mouse (46). We also found that the IFN-γ receptor and STAT1 are critical for the development of skin depigmentation as well (in press and unpublished results). IFN-γ signals through the IFN-γ receptor, which recruits Janus Kinases (JAKs) to transduce the signal. JAKs phosphorylate STAT1, a transcription factor that then translocates to the nucleus to induce transcription of IFN-γ-inducible genes, including CXCL10 (Figure 2)(47). Keratinocytes are the principal cellular source of CXCL10 in the epidermis during disease progression (in press). Intriguingly, blocking CXCL10 in our mouse model can both prevent vitiligo as well as restore pigmentation in mice with established disease (46), providing support for targeting this pathway as a treatment strategy.
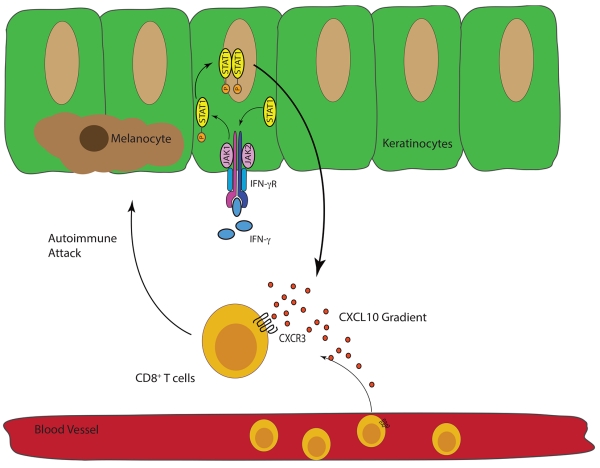
“The IFN-γ-STAT1-CXCL10 Axis Drives Melanocyte Destruction.”
IFNγ signals through the IFN-γ receptor (IFN-γR), which then requires JAK1 and JAK2 to phosphorylate the transcription factor STAT-1. Phosphorylated STAT1 homodimerizes and then translocates into the nucleus where IFN-γ-dependent genes, including CXCL9 and CXCL10, are transcribed(47). Autoreactive CXCR3-expressing CD8+ T cells follow these ligands to the skin where they kill melanocytes(46).
The importance of this pathway has been supported by translational studies of other groups (48-50) and by clinical studies in which the drugs that target this signaling axis were tested as treatments of vitiligo patients (51,52). Wang and colleagues tracked the levels of serum CXCL9 and CXCL10 in a cohort of patients and found that CXCL10 correlated with disease severity and inversely with treatment response (50), underpinning its role in human disease pathogenesis. Inhibitors of JAK/STAT signaling, tofacitinib and ruxolitinib, have shown promising results in the repigmentation of two patients with vitiligo (51,52), further validating the importance of this signaling pathway in vitiligo pathogenesis. Moreover, HMG-CoA reductase inhibitors have been shown to inhibit IFN-γ-dependent STAT1 signaling in vitro (53), suggesting statins could be used to inhibit the IFN-γ-STAT1-CXCL10 axis. One patient repigmented shortly after receiving high-dose simvastatin (54), and simvastatin both prevented depigmentation and promoted repigmentation in our mouse model, although its exact mechanism remains undetermined (55). However a small, randomized, placebo-controlled trial did not confirm efficacy in vitiligo patients [in press]. This may be a consequence of potential toxicity and consequent limited dosing of simvastatin in humans, which is not a limitation in mice.
The Roles of Other Cell Types Involved Are Poorly Defined
Additional cell types may also play a role in active disease, but their roles remain less defined than CD8+ T cells. NK, CD11b+CD11c+ cells, and macrophages infiltrate vitiligo lesions (37,38,56,57). Transcriptome and flow analysis of human skin revealed an infiltration of NK cells in lesional and non-lesional skin (56) which, in light of the recent discovery of NK-mediated killing of stressed melanocytes (36), could allow for a larger role in pathogenesis than previously thought. CD11b+CD11c+ cells appear in human and murine vitiligo lesional skin (57) and macrophages are also present in lesional skin (36,37), although the exact roles of these cells remain undefined. Stressed melanocytes release HSP70i (34) as well as exosomes of unknown content (31); it is possible that exosomes contain HSP70i as well as other potential danger associated molecular patterns (DAMPs) that activate nearby cell types (4,20,36).
