
면역세포는 pathogen을 패턴으로 인식한다.pattern recognition receptor PRR
조직에 pathogen associated molecular pattern(PMAP)가 있으면 PRR과 결합한다.
평소 염증을 일으키는 식사(음식과민증, 히스타민 과민증, 알레르기 음식)를 하면
PAMP가 많아지고
PRR은 혼란에 빠진다
그 과정에 중요한 면역세포 활성인자, 염증전사인자 NF-kB가 있다. .
Cell Res. 2011 Feb; 21(2): 223–244.
Published online 2011 Jan 18. doi: 10.1038/cr.2011.13
PMCID: PMC3193440
PMID: 21243012
NF-κB in immunobiology
Matthew S Hayden1 and Sankar Ghosh1,*
Author information Copyright and License information PMC Disclaimer
Abstract
NF-κB was first discovered and characterized 25 years ago as a key regulator of inducible gene expression in the immune system. Thus, it is not surprising that the clearest biological role of NF-κB is in the development and function of the immune system. Both innate and adaptive immune responses as well as the development and maintenance of the cells and tissues that comprise the immune system are, at multiple steps, under the control of the NF-κB family of transcription factors. Although this is a well-studied area of NF-κB research, new and significant findings continue to accumulate. This review will focus on these areas of recent progress while also providing a broad overview of the roles of NF-κB in mammalian immunobiology.
NF-κB는
면역계에서 유도성 유전자 발현의 핵심 조절자로서
25년 전에 처음 발견되어 그 특성을 규명했습니다.
따라서
NF-κB의 가장 명확한 생물학적 역할이
면역 체계의 발달과 기능에 있다는 것은 놀라운 일이 아닙니다.
선천성 및 적응성 면역 반응과 면역 체계를 구성하는 세포와
조직의 발달 및 유지 모두
여러 단계에서 NF-κB 전사 인자 계열의 제어를 받습니다.
이 분야는
NF-κB 연구에서 잘 연구된 분야이지만
새롭고 중요한 연구 결과가 계속 축적되고 있습니다.
이 리뷰에서는
포유류 면역생물학에서
NF-κB의 역할에 대한 광범위한 개요를 제공하면서
최근 진전된 이러한 영역에 초점을 맞출 것입니다.
Keywords: NF-κB, TLR, TCR, BCR, lymphoid organogenesis, inflammation, hematopoiesis
Introduction
As discussed in the introduction to the January special issue of Cell Research, the discovery and characterization of the NF-κB family of transcription factors resulted from studies in two major areas of research: immunology and cancer biology. Although the role of NF-κB in cancer biology is becoming progressively better established, historically much of our current knowledge of NF-κB resulted from efforts directed at understanding its role in the regulation and function of the immune response. This review will attempt to provide a comprehensive discussion of the diverse functions of NF-κB in immunobiology and update our previous efforts in reviewing the far-reaching role of NF-κB in this area of biology 1, 2, 3.
The mammalian immune response comprises various mechanisms by which the physiological and functional integrity of the host is maintained in the face of microbial insult. The bone marrow-derived cells of the immune system are chiefly, though by no means solely, responsible for performing these functions. Thus, we begin by reviewing the role of NF-κB in the development of these hematopoietic cells that mediate both innate and adaptive immune responses. With the relevant cell types and tissues in place, the immune response is triggered by host recognition of foreign pathogens. Our knowledge in this area of the immune response has expanded rapidly in the past decade. Pathogen recognition is followed by pathogen clearance, a highly variable response that may draw upon coordinated responses at the level of cell, tissue, and the organism. Finally, the immune response must be resolved, damage must be repaired and, whenever possible, the triggering insult remembered for future immunity.
In mammals, the NF-κB family is composed of five related transcription factors: p50, p52, p65 (also RelA), c-Rel, and RelB (Figure 1). These transcription factors share an N-terminal DNA-binding/dimerization domain, known as the Rel homology domain, through which they can form homo- and heterodimers. NF-κB dimers can bind to a variety of related target DNA sequences called κB sites to modulate gene expression. RelB, c-Rel, and p65 contain C-terminal transcription activation domains (TADs) that enable co-activator recruitment and target gene expression. As they lack TADs, p50 and p52 can activate transcription by forming heterodimers with p65, c-Rel, or RelB, or by recruiting other TAD-containing proteins. However, as homodimers lacking the ability to drive transcription, they can repress transcription on binding to DNA.
Cell Research 1월 특별호 소개에서 설명한 것처럼,
NF-κB 전사인자 계열의 발견과 특성 분석은
면역학과 암 생물학이라는 두 가지 주요 연구 분야의 연구를 통해 이루어졌습니다.
암 생물학에서
NF-κB의 역할이 점차 더 잘 밝혀지고 있지만,
역사적으로 현재 NF-κB에 대한 지식의 대부분은
면역 반응의 조절과 기능에 대한 역할을 이해하려는 노력에서 비롯된 것입니다.
이 리뷰에서는
면역 생물학에서 NF-κB의 다양한 기능에 대한 포괄적인 논의를 제공하고
이 생물학 영역에서 NF-κB의 광범위한 역할을 검토한 이전의 노력을 업데이트하려고 합니다 1, 2, 3.
포유류의 면역 반응은
미생물의 공격에 맞서
숙주의 생리적, 기능적 무결성을 유지하는
다양한 메커니즘으로 구성됩니다.
면역 체계의 골수 유래 세포가
이러한 기능을 주로 담당하지만 전적으로 담당하는 것은 아닙니다.
따라서
선천성 및 후천성 면역 반응을 매개하는
이러한 조혈 세포의 발달에서 NF-κB의 역할을 검토하는 것부터 시작합니다.
관련 세포 유형과 조직이 제자리에 있으면
외부 병원체를 숙주가 인식함으로써
면역 반응이 촉발됩니다.
면역 반응의 이 분야에 대한 지식은
지난 10년 동안 급속도로 확장되었습니다.
병원체 인식 후에는
세포, 조직 및 유기체 수준에서 조정된 반응에 의존할 수 있는
매우 가변적인 반응인 병원체 제거가 이어집니다.
마지막으로
면역 반응이 해결되고
손상이 복구되어야 하며,
가능하면 향후 면역을 위해 유발된 모욕이 기억되어야 합니다.
포유류에서 NF-κB 계열은
p50, p52, p65(RelA라고도 함), c-Rel, RelB 등
5개의 관련 전사인자로 구성됩니다(그림 1).
이러한
전사 인자들은 Rel 상동 도메인이라고 하는
N-말단 DNA 결합/이합체화 도메인을 공유하며,
이를 통해 동형 및 이종 이합체를 형성할 수 있습니다.
NF-κB 이합체는
κB 사이트라고 하는 다양한 관련 표적 DNA 서열에 결합하여
유전자 발현을 조절할 수 있습니다.
RelB, c-Rel 및 p65에는
보조 인자 모집 및 표적 유전자 발현을 가능하게 하는
C-말단 전사 활성화 도메인(TAD)이 포함되어 있습니다.
TAD가 부족하기 때문에
p50과 p52는 p65, c-Rel 또는 RelB와 이이합체를 형성하거나
다른 TAD 함유 단백질을 모집하여 전사를 활성화할 수 있습니다.
그러나
전사 능력이 부족한 동이합체는
DNA에 결합할 때 전사를 억제할 수 있습니다.
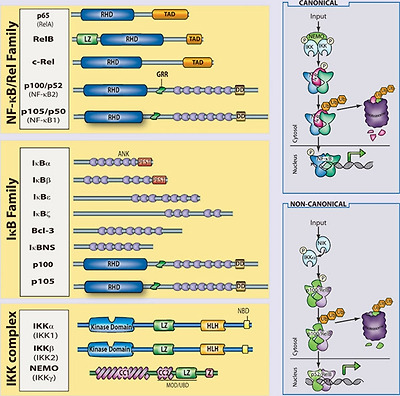
The mammalian NF-κB, IκB and IKK protein families.
Relevant domains typifying each protein family are indicated and alternative nomenclatures are provided in parenthesis. The precursor proteins p100 and p105 function as both IκB and, when processed by the proteasome, NF-κB family members. Highly simplified schematics of canonical and non-canonical NF-κB pathways are shown. In the canonical pathway NEMO-containing IKK complexes are activated and induce phosphorylation and degradation of IκBα leading to the release of NF-κB dimers including p65:p50 dimers. In the non-canonical pathway, NEMO-independent activation of IKKα is mediated by the upstream kinase NIK. IKKα, with NIK, induces phosphorylation and processing of p100 to p52 resulting in the activation of predominantly p52:RelB complexes. Although they are illustrated separately here, as discussed in the text, signaling through these two pathways often is initiated by the same receptor – e.g., LTβR or CD40. (ANK, ankyrin-repeat domain; DD, death domain; RHD, REL homology domain; TAD, transactivation domain; LZ, leucine-zipper domain; GRR, glycine-rich region; HLH, helix-loop-helix domain; Z, zinc-finger domain; CC, coiled-coil domain; NBD, NEMO-binding domain; MOD/UBD, minimal oligomerization domain/ubiquitin-binding domain; PEST, proline, glutamic acid, serine, and threonine rich.)
포유류의 NF-κB, IκB 및 IKK 단백질 계열.
각 단백질군을 대표하는 관련 도메인이 표시되어 있으며 괄호 안에 대체 명명법이 제공됩니다.
전구체 단백질 p100 및 p105는
프로테아좀에 의해 처리될 때
IκB 및 NF-κB 계열의 구성원으로 기능합니다.
표준 및 비표준 NF-κB 경로의 매우 단순화된 모식도가 나와 있습니다.
정식 경로에서는
NEMO 함유 IKK 복합체가 활성화되어
IκBα의 인산화 및 분해를 유도하여 p65:p50 이합체를 포함한
NF-κB 이합체의 방출을 유도합니다.
비정식적 경로에서 IKKα의 NEMO 독립적 활성화는
업스트림 키나아제 NIK에 의해 매개됩니다.
IKKα는 NIK와 함께 p100에서 p52로의 인산화 및 처리를 유도하여
주로 p52:RelB 복합체를 활성화합니다.
여기서는 별도로 설명하지만, 본문에서 설명한 것처럼 이 두 경로를 통한 신호 전달은 종종 동일한 수용체(예, LTβR 또는 CD40. (ANK, 안키린 반복 도메인; DD, 사멸 도메인; RHD, REL 상동성 도메인; TAD, 거래 활성화 도메인; LZ, 류신 지퍼 도메인; GRR, 글리신 풍부 영역; HLH, 나선 루프 헬릭스 도메인; Z, 아연-핑거 도메인; CC, 코일-코일 도메인; NBD, NEMO 결합 도메인; MOD/UBD, 최소 올리고머화 도메인/유비퀴틴 결합 도메인; PEST, 프롤린, 글루탐산, 세린 및 트레오닌 풍부 영역. )
In most cells, NF-κB complexes are inactive, residing predominantly in the cytoplasm in a complex with inhibitory IκB proteins (IκBα, IκBβ, IκBɛ, IκBζ, p100, p105, Bcl3, IκBns; Figure 1). When signaling pathways are activated, the IκB protein is degraded and NF-κB dimers enter the nucleus to modulate target gene expression. In almost all cases, the common step in this process is mediated by the IκB kinase (IKK) complex, which phosphorylates IκB and targets it for proteasomal degradation (see Liu and Chen, Cell Res 2011; 21:6–21). The IKK complex consists of two catalytically active kinases, IKKα (IKK1) and IKKβ (IKK2) and a regulatory scaffolding protein, NEMO (IKKγ Figure 1). These are the basic players in the NF-κB pathway; however, there is considerable complexity in how the individual proteins function and how they coordinate a nuanced NF-κB response (see Shih et al., Cell Res 2011; 21:86–102). The details of individual signaling pathways, the mechanism of action of upstream signaling intermediates, the role of various post-translational modifications, and cross-talk between NF-κB and other signaling pathways are covered in depth elsewhere in this special issue. Here, we focus on the function of NF-κB and the signaling pathways that regulate it in the mammalian immune response.
대부분의 세포에서
NF-κB 복합체는 비활성 상태이며,
주로 세포질에 억제성 IκB 단백질(IκBα, IκBβ, IκBɛ, IκBζ, p100, p105, Bcl3, IκBns, 그림 1)과
복합체 형태로 존재합니다.
신호 경로가 활성화되면
IκB 단백질이 분해되고
NF-κB 이합체가 핵으로 들어가
표적 유전자 발현을 조절합니다.
거의 모든 경우에서
이 과정의 공통 단계는
IκB 키나아제(IKK) 복합체에 의해 매개되며,
이 복합체는 IκB를 인산화하여 프로테아좀 분해를 표적으로 삼습니다(Liu and Chen, Cell Res 2011; 21:6-21 참조).
IKK 복합체는
두 개의 촉매 활성 키나아제인 IKKα(IKK1)와 IKKβ(IKK2)와
조절 스캐폴딩 단백질인 NEMO(IKKγ 그림 1)로 구성됩니다.
이들은 NF-κB 경로의 기본적인 역할을 하지만
개별 단백질이 어떻게 기능하고
미묘한 NF-κB 반응을 어떻게 조정하는지는 상당히 복잡합니다(Shih 등, Cell Res 2011; 21:86-102 참조).
개별 신호 경로의 세부 사항,
상류 신호 중간체의 작용 메커니즘,
다양한 번역 후 변형의 역할,
NF-κB와 다른 신호 경로 간의 교차 신호에 대해서는
이 특집호의 다른 곳에서 자세히 다룹니다.
여기서는
포유류 면역 반응에서
NF-κB의 기능과 이를 조절하는 신호 전달 경로에 초점을 맞춥니다.
Development of the immune system
The mammalian immune system consists of a functionally linked group of anatomically disparate tissues and cell types. The dispersed cellular components of the immune system that arise from the bone marrow receive much of the attention in immunology and the study of NF-κB has likewise focused on these cells. However, lymphoid organs that facilitate coordination and dissemination of immune responses carried out by immune cells are also key sites of NF-κB function. Therefore, while this section is largely concerned with the role of NF-κB in hematopoiesis, the role of NF-κB in lymphoid organogenesis is also discussed briefly.
포유류의 면역 체계는
해부학적으로 서로 다른 조직과 세포 유형이
기능적으로 연결된 그룹으로 구성되어 있습니다.
면역학에서는
골수에서 발생하는 면역계의 분산된 세포 성분이 많은 관심을 받고 있으며,
NF-κB에 대한 연구도 마찬가지로
이러한 세포에 초점을 맞추고 있습니다.
그러나
면역 세포가 수행하는 면역 반응의 조정과 전파를 촉진하는 림프 기관도
NF-κB 기능의 핵심 부위입니다.
따라서
이 섹션에서는 주로
조혈에서 NF-κB의 역할에 대해 다루지만
림프 기관 생성에서 NF-κB의 역할도 간략하게 설명합니다.
NF-κB and lymphoid organogenesis
NF-κB plays an important role in the development and function of primary (bone marrow, thymus) and secondary (lymph nodes, Peyer's patches, mucosal-associated lymphoid tissue, and the spleen) lymphoid tissues. There is clearly a role for NF-κB in the development and regulation of bone, and we refer the reader to the excellent review on the subject in this special issue (see Novack, Cell Res 2011; 21:169–182). The role of NF-κB in thymic architecture, originally thought to be limited to the important role of RelB in thymic architecture 4, has become clearer in terms of the development of medullary thymic epithelial cells 5, 6, 7, but still remains to be more fully elucidated 8. The secondary lymphoid tissues facilitate maintenance and activation of mature lymphocytes by providing an environment within which the interaction of lymphocytes and other leukocytes can be carefully orchestrated 9. The role of NF-κB in this process is now well appreciated. Multiple NF-κB knockouts exhibit defects in some aspect of secondary lymphoid organ development.
The initial events of lymphoid organogenesis involve the association of lymphotoxin (LT)α1β2-expressing hematopoietic cells and vascular cell adhesion molecule-1 (VCAM-1)-expressing stromal cells 10. This interaction initiates a positive feedback loop through NF-κB (Figure 2). LTα1β2, receptor activator of NF-κB ligand (RANKL), and TNFα are known to activate NF-κB, and figure prominently in lymphorganogenesis 10 (Figure 2). Also, mediators of lymphoid organogenesis and homeostasis, such as adhesion molecules (e.g., intercellular adhesion molecule, VCAM, peripheral node addressin, glycosylation-dependent cell adhesion molecule-1, and mucosal addressin cellular adhesion molecule (MadCAM)), cytokines (e.g., TNFα and LTα1β2) and organogenic chemokines (e.g., CXCL12 (GRO/MIP-2), CXCL13 (BLC), CCL19 (ELC), and CCL21 (SLC)), are regulated by NF-κB.
NF-κB와 림프 조직 형성
NF-κB는
일차(골수, 흉선) 및
이차(림프절, 페이어 패치, 점막 관련 림프 조직 및 비장) 림프 조직의 발달과 기능에
중요한 역할을 합니다.
뼈의 발달과 조절에서
NF-κB의 역할은 분명히 존재하며,
이번 특집호에서 이 주제에 대한 훌륭한 리뷰를 참조하시기 바랍니다(Novack, Cell Res 2011; 21:169-182 참조).
흉선 구조에서 NF-κB의 역할은
원래 흉선 구조에서 RelB의 중요한 역할에 국한된 것으로 생각되었지만4,
수질 흉선 상피 세포 5, 6, 7의 발달 측면에서 더 명확해졌지만
여전히 더 완전히 밝혀지지 않은 상태입니다8.
2차 림프 조직은
림프구와 다른 백혈구의 상호 작용이 신중하게 조율될 수 있는 환경을 제공함으로써
성숙한 림프구의 유지와 활성화를 촉진합니다 9.
이 과정에서
NF-κB의 역할은 이제 잘 알려져 있습니다.
다수의 NF-κB 녹아웃은
이차 림프 기관 발달의 일부 측면에서 결함을 나타냅니다.
림프 기관 발생의 초기 사건에는 림포톡신(LT)α1β2 발현 조혈 세포와 혈관 세포 부착 분자-1(VCAM-1)을 발현하는 기질 세포의 연관성이 포함됩니다10. 이러한 상호 작용은 NF-κB를 통해 긍정적인 피드백 루프를 시작합니다(그림 2). LTα1β2, NF-κB 리간드 수용체 활성화제(RANKL), TNFα는 NF-κB를 활성화하는 것으로 알려져 있으며 림프구 생성에서 두드러지게 나타납니다 10 (그림 2). 또한 접착 분자(예: 세포 간 접착 분자, VCAM, 말초 노드 어드레신, 글리코실화 의존 세포 접착 분자-1, 점막 어드레신 세포 접착 분자(MadCAM) 등), 사이토카인(예:, TNFα 및 LTα1β2) 및 유기성 케모카인(예: CXCL12(GRO/MIP-2), CXCL13(BLC), CCL19(ELC), CCL21(SLC))은 NF-κB의 조절을 받습니다.

NF-κB in lymphoid organogenesis.
NF-κB regulates key components of a positive feedback loop between hematopoietic and stromal cells. LTα1β2-expressing hematopoietic cells induce production of VCAM-1 through the canonical NF-κB pathway and chemokines through the non-canonical pathway in LTβR-expressing stromal cells. Stromal expression of chemokines induces the upregulation of integrins (α4β1) on hematopoietic cells resulting in increased recruitment of LTα1β2-expressing cells, and increased signaling through stromal LTβR. RANKL signaling through RANK on the hematopoietic cell leads to activation of NF-κB, and further upregulation of LTα1β2.
림프 조직 형성에서 NF-κB.
NF-κB는
조혈 세포와 기질 세포 사이의
긍정적인 피드백 루프의 핵심 구성 요소를 조절합니다.
LTα1β2 발현 조혈 세포는
표준 NF-κB 경로를 통해 VCAM-1의 생성을 유도하고, L
TβR 발현 기질 세포에서는 비표준 경로를 통해
케모카인의 생성을 유도합니다. 케
모카인의 기질 발현은
조혈 세포에서 인테그린(α4β1)의 상향 조절을 유도하여
LTα1β2 발현 세포의 모집을 증가시키고,
기질 LTβR을 통한 신호 전달을 증가시킵니다.
조혈 세포에서 RANK를 통한 RANKL 신호는
NF-κB의 활성화와 LTα1β2의 추가 상향 조절로 이어집니다.
Signaling through TNFR1, LTβR, and RANK activates p65-containing complexes and, hence, it is not surprising that rela−/−/tnfr1−/− double-knockout mice lack Peyer's patches and lymph nodes, and exhibit a disorganized spleen 11. The requirement for p65 in the development of these tissues is attributable to its function in stromal cells, and is likely because of a combination of effects: regulation of apoptosis (e.g., that induced by TNF); regulation of expression of organogenic factors, such as VCAM and LTα1β2; and enhancement of the non-canonical p52/RelB NF-κB pathway. The non-canonical NF-κB pathway has a well-established role in secondary lymphoid organ development. Deletion of components of the non-canonical NF-κB signaling pathway, including NIK, IKKα, LT, and RANK, all lead to severe defects in secondary lymphoid organogenesis 12, 13, 14, 15, 16, 17. The target of the non-canonical pathway, the p52/RelB dimer, is the primary transcriptional regulator of key organogenic factors including CXCL12, CXCL13, CCL19, CCL21, and MadCAM-1 18. Thus, the p52 single knockout lacks normal B-cell follicles, germinal centers, and Peyer's patches 19, 20, 21, while RelB knockouts lack Peyer's patches and exhibit compromised lymphnode development 18.
Splenic architecture is crucial for B-cell development as well as for the initiation and maturation of B-cell responses. The spleen is divided functionally and histologically into white and red pulp zones. Macrophages in the red pulp are responsible for destroying damaged erythrocytes, while the white pulp is populated by splenic lymphocytes organized into B-cell follicles and T-cell zones. Yet splenic architecture is also dynamic, as exemplified by the formation of germinal centers during the initiation and maturation of B-cell responses. As with other secondary lymphoid organs, NF-κB figures prominently in the development and maintenance of splenic architecture. Mice in which p65 has been deleted exhibit defects in both static and dynamic splenic architecture 11. Deletion of RelB, NIK, or IKKα leads to defects in splenic architecture, similar to those of ltbr−/− spleens 12, 13. Similar to p65/TNFR1 knockouts, mice with a defective non-canonical pathway fail to segregate B cell/T cell zones and they fail to form germinal centers following immunization. Marginal zone macrophages, which line the border between red and white pulp areas, are also absent or disorganized in RelB, p52, NIK, or IKKα knockouts 21, 22. Recent work has highlighted the role of LTα1β2 in the development and maintenance of the marginal sinus architecture, suggesting that the role of NF-κB in splenic architecture should perhaps be revisited with an eye toward an analysis of endothelial function. Some splenic defects are also attributable to effects on hematopoietic cells, most notably of the myeloid lineages.
In summary, both the canonical and non-canonical NF-κB pathways are required for the development of most secondary lymphoid organs (Figure 2). However, the non-canonical pathway, as assessed by examining mice deficient for IKKα, p52, NIK, or RelB, is especially important both during organogenesis and for maintenance of splenic architecture. Recent advances suggest that NF-κB signaling in endothelial cells, which are crucial regulators of cellular movement into and out of lymphoid organs, should be better characterized. The functional consequences of defects in these processes are severe and have direct ramifications for the ability of host to mount a robust immune response. As such, the function of NF-κB in the development and regulation of these organs represents an important facet of the role of NF-κB in immunobiology.
TNFR1, LTβR 및 RANK를 통한 신호는 p65 함유 복합체를 활성화하므로, rela-/-/tnfr1-/- 이중 녹아웃 마우스가 페이어 패치와 림프절이 부족하고 비장 조직이 무질서한 것은 놀라운 일이 아닙니다 11. 이러한 조직의 발달에서 p65의 필요성은 기질 세포에서의 기능에 기인하며, 이는 세포 사멸 조절(예: TNF에 의해 유도되는 세포 사멸), VCAM 및 LTα1β2와 같은 유기성 인자의 발현 조절, 비규범적 p52/RelB NF-κB 경로의 강화 등 복합적인 효과 때문일 수 있습니다. 비정규적 NF-κB 경로는 이차 림프 기관 발달에서 잘 확립된 역할을 합니다. 비규범적 NF-κB 신호 경로의 구성 요소인 NIK, IKKα, LT 및 RANK를 포함한 모든 구성 요소의 결실은 이차 림프 기관 발생 12, 13, 14, 15, 16, 17에 심각한 결함을 초래합니다. 비정식 경로의 표적인 p52/RelB 이합체는 CXCL12, CXCL13, CCL19, CCL21 및 MadCAM-1 18을 포함한 주요 기관 생성 인자의 주요 전사 조절 인자입니다. 따라서 p52 단일 녹아웃은 정상적인 B세포 모낭, 배아 중심, 페이어 패치 19, 20, 21이 결여된 반면, RelB 녹아웃은 페이어 패치가 결여되고 림프절 발달이 손상된 것으로 나타났습니다 18.
비장 구조는 B세포 발달뿐만 아니라 B세포 반응의 시작과 성숙에도 매우 중요합니다. 비장은 기능적, 조직학적으로 백질과 적색 펄프 영역으로 나뉩니다. 적색 펄프의 대식세포는 손상된 적혈구를 파괴하는 역할을 하며, 백색 펄프는 B세포 여포와 T세포 영역으로 구성된 비장 림프구로 채워져 있습니다. 그러나 비장 구조는 B세포 반응이 시작되고 성숙하는 동안 배아 중심이 형성되는 것에서 알 수 있듯이 역동적입니다. 다른 이차 림프 기관과 마찬가지로 비장 구조의 발달과 유지에 NF-κB가 두드러지게 관여합니다. p65가 결손된 마우스는 정적 및 동적 비장 구조 모두에서 결함이 나타납니다 11. RelB, NIK 또는 IKKα의 결실은 ltbr-/- 비장 12, 13과 유사한 비장 구조의 결함으로 이어집니다. p65/TNFR1 녹아웃과 유사하게, 비표준 경로에 결함이 있는 마우스는 B 세포/T 세포 영역 분리에 실패하고 면역 접종 후 생식 중심을 형성하지 못합니다. 적색 펄프 영역과 백색 펄프 영역의 경계를 이루는 한계 영역 대식세포도 RelB, p52, NIK 또는 IKKα 녹아웃 21, 22에서는 존재하지 않거나 조직화되지 않습니다. 최근 연구에서는 변연동 구조의 발달과 유지에 있어 LTα1β2의 역할이 강조되었으며, 이는 비장 구조에서 NF-κB의 역할을 내피 기능 분석에 초점을 맞추어 재검토해야 함을 시사합니다. 일부 비장 결함은 조혈 세포, 특히 골수 계통의 조혈 세포에 미치는 영향에 기인하기도 합니다.
요약하면,
대부분의 이차 림프 기관의 발달에는
표준 및 비표준 NF-κB 경로가 모두 필요합니다(그림 2).
그러나 IKKα, p52, NIK 또는 RelB가 결핍된 생쥐를 검사하여 평가한 결과, 비표준 경로는 장기 발생과 비장 구조 유지 모두에 특히 중요합니다. 최근의 발전은 림프 기관 안팎으로 세포 이동을 조절하는 중요한 조절자인 내피 세포의 NF-κB 신호가 더 잘 특성화되어야 한다는 것을 시사합니다. 이러한 과정의 결함으로 인한 기능적 결과는 심각하며 숙주가 강력한 면역 반응을 일으키는 능력에 직접적인 영향을 미칩니다. 따라서 이러한 기관의 발달과 조절에 있어 NF-κB의 기능은 면역 생물학에서 NF-κB의 역할의 중요한 측면을 나타냅니다.
NF-κB and hematopoiesis
The immune system includes cells of the lymphoid and myeloid lineages: T cells, B cells, monocytes, macrophages, dendritic cells (DCs), natural killer (NK) cells, basophils, eosinophils, neutrophils, and mast cells (Figure 3). These bone marrow-derived cells are the core constituents of both the innate and adaptive immune responses. Proliferation, differentiation, and apoptosis are the defining characteristic of hematopoiesis, and NF-κB participates in the regulation of each of these processes. While the study of the role of NF-κB in development and homeostasis of hematopoetic cells has focused largely on B cell and T lymphocytes, it is likely that the NF-κB pathway is also important for the development of NK cells, DCs, monocytes, granulocytes, and other cellular components of the immune system. In general, an anti-apoptotic and pro-proliferative role for NF-κB is invoked in the hematopoietic system (Figure 3). Indeed, deletion of the kinase TAK1, an upstream component of both NF-κB and AP1 pathways, results in decreased antiapoptotic gene expression, hematopoietic stem cell apoptosis, and failure of hemtopoiesis 23. However, in actuality, the situation is much more complex. For example, ikba−/−ikbe−/− cells, which have elevated NF-κB activation are broadly defective in myelopoiesis and exhibit defects in NK cells and lymphoid lineages 24, 25. Thus, NF-κB function must be interrogated at each point of cellular differentiation in order to fully appreciate its contribution to hematopoiesis. Greater insight into the majority of events that lead to the development of immune cells, awaits both further characterization of the developmental processes and generation of better genetic tools to examine the consequences of perturbing NF-κB function.
NF-κB와 조혈작용
면역계에는
림프계 및 골수계 세포가 포함됩니다:
T세포, B세포, 단핵구,
대식세포, 수지상세포(DC), 자연살해(NK) 세포,
호염기구, 호산구, 호중구, 비만세포 등이 있습니다(그림 3).
골수 유래 세포는
선천성 면역 반응과 후천성 면역 반응의 핵심 구성 요소입니다.
증식, 분화, 세포 사멸은
조혈의 주요 특징이며,
NF-κB는 이러한 각 과정의 조절에 관여합니다.
조혈 세포의 발달과 항상성 유지에서
NF-κB의 역할에 대한 연구는
주로 B 세포와 T 림프구에 집중되어 왔지만,
NK 세포, DC, 단핵구, 과립구 및 면역계의 다른 세포 구성 요소의 발달에도
NF-κB 경로가 중요할 가능성이 높습니다.
일반적으로
NF-κB는 조혈 시스템에서
항세포사멸 및 증식 촉진 역할을 합니다(그림 3).
실제로
NF-κB와 AP1 경로의 상류 구성 요소인
키나아제 TAK1이 결핍되면
항세포사멸 유전자 발현이 감소하고
조혈 줄기세포가 세포 사멸하며 조혈이 실패합니다23.
그러나 실제로는 상황이 훨씬 더 복잡합니다.
예를 들어,
NF-κB 활성화가 높은 ikba-/-ikbe-/- 세포는
광범위하게 골수 생성에 결함이 있으며
NK 세포와 림프계 혈통에서 결함을 나타냅니다24, 25.
따라서
조혈에 대한 NF-κB의 기여를 완전히 이해하려면
세포 분화의 각 시점에서 NF-κB 기능을 조사해야 합니다.
면역 세포의 발달로 이어지는 대부분의 사건에 대한
더 큰 통찰력은 발달 과정의 추가 특성화와
NF-κB 기능 교란의 결과를 조사할 수 있는
더 나은 유전적 도구의 생성을 기다리고 있습니다.
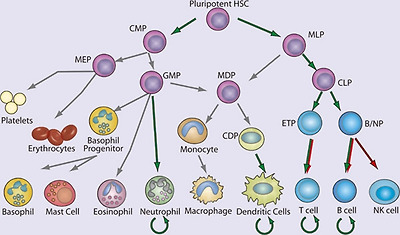
NF-κB in hematopoiesis.
Red arrows indicate stages in which NF-κB activation is thought to contribute negatively and green arrows indicate a positive function in the development of the indicated lineages. Curved arrows indicate examples in which NF-κB contributes to the survival of cell population, either in the resting state or during immune responses. Gray arrows indicate developmental events for which NF-κB plays no role or for which the role of NF-κB has not been clearly demonstrated. (HSC, hematopoietic stem cell; CMP, common myeloid progenitor; MLP, myeloid/lymphoid progenitor; MEP, megakaryocyte erythrocyte progenitor; GMP, granulocyte monocyte progenitor; MDP, macrophage dendritic cell progenitor; CDP, common dendritic cell progenitor; CLP, common lymphoid progenitor; ETP, early thymic precursor; and B/NP, B-cell natural killer cell progenitor).
조혈에서 NF-κB.
빨간색 화살표는
NF-κB 활성화가 부정적으로 기여하는 것으로 생각되는 단계를 나타내고
녹색 화살표는 표시된 계통의 발달에 긍정적인 기능을 하는 단계를 나타냅니다.
곡선 화살표는
휴식 상태 또는 면역 반응 중에
NF-κB가 세포 집단의 생존에 기여하는 예를 나타냅니다.
회색 화살표는
NF-κB가 역할을 하지 않거나
NF-κB의 역할이 명확하게 입증되지 않은 발달 사건을 나타냅니다.
(HSC, 조혈 줄기세포; CMP, 공통 골수 전구세포; MLP, 골수/림프 전구세포; MEP, 거핵구 적혈구 전구세포; GMP, 과립구 단핵구 전구세포; MDP, 대식세포 수지상세포 전구세포; CDP, 일반 수지상세포 전구세포; CLP, 일반 림프구 전구세포; ETP, 초기 흉선 전구세포; 및 B/NP, B세포 자연살해세포 전구세포).
NF-κB in non-lymphocyte hematopoiesis
Although most, if not all cells of the body can exhibit some innate immune function, the ability to recognize microbes and initiate an antimicrobial response, we focus here only on the development of certain hematopoietic, non-lymphoid components of the innate immune system. In general, our understanding of the developmental origin of these innate cells lags far behind that of lymphocytes. Yet recent progress in characterizing the developmental process for several myeloid lineages should provide a better framework within which the contribution of NF-κB can be better assessed in the future.
Significant progress has been made recently in understanding DC development, including the characterization of a common DC progenitor 26, 27, 28, yet the contribution of NF-κB to early steps of DC lineage commitment remains poorly understood. The most notable contribution of NF-κB in this area is the requirement for RelB in development of the CD8α− DCs 4, 29, 30, 31. Although single knockouts of other NF-κB genes do not result in deficiencies in DC development, deletion of both p65 and p50 has a profound effect 32, 33. Again, however, the limitations of the experimental systems used to examine the contribution of NF-κB, particularly with regard to the need to either delete TNFR1 or utilize fetal liver transfer experiments, should be underscored. In the periphery, NF-κB is clearly required for DC maturation. Loss of IKKβ or inhibition of the IKK complex using a cell permeable peptide prevents DC maturation and antigen-presenting cell (APC) function 34, 35. In addition, peripheral differentiation and function of myeloid DCs, e.g., monocyte-derived DCs, is significantly impaired by NF-κB inhibition 36. DCs in the periphery survive for a short period of time following activation, but their survival can be prolonged by CD40L expression on T cells. CD40L activates both the canonical and non-canonical NF-κB pathways, and hence DCs deficient in both p50 and c-Rel, or DCs overexpressing a mutant super-repressor form of IκBα, demonstrate significantly decreased survival 32, 37. To date, the specific targets of NF-κB in DC development and, indeed, the role of NF-κB at individual stages of DC development, remain quite poorly characterized.