T regulatory cells (Tregs) are important in controlling inflammation, including many different autoimmune diseases (58). Human studies report dysfunctional Treg responses in vitiligo patients compared to controls, but have not led to a consensus as to where the defect lies, in Treg number, skin homing capacity, or function (59-65). A recent study found that human Tregs control melanocyte-specific, CD8+ T cell responses through induction of CTLA4 in vitro, and that melanocyte-specific, CD8+ T cells exhibit an unregulated immunophenotype in vitiligo patients compared to controls (66). Mouse studies implicate an important role for Tregs as well (44,45), and future translational studies directly in the skin may better reveal their functional mechanism of regulation during disease evolution and maintenance of tolerance.
Resident cells of the skin also have emerging, but as yet undefined, roles in vitiligo. Much of the previous work has focused on melanocytes as a precipitating source of stress, DAMPs, and antigen, but the roles of neighboring cell types, like keratinocytes and Langerhans cells, are emerging. In the recent model of chemical induced vitiligo, Langerhans cells were dispensable (36), but this does not preclude additional roles during active vitiligo. Langerhans cells and dermal dendritic cells have distinct, sometimes antagonistic roles in regulating and promoting immune responses (67-69). Keratinocytes, which make up the majority of the epidermis, are hypothesized to play an important role in promoting T cell recruitment and inflammation in psoriasis (70). A recent study from our lab suggests keratinocytes and their ability to make CXCL10 are required for disease progression (in press). It remains unknown if keratinocytes play a similar role during initiation or in detecting melanocyte stress.
Summary: Realizing a Complete Model of Vitiligo Pathogenesis through Translational Research
In summary, the past three decades have uncovered many mechanistic details in vitiligo pathogenesis. Genetic studies of vitiligo patients reveal that it is principally an autoimmune disease, although melanocytes may ignite the entire process after crossing a certain threshold level of stress. ROS or ER stress may activate nearby innate myeloid or stromal skin populations, which in turn recruit T cells that kill melanocytes (Figure 3). Perhaps the most important details to be worked out revolve around how melanocytes signal stress in vivo, how it is interpreted by the immune system, and how this leads to disruption of immune tolerance. The mechanisms of T cell-mediated melanocyte killing are more clearly defined: CXCR3-expressing melanocyte-specific CD8+ T cells follow the chemokine CXCL10 to infiltrate the skin and kill melanocytes (46). Targeting aspects of this pathway may lead to better treatments, but more can still be worked out to fully detail what directs these T cells once they infiltrate the skin. What are the roles of the additional infiltrating cell types previously reported? How do melanocytes communicate cellular stress to the immune system? Can the IFN-γ-STAT1-CXCL10 and cellular stress pathways be effectively targeted to halt vitiligo?
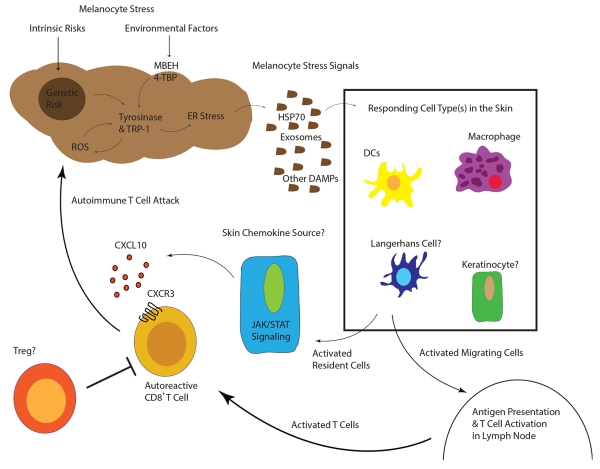
“Working Model of Vitiligo Pathogenesis”
Inherited genetic risk (HLA (11)(10), XBP1 (16), TYR (13), OCA2 (15), M1CR1 (15)) and environmental insults (MBEH and 4-TBP) induce a state of melanocyte stress, exemplified by ER stress. Stressed melanocytes signal to local innate and resident skin cell types via exosomes containing antigen and DAMPs, soluble HSP70, and/or other factors (30-34). Responding cell types are activated by these signals and some may migrate to the draining lymph nodes where they activate T cells. Other responding cells in the skin secrete chemokines to recruit autoreactive T cells, which are directly responsible for killing melanocytes. In active disease, one or more cell types may respond to IFN-γ and secrete CXCL10 to recruit T cells to the skin where melanocytes reside.