Genetic models of NF-κB deficiency and activation have revealed other roles for NF-κB in the myeloid lineage. Of the non-lymphocyte lineages, neutrophil development is perhaps best characterized in terms of the role of NF-κB. There remains, however, significant gaps in both our understanding of neutrophil development, and more so the role of NF-κB in this process. IκBα-knockout mice, which have elevated classical NF-κB activity, display robust granulocytosis 38. RelB-deficient mice exhibit an inflammatory phenotype with a marked increase in peripheral neutrophil numbers. Mice lacking c-Rel and p50, and heterozygous for p65, also exhibit neutrophilia 39. This increase in peripheral neutrophil numbers resulting from deletion of NF-κB appears to be related to both increased development and increased egress from the bone marrow 39. In mature neutrophils, the anti-apoptotic signal provided by NF-κB is crucial. In sharp contrast to lymphocytes, which are relatively long-lived in the absence of activation, neutrophils normally exhibit a very short lifespan but require protection from apoptosis during inflammation when they provide effector function. Neutrophils can respond by activating NF-κB in response to numerous pro-inflammatory stimuli, and the activated NF-κB can increase neutrophil survival 40. Although neutrophils are capable of activating NF-κB in response to many pro-inflammatory stimuli 41 they lack p52 and RelB 42, which are crucial for the maintenance of long-lived lymphocytes. Thus, in neutrophils the role of NF-κB is selective and cell type specific.
비림프구 조혈에서의 NF-κB
신체의 모든 세포가 미생물을 인식하고 항균 반응을 일으키는 능력인 선천성 면역 기능을 발휘할 수는 없지만, 여기서는 선천성 면역 체계의 특정 조혈 비림프 구성 요소의 발달에만 초점을 맞추고자 합니다. 일반적으로 이러한 선천성 세포의 발달 기원에 대한 우리의 이해는 림프구에 비해 훨씬 뒤쳐져 있습니다. 그러나 최근 여러 골수성 계통의 발달 과정을 특성화하는 데 있어 진전이 이루어짐에 따라 향후 NF-κB의 기여도를 더 잘 평가할 수 있는 더 나은 프레임워크가 제공될 것입니다.
최근 일반적인 DC 전구세포 26, 27, 28의 특성 규명을 포함하여 DC 발달을 이해하는 데 상당한 진전이 있었지만, DC 계통의 초기 단계에 대한 NF-κB의 기여는 여전히 제대로 이해되지 않고 있습니다. 이 분야에서 NF-κB의 가장 주목할 만한 기여는 CD8α- DC 4, 29, 30, 31의 발달에 RelB가 필요하다는 것입니다. 다른 NF-κB 유전자의 단일 녹아웃은 DC 발달에 결핍을 초래하지 않지만, p65와 p50의 결실은 중대한 영향을 미칩니다 32, 33. 그러나 NF-κB의 기여도를 조사하는 데 사용되는 실험 시스템의 한계, 특히 TNFR1을 삭제하거나 태아 간 전이 실험을 활용해야 할 필요성과 관련하여 다시 한 번 강조해야 합니다. 말초에서 NF-κB는 DC 성숙에 분명히 필요합니다. IKKβ의 손실 또는 세포 투과성 펩타이드를 사용한 IKK 복합체의 억제는 DC 성숙과 항원 제시 세포(APC) 기능을 방해합니다 34, 35. 또한, 단핵구 유래 DC와 같은 골수성 DC의 말초 분화 및 기능은 NF-κB 억제로 인해 현저히 손상됩니다 36. 말초의 DC는 활성화 후 짧은 기간 동안 생존하지만, T 세포의 CD40L 발현에 의해 생존이 연장될 수 있습니다. CD40L은 정식 및 비정식 NF-κB 경로를 모두 활성화하므로 p50과 c-Rel이 모두 결핍된 DC 또는 돌연변이 슈퍼억제자 형태의 IκBα를 과발현하는 DC는 생존율이 현저히 감소합니다32, 37. 현재까지 DC 발달에서 NF-κB의 특정 표적과 실제로 DC 발달의 개별 단계에서 NF-κB의 역할은 아직 제대로 규명되지 않은 상태입니다.
NF-κB 결핍 및 활성화에 대한 유전자 모델은 골수 계통에서 NF-κB의 다른 역할을 밝혀냈습니다. 비림프구 계통 중 호중구 발달은 아마도 NF-κB의 역할 측면에서 가장 잘 특징지어질 수 있습니다. 그러나 호중구 발달에 대한 이해와 이 과정에서 NF-κB의 역할에 대한 이해에는 상당한 격차가 남아 있습니다. 고전적인 NF-κB 활성이 높아진 IκBα 녹아웃 마우스는 강력한 과립구증 38을 나타냅니다. RelB 결핍 마우스는 말초 호중구 수가 현저하게 증가하는 염증성 표현형을 보입니다. c-Rel과 p50이 결핍되고 p65에 이형접합을 보이는 마우스도 호중구 증가증을 나타냅니다39. NF-κB의 결핍으로 인한 말초 호중구 수의 증가는 골수 39의 발달 증가 및 배출 증가와 관련이 있는 것으로 보입니다. 성숙한 호중구에서는 NF-κB가 제공하는 항세포사멸 신호가 매우 중요합니다. 활성화되지 않을 때 상대적으로 수명이 긴 림프구와 대조적으로 호중구는 일반적으로 수명이 매우 짧지만 염증 중에 이펙터 기능을 제공할 때 세포 사멸로부터 보호해야 합니다. 호중구는 수많은 전 염증 자극에 반응하여 NF-κB를 활성화하여 대응할 수 있으며, 활성화된 NF-κB는 호중구 생존을 증가시킬 수 있습니다 40. 호중구는 많은 전염증성 자극에 반응하여 NF-κB를 활성화할 수 있지만41 수명이 긴 림프구의 유지에 중요한 p52 및 RelB 42가 부족합니다. 따라서 호중구에서 NF-κB의 역할은 선택적이고 세포 유형에 따라 특이적입니다.
NF-κB in lymphopoiesis
Development of T and B cells has, historically, been the subject of much greater scrutiny than the development of cells of the myeloid lineages. NF-κB has been examined in many aspects of lymphopoiesis and found to be vital for the development and function of adaptive immune cells 43 (Figure 4). Despite their potential longevity in the periphery, lymphocyte development is characterized by abundant apoptosis. As a consequence, the anti-apoptotic properties of NF-κB play a key role in lymphopoeisis. Indeed, in many instances the requirement for NF-κB can be overcome by transgenic expression of the anti-apoptotic factor Bcl-2 44. Indeed, the role of NF-κB in B cell development was recently re-examined 45. Although NF-κB was expendable in cells undergoing rearrangement of the eponymous Igκ loci, NF-κB was required to protect the resulting cells from apoptosis, and for these cells to progress to rearrangement of the Igλ locus. This deficit in NF-κB function could be circumvented through overexpression of Bcl-2 45. The necessity of NF-κB for lymphopoesis is strikingly illustrated in human genetic diseases in which the gene encoding NEMO is inactivated by mutation. Because the NEMO gene is located on the X chromosome, it is usually subject to random inactivation in individual cells in females. However, in female patients who are heterozygous for a mutant version of NEMO, all peripheral lymphocytes possess an intact NEMO gene, rather than the 50% predicted by random inactivation, suggesting that in the absence of NEMO-dependent NF-κB signaling, B and T cells fail to develop.
림프구 생성에 관여하는 NF-κB
역사적으로 T세포와 B세포의 발달은 골수 계통의 세포 발달보다 훨씬 더 많은 조사의 대상이 되어 왔습니다. NF-κB는 림프 생성의 여러 측면에서 조사되어 왔으며 적응 면역 세포의 발달과 기능에 필수적인 것으로 밝혀졌습니다43 (그림 4). 림프구는 말초에서 잠재적으로 오래 살 수 있지만, 림프구 발달은 풍부한 세포 사멸이 특징입니다. 결과적으로 NF-κB의 항세포사멸 특성은 림프구 증식에 중요한 역할을 합니다. 실제로 많은 경우 항세포사멸 인자인 Bcl-2 44의 형질전환 발현을 통해 NF-κB의 필요성을 극복할 수 있습니다. 실제로 B세포 발달에서 NF-κB의 역할은 최근 재조명되었습니다 45. 시조 Igκ 유전자좌의 재배열을 겪는 세포에서 NF-κB는 소모적이지만, 그 결과 세포를 세포 사멸로부터 보호하고 이러한 세포가 Igλ 유전자좌의 재배열로 진행하기 위해서는 NF-κB가 필요했습니다. 이러한 NF-κB 기능의 결핍은 Bcl-2 45의 과발현을 통해 우회할 수 있습니다. 림프구 감소증에 대한 NF-κB의 필요성은 NEMO를 암호화하는 유전자가 돌연변이에 의해 비활성화되는 인간 유전 질환에서 극명하게 드러납니다. NEMO 유전자는 X 염색체에 위치하기 때문에 일반적으로 여성의 개별 세포에서 무작위로 비활성화될 수 있습니다. 그러나 돌연변이 버전의 NEMO를 이형접합하는 여성 환자의 경우 모든 말초 림프구가 무작위 비활성화에 의해 예측된 50%가 아닌 온전한 NEMO 유전자를 보유하고 있으며, 이는 NEMO 의존성 NF-κB 신호가 없는 경우 B 및 T 세포가 발달하지 못함을 시사합니다.
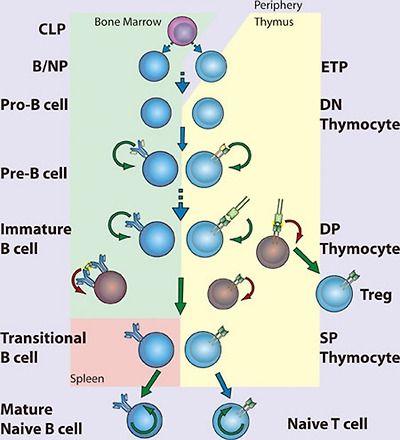
NF-κB in lymphopoiesis.
NF-κB plays a pro-survival role in common lymphoid precursor (CLP) cells which give rise to B- and T-cell lineages. B-cell development occurs in the bone marrow, where NF-κB protects pre-B cells from pro-apoptotic stimuli including TNFα. Signaling to NF-κB through the pre-B cell receptor mediates survival of Pre-B cells, which then undergo light chain recombination to produce a functional B cell receptor. NF-κB provides a necessary pro-survival signal during Igλ but not Igκ rearrangement. Expression of BCR leads to NF-κB-dependent differentiation into immature B cells. High levels of BCR signaling, i.e., through recognition of self-antigen, results in negative selection through the loss of NF-κB activity. Transitional B cells exit the bone marrow and migrate to the spleen, where they mature and differentiate, a process that also requires NF-κB. T-cell development occurs following migration of precursor cells into the thymus. Stimulation of NF-κB through pre-TCRα provides a pro-survival signal allowing recombination of the TCR α chain and maturation to the double-positive (DP) stage. Optimal signaling through the TCRα/β complex induces NF-κB-dependent survival pathways, while a failure to signal or high level signaling results in death by neglect or negative selection, respectively. Intermediate high NF-κB activation facilitates intrathymic regulatory T cell (Treg) development. NF-κB activity is required for the maintenance of long-lived B and T cells. (CLP, common lymphoid progenitor; ETP, early thymic progenitor; DN, double negative – CD4−CD8−; DP, double positive – CD4+CD8+; SP, single positive – either CD4+CD8− or CD4−CD8+)
림프구 생성에 관여하는 NF-κB.
NF-κB는 B세포와 T세포 계통을 생성하는 공통 림프 전구체(CLP) 세포에서 생존을 촉진하는 역할을 합니다. B세포 발달은 골수에서 일어나며, NF-κB는 TNFα를 포함한 세포 사멸 촉진 자극으로부터 예비 B세포를 보호합니다. 프리-B 세포 수용체를 통한 NF-κB로의 신호 전달은 프리-B 세포의 생존을 매개하며, 프리-B 세포는 경쇄 재조합을 거쳐 기능적인 B 세포 수용체를 생성합니다. NF-κB는 Igλ 동안에는 필요한 생존 신호를 제공하지만 Igκ 재배열에는 필요하지 않습니다. BCR의 발현은 미성숙 B 세포로의 NF-κB 의존적 분화로 이어집니다. 높은 수준의 BCR 신호, 즉 자가 항원 인식을 통한 신호는 NF-κB 활성의 상실을 통한 음성 선택을 초래합니다. 과도기 B 세포는 골수에서 나와 비장으로 이동하여 성숙하고 분화하는데, 이 과정에도 NF-κB가 필요합니다. T세포 발달은 전구세포가 흉선으로 이동한 후 발생합니다. 전-TCRα를 통한 NF-κB의 자극은 TCR α 사슬의 재결합과 이중 양성(DP) 단계로의 성숙을 가능하게 하는 생존 촉진 신호를 제공합니다. TCRα/β 복합체를 통한 최적의 신호는 NF-κB 의존적 생존 경로를 유도하는 반면, 신호가 없거나 신호 수준이 높으면 각각 방치 또는 음성 선택에 의한 사멸을 초래합니다. 중간 정도의 높은 NF-κB 활성화는 세포 내 조절 T 세포(Treg) 발달을 촉진합니다. NF-κB 활성은 수명이 긴 B세포와 T세포의 유지에 필요합니다. (CLP, 공통 림프 전구세포; ETP, 초기 흉선 전구세포; DN, 이중 음성 - CD4-CD8-; DP, 이중 양성 - CD4+CD8+; SP, 단일 양성 - CD4+CD8- 또는 CD4-CD8+).
The effects of NEMO inactivation in both mice and humans solidify the role of NF-κB in lymphopoiesis, although the details by which NF-κB functions in this process remain obscure. NF-κB plays diverse roles in lymphocyte development that can be grouped according to timing – that is, before, during, or after pre-antigen receptor signaling. Although no single NF-κB-subunit-knockout mouse has as severe a phenotype as NEMO knockouts with regard to the generation of mature lymphocytes, double knockouts of Rel proteins confirm the essential anti-apoptotic function of NF-κB. For example, loss of both p50 and p65, or both p65 and c-Rel, terminates lymphopoiesis before expression of the pre-antigen receptors 46, 47, suggesting that NF-κB regulates anti-apoptotic factors required for early lymphoid cell survival in response to pro-apoptotic stimuli (Figure 4). In fact, hematopoietic stem cells can activate NF-κB in response to TNFα, and in these cells NF-κB acts as a pro-survival factor 48. The role of NF-κB in early lymphocyte development seems clear – expression of either pre-T-cell receptor (pre-TCR) 49 or pre-BCR (pre-B-cell receptor) coincides with increasing NF-κB activity and induction of anti-apoptotic signals through NF-κB 50, 51. However, it remains unclear how NF-κB is activated downstream of the pre-AgR as the nature of the signaling pathway at play remains almost entirely uncharacterized. Whether NF-κB contributes to lineage choice made at these early stages or merely promotes survival is also unknown.
The extent of NF-κB activation serves as a rheostat in the selection of DP (double positive; CD4+CD8+) thymocytes (Figure 4). TCR-mediated NF-κB activation follows binding to peptide:major histocompatibility complex (MHC). A thymocyte that expresses a TCR that cannot bind MHC succumbs to “death by neglect”, whereas those that bind peptide:MHC are either positively or negatively selected depending on the strength of signaling. Thymocytes that bind self-peptide:MHC with very high affinity, are likely to be self-reactive, and hence are deleted through negative selection. Thus, only DP thymocytes that recognize self-peptide:MHC and signal within a defined range are positively selected and become single-positive (SP) T cells. During negative selection, NF-κB facilitates the induction of apoptosis following high affinity TCR ligation 52, perhaps by facilitating expression of pro-apoptotic genes and consequent sensitization to pro-apoptotic signals 53. The role of NF-κB in positive selection of thymocytes is more in keeping with the better-established role of NF-κB as an inducer of anti-apoptotic genes. Unlike in thymocytes, however, NF-κB functions as a pro-survival factor during negative selection of B cells. Immature B cells display constitutive NF-κB activity that is downregulated following BCR ligation 54 (Figure 4). Decreased NF-κB activity might then sensitize these cells to pro-apoptotic signals. Interestingly, some signaling components required for NF-κB activation in mature B and T cells can be genetically disrupted without affecting their development, suggesting that pathways leading to activation of NF-κB in developing B or T cells differ significantly from the pathways engaged following AgR ligation in mature lymphocytes.
Following positive and negative selection, DP thymocytes must make a lineage commitment as SP thymocytes (CD4+CD8− or CD4−CD8+) and thereafter emigrate from the thymus. This process requires NF-κB as deletion of NEMO in cd4-Cre mice results in loss of mature peripheral T cells 55. Equivalent deletion of the upstream kinase TAK1 has a similar outcome 56. The exact nature of NF-κB pathway requirement is somewhat unclear, as ikkb−/− chimeras, or cd4-Cre IKKβ conditional knockouts, are not defective in the production of naive T cells 55, 57. Further, the contribution of NF-κB to CD8 and CD4 lineages is not equivalent. CD8 SP cells have significantly higher levels of NF-κB activity than CD4 SP thymocytes 49, 58, yet the anti-apoptotic factor Bcl-2 is more highly expressed in CD4 than CD8 cells. Therefore, CD8 SP thymocytes are dependent on NF-κB for survival, while CD4 SP thymocytes are not 58. NF-κB is, however, clearly important in CD4 SP cell development, and forced activation of NF-κB in CD4 SP cells results in negative selection 58.
In addition to peripheral maturation and differentiation of T cells into various subsets, thymic differentiation into both TH17 cells and regulatory T cells (Tregs) is known to occur. The transcriptional control of TH17 cell differentiation and the role of thymic TH17 remain relatively obscure. One recent report has described an important role for IκBζ in TH17 differentiation 59. In this capacity, IκBζ acts with the nuclear orphan receptors RORγ and RORα to promote IL17A gene expression. Beyond this tangential role in TH17 development, a TH17-intrinsic role for NF-κB has been minimally interrogated. Instead the role of NF-κB in Treg development 60 is somewhat better understood. The differentiation of thymocytes into Foxp3/CD25-positive Tregs occurs in the thymus and depends on NF-κB. It is thought that NF-κB acts as a pioneer transcription factor enhancing accessibility of the foxp3 locus 61, 62, 63, 64.
Immature B cells complete development into either follicular or marginal zone B cells after exiting the bone marrow as transitional B cells. NF-κB-regulated expression of pro-survival factors is important to these final steps of B-cell development 65. Signaling through the TNFR superfamily member BAFFR is crucial for transitional B-cell survival through induction of anti-apoptotic Bcl-2 family members 66, 67, 68. Thus, BAFF-knockout mice exhibit a complete failure of transitional B-cell maturation, which mirrors that seen in Bcl-XL-knockout mice 66, 69, 70. BAFF activates both non-canonical and canonical NF-κB pathways, though the former is principally responsible for the anti-apoptotic function in transitional B cells. Nevertheless, deficiency in NEMO, IKKα, or IKKβ decreases the numbers of mature B cells 57, 71, 72. Likewise, p50/p52 or p65/c-Rel double-knockout progenitor cells are defective in their ability to mature beyond the transitional B-cell stage 47, 73. A requirement for both the canonical and non-canonical NF-κB pathways may explain why deletion of p50 and p52 produces a more complete block in B-cell development than loss of p65 and c-Rel. Thus, only those knockouts that target both the canonical and non-canonical NF-κB pathways have an effect that approximates the phenotype seen in BAFF or Bcl-XL deficiency. Constitutive hyperactivation of the canonical pathway can circumvent BAFF-R deletion 74, yet it remains unclear whether the canonical pathway merely supports the non-canonical pathway 75, or has an independent function.
생쥐와 사람 모두에서 NEMO 비활성화의 효과는 림프구 생성에서 NF-κB의 역할을 확고히 하지만, 이 과정에서 NF-κB가 어떻게 기능하는지는 아직 명확하게 밝혀지지 않았습니다. NF-κB는 시기, 즉 항원 수용체 신호 전달 전, 전달 중 또는 전달 후에 따라 림프구 발달에서 다양한 역할을 수행합니다. 성숙한 림프구 생성과 관련하여 단일 NF-κB 서브유닛 녹아웃 마우스는 NEMO 녹아웃 마우스만큼 심각한 표현형을 가지고 있지 않지만, Rel 단백질의 이중 녹아웃은 NF-κB의 필수적인 항세포사멸 기능을 확인시켜 줍니다. 예를 들어, p50과 p65 또는 p65와 c-Rel이 모두 손실되면 전항원 수용체 46, 47이 발현되기 전에 림프구 생성이 종료되며, 이는 NF-κB가 세포 사멸 자극에 대한 반응으로 초기 림프구 세포 생존에 필요한 항세포 사멸 인자를 조절한다는 것을 시사합니다(그림 4). 실제로 조혈 줄기세포는 TNFα에 반응하여 NF-κB를 활성화할 수 있으며, 이러한 세포에서 NF-κB는 생존 촉진 인자로 작용합니다48. 초기 림프구 발달에서 NF-κB의 역할은 분명해 보입니다. 전-T세포 수용체(pre-TCR) 49 또는 전-B세포 수용체(pre-BCR)의 발현은 NF-κB 활성 증가와 NF-κB를 통한 항자살 신호의 유도 50, 51와 일치합니다. 그러나 작용하는 신호 경로의 특성이 거의 완전히 밝혀지지 않았기 때문에 NF-κB가 pre-AgR의 하류에서 어떻게 활성화되는지는 여전히 불분명합니다. NF-κB가 이러한 초기 단계에서 이루어지는 계통 선택에 기여하는지 아니면 단순히 생존을 촉진하는지에 대해서도 알려져 있지 않습니다.
NF-κB 활성화 정도는 DP(이중 양성, CD4+CD8+) 흉선세포의 선택에 있어 가변 조절기 역할을 합니다(그림 4). TCR 매개 NF-κB 활성화는 펩타이드:주요 조직적합성 복합체(MHC)와의 결합을 따릅니다. MHC와 결합할 수 없는 TCR을 발현하는 흉선세포는 '방치에 의한 사멸'에 굴복하는 반면, 펩타이드:MHC와 결합하는 흉선세포는 신호의 강도에 따라 양성 또는 음성으로 선택됩니다. 자기 펩타이드:MHC와 매우 높은 친화력으로 결합하는 흉선세포는 자기 반응성이 높기 때문에 음성 선택을 통해 삭제될 가능성이 높습니다. 따라서 자기 펩타이드:MHC를 인식하고 정의된 범위 내에서 신호를 보내는 DP 흉선세포만 양성 선택되어 단일 양성(SP) T 세포가 됩니다. 음성 선택 과정에서 NF-κB는 세포사멸 유도 유전자의 발현을 촉진하고 결과적으로 세포사멸 신호에 대한 감작을 촉진함으로써 52 고친화성 TCR 결합에 따른 세포사멸 유도를 용이하게 합니다 53. 흉선세포의 양성 선택에서 NF-κB의 역할은 항세포사멸 유전자의 유도자로서 잘 정립된 NF-κB의 역할과 더 일치합니다. 그러나 흉선세포에서와 달리 NF-κB는 B 세포의 음성 선택 시에는 생존을 촉진하는 인자로 기능합니다. 미성숙 B 세포는 BCR 결합 54 이후 하향 조절되는 구성적 NF-κB 활성을 나타냅니다(그림 4). 감소된 NF-κB 활성은 이러한 세포를 세포 사멸 촉진 신호에 민감하게 만들 수 있습니다. 흥미롭게도 성숙한 B세포와 T세포에서 NF-κB 활성화에 필요한 일부 신호 성분은 발달에 영향을 미치지 않고 유전적으로 파괴될 수 있으며, 이는 발달 중인 B세포 또는 T세포에서 NF-κB 활성화로 이어지는 경로가 성숙한 림프구에서 AgR 연결 후 관여하는 경로와 크게 다르다는 것을 시사합니다.
양성 및 음성 선택 후, DP 림프구는 SP 림프구(CD4+CD8- 또는 CD4-CD8+)로서 계통적 약속을 한 후 흉선에서 이동해야 합니다. 이 과정에는 NF-κB가 필요한데, cd4-Cre 마우스에서 NEMO를 결실하면 성숙한 말초 T 세포가 손실되기 때문입니다 55. 상류 키나아제 TAK1을 동등하게 결실시킨 경우에도 비슷한 결과가 나타납니다 56. ikkb-/- 키메라 또는 cd4-Cre IKKβ 조건부 녹아웃은 순진한 T 세포의 생성에 결함이 없기 때문에 NF-κB 경로 요구의 정확한 본질은 다소 불분명합니다55, 57. 또한 CD8 및 CD4 계통에 대한 NF-κB의 기여도는 동일하지 않습니다. CD8 SP 세포는 CD4 SP 흉선세포 49, 58보다 훨씬 더 높은 수준의 NF-κB 활성을 가지지만, 항세포사멸 인자 Bcl-2는 CD8 세포보다 CD4에서 더 많이 발현됩니다. 따라서 CD8 SP 흉선세포는 생존을 위해 NF-κB에 의존하는 반면 , CD4 SP 흉선세포는 그렇지 않습니다. 그러나 NF-κB는 CD4 SP 세포 발달에 분명히 중요하며, CD4 SP 세포에서 NF-κB를 강제로 활성화하면 음성 선택 58이 발생합니다.
말초 성숙과 다양한 하위 집합으로의 T 세포 분화 외에도 흉선 분화는 TH17 세포와 조절 T 세포(Treg)로 모두 일어나는 것으로 알려져 있습니다. TH17 세포 분화의 전사 조절과 흉선 TH17의 역할은 비교적 잘 알려지지 않았습니다. 최근 한 보고서에서는 TH17 분화에서 IκBζ의 중요한 역할에 대해 설명했습니다59. 이러한 역할에서 IκBζ는 핵 고아 수용체 RORγ 및 RORα와 함께 작용하여 IL17A 유전자 발현을 촉진합니다. TH17 발달에서 이러한 접선적 역할 외에도 NF-κB의 TH17 내재적 역할은 거의 규명되지 않았습니다. 대신 Treg 발달에서 NF-κB의 역할 60은 다소 더 잘 이해되고 있습니다. 흉선세포가 Foxp3/CD25 양성 Treg로 분화되는 것은 흉선에서 일어나며 NF-κB에 의존합니다. NF-κB는 Foxp3 유전자좌 61, 62, 63, 64의 접근성을 향상시키는 선구자 전사 인자로 작용하는 것으로 생각됩니다.
미성숙 B 세포는 과도기 B 세포로 골수에서 나온 후 여포 또는 변연 영역 B 세포로 발달을 완료합니다. 이러한 B세포 발달의 마지막 단계에서는 NF-κB에 의해 조절되는 생존 인자의 발현이 중요합니다 65. TNFR 수퍼패밀리 구성원인 BAFFR을 통한 신호는 항세포사멸 Bcl-2 패밀리 구성원 66, 67, 68의 유도를 통한 과도기적 B세포 생존에 매우 중요합니다. 따라서 BAFF-녹아웃 마우스는 과도기적 B세포 성숙이 완전히 실패하며, 이는 Bcl-XL-녹아웃 마우스 66, 69, 70에서 볼 수 있는 것과 유사합니다. BAFF는 비규범적 및 규범적 NF-κB 경로를 모두 활성화하지만, 전자는 주로 전이성 B 세포의 항자멸사 기능을 담당합니다. 그럼에도 불구하고 NEMO, IKKα 또는 IKKβ가 결핍되면 성숙한 B 세포 57, 71, 72의 수가 감소합니다. 마찬가지로, p50/p52 또는 p65/c-Rel 이중 녹아웃 전구세포는 과도기적 B 세포 단계 47, 73을 넘어 성숙하는 능력에 결함이 있습니다. 정식 및 비정식 NF-κB 경로 모두에 대한 요구 사항은 p50 및 p52의 결실이 p65 및 c-Rel의 손실보다 B세포 발달에서 더 완전한 차단을 생성하는 이유를 설명할 수 있습니다. 따라서 표준 및 비표준 NF-κB 경로를 모두 표적으로 하는 녹아웃만이 BAFF 또는 Bcl-XL 결핍에서 보이는 표현형과 유사한 효과를 나타냅니다. 정식 경로의 구성적 과활성화는 BAFF-R 결실 74을 우회할 수 있지만, 정식 경로가 비정식 경로 75를 단순히 지원하는 것인지 아니면 독립적인 기능을 하는지는 아직 불분명합니다.
NF-κB in innate immunity
Pattern recognition receptors
Recognizing pathogens is an absolute requirement for the initiation of effector functions. Mammals express several diverse classes of pattern recognition receptors (PRRs) that are dedicated to this purpose. These PPRs include transmembrane Toll-like receptors (TLRs), cytoplasmic Nod-like receptors (NLR) and RIG-I like receptors (RLR), scavenger receptors, C-type lectins, and the complement system. Although epithelial cells are frequently the first to encounter pathogens, they are also constantly exposed to non-pathogenic microbes. Therefore, while a variety of PRRs are differentially expressed in epidermis, gut, pulmonary, urinary, and reproductive epithelium, their expression and responsiveness is tightly controlled. Instead sentinel cells of the innate immune system, particularly tissue resident DCs and macrophages, express a more complete complement of PRRs, and exhibit a higher propensity to signal on exposure to pathogens.
TLRs are evolutionarily conserved pattern recognition receptors that recognize unique, essential molecules characteristic of various classes of microbes 76. The 11 characterized mammalian TLRs have varied tissue distribution and serve as recognition receptors for pathogen-associated molecular patterns (PAMPs) present on bacteria, viruses, fungi, and parasites. Perhaps because of the modular nature of the TLR extracellular domain, which consists of multiple leucine-rich repeats (LRRs), these receptors are capable of recognizing more than one microbial molecule (Figure 5). Heterodimerization of TLRs further expands the repertoire of recognized molecules. In general, the ability of TLRs to distinguish between pathogen types is translated into appropriate innate and adaptive responses through the selective activation of NF-κB, IRFs, and other inducible transcription factors. TLR signaling pathways are complex and have been extensively reviewed elsewhere 2, 77, 78. In general, TLR signaling is often divided into MyD88-dependent and TRIF-dependent pathways: MyD88 signaling predominantly leads to the activation of NF-κB, while TRIF signaling leads to both IRF3 and, to a lesser extent, NF-κB activation.
선천성 면역의 NF-κB
패턴 인식 수용체
병원균을 인식하는 것은
이펙터 기능을 시작하기 위한 절대적인 요건입니다.
포유류는
이러한 목적에 전념하는
여러 종류의 패턴 인식 수용체(PRR)를 다양하게 발현합니다.
이러한 PRR에는
막 통과 톨 유사 수용체(TLR),
세포질 놋 유사 수용체(NLR) 및
RIG-I 유사 수용체(RLR),
청소부 수용체,
C형 렉틴 및 보체 시스템이 포함됩니다.
상피 세포는
병원균을 가장 먼저 접하는 경우가 많지만,
비병원성 미생물에도 지속적으로 노출됩니다.
따라서
표피, 장, 폐, 비뇨기 및 생식 상피에서
다양한 PRR이 차별적으로 발현되지만,
그 발현과 반응성은 엄격하게 제어됩니다.
대신 선천 면역계의 감시 세포,
특히 조직에 상주하는 DC와 대식세포는
더 완전한 PRR을 발현하며
병원균에 노출되면 신호를 보내는 경향이 더 높습니다.
TLR은
진화적으로 보존된 패턴 인식 수용체로,
다양한 종류의 미생물에서 특징적인 고유하고 필수적인 분자를 인식합니다 76.
11개의 특징적인 포유류 TLR은
조직 분포가 다양하며
박테리아, 바이러스, 곰팡이, 기생충에 존재하는 병원체 관련 분자 패턴(PAMP)을
인식하는 수용체 역할을 합니다.
여러 개의 류신 풍부 반복(LRR)으로 구성된
TLR 세포 외 도메인의 모듈식 특성으로 인해
이러한 수용체는 하나 이상의 미생물 분자를 인식할 수 있습니다(그림 5).
TLR의 이합체화는
인식하는 분자의 레퍼토리를 더욱 확장합니다.
일반적으로 병원체 유형을 구별하는 TLR의 능력은
NF-κB, IRF 및 기타 유도성 전사 인자의 선택적 활성화를 통해
적절한 선천적 및 적응적 반응으로 전환됩니다.
TLR 신호 경로는 복잡하며
다른 곳에서 광범위하게 검토된 바 있습니다 2, 77, 78.
일반적으로 TLR 신호 전달은
MyD88 의존 경로와 TRIF 의존 경로로 나뉩니다:
MyD88 신호는
주로 NF-κB의 활성화를 유도하는 반면,
TRIF 신호는 IRF3와 NF-κB 활성화를 모두 유도합니다.
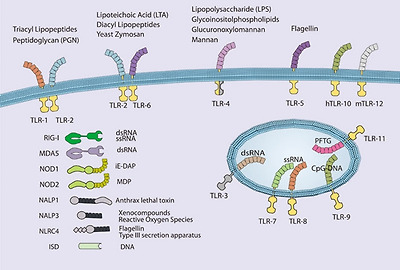
Pattern recognition receptors and their cognate ligands.
TLRs 3, 7, 8, 9 and 11 have been reported to exhibit endosomal or intracellular localization while NOD1, NOD2, RIG-I, MDA-5, NALP1, NALP3, NLRC4, and the intracellular DNA sensor (ISD) function in the cytoplasm. Only a partial list of ligands or classes of ligands for each receptor is given.
In recent years, there has been significant activity in characterization of cytoplasmic PRRs, particularly those recognizing viral nucleic acids. These efforts followed the initial description of nucleotide oligomerization domain proteins (NOD), NOD1 and NOD2. NLRs, of which NOD1 and NOD2 are the best-known examples, are characterized by LRRs and NODs. NOD1, NOD2, and IPAF have CARD domains and can signal to NF-κB (see Blonska and Lin, Cell Res 2011; 21:55-70). NOD1 recognizes a peptidoglycan containing meso-diaminopimelic acid and induces NF-κB through a canonical pathway that includes activation of IKKβ. NOD2 recognizes muramyl dipeptide (MDP), a ubiquitous component of nearly all bacterial cell walls. The CARD-containing kinase RIP2 is required for NF-κB activation. The function of RIP2 tyrosine kinase activity is unclear, although it was recently shown that kinase inhibitors could prevent MDP-induced cytokine responses 79. Although RIP2 binds to NEMO, it is not thought that RIP2 tyrosine kinase activity mediates IKK phosphorylation directly. Instead it has been proposed that RIP2 directly mediates activation of the IKK complex through NEMO binding and induced proximity 80.