Directly testing human skin may be the answer to some of these questions. Mouse models and cell-based systems have helped to identify these pathways, and many translational studies were first initiated in mice or cell-based systems; however, the earliest observations of intrinsic stress, chemical induced vitiligo, and T cell-mediated melanocyte killing came from studying human skin. Vitiligo, in contrast to other autoimmune diseases, is uniquely amenable to translational research, as the target organ, the skin, is easily accessible. It is a disease that progresses over many years, allowing researchers the time to assess these tissues, under both current and novel therapies as well as exacerbating factors such as MBEH. Through such translational efforts, a complete working model of vitiligo pathogenesis can be realized and new therapeutic avenues can be explored, such that the needs of this patient population are finally met. In addition, complex interactions that balance autoimmune inflammation with mechanisms of immune tolerance can be more clearly defined. As such, we suspect that this work will directly inform that of other organ-specific autoimmune diseases, potentially supporting human vitiligo as a relevant model for diseases that are more difficult to study in humans, such as type 1 diabetes and multiple sclerosis.
Highlights
Genetic risk and environmental factors contribute to cellular stress in melanocytes
Activation of innate pathways is a direct result of melanocyte stress
The IFNγ-STAT1-CXCL10 signaling axis drives vitiligo progression
Translational research in vitiligo is a powerful tool to understand autoimmunity
Acknowledgements
JEH is supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases, part of the NIH, under award number AR061473, and a research grant from the Dermatology Foundation Stiefel Scholar Award.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
1. Picardo M, Dell’Anna ML, Ezzedine K, Hamzavi I, Harris JE, Parsad D, Taieb A. Vitiligo. Nature Reviews Disease Primers. 2015;1:15011. [PubMed] [Google Scholar]
2. Ezzedine K, Eleftheriadou V, Whitton M, van Geel N. Vitiligo. Lancet. 2015;386(9988):74–84. [PubMed] [Google Scholar]
3. Ezzedine K, Sheth V, Rodrigues M, Eleftheriadou V, Harris JE, Hamzavi IH, Pandya AG, Vitiligo Working Group Vitiligo is not a cosmetic disease. Journal of the American Academy of Dermatology. 2015;73(5):883–5. [PubMed] [Google Scholar]
4. Richmond JM, Frisoli ML, Harris JE. Innate immune mechanisms in vitiligo: Danger from within. Current Opinion in Immunology. 2013;25(6):676–82. [PMC free article] [PubMed] [Google Scholar]
5. Rashighi M, Harris JE. Interfering with the IFN-gamma/CXCL10 pathway to develop new targeted treatments for vitiligo. Annals of Translational Medicine. 2015;3(21):343. [PMC free article] [PubMed] [Google Scholar]
6. Cunliffe WJ, Hall R, Newell DJ, Stevenson CJ. Vitiligo, thyroid disease and autoimmunity. British Journal of Dermatology. 1968;80(3):135–9. [PubMed] [Google Scholar]
7. Spritz RA. Modern vitiligo genetics sheds new light on an ancient disease. J Dermatol. 2013;40(5):310–8. [PMC free article] [PubMed] [Google Scholar]
8. Alkhateeb A, Fain PR, Thody A, Bennett DC, Spritz RA. Epidemiology of vitiligo and associated autoimmune diseases in Caucasian probands and their families. Pigement Cell & Melanoma Research. 2003;16(3):208–14. [PubMed] [Google Scholar]
9. Gill L, Zarbo A, Isedeh P, Jacobsen G, Lim HW, Hamzavi I. Comorbid autoimmune diseases in patients with vitiligo: A cross-sectional study. Journal of the American Academy of Dermatology. 2016;74(2):295–302. [PubMed] [Google Scholar]
10** Hayashi M, Jin Y, Yorgov D, Santorico SA, Hagman J, Ferrara TM, Jones KL, Cavalli G, Dinarello CA, Spritz RA. Autoimmune vitiligo is associated with gain-of- function by a transcriptional regulator that elevates expression of HLA-A * 02 : 01 in vivo. PNAS. 2016;113(5):1357–62. [PMC free article] [PubMed] [Google Scholar] **This vitiligo functional genomics study found that the HLA-A*02:01 risk allele is associated with enhanced expression of class I HLA on the cell surface, allowing for increased presentation of melanocyte specific antigens. These findings underscore the contribution of this HLA risk allele to developing tissue-specific autoimmunity.