Retinoic acid inducible gene I (RIG-I) and melanoma differentiation-associated gene 5 (MDA5) are DExD/H-box RNA helicase-containing cytoplasmic proteins that bind directly to dsRNA 81, 82, 83. The details of RNA binding to RIG-I and MDA5 continue to be the subject of intense research. Thus, it was thought that RIG-I recognizes dsRNA replicative intermediates of negative stranded single-stranded RNA (ssRNA) viruses, while MDA5 responds to long dsRNA replicative intermediates of plus strand ssRNA viruses 84, 85, 86, 87. Indeed, the structure of RIG-I bound to short dsRNA products has recently been solved 88, 89. RIG-I has also recently been implicated in the recognition of DNA viruses by binding short dsRNAs generated by RNA polIII 90. However, it has been shown that RIG-I can directly recognize ssRNA genomes containing 5′ triphosphates, and suggested that for negative stranded RNA viruses, this may be the relevant PAMP 91. On binding to either the viral genome, viral replication intermediate, or RNA product, RIG-I, and MDA5 induce the activation of IRF3 and NF-κB. The CARD-containing protein MAVS (also known as CARDIF, IPS1, and VISA) mediates signaling from RIG-I 92, 93, 94, 95. RIG-I and MDA5 differentially recognize various groups of RNA viruses and are independently required for robust antiviral responses 86. Therefore, together with TLRs, RLRs provide an additional mechanism of sensing viral infection and mediating type-I IFN (IFN-α/β) responses.
Pathogen recognition in innate immunity
Pathogens recognized by PRRs can be categorized as bacterial, viral, or eukaryotic, and each of these categories has several well-described PAMPs 96. Both in terms of accessibility and uniqueness to prokaryotes, the bacterial cell well is the most abundant source of PAMPs for TLRs and other PRRs. Lipoproteins, glycolipids, and protein components of bacterial cell wells have all been shown to function as PAMPs. The TLR4 ligand LPS, a glycolipid component of the outer membrane of Gram-negative bacteria, is the most thoroughly studied PAMP and the most potent TLR ligand known. Trace amounts of LPS activates the innate immune system via TLR4, leading to the production of numerous proinflammatory mediators, such as TNFα, IL-1, and IL-6. TLR4-mediated responses to LPS require CD14 and MD-2.
Conserved differences in bacterial nucleic acid structures can also be recognized by the innate immune system. TLR9 recognizes bacterial DNA containing unmethylated CpG motifs, and TLR9-deficient mice are not responsive to CpG DNA challenge 97. The low frequency and high rate of methylation of CpG motifs prevent recognition of mammalian DNA by TLR9 under physiological circumstances. A recent report indicated that the intracellular, endosomal restriction of TLR9 is critical for discriminating between self and non-self DNA, as host DNA, unlike microbial DNA, does not usually enter the endosomal compartment 98.
Nucleic acids are also key viral PAMPs, and are recognized by TLRs 3, 7, 8, and 9, as well as by RLRs. Signaling from viral PAMPs results in the activation of NF-κB and IRFs, which cooperatively mediate the production of IFN-α/β 99, 100. TLR3 recognizes dsRNA, a common viral replicative intermediate 101. TLR7 and TLR8, initially found to recognize synthetic antiviral compounds 102, 103, recognize guanosine- or uridine-rich ssRNA derived from RNA viruses 104, 105, 106. Interestingly, mammalian RNA is significantly less stimulatory than bacterial RNA, suggesting that nucleoside modifications facilitate discrimination between endogenous and pathogen RNA 107. In addition, subcellular localization of TLR7 and TLR8 to endocytic compartments, and limited constitutive cell type expression, may also facilitate self/non-self discrimination by minimizing exposure to host RNA. TLR9 recognizes viral CpG sequences and induces the induction of IFN-α 108, 109, 110. However, as membrane restriction prevents TLRs from sampling the cytosol where much of the viral lifecycle occurs, cytosolic PRRs provide comprehensive innate immune recognition. Thus, TLR3 and the adaptor TRIF are not required for viral induction of type I IFN in many tissues and cell types 111, although specialized innate antiviral cells, such as plasmacytoid DCs, rely more heavily on TLR mediated recognition of viral nucleic acids 112. Recognition of cytoplasmic dsDNA leading to NF-κB activation and type I interferon production has also been reported 113, 114. Although several candidates have been found, the relevant receptor has not yet been conclusively identified 115. This receptor(s) is predicted to be important for type I IFN production in response to viruses and intracellular pathogens, such as Listeria sp.
Although research on the recognition of PAMP/PRR pairs is an ongoing process of discovery, defective responses of MyD88-deficient cells indicate that many fungal and parasite species are capable of activating TLR pathways. TLR4 has been shown to recognize Aspergillus hyphae 116, and Cryptococcus neoformans capsular polysaccharide 117. TLR2 and TLR6 are required for recognition of yeast zymosan, while TLR4 is thought to recognize certain yeast mannans. The identification of parasite PAMPs has been more elusive, and their existence is somewhat controversial. Nevertheless, TLR2 heterodimers recognize various parasite GPI-anchored proteins and glycoinositolphospholipids from the parasitic protozoa Trypanosoma cruzi 118. TLR9 was initially reported to recognize the malarial pigment hemozoin, a byproduct of heme metabolism in infected erythrocytes 119, although subsequent studies suggest that DNA associated with hemozoin is the relevant TLR9 ligand 120. Instead, hemozoin may be recognized by the NLRP3 inflammasome 121. TLR11 recognizes a profilin-like protein that is conserved in apicomplexan parasites including Toxoplasma gondii 122. Recognition of helminths remains an area of controversy 123. In particular, it remains unclear how helminth PAMPs might preferentially induce TH2 responses that are characteristic of the immune responses to this class of pathogen. Direct skewing of adaptive responses by the mechanism of innate recognition is suggested to occur in other scenerios as well 124. For example, C-type lectins can directly affect TH activation and differentiantion 125, thus recognition of C. albicans α-mannans by dectin-2 induces a protective TH17-predominant response 126. Therefore, whether through cell type or PRR specificity, the recognition of specific PAMPS can shape the nature of the subsequent innate and adaptive immune responses.
Immediate anti-microbial responses
PAMP recognition initiates a complex series of events: the first is the mounting of immediate antimicrobial responses at the cellular level. This effective and evolutionarily conserved function of PRRs is dependent on activation of NF-κB gene programs. In particular, much of the early innate response has been demonstrated to depend on the canonical NF-κB pathway. Thus, rela−/−/ tnfr1−/− and ikbkb−/−/tnfr1−/− double-knockout mice have increased susceptibility to bacterial infection 57, 127, 128, 129. MEFs from Nemo-deficent mice do not exhibit NF-κB activation by LPS or IL-1 130. Therefore, activation of NF-κB responsive genes by the innate immune system depends on NEMO and likely progresses through the canonical NF-κB signaling pathway. Early anti-microbial effectors include NF-κB-dependent expression of defensins, which are cationic peptides that exert direct bactericidal activity by inducing membrane permeabilization. The classic mediators of defensin release are small intestinal Paneth cells, which secrete α-defensins into the intestinal lumen following exposure to bacterial PAMPs 131. The production of anti-microbial nitrogen and oxygen species, which are acutely toxic to a variety of microbes, augments the activity of anti-microbial peptides. Production of nitric oxide (NO) is mediated in part by inducible NO synthase (iNOS), which is coordinately regulated by NF-κB and STAT transcription factors 132.
Inflammation
Inflammation begins with epithelial or stromal cells of the infected tissue or tissue resident hematopoietic cells such as mast cells or DCs recognizing an inflammatory stimulus and propagating pro-inflammatory signals. These signals lead to the recruitment and activation of effector cells, initially neutrophils and later macrophages and other leukocytes, resulting in the tissue changes characteristic of inflammation – rubor, calor, dolor, and tumor (redness, heat, pain, and swelling, respectively). There is a vast amount of literature that correlates NF-κB activation with inflammation in a wide array of diseases and animal models. There are, likewise, numerous studies using gene targeting and inhibitors of NF-κB that have established the causative role of NF-κB in inflammatory processes.
NF-κB is responsible for the transcription of the genes encoding many pro-inflammatory cytokines and chemokines. The pathway from pathogen recognition to pro-inflammatory cytokine production demonstrates a particular reliance on NF-κB. The immediate targets of NF-κB-dependent pro-inflammatory cytokines, such as TNFα, tend to be receptors that, in turn, activate NF-κB. Therefore, NF-κB is crucial to the propagation and elaboration of cytokine responses. TNFα is particularly important for both local and systemic inflammation, and it is a potent and well-studied inducer of NF-κB. One important early target of these effectors is the vascular endothelium. Changes in vascular endothelial cells both recruit circulating leukocytes and provide them with a means of exiting the vasculature into the infected tissue. NF-κB regulates the expression of adhesion molecules, both on leukocytes and on endothelial cells, which allow the extravasation of leukocytes from the circulation to the site of infection 133. Thus, in the absence of p65, the recruitment of circulating leukocytes to sites of inflammation is impaired 127.
Recruited neutrophils are the key mediators of local inflammation and, as mentioned above, NF-κB is important for their survival and function under relatively toxic conditions 134. NF-κB is important for the production of the enzymes that generate prostaglandins and reactive oxygen species (e.g., iNOS and Cox, both NF-κB target genes) and may, furthermore, be involved in the signaling induced by prostaglandins 135, 136. NF-κB has also been implicated in the response to leukotrienes, which similar to prostaglandins are short-lived paracrine effectors. For example, leukotriene-induced IL-8 production appears to be dependent on rapid activation of NF-κB 137. Finally, matrix metalloporteinases are also crucial mediators of local inflammation and leukocyte chemotaxtis, and their expression is also regulated by NF-κB 138, 139, 140.
Resolution of inflammation and subsequent tissue repair is a crucial event, and its failure is a common source of pathology. Resolution of inflammation is an active process that is as complex as the inflammatory response itself, and involves numerous pathways that are not all directly relevant to NF-κB 141. Macrophages are key regulators of both inflammation and its resolution, and NF-κB plays a key role in the instruction of macrophage responses. Macrophage differentiation into pro-inflammatory (M1) or anti-inflammatory (M2) cells depends both on cell intrinsic NF-κB pathway activation as well as the local cytokine milieu, which is strongly influenced by NF-κB.
During acute inflammation, there are multiple negative feedback pathways that help to rein in inflammatory responses 3. It has long been known that cells, such as macrophages, become resistant to repeated pro-inflammatory stimuli 142. The IκB family protein Bcl-3 can function both as an inhibitor and mediator of NF-κB transcriptional programs 2. Following LPS stimulation, Bcl-3 is induced and binds p50 dimers, with which it can repress the expression of a subset of NF-κB target genes. In this manner, Bcl-3 mediates the selective inhibition of repeated LPS responses in macrophages 143. Although clearly not the only mechanism, induced transcriptional repression by Bcl-3/p50 likely contributes to LPS-tolerance. Furthermore by selectively affecting chromatin remodeling, Bcl-3 mediates repression of pro-inflammatory genes, and also facilitates the expression of the anti-inflammatory gene IL-10. Thus, it appears that Bcl-3 acts appropriately in the regulation of genes that are categorized as either “tolerizable or “non-tolerizable” 144. NF-κB p50 appears to be capable of mediating anti-inflammatory responses under several additional circumstances as well. For example, p50 negatively regulates IFNγ production by and proliferation of NK cells 145. Also, expression levels of p50 in M2-like tumor associated macrophages (TAMs) regulate the balance between anti- and pro-inflammatory functions. Thus, it was shown that TAMs overexpress p50 and that deletion of p50 renders these macrophages pro-inflammatory 146.
Inhibition of NF-κB during the resolution phase of inflammation can prolong the inflammatory process and prevent proper tissue repair 147. Furthermore, IKKα-deficient mice display increased inflammatory responses in models of local and systemic inflammation 148, and show increased production of pro-inflammatory chemokines and cytokines 148, 149. Rather unexpectedly, IKKα-deficient mice and embryonic-liver-derived macrophages from IKKα-deficient mice exhibit augmented inflammatory responses compared with wild-type mice 149. The mechanisms of IKKα-mediated repression of transcriptional responses seem to be through effects on the level of nuclear p65 and c-Rel 148. Although there are significant differences between these two reports with regard to the proposed mechanism of action, both, nevertheless, observe the same biological consequence of IKKα deficiency in macrophages. Furthermore, recent work has suggested that IKKβ also exhibits some anti-inflammatory capacity in macrophages. First, IKKβ suppresses the secretion of the potent pro-inflammatory cytokine IL-1β 150. Second, IKKβ is associated with the maintenance of an anti-inflammatory M2-like phenotype in TAMs 151. Third, IKKβ is thought to inhibit M1 macrophage function by interfering with the STAT pathway during infection 152.
Initiation of adaptive responses
The innate immune system invokes potent anti-microbial activities and maintains host protection under many settings. Nevertheless, initiating the adaptive immune response remains a crucial step for robust and durable pathogen clearance. The process of information transfer from the innate to adaptive immune system is mediated by activation and maturation of APCs. The nature of the PAMP and the signaling pathways activated by the cognate PRR, as well as the context within which pathogen recognition occurs, all influence the manner in which the APC instructs T and B cell responses.
DC maturation mediated by pathogen recognition is crucial for the initiation of the adaptive immune response. To activate naive T cells, DCs must alter their chemokine receptor expression to enable migration into lymphoid tissues. During activation DCs fine-tune their antigen processing machinery such that the presentation of pathogen epitopes is favored. Finally, the expression of co-stimulatory molecules including B7.1/B7.2 (or CD80/CD86) is upregulated. These co-stimulatory molecules ligate the co-stimulatory molecule CD28, thus providing the second signal necessary to induce full T-cell activation. The response to pathogens is tailored on the basis of the distribution of PRRs in different cell types, and the ability of different cell types to, in turn, interact with T cells in a biasing manner 153.
There continues to be debate surrounding how PRRs differentially induce TH1 versus TH2 responses. Maturation of DCs following viral infection depends on nucleic acid-binding PRRs 154, 155. Plasmacytoid DCs, which are robust inducers of interferon during viral infection, express viral PRRs including elevated expression of TLR7 and TLR9. Cytoplasmic nucleic acid-sensing PRRs, RLRs, are expressed more broadly, although they are further induced by type-I interferons, and they may, therefore, provide crucial amplification of antiviral responses following virus recognition by TLRs. While studies of anti-viral responses suggest that both cell type-specific distribution of PRRs, and selective activation of signaling pathways by certain PAMPs are key determinants of TH1 versus TH2 responses, the situation is less clear for other pathogens. It appears that multiple mechanisms converge in shaping how APCs interact with the adaptive immune system.
Role of NF-κB in the adaptive response
T-cell responses mediated by NF-κB
T-cell activation, differentiation, proliferation, and effector function are all influenced by gene programs regulated by NF-κB. The majority of effort directed at understanding the role of NF-κB in T cell activation has focused on CD4+ T cells (Figures 6 and and7).7). On the whole, the historical lack of CD8+ T-cell conditional knockouts and the selective loss of CD8 cells in the absence of functional NF-κB have impeded thorough characterization of the role of NF-κB in these cells. Nevertheless, there continues to be progress in this area and, in fact, there has been a resurgence of interest in recent years. CD4+ T cells have long been known to undergo differentiation into either TH1 or TH2 subsets depending on the cytokines present during activation 156. However, an appreciation of the ability of CD4+ helper T cells to differentiate into additional effector cell types, induced regulatory T cells, pro-inflammatory TH17 cells, TH22 cells TH9 cells, has prompted a fresh look at the contribution of NF-κB to T cell differentiation and effector function (Figure 7).

NF-κB in T cell activation.
NF-κB participates in the maintenance, activation, differentiation and proliferation of naive T cells. Tonic TCR stimulation promotes T cell maintenance, of both memory and naive cells, through NF-κB activation. Activation occurs when the naive T cell recognizes its cognate antigen presented by an activated APC expressing both peptide:MHC and B7 family co-stimulatory molecules. NF-κB-dependent proliferation and differentiation ensue and are influenced by the local cytokine milieu. NF-κB supports proliferation, differentiation and survival as indicated (green arrows).

TH cell differentiation. NF-κB participates in differentiation of several TH cell types following activation of naive CD4+ TH cells. Differentiation pathways in which NF-κB is implicated are indicated with a green arrow. Key transcription factors in each differentiation pathway are indicated above each arrow, while cytokine responsible for skewing TH cells toward a given pathway are indicated below each arrow. Additional cytokines and transcription factors implicated in several of the pathways are not depicted.
Activation of naive T cells requires antigen-specific and co-stimulatory signaling provided by activated APCs. Binding of the TCR to its cognate peptide presented in the binding cleft of MHC supplies the antigen-specific signal, while co-stimulatory signaling results from ligation of CD28 by B7 molecules expressed on activated APCs. Activated naive T-cells proliferate rapidly while simultaneously differentiating into effector cells (Figure 6). In the case of CD4+ T cells, proliferation leads to differentiation into immature effector cells, TH0, which subsequently differentiate into TH1, TH2, TH9, TH17, TH22, or inducible Treg (iTreg) cells depending on the cytokine milieu.
Stimulation of T cells through TCR and CD28 results, initially, in activation of p65-containing NF-κB complexes followed by delayed and sustained activation of c-Rel complexes 157, 158, 159, 160. Rapidly proliferating activated T cells rely on NF-κB activity for protection from apoptosis as well as for the production of cytokines, especially IL-2, supporting proliferation and differentiation (Figure 6). As expected, inhibition of NF-κB in activated T cells facilitates progression toward activation-induced cell death (AICD) or apoptosis 161, 162, and stimulation of p65-deficient naive T cells induces cell death 163. In contrast, T cells lacking c-Rel do not undergo apoptosis, but nevertheless, fail to proliferate in response to typical mitogenic stimuli 164. T cells, in which p105 cannot undergo inducible processing to p50 also fail to proliferate normally 165. In vitro, c-Rel is needed for production of IL-2 and therefore, for proliferation, but this need is not recapitulated in vivo 166. Interestingly, c-Rel-deficient T cells appear to have a defect in TH1 proliferation and production of IFNγ, indicating a selective role for NF-κB family members in TH1/TH2 differentiation, independent of that mediated by the innate response.