11** Cavalli G, Hayashi M, Jin Y, Yorgov D, Santorico SA, Holcomb C, Rastrou M, Erlich H, Tengesdal IW, Dagna L, et al. MHC class II super-enhancer increases surface expression of HLA-DR and HLA-DQ and affects cytokine production in autoimmune vitiligo. PNAS. 2016;113(5):1363–8. [PMC free article] [PubMed] [Google Scholar] ** Similar to the above study, this vitiligo functional genomics study found that the HLA-DR and HLA-DQ risk alleles are associated with enhanced expression of class II HLA on the cell surface due to alteration of the MHC class II super-enhancer. PBMCs with enhanced HLA expression were more responsive to inflammatory triggers, thus supporting the hypothesis that genetic risk promotes loss of immune tolerance through enhanced inflammation.
12. Levandowski CB, Mallioux CM, Ferrara TM, Gowan K, Ben S, Jin Y, McFann KK, Holland PJ, Fain PR, Dinarello CA, Spritz RA. NLRP1 haplotypes associated with vitiligo and autoimmunity increase interleukin-1beta processing via the NLRP1 inflammasome. PNAS. 2013;110(8):2952–6. [PMC free article] [PubMed] [Google Scholar]
13. Spritz RA. The genetics of generalized vitiligo: autoimmune pathways and an inverse relationship with malignant melanoma. Genome Med. 2010;2(10):78. [PMC free article] [PubMed] [Google Scholar]
14. Yamshchikov GV, Mullins DW, Ogino T, Thompson L, Presley J, Galavotti H, Aquila W, Deacon D, Patterson JW, Engelhard VH, et al. Sequential immune escape and shifting of T Cell responses in a long-term survivor of melanoma. Journal of Immunology. 2005;174:6863–71. [PubMed] [Google Scholar]
15. Jin Y, Birlea SA, Fain PR, Ferrara TM, Ben S, Riccardi SL, Cole JB, Gowan K, Holland PJ, Bennett DC, et al. Genome-wide association analyses identify 13 new susceptibility loci for generalized vitiligo. Nature Genetics. 2012;44(6):676–80. [PMC free article] [PubMed] [Google Scholar]
16. Ren Y, Yang S, Xu S, Gao M, Huang W, Gao T, Huang W, Gao T, Fang Q, Quan C, Zhang C, Sun L, et al. Genetic variation of promoter sequence modulates XBP1 expression and genetic risk for vitiligo. PLoS Genetics. 2009;5(6):e1000523. [PMC free article] [PubMed] [Google Scholar]
17. Lee AH, Iwakoshi NN, Glimcher LH. Xbp-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Molecular and Cellular Biology. 2003;23(21):7448–59. [PMC free article] [PubMed] [Google Scholar]
18. Cubillos-Ruiz JR, Silberman PC, Rutkowski MR, Chopra S, Perales-Puchalt A, Song M, Zhang S, Bettigole SE, Gupta D, Holcomb K, et al. ER stress sensor XBP1 controls anti-tumor immunity by disrupting dendritic cell homeostasis. Cell. 2015;161(7):1527–38. [PMC free article] [PubMed] [Google Scholar]
19. Kaser A, Lee AH, Franke A, Glickman JN, Zeissig S, Tilg H, Nieuwenhuis EE, Higgins DE, Schreiber S, Glimcher LH, Blumberg RS. XBP1 links ER stress to intestinal inflammation and confers genetic risk for human inflammatory bowel disease. Cell. 2008;134(5):743–56. [PMC free article] [PubMed] [Google Scholar]
20. Harris JE. Cellular stress and innate inflammation in organ-specific autoimmunity: lessons learned from vitiligo. Immunological Reviews. 2015;269(1):11–25. [PMC free article] [PubMed] [Google Scholar]
21. Schallreuter KU, Moore J, Wood JM, Beazley WD, Gaze DC, Tobin DJ, Marshal HS, Panske A, Panzig E, Hibberts NA. In vivo and in vitro evidence for hydrogen peroxide (H2O2) accumulation in the epidermis of patients with vitililigo and its successful removal by a UVB-activated pseudocatalase. Journal of Investigative Dermatology Symposium Proceedings. 1999;4(1):91–6. [PubMed] [Google Scholar]
22. Boissy RE, Liu Y, Medrano EE, Nordlund JJ. Structural aberration of the rough endoplasmic reticulum and melanosome compartmentalization in long-term cultures of melanocytes. Journal of Investigative Dermatology. 1991;97(3):395–404. [PubMed] [Google Scholar]