Multiple transcriptional activators and repressors regulate expression of IL-2. Amongst these, members of the NF-κB family play multiple roles. In naive T cells, which do not express IL-2, repressive p50 homodimers are found associated with the IL-2 promoter 167. Failure of T-cell proliferative responses in c-Rel-knockout mice is attributable to a failure to produce IL-2 164. In naive T cells, c-Rel is responsible for mediating chromatin remodeling across the IL-2 locus following CD3/CD28 co-stimulation 158. Thus, naive T cells can be primed by activation of c-Rel by pro-inflammatory cytokines, such as those elicited following stimulation with TLR ligands 157. Priming, which requires NF-κB activation, results in a more robust response to CD3/CD28 co-stimulation 168. Conversely, overexpression of p65 with c-Jun can overcome the requirement for co-stimulation in naive T cells 169.
TH0 differentiation into TH1, TH2, TH9, TH17, Treg effector cells depends on the induction of specific transcription factors, namely T-bet, GATA3, PU.1, RORγt, and Foxp3, respectively (Figure 7). There is increasing evidence that NF-κB family members are key arbitrators of the decision. For example, mice lacking p50 are unable to mount an asthma-like, airway TH2 response 170. Indeed, p50-deficient T cells fail to induce GATA3 expression during T-cell stimulation under TH2 differentiating conditions. These results suggest a transcriptional role for p50, and it was subsequently found that Bcl-3-deficient T cells also fail to undergo TH2 differentiation, suggesting that p50/Bcl-3 complexes are crucial for TH2 differentiation 171. Conversely, the same authors found that RelB-deficient T cells are deficient in TH1 differentiation due to decreased expression of T-bet. RelB may mediate upregulation of STAT4, which is responsible for T-bet induction downstream of IFNγ. Therefore, it appears that NF-κB activation during TCR stimulation may render cells competent for both proliferative and differentiating stimuli. As a corollary, NF-κB transactivation of the IL-2 gene is repressed as TH cells differentiate into TH1 or TH2 and become dependent on TH1 and TH2 cytokines (e.g., IFNγ and IL-4). It is thought that direct binding of T-bet to p65 that is associated with the IL-2 gene enhancer, may mediate the repression of IL-2 production in TH1 cells 172. In TH2 cells the lack of IL-2 transcription may be because of the decreased levels of p65 activation 173. As discussed above, several groups recently implicated NF-κB, and specifically c-Rel, in the regulation of Foxp3 expression and Treg development 61, 62, 63, 174. The relative contribution of c-Rel to iTreg development still requires further clarification. While a recent report demonstrated that IκBζ cooperates with ROR and is required for TH17 differentiation 59, the more general contribution of NF-κB in this process, either in the thymus or periphery, remains unclear. While the role of NF-κB in TH9 cells has yet to be interrogated, there are interesting clues suggesting an important role for NF-κB. Recently, PU.1 was reported to be the transcription factor responsible for TH9 differentiation 175. In unrelated recent work on acute myelogenous leukemia, it was noted that NF-κB regulates the transcription of PU.1 176. Furthermore, it has been suggested that IL-9 expression in T cells is regulated by NF-κB 177. Thus, it would seem reasonable to speculate that NF-κB may play an important role in TH9 differentiation (Figure 7). The transcription factors responsible for the differentiation of TH22 cells have not yet been identified. However, the possible role of TNFα as a TH22 differentiating cytokine is suggestive that NF-κB may be an important mediator of this differentiation process as well. In summary, NF-κB participates in both the initial T cell responses by supporting proliferation and regulating apoptosis, and is increasingly being shown to have an important role in the regulation of TH differentiation.
B-cell responses mediated by NF-κB
There are many parallels in the functions of NF-κB in T and B cells. NF-κB acts to support proliferation, regulate apoptosis, and control the processes of differentiation and maturation in B cells (Figure 8). B-cell responses are classified into two groups: thymus-dependent (TD) or thymus-independent (TI). In TD response follicular B cells require co-stimulatory signaling from TH cells expressing CD40L and cytokines, such as IL-4. These initial steps trigger germinal center formation, in which somatic hypermutation, isotype switching, and plasma cell differentiation occur. Thus, in individuals with a mutation in CD40L B cells fail to undergo class switch recombination in response to T-dependent antigens 178. Signaling through CD40 activates both canonical and non-canonical NF-κB pathways, although several lines of evidence suggest that the non-canonical pathway is not required in the response to TD antigens. Thus, B cells lacking p52 are fully capable of appropriate TD antigen responses when transferred 21, and B cells from relB−/− mice, although crippled in their proliferative response, undergo normal IgM secretion and class switching 179. In contrast to these results, B cells lacking either functional NIK or IKKα, exhibit defective responses 180, 181. It remains unclear whether the role of NIK and IKKα in this context is to induce p100 processing (non-canonical pathway) or to contribute to activation of the canonical pathway. Although both pathways are triggered by CD40 ligation, the current data suggest that canonical pathway activation is of great importance.
NF-κB가 매개하는 B세포 반응
T세포와 B세포에서
NF-κB의 기능에는 많은 유사점이 있습니다.
NF-κB는 B
세포의 증식을 지원하고
세포 사멸을 조절하며 분화 및 성숙 과정을 제어하는 역할을 합니다(그림 8).
B 세포 반응은 흉선 의존성(TD) 또는 흉선 독립성(TI)의 두 그룹으로 분류됩니다.
TD 반응에서 여포성 B 세포는 CD40L을 발현하는 TH 세포와 IL-4와 같은 사이토카인의 공동 자극 신호가 필요합니다. 이러한 초기 단계는 체세포 과돌연변이, 이소타입 전환 및 혈장 세포 분화가 발생하는 생식 중심 형성을 촉발합니다. 따라서 CD40L B 세포에 돌연변이가 있는 개인은 T 의존성 항원 178에 대한 반응으로 클래스 스위치 재조합을 거치지 못합니다. CD40을 통한 신호는 표준 및 비표준 NF-κB 경로를 모두 활성화하지만, 여러 증거에 따르면 TD 항원에 대한 반응에서 비표준 경로가 필요하지 않다고 합니다. 따라서 p52가 결여된 B 세포는 이식 시 적절한 TD 항원 반응을 충분히 수행할 수 있으며 21, relB-/- 마우스의 B 세포는 증식 반응에 장애가 있지만 정상적인 IgM 분비와 클래스 전환을 거칩니다 179. 이러한 결과와 대조적으로, 기능적 NIK 또는 IKKα가 결여된 B 세포는 결함 반응을 보입니다 180, 181. 이러한 맥락에서 NIK와 IKKα의 역할이 p100 처리(비표준 경로)를 유도하는 것인지 아니면 표준 경로의 활성화에 기여하는 것인지는 아직 불분명합니다. 두 경로 모두 CD40 결합에 의해 촉발되지만, 현재 데이터는 정식 경로 활성화가 매우 중요하다는 것을 시사합니다.

NF-κB in B cell activation.
NF-κB participates in the maintenance, activation, differentiation and proliferation of naive B cells. Tonic BCR and cytokine, BAFF, stimulation promotes naive B cell maintenance through NF-κB activation. Activation occurs when the naïve B cell recognizes its cognate antigen and receives co-stimulatory signaling (CD40L) from an activated TH cell within the germinal center. NF-κB-dependent proliferation and differentiation ensue and are coupled to BCR affinity maturation. NF-κB signaling in B cells expressing selected BCRs results in class switch recombination and differentiation into either memory B cells or plasma cells. NF-κB supports proliferation, differentiation and survival as indicated (green arrows).
Evidence also supports a role for the canonical pathway in class switch recombination. In contrast to B cells lacking RelB or p52, those from rela−/− mice exhibit markedly diminished class switching, despite a modest loss of lymphocyte proliferation following various stimuli 182. Likewise, c-Rel-deficient mice fail to generate protective humoral immune responses demonstrating the requirement for c-Rel in class switch recombination 164, 183, 184. B cells from nfbk1−/− mice exhibit decreased proliferation in response to mitogenic stimulation, and p50/p65 double-knockout B cells exhibit greater defects in proliferation and class switching 185, 186. Therefore, B cell proliferation and the TD B-cell maturation response are dependent on the canonical NF-κB pathway.
TI antigens are those that have an intrinsic ability to initiate B-cell responses in the absence of T-cell help. TI responses are carried out by marginal zone and CD5+ B cells. TI antigens function by acting as both antigen and B-cell mitogens, for example antigens that are PRR ligands or antigens with high avidity that are capable of crosslinking the BCR through repetitive structural features. In such cases it is expected that B-cell responses are more dependent on members of the canonical pathway that have well-documented roles in TLR and BCR signaling. For example, c-Rel-deficient B cells are highly sensitive to apoptosis following BCR cross-linking 187, 188, 189. As mentioned above, p50 and p50/RelA double-knockout B cells are deficient in responses to TI stimulation. Likewise, IKKβ-deficient B cells fail to mount TI or TD responses 190. These IKKβ-deficient B cells also exhibit increased spontaneous apoptosis, supporting that NF-κB is important in survival of B cells.
NF-κB and lymphocyte longevity
Lymphocyte homeostasis reflects balanced lymphocyte turnover and lymphopoiesis. Turnover of mature lymphocytes is complex as it depends upon both activation induced and homeostatic proliferation, and cell death. Lymphocyte survival is mediated through tonic stimulation provided by both the antigen receptor and certain cytokine receptors. As such, NF-κB can be assumed to play an important role in lymphocyte homeostasis and this assumption is supported by various genetic models.
T cells can be quite long-lived and naive T cells require continued contact with MHC:self-peptides for survival. These self-peptide complexes are most likely expressed on lymphoid DCs and generate a tonic low grade TCR signal. Survival of memory cells, on the other hand, is independent of continued contact with self-peptide:MHC complexes. B cells are formed at a far higher rate than T cells and therefore, B cells also undergo a significantly higher rate of turnover. Nonetheless, B cells too require maintenance signals to achieve peripheral homeostasis. The antigen receptor on B cells provides a basal level of signaling, albeit independent of the presence of antigen, which is required for maintenance of mature B cells. Thus, deletion of the BCR from mature B cells results in a complete loss of the peripheral B cell pool 191. The assumption is that BCR provides tonic activation of the NF-κB pathway resulting in the expression of anti-apoptotic proteins. Consistent with this assumption, loss of IKKβ, NEMO, or components of the CBM complex in mature B cells also results in complete loss of peripheral B cells 71, 190, 192. Furthermore, B cells from rela−/−, nfkb1−/−, nfkb2−/−, and c-rel−/− mice all have increased sensitivity to apoptosis and/or decreased survival ex vivo 67, 188, 193. In addition to contributing directly, it is thought that low level NF-κB activation in B cells contributes to survival through upregulation of p100, which is necessary for BAFFR-induced activation of the alternative pathway 67, 75, 166.
Several studies have implicated BAFFR in this aspect of the B lymphocyte survival 194, suggesting that the non-canonical NF-κB pathway is also relevant to B-cell survival. Consistent with this idea, constitutive activation of NIK or IKKα can circumvent the requirement for B cell BAFFR 74, 195. Conversely, B cells lacking IKKα exhibit defective survival 72, 196. The relevant targets of the non-canonical pathway downstream of BAFFR are thought to be Bcl-2 family members, e.g., the anti-apoptotic factor A1 187 or Bcl-XL, which are known to be regulated by p52/RelB-containing complexes, and are required for the maintenance of mature B cells. Indeed, Bcl-XL can complement B cells with a mutant BAFFR 197. Thus, tonic BCR stimulation, through upregulation of p100, may cooperate with BAFFR ligation and alternative pathway activation, to upregulate anti-apoptotic Bcl2 family members and mediate maintenance and survival of mature B cells.
Concluding remarks
The areas covered in the current review necessarily represent a subset of those in which NF-κB is a crucial regulator of immune responses. Recent work has highlighted the importance of NF-κB in the maintenance of epithelium barriers 198; as a regulator of inflammation associated with obesity and hyperlipidemia; and as a key factor in the development of inflammatory diseases and cancer. Our understanding of the immune system continues to improve, and coordinately our recognition of the many roles played by the NF-κB family of transcription factors also grows. As one example, the expanding universe of T-cell subsets opens up a new area for the analysis of the role of NF-κB in T-cell differentiation and effector function. It will be intriguing to see how NF-κB contributes to the development of TH9, TH17, TH22, and iTreg/TH3 cells. Deciphering the role of NF-κB in the effector function of these recently described cell types should also prove to be an area of great interest, particularly given the growing evidence for dysregulation of these cell types in autoimmune and inflammatory disease. There is much that remains unknown about the contributions of NF-κB in early and non-lymphoid hematopoiesis; in responses to danger signaling and allergens; in the coordination of pathogen-specific adaptive immune responses by the innate immune system; and in the differentiation and effector responses of both lymphoid and non-lymphoid innate immune cells. Thus moving forward, as in the past, much of the work done to understand the physiological and pathophysiological role of NF-κB will continue to focus on the role of this transcription factor family in Immunobiology.
이번 리뷰에서 다루는 분야는
NF-κB가 면역 반응의 중요한 조절자인 분야 중 일부에 해당합니다.
최근 연구에서는
상피 장벽의 유지, 비만 및 고지혈증과 관련된 염증 조절, 염증성 질환 및 암 발생의 핵심 인자로서
NF-κB의 중요성이 강조되었습니다 198.
면역 체계에 대한 이해는 계속 향상되고 있으며,
이에 따라 NF-κB 전사 인자 계열이 수행하는
다양한 역할에 대한 인식도 함께 증가하고 있습니다.
한 가지 예로,
T세포 하위 집합의 세계가 확장되면서
T세포 분화 및 이펙터 기능에서 NF-κB의 역할을 분석할 수 있는
새로운 영역이 열리고 있습니다.
NF-κB가
TH9, TH17, TH22 및 iTreg/TH3 세포의 발달에 어떻게 기여하는지 알아보는 것은 흥미로울 것입니다.
특히 자가면역 및 염증성 질환에서
이러한 세포 유형의 조절 장애에 대한 증거가 증가하고 있는 상황에서
최근에 설명한 이러한 세포 유형의 이펙터 기능에서
NF-κB의 역할을 해독하는 것도 큰 관심 분야가 될 것입니다.
초기 및 비림프계 조혈, 위험 신호 및 알레르겐에 대한 반응,
선천 면역계에 의한 병원체 특이적 적응 면역 반응의 조정,
림프계 및 비림프계 선천 면역 세포의 분화 및 이펙터 반응에서
NF-κB의 기여에 대해 아직 밝혀지지 않은 것이 많습니다.
따라서
과거와 마찬가지로 앞으로도
NF-κB의 생리적 및 병태생리학적 역할을 이해하기 위한 많은 연구가
면역생물학에서 이 전사 인자 계열의 역할에 초점을 맞출 것입니다.
Acknowledgments
Research in the authors' laboratory was supported by grants from the National Institutes of Health (to SG).
References
- Hayden MS, Ghosh S. Signaling to NF-kappaB. Genes Dev. 2004;18:2195–2224. [PubMed] [Google Scholar]
- Hayden MS, West AP, Ghosh S. NF-kappaB and the immune response. Oncogene. 2006;25:6758–6780. [PubMed] [Google Scholar]
- Ghosh S, Hayden MS. New regulators of NF-kappaB in inflammation. Nat Rev Immunol. 2008;8:837–848. [PubMed] [Google Scholar]
- Burkly L, Hession C, Ogata L, et al. Expression of relB is required for the development of thymic medulla and dendritic cells. Nature. 1995;373:531–536. [PubMed] [Google Scholar]
- Irla M, Hugues S, Gill J, et al. Autoantigen-specific interactions with CD4+ thymocytes control mature medullary thymic epithelial cell cellularity. Immunity. 2008;29:451–463. [PubMed] [Google Scholar]
- Hikosaka Y, Nitta T, Ohigashi I, et al. The cytokine RANKL produced by positively selected thymocytes fosters medullary thymic epithelial cells that express autoimmune regulator. Immunity. 2008;29:438–450. [PubMed] [Google Scholar]
- Akiyama T, Shimo Y, Yanai H, et al. The tumor necrosis factor family receptors RANK and CD40 cooperatively establish the thymic medullary microenvironment and self-tolerance. Immunity. 2008;29:423–437. [PubMed] [Google Scholar]
- Anderson G, Jenkinson EJ, Rodewald HR. A roadmap for thymic epithelial cell development. Eur J Immunol. 2009;39:1694–1699. [PubMed] [Google Scholar]
- Ruddle NH, Akirav EM. Secondary lymphoid organs: responding to genetic and environmental cues in ontogeny and the immune response. J Immunol. 2009;183:2205–2212. [PMC free article] [PubMed] [Google Scholar]
- van de Pavert SA, Mebius RE. New insights into the development of lymphoid tissues. Nat Rev Immunol. 2010;10:664–674. [PubMed] [Google Scholar]
- Alcamo E, Hacohen N, Schulte LC, Rennert PD, Hynes RO, Baltimore D. Requirement for the NF-kappaB family member RelA in the development of secondary lymphoid organs. J Exp Med. 2002;195:233–244. [PMC free article] [PubMed] [Google Scholar]
- Miyawaki S, Nakamura Y, Suzuka H, et al. A new mutation, aly, that induces a generalized lack of lymph nodes accompanied by immunodeficiency in mice. Eur J Immunol. 1994;24:429–434. [PubMed] [Google Scholar]
- Koike R, Nishimura T, Yasumizu R, et al. The splenic marginal zone is absent in alymphoplastic aly mutant mice. Eur J Immunol. 1996;26:669–675. [PubMed] [Google Scholar]
- Shinkura R, Kitada K, Matsuda F, et al. Alymphoplasia is caused by a point mutation in the mouse gene encoding Nf-kappa b-inducing kinase. Nat Genet. 1999;22:74–77. [PubMed] [Google Scholar]
- Mebius RE. Organogenesis of lymphoid tissues. Nat Rev Immunol. 2003;3:292–303. [PubMed] [Google Scholar]
- Bonizzi G, Karin M. The two NF-kappaB activation pathways and their role in innate and adaptive immunity. Trends Immunol. 2004;25:280–288. [PubMed] [Google Scholar]
- Dougall WC, Glaccum M, Charrier K, et al. RANK is essential for osteoclast and lymph node development. Genes Dev. 1999;13:2412–2424. [PMC free article] [PubMed] [Google Scholar]
- Yilmaz ZB, Weih DS, Sivakumar V, Weih F. RelB is required for Peyer's patch development: differential regulation of p52-RelB by lymphotoxin and TNF. EMBO J. 2003;22:121–130. [PMC free article] [PubMed] [Google Scholar]
- Paxian S, Merkle H, Riemann M, et al. Abnormal organogenesis of Peyer's patches in mice deficient for NF-kappaB1, NF-kappaB2, and Bcl-3. Gastroenterology. 2002;122:1853–1868. [PubMed] [Google Scholar]
- Caamano JH, Rizzo CA, Durham SK, et al. Nuclear factor (NF)-kappa B2 (p100/p52) is required for normal splenic microarchitecture and B cell-mediated immune responses. J Exp Med. 1998;187:185–196. [PMC free article] [PubMed] [Google Scholar]
Cell Res. 2011 Feb; 21(2): 223–244.
Published online 2011 Jan 18. doi: 10.1038/cr.2011.13
PMCID: PMC3193440
PMID: 21243012
NF-κB in immunobiology
Matthew S Hayden1 and Sankar Ghosh1,*
Author information Copyright and License information PMC Disclaimer
Abstract
NF-κB was first discovered and characterized 25 years ago as a key regulator of inducible gene expression in the immune system. Thus, it is not surprising that the clearest biological role of NF-κB is in the development and function of the immune system. Both innate and adaptive immune responses as well as the development and maintenance of the cells and tissues that comprise the immune system are, at multiple steps, under the control of the NF-κB family of transcription factors. Although this is a well-studied area of NF-κB research, new and significant findings continue to accumulate. This review will focus on these areas of recent progress while also providing a broad overview of the roles of NF-κB in mammalian immunobiology.
Keywords: NF-κB, TLR, TCR, BCR, lymphoid organogenesis, inflammation, hematopoiesis
Introduction
As discussed in the introduction to the January special issue of Cell Research, the discovery and characterization of the NF-κB family of transcription factors resulted from studies in two major areas of research: immunology and cancer biology. Although the role of NF-κB in cancer biology is becoming progressively better established, historically much of our current knowledge of NF-κB resulted from efforts directed at understanding its role in the regulation and function of the immune response. This review will attempt to provide a comprehensive discussion of the diverse functions of NF-κB in immunobiology and update our previous efforts in reviewing the far-reaching role of NF-κB in this area of biology 1, 2, 3.
The mammalian immune response comprises various mechanisms by which the physiological and functional integrity of the host is maintained in the face of microbial insult. The bone marrow-derived cells of the immune system are chiefly, though by no means solely, responsible for performing these functions. Thus, we begin by reviewing the role of NF-κB in the development of these hematopoietic cells that mediate both innate and adaptive immune responses. With the relevant cell types and tissues in place, the immune response is triggered by host recognition of foreign pathogens. Our knowledge in this area of the immune response has expanded rapidly in the past decade. Pathogen recognition is followed by pathogen clearance, a highly variable response that may draw upon coordinated responses at the level of cell, tissue, and the organism. Finally, the immune response must be resolved, damage must be repaired and, whenever possible, the triggering insult remembered for future immunity.
In mammals, the NF-κB family is composed of five related transcription factors: p50, p52, p65 (also RelA), c-Rel, and RelB (Figure 1). These transcription factors share an N-terminal DNA-binding/dimerization domain, known as the Rel homology domain, through which they can form homo- and heterodimers. NF-κB dimers can bind to a variety of related target DNA sequences called κB sites to modulate gene expression. RelB, c-Rel, and p65 contain C-terminal transcription activation domains (TADs) that enable co-activator recruitment and target gene expression. As they lack TADs, p50 and p52 can activate transcription by forming heterodimers with p65, c-Rel, or RelB, or by recruiting other TAD-containing proteins. However, as homodimers lacking the ability to drive transcription, they can repress transcription on binding to DNA.
The mammalian NF-κB, IκB and IKK protein families. Relevant domains typifying each protein family are indicated and alternative nomenclatures are provided in parenthesis. The precursor proteins p100 and p105 function as both IκB and, when processed by the proteasome, NF-κB family members. Highly simplified schematics of canonical and non-canonical NF-κB pathways are shown. In the canonical pathway NEMO-containing IKK complexes are activated and induce phosphorylation and degradation of IκBα leading to the release of NF-κB dimers including p65:p50 dimers. In the non-canonical pathway, NEMO-independent activation of IKKα is mediated by the upstream kinase NIK. IKKα, with NIK, induces phosphorylation and processing of p100 to p52 resulting in the activation of predominantly p52:RelB complexes. Although they are illustrated separately here, as discussed in the text, signaling through these two pathways often is initiated by the same receptor – e.g., LTβR or CD40. (ANK, ankyrin-repeat domain; DD, death domain; RHD, REL homology domain; TAD, transactivation domain; LZ, leucine-zipper domain; GRR, glycine-rich region; HLH, helix-loop-helix domain; Z, zinc-finger domain; CC, coiled-coil domain; NBD, NEMO-binding domain; MOD/UBD, minimal oligomerization domain/ubiquitin-binding domain; PEST, proline, glutamic acid, serine, and threonine rich.)
In most cells, NF-κB complexes are inactive, residing predominantly in the cytoplasm in a complex with inhibitory IκB proteins (IκBα, IκBβ, IκBɛ, IκBζ, p100, p105, Bcl3, IκBns; Figure 1). When signaling pathways are activated, the IκB protein is degraded and NF-κB dimers enter the nucleus to modulate target gene expression. In almost all cases, the common step in this process is mediated by the IκB kinase (IKK) complex, which phosphorylates IκB and targets it for proteasomal degradation (see Liu and Chen, Cell Res 2011; 21:6–21). The IKK complex consists of two catalytically active kinases, IKKα (IKK1) and IKKβ (IKK2) and a regulatory scaffolding protein, NEMO (IKKγ Figure 1). These are the basic players in the NF-κB pathway; however, there is considerable complexity in how the individual proteins function and how they coordinate a nuanced NF-κB response (see Shih et al., Cell Res 2011; 21:86–102). The details of individual signaling pathways, the mechanism of action of upstream signaling intermediates, the role of various post-translational modifications, and cross-talk between NF-κB and other signaling pathways are covered in depth elsewhere in this special issue. Here, we focus on the function of NF-κB and the signaling pathways that regulate it in the mammalian immune response.
Development of the immune system
The mammalian immune system consists of a functionally linked group of anatomically disparate tissues and cell types. The dispersed cellular components of the immune system that arise from the bone marrow receive much of the attention in immunology and the study of NF-κB has likewise focused on these cells. However, lymphoid organs that facilitate coordination and dissemination of immune responses carried out by immune cells are also key sites of NF-κB function. Therefore, while this section is largely concerned with the role of NF-κB in hematopoiesis, the role of NF-κB in lymphoid organogenesis is also discussed briefly.
NF-κB and lymphoid organogenesis
NF-κB plays an important role in the development and function of primary (bone marrow, thymus) and secondary (lymph nodes, Peyer's patches, mucosal-associated lymphoid tissue, and the spleen) lymphoid tissues. There is clearly a role for NF-κB in the development and regulation of bone, and we refer the reader to the excellent review on the subject in this special issue (see Novack, Cell Res 2011; 21:169–182). The role of NF-κB in thymic architecture, originally thought to be limited to the important role of RelB in thymic architecture 4, has become clearer in terms of the development of medullary thymic epithelial cells 5, 6, 7, but still remains to be more fully elucidated 8. The secondary lymphoid tissues facilitate maintenance and activation of mature lymphocytes by providing an environment within which the interaction of lymphocytes and other leukocytes can be carefully orchestrated 9. The role of NF-κB in this process is now well appreciated. Multiple NF-κB knockouts exhibit defects in some aspect of secondary lymphoid organ development.
The initial events of lymphoid organogenesis involve the association of lymphotoxin (LT)α1β2-expressing hematopoietic cells and vascular cell adhesion molecule-1 (VCAM-1)-expressing stromal cells 10. This interaction initiates a positive feedback loop through NF-κB (Figure 2). LTα1β2, receptor activator of NF-κB ligand (RANKL), and TNFα are known to activate NF-κB, and figure prominently in lymphorganogenesis 10 (Figure 2). Also, mediators of lymphoid organogenesis and homeostasis, such as adhesion molecules (e.g., intercellular adhesion molecule, VCAM, peripheral node addressin, glycosylation-dependent cell adhesion molecule-1, and mucosal addressin cellular adhesion molecule (MadCAM)), cytokines (e.g., TNFα and LTα1β2) and organogenic chemokines (e.g., CXCL12 (GRO/MIP-2), CXCL13 (BLC), CCL19 (ELC), and CCL21 (SLC)), are regulated by NF-κB.
NF-κB in lymphoid organogenesis. NF-κB regulates key components of a positive feedback loop between hematopoietic and stromal cells. LTα1β2-expressing hematopoietic cells induce production of VCAM-1 through the canonical NF-κB pathway and chemokines through the non-canonical pathway in LTβR-expressing stromal cells. Stromal expression of chemokines induces the upregulation of integrins (α4β1) on hematopoietic cells resulting in increased recruitment of LTα1β2-expressing cells, and increased signaling through stromal LTβR. RANKL signaling through RANK on the hematopoietic cell leads to activation of NF-κB, and further upregulation of LTα1β2.
Signaling through TNFR1, LTβR, and RANK activates p65-containing complexes and, hence, it is not surprising that rela−/−/tnfr1−/− double-knockout mice lack Peyer's patches and lymph nodes, and exhibit a disorganized spleen 11. The requirement for p65 in the development of these tissues is attributable to its function in stromal cells, and is likely because of a combination of effects: regulation of apoptosis (e.g., that induced by TNF); regulation of expression of organogenic factors, such as VCAM and LTα1β2; and enhancement of the non-canonical p52/RelB NF-κB pathway. The non-canonical NF-κB pathway has a well-established role in secondary lymphoid organ development. Deletion of components of the non-canonical NF-κB signaling pathway, including NIK, IKKα, LT, and RANK, all lead to severe defects in secondary lymphoid organogenesis 12, 13, 14, 15, 16, 17. The target of the non-canonical pathway, the p52/RelB dimer, is the primary transcriptional regulator of key organogenic factors including CXCL12, CXCL13, CCL19, CCL21, and MadCAM-1 18. Thus, the p52 single knockout lacks normal B-cell follicles, germinal centers, and Peyer's patches 19, 20, 21, while RelB knockouts lack Peyer's patches and exhibit compromised lymphnode development 18.
Splenic architecture is crucial for B-cell development as well as for the initiation and maturation of B-cell responses. The spleen is divided functionally and histologically into white and red pulp zones. Macrophages in the red pulp are responsible for destroying damaged erythrocytes, while the white pulp is populated by splenic lymphocytes organized into B-cell follicles and T-cell zones. Yet splenic architecture is also dynamic, as exemplified by the formation of germinal centers during the initiation and maturation of B-cell responses. As with other secondary lymphoid organs, NF-κB figures prominently in the development and maintenance of splenic architecture. Mice in which p65 has been deleted exhibit defects in both static and dynamic splenic architecture 11. Deletion of RelB, NIK, or IKKα leads to defects in splenic architecture, similar to those of ltbr−/− spleens 12, 13. Similar to p65/TNFR1 knockouts, mice with a defective non-canonical pathway fail to segregate B cell/T cell zones and they fail to form germinal centers following immunization. Marginal zone macrophages, which line the border between red and white pulp areas, are also absent or disorganized in RelB, p52, NIK, or IKKα knockouts 21, 22. Recent work has highlighted the role of LTα1β2 in the development and maintenance of the marginal sinus architecture, suggesting that the role of NF-κB in splenic architecture should perhaps be revisited with an eye toward an analysis of endothelial function. Some splenic defects are also attributable to effects on hematopoietic cells, most notably of the myeloid lineages.
In summary, both the canonical and non-canonical NF-κB pathways are required for the development of most secondary lymphoid organs (Figure 2). However, the non-canonical pathway, as assessed by examining mice deficient for IKKα, p52, NIK, or RelB, is especially important both during organogenesis and for maintenance of splenic architecture. Recent advances suggest that NF-κB signaling in endothelial cells, which are crucial regulators of cellular movement into and out of lymphoid organs, should be better characterized. The functional consequences of defects in these processes are severe and have direct ramifications for the ability of host to mount a robust immune response. As such, the function of NF-κB in the development and regulation of these organs represents an important facet of the role of NF-κB in immunobiology.
NF-κB and hematopoiesis
The immune system includes cells of the lymphoid and myeloid lineages: T cells, B cells, monocytes, macrophages, dendritic cells (DCs), natural killer (NK) cells, basophils, eosinophils, neutrophils, and mast cells (Figure 3). These bone marrow-derived cells are the core constituents of both the innate and adaptive immune responses. Proliferation, differentiation, and apoptosis are the defining characteristic of hematopoiesis, and NF-κB participates in the regulation of each of these processes. While the study of the role of NF-κB in development and homeostasis of hematopoetic cells has focused largely on B cell and T lymphocytes, it is likely that the NF-κB pathway is also important for the development of NK cells, DCs, monocytes, granulocytes, and other cellular components of the immune system. In general, an anti-apoptotic and pro-proliferative role for NF-κB is invoked in the hematopoietic system (Figure 3). Indeed, deletion of the kinase TAK1, an upstream component of both NF-κB and AP1 pathways, results in decreased antiapoptotic gene expression, hematopoietic stem cell apoptosis, and failure of hemtopoiesis 23. However, in actuality, the situation is much more complex. For example, ikba−/−ikbe−/− cells, which have elevated NF-κB activation are broadly defective in myelopoiesis and exhibit defects in NK cells and lymphoid lineages 24, 25. Thus, NF-κB function must be interrogated at each point of cellular differentiation in order to fully appreciate its contribution to hematopoiesis. Greater insight into the majority of events that lead to the development of immune cells, awaits both further characterization of the developmental processes and generation of better genetic tools to examine the consequences of perturbing NF-κB function.
NF-κB in hematopoiesis. Red arrows indicate stages in which NF-κB activation is thought to contribute negatively and green arrows indicate a positive function in the development of the indicated lineages. Curved arrows indicate examples in which NF-κB contributes to the survival of cell population, either in the resting state or during immune responses. Gray arrows indicate developmental events for which NF-κB plays no role or for which the role of NF-κB has not been clearly demonstrated. (HSC, hematopoietic stem cell; CMP, common myeloid progenitor; MLP, myeloid/lymphoid progenitor; MEP, megakaryocyte erythrocyte progenitor; GMP, granulocyte monocyte progenitor; MDP, macrophage dendritic cell progenitor; CDP, common dendritic cell progenitor; CLP, common lymphoid progenitor; ETP, early thymic precursor; and B/NP, B-cell natural killer cell progenitor).
NF-κB in non-lymphocyte hematopoiesis
Although most, if not all cells of the body can exhibit some innate immune function, the ability to recognize microbes and initiate an antimicrobial response, we focus here only on the development of certain hematopoietic, non-lymphoid components of the innate immune system. In general, our understanding of the developmental origin of these innate cells lags far behind that of lymphocytes. Yet recent progress in characterizing the developmental process for several myeloid lineages should provide a better framework within which the contribution of NF-κB can be better assessed in the future.
Significant progress has been made recently in understanding DC development, including the characterization of a common DC progenitor 26, 27, 28, yet the contribution of NF-κB to early steps of DC lineage commitment remains poorly understood. The most notable contribution of NF-κB in this area is the requirement for RelB in development of the CD8α− DCs 4, 29, 30, 31. Although single knockouts of other NF-κB genes do not result in deficiencies in DC development, deletion of both p65 and p50 has a profound effect 32, 33. Again, however, the limitations of the experimental systems used to examine the contribution of NF-κB, particularly with regard to the need to either delete TNFR1 or utilize fetal liver transfer experiments, should be underscored. In the periphery, NF-κB is clearly required for DC maturation. Loss of IKKβ or inhibition of the IKK complex using a cell permeable peptide prevents DC maturation and antigen-presenting cell (APC) function 34, 35. In addition, peripheral differentiation and function of myeloid DCs, e.g., monocyte-derived DCs, is significantly impaired by NF-κB inhibition 36. DCs in the periphery survive for a short period of time following activation, but their survival can be prolonged by CD40L expression on T cells. CD40L activates both the canonical and non-canonical NF-κB pathways, and hence DCs deficient in both p50 and c-Rel, or DCs overexpressing a mutant super-repressor form of IκBα, demonstrate significantly decreased survival 32, 37. To date, the specific targets of NF-κB in DC development and, indeed, the role of NF-κB at individual stages of DC development, remain quite poorly characterized.