23. Puri N, Mojamdar M, Ramajah A. In vitro growth characteristics of melanocytes obtained from adult normal and vitiligo subjects. Journal of Investigative Dermatology. 1987;88(4):434–8. [PubMed] [Google Scholar]
24. Puri N, Mojamdar M, Ramajah A. Growth defects of melanocytes in culture from vitiligo subjects are spontaneously corrected in vivo in repigmenting subjects and can be partially corrected by the addition of fibroblast-derived growth factors in vitro. Archives of Dermatological Research. 1989;281(3):178–84. [PubMed] [Google Scholar]
25. Medrano EE, Nordlund JJ. Successful culture of adult human melanocytes obtained from normal and vitiligo donors. Journal of Investigative Dermatology. 1990;95(4):441–5. [PubMed] [Google Scholar]
26. Jimbow K, Chen H, Park JS, Thomas PD. Increased sensitivity of melanocytes to oxidative stress and abnormal expression of tyrosinase-related protein in vitiligo. British Journal of Dermatology. 2001;144(1):55–65. [PubMed] [Google Scholar]
27. Meyskens FL, Farmer P, Fruehauf JP. Redox regulation in human melanocytes and melanoma. Pigment cell & Melanoma Research. 2001;14(3):148–54. [PubMed] [Google Scholar]
28. Maresca V, Roccella M, Roccella F, Camera E, Del Porto G, Passi S, Grammatico P, Picardo M. Increased sensitivity to peroxidative agents as a possible pathogenic factor of melanocyte damage in vitiligo. Journal of Investigative Dermatology. 1997;109(3):310–3. [PubMed] [Google Scholar]
29. Oliver EA, Schwartz L, Warren L. Occupational Leukoderma. JAMA. 1937;113:927–8. [Google Scholar]
30. Mosher DB, Parrish JA, Fitzpatrick TB. Monobenzylether of hydroquinone: a retrospective study of treatment of 18 vitiligo patients and review of the literature. British Journal of Dermatology. 1977;97(6):669–79. [PubMed] [Google Scholar]
31. van den Boorn JG, Picavet DI, van Swieten PF, van Veen HA, Konijnenberg D, van Veelen PA, van Capel T, Jong EC, Reits EA, Drijfhout JW, et al. Skin-depigmenting agent monobenzone induces potent T-cell autoimmunity toward pigmented cells by tyrosinase haptenation and melanosome autophagy. The Journal of Investigative Dermatology. 2011;131(6):1240–51. [PubMed] [Google Scholar]
32. Yang F, Sarangarajan R, Le Poole IC, Medrano EE, Boissy RE. The cytotoxicity and apoptosis induced by 4-tertiary butylphenol in human melanocytes are independent of tyrosinase activity. Journal of Investigative Dermatology. 2000;114(1):157–64. [PubMed] [Google Scholar]
33. Toosi S, Orlow SJ, Manga P. Vitiligo-inducing phenols activate the unfolded protein response in melanocytes resulting in upregulation of IL6 and IL8. Journal of Investigative Dermatology. 2012;132(11):2601–9. [PMC free article] [PubMed] [Google Scholar]
34. Kroll TM, Bommiasamy H, Boissy RE, Hernandez C, Nickoloff BJ, Mestril R, Le Poole IC. 4-Tertiary butyl phenol exposure sensitizes human melanocytes to dendritic cell-mediated killing: relevance to vitiligo. The Journal of Investigative Dermatology. 2005;124(4):798–806. [PMC free article] [PubMed] [Google Scholar]
35. Mosenson JA, Flood K, Klarquist J, Eby JM, Koshoffer A, Boissy RE, Overbeck A, Tung RC, Le Poole IC. Preferential secretion of inducible HSP70 by vitiligo melanocytes under stress. Pigment Cell & Melanoma Research. 2014;27(2):209–20. [PMC free article] [PubMed] [Google Scholar]
36* van den Boorn JG, Jakobs C, Hagen C, Penn M, Luiten RM, Melief CJ, Tuting T, Garbi N, Hartmann G, Hornung V. Inflammasome-dependent induction of adaptive NK memory. Immunity. 2016;44(6):1406–21. [PubMed] [Google Scholar] * This study describes the activation of an NK memory population that targets and kills melanocytes exposed to the inducing phenol MBEH. These cells were dependent on the prior activation of macrophages and the NLRP3 inflammasome. It addresses how cellular stress precipitates organ-specific autoimmunity through activation of the innate immune system.