Genetic models of NF-κB deficiency and activation have revealed other roles for NF-κB in the myeloid lineage. Of the non-lymphocyte lineages, neutrophil development is perhaps best characterized in terms of the role of NF-κB. There remains, however, significant gaps in both our understanding of neutrophil development, and more so the role of NF-κB in this process. IκBα-knockout mice, which have elevated classical NF-κB activity, display robust granulocytosis 38. RelB-deficient mice exhibit an inflammatory phenotype with a marked increase in peripheral neutrophil numbers. Mice lacking c-Rel and p50, and heterozygous for p65, also exhibit neutrophilia 39. This increase in peripheral neutrophil numbers resulting from deletion of NF-κB appears to be related to both increased development and increased egress from the bone marrow 39. In mature neutrophils, the anti-apoptotic signal provided by NF-κB is crucial. In sharp contrast to lymphocytes, which are relatively long-lived in the absence of activation, neutrophils normally exhibit a very short lifespan but require protection from apoptosis during inflammation when they provide effector function. Neutrophils can respond by activating NF-κB in response to numerous pro-inflammatory stimuli, and the activated NF-κB can increase neutrophil survival 40. Although neutrophils are capable of activating NF-κB in response to many pro-inflammatory stimuli 41 they lack p52 and RelB 42, which are crucial for the maintenance of long-lived lymphocytes. Thus, in neutrophils the role of NF-κB is selective and cell type specific.
NF-κB in lymphopoiesis
Development of T and B cells has, historically, been the subject of much greater scrutiny than the development of cells of the myeloid lineages. NF-κB has been examined in many aspects of lymphopoiesis and found to be vital for the development and function of adaptive immune cells 43 (Figure 4). Despite their potential longevity in the periphery, lymphocyte development is characterized by abundant apoptosis. As a consequence, the anti-apoptotic properties of NF-κB play a key role in lymphopoeisis. Indeed, in many instances the requirement for NF-κB can be overcome by transgenic expression of the anti-apoptotic factor Bcl-2 44. Indeed, the role of NF-κB in B cell development was recently re-examined 45. Although NF-κB was expendable in cells undergoing rearrangement of the eponymous Igκ loci, NF-κB was required to protect the resulting cells from apoptosis, and for these cells to progress to rearrangement of the Igλ locus. This deficit in NF-κB function could be circumvented through overexpression of Bcl-2 45. The necessity of NF-κB for lymphopoesis is strikingly illustrated in human genetic diseases in which the gene encoding NEMO is inactivated by mutation. Because the NEMO gene is located on the X chromosome, it is usually subject to random inactivation in individual cells in females. However, in female patients who are heterozygous for a mutant version of NEMO, all peripheral lymphocytes possess an intact NEMO gene, rather than the 50% predicted by random inactivation, suggesting that in the absence of NEMO-dependent NF-κB signaling, B and T cells fail to develop.
NF-κB in lymphopoiesis. NF-κB plays a pro-survival role in common lymphoid precursor (CLP) cells which give rise to B- and T-cell lineages. B-cell development occurs in the bone marrow, where NF-κB protects pre-B cells from pro-apoptotic stimuli including TNFα. Signaling to NF-κB through the pre-B cell receptor mediates survival of Pre-B cells, which then undergo light chain recombination to produce a functional B cell receptor. NF-κB provides a necessary pro-survival signal during Igλ but not Igκ rearrangement. Expression of BCR leads to NF-κB-dependent differentiation into immature B cells. High levels of BCR signaling, i.e., through recognition of self-antigen, results in negative selection through the loss of NF-κB activity. Transitional B cells exit the bone marrow and migrate to the spleen, where they mature and differentiate, a process that also requires NF-κB. T-cell development occurs following migration of precursor cells into the thymus. Stimulation of NF-κB through pre-TCRα provides a pro-survival signal allowing recombination of the TCR α chain and maturation to the double-positive (DP) stage. Optimal signaling through the TCRα/β complex induces NF-κB-dependent survival pathways, while a failure to signal or high level signaling results in death by neglect or negative selection, respectively. Intermediate high NF-κB activation facilitates intrathymic regulatory T cell (Treg) development. NF-κB activity is required for the maintenance of long-lived B and T cells. (CLP, common lymphoid progenitor; ETP, early thymic progenitor; DN, double negative – CD4−CD8−; DP, double positive – CD4+CD8+; SP, single positive – either CD4+CD8− or CD4−CD8+)
The effects of NEMO inactivation in both mice and humans solidify the role of NF-κB in lymphopoiesis, although the details by which NF-κB functions in this process remain obscure. NF-κB plays diverse roles in lymphocyte development that can be grouped according to timing – that is, before, during, or after pre-antigen receptor signaling. Although no single NF-κB-subunit-knockout mouse has as severe a phenotype as NEMO knockouts with regard to the generation of mature lymphocytes, double knockouts of Rel proteins confirm the essential anti-apoptotic function of NF-κB. For example, loss of both p50 and p65, or both p65 and c-Rel, terminates lymphopoiesis before expression of the pre-antigen receptors 46, 47, suggesting that NF-κB regulates anti-apoptotic factors required for early lymphoid cell survival in response to pro-apoptotic stimuli (Figure 4). In fact, hematopoietic stem cells can activate NF-κB in response to TNFα, and in these cells NF-κB acts as a pro-survival factor 48. The role of NF-κB in early lymphocyte development seems clear – expression of either pre-T-cell receptor (pre-TCR) 49 or pre-BCR (pre-B-cell receptor) coincides with increasing NF-κB activity and induction of anti-apoptotic signals through NF-κB 50, 51. However, it remains unclear how NF-κB is activated downstream of the pre-AgR as the nature of the signaling pathway at play remains almost entirely uncharacterized. Whether NF-κB contributes to lineage choice made at these early stages or merely promotes survival is also unknown.
The extent of NF-κB activation serves as a rheostat in the selection of DP (double positive; CD4+CD8+) thymocytes (Figure 4). TCR-mediated NF-κB activation follows binding to peptide:major histocompatibility complex (MHC). A thymocyte that expresses a TCR that cannot bind MHC succumbs to “death by neglect”, whereas those that bind peptide:MHC are either positively or negatively selected depending on the strength of signaling. Thymocytes that bind self-peptide:MHC with very high affinity, are likely to be self-reactive, and hence are deleted through negative selection. Thus, only DP thymocytes that recognize self-peptide:MHC and signal within a defined range are positively selected and become single-positive (SP) T cells. During negative selection, NF-κB facilitates the induction of apoptosis following high affinity TCR ligation 52, perhaps by facilitating expression of pro-apoptotic genes and consequent sensitization to pro-apoptotic signals 53. The role of NF-κB in positive selection of thymocytes is more in keeping with the better-established role of NF-κB as an inducer of anti-apoptotic genes. Unlike in thymocytes, however, NF-κB functions as a pro-survival factor during negative selection of B cells. Immature B cells display constitutive NF-κB activity that is downregulated following BCR ligation 54 (Figure 4). Decreased NF-κB activity might then sensitize these cells to pro-apoptotic signals. Interestingly, some signaling components required for NF-κB activation in mature B and T cells can be genetically disrupted without affecting their development, suggesting that pathways leading to activation of NF-κB in developing B or T cells differ significantly from the pathways engaged following AgR ligation in mature lymphocytes.
Following positive and negative selection, DP thymocytes must make a lineage commitment as SP thymocytes (CD4+CD8− or CD4−CD8+) and thereafter emigrate from the thymus. This process requires NF-κB as deletion of NEMO in cd4-Cre mice results in loss of mature peripheral T cells 55. Equivalent deletion of the upstream kinase TAK1 has a similar outcome 56. The exact nature of NF-κB pathway requirement is somewhat unclear, as ikkb−/− chimeras, or cd4-Cre IKKβ conditional knockouts, are not defective in the production of naive T cells 55, 57. Further, the contribution of NF-κB to CD8 and CD4 lineages is not equivalent. CD8 SP cells have significantly higher levels of NF-κB activity than CD4 SP thymocytes 49, 58, yet the anti-apoptotic factor Bcl-2 is more highly expressed in CD4 than CD8 cells. Therefore, CD8 SP thymocytes are dependent on NF-κB for survival, while CD4 SP thymocytes are not 58. NF-κB is, however, clearly important in CD4 SP cell development, and forced activation of NF-κB in CD4 SP cells results in negative selection 58.
In addition to peripheral maturation and differentiation of T cells into various subsets, thymic differentiation into both TH17 cells and regulatory T cells (Tregs) is known to occur. The transcriptional control of TH17 cell differentiation and the role of thymic TH17 remain relatively obscure. One recent report has described an important role for IκBζ in TH17 differentiation 59. In this capacity, IκBζ acts with the nuclear orphan receptors RORγ and RORα to promote IL17A gene expression. Beyond this tangential role in TH17 development, a TH17-intrinsic role for NF-κB has been minimally interrogated. Instead the role of NF-κB in Treg development 60 is somewhat better understood. The differentiation of thymocytes into Foxp3/CD25-positive Tregs occurs in the thymus and depends on NF-κB. It is thought that NF-κB acts as a pioneer transcription factor enhancing accessibility of the foxp3 locus 61, 62, 63, 64.
Immature B cells complete development into either follicular or marginal zone B cells after exiting the bone marrow as transitional B cells. NF-κB-regulated expression of pro-survival factors is important to these final steps of B-cell development 65. Signaling through the TNFR superfamily member BAFFR is crucial for transitional B-cell survival through induction of anti-apoptotic Bcl-2 family members 66, 67, 68. Thus, BAFF-knockout mice exhibit a complete failure of transitional B-cell maturation, which mirrors that seen in Bcl-XL-knockout mice 66, 69, 70. BAFF activates both non-canonical and canonical NF-κB pathways, though the former is principally responsible for the anti-apoptotic function in transitional B cells. Nevertheless, deficiency in NEMO, IKKα, or IKKβ decreases the numbers of mature B cells 57, 71, 72. Likewise, p50/p52 or p65/c-Rel double-knockout progenitor cells are defective in their ability to mature beyond the transitional B-cell stage 47, 73. A requirement for both the canonical and non-canonical NF-κB pathways may explain why deletion of p50 and p52 produces a more complete block in B-cell development than loss of p65 and c-Rel. Thus, only those knockouts that target both the canonical and non-canonical NF-κB pathways have an effect that approximates the phenotype seen in BAFF or Bcl-XL deficiency. Constitutive hyperactivation of the canonical pathway can circumvent BAFF-R deletion 74, yet it remains unclear whether the canonical pathway merely supports the non-canonical pathway 75, or has an independent function.
NF-κB in innate immunity
Pattern recognition receptors
Recognizing pathogens is an absolute requirement for the initiation of effector functions. Mammals express several diverse classes of pattern recognition receptors (PRRs) that are dedicated to this purpose. These PPRs include transmembrane Toll-like receptors (TLRs), cytoplasmic Nod-like receptors (NLR) and RIG-I like receptors (RLR), scavenger receptors, C-type lectins, and the complement system. Although epithelial cells are frequently the first to encounter pathogens, they are also constantly exposed to non-pathogenic microbes. Therefore, while a variety of PRRs are differentially expressed in epidermis, gut, pulmonary, urinary, and reproductive epithelium, their expression and responsiveness is tightly controlled. Instead sentinel cells of the innate immune system, particularly tissue resident DCs and macrophages, express a more complete complement of PRRs, and exhibit a higher propensity to signal on exposure to pathogens.
TLRs are evolutionarily conserved pattern recognition receptors that recognize unique, essential molecules characteristic of various classes of microbes 76. The 11 characterized mammalian TLRs have varied tissue distribution and serve as recognition receptors for pathogen-associated molecular patterns (PAMPs) present on bacteria, viruses, fungi, and parasites. Perhaps because of the modular nature of the TLR extracellular domain, which consists of multiple leucine-rich repeats (LRRs), these receptors are capable of recognizing more than one microbial molecule (Figure 5). Heterodimerization of TLRs further expands the repertoire of recognized molecules. In general, the ability of TLRs to distinguish between pathogen types is translated into appropriate innate and adaptive responses through the selective activation of NF-κB, IRFs, and other inducible transcription factors. TLR signaling pathways are complex and have been extensively reviewed elsewhere 2, 77, 78. In general, TLR signaling is often divided into MyD88-dependent and TRIF-dependent pathways: MyD88 signaling predominantly leads to the activation of NF-κB, while TRIF signaling leads to both IRF3 and, to a lesser extent, NF-κB activation.
Pattern recognition receptors and their cognate ligands. TLRs 3, 7, 8, 9 and 11 have been reported to exhibit endosomal or intracellular localization while NOD1, NOD2, RIG-I, MDA-5, NALP1, NALP3, NLRC4, and the intracellular DNA sensor (ISD) function in the cytoplasm. Only a partial list of ligands or classes of ligands for each receptor is given.
In recent years, there has been significant activity in characterization of cytoplasmic PRRs, particularly those recognizing viral nucleic acids. These efforts followed the initial description of nucleotide oligomerization domain proteins (NOD), NOD1 and NOD2. NLRs, of which NOD1 and NOD2 are the best-known examples, are characterized by LRRs and NODs. NOD1, NOD2, and IPAF have CARD domains and can signal to NF-κB (see Blonska and Lin, Cell Res 2011; 21:55-70). NOD1 recognizes a peptidoglycan containing meso-diaminopimelic acid and induces NF-κB through a canonical pathway that includes activation of IKKβ. NOD2 recognizes muramyl dipeptide (MDP), a ubiquitous component of nearly all bacterial cell walls. The CARD-containing kinase RIP2 is required for NF-κB activation. The function of RIP2 tyrosine kinase activity is unclear, although it was recently shown that kinase inhibitors could prevent MDP-induced cytokine responses 79. Although RIP2 binds to NEMO, it is not thought that RIP2 tyrosine kinase activity mediates IKK phosphorylation directly. Instead it has been proposed that RIP2 directly mediates activation of the IKK complex through NEMO binding and induced proximity 80.
Retinoic acid inducible gene I (RIG-I) and melanoma differentiation-associated gene 5 (MDA5) are DExD/H-box RNA helicase-containing cytoplasmic proteins that bind directly to dsRNA 81, 82, 83. The details of RNA binding to RIG-I and MDA5 continue to be the subject of intense research. Thus, it was thought that RIG-I recognizes dsRNA replicative intermediates of negative stranded single-stranded RNA (ssRNA) viruses, while MDA5 responds to long dsRNA replicative intermediates of plus strand ssRNA viruses 84, 85, 86, 87. Indeed, the structure of RIG-I bound to short dsRNA products has recently been solved 88, 89. RIG-I has also recently been implicated in the recognition of DNA viruses by binding short dsRNAs generated by RNA polIII 90. However, it has been shown that RIG-I can directly recognize ssRNA genomes containing 5′ triphosphates, and suggested that for negative stranded RNA viruses, this may be the relevant PAMP 91. On binding to either the viral genome, viral replication intermediate, or RNA product, RIG-I, and MDA5 induce the activation of IRF3 and NF-κB. The CARD-containing protein MAVS (also known as CARDIF, IPS1, and VISA) mediates signaling from RIG-I 92, 93, 94, 95. RIG-I and MDA5 differentially recognize various groups of RNA viruses and are independently required for robust antiviral responses 86. Therefore, together with TLRs, RLRs provide an additional mechanism of sensing viral infection and mediating type-I IFN (IFN-α/β) responses.
Pathogen recognition in innate immunity
Pathogens recognized by PRRs can be categorized as bacterial, viral, or eukaryotic, and each of these categories has several well-described PAMPs 96. Both in terms of accessibility and uniqueness to prokaryotes, the bacterial cell well is the most abundant source of PAMPs for TLRs and other PRRs. Lipoproteins, glycolipids, and protein components of bacterial cell wells have all been shown to function as PAMPs. The TLR4 ligand LPS, a glycolipid component of the outer membrane of Gram-negative bacteria, is the most thoroughly studied PAMP and the most potent TLR ligand known. Trace amounts of LPS activates the innate immune system via TLR4, leading to the production of numerous proinflammatory mediators, such as TNFα, IL-1, and IL-6. TLR4-mediated responses to LPS require CD14 and MD-2.
Conserved differences in bacterial nucleic acid structures can also be recognized by the innate immune system. TLR9 recognizes bacterial DNA containing unmethylated CpG motifs, and TLR9-deficient mice are not responsive to CpG DNA challenge 97. The low frequency and high rate of methylation of CpG motifs prevent recognition of mammalian DNA by TLR9 under physiological circumstances. A recent report indicated that the intracellular, endosomal restriction of TLR9 is critical for discriminating between self and non-self DNA, as host DNA, unlike microbial DNA, does not usually enter the endosomal compartment 98.
Nucleic acids are also key viral PAMPs, and are recognized by TLRs 3, 7, 8, and 9, as well as by RLRs. Signaling from viral PAMPs results in the activation of NF-κB and IRFs, which cooperatively mediate the production of IFN-α/β 99, 100. TLR3 recognizes dsRNA, a common viral replicative intermediate 101. TLR7 and TLR8, initially found to recognize synthetic antiviral compounds 102, 103, recognize guanosine- or uridine-rich ssRNA derived from RNA viruses 104, 105, 106. Interestingly, mammalian RNA is significantly less stimulatory than bacterial RNA, suggesting that nucleoside modifications facilitate discrimination between endogenous and pathogen RNA 107. In addition, subcellular localization of TLR7 and TLR8 to endocytic compartments, and limited constitutive cell type expression, may also facilitate self/non-self discrimination by minimizing exposure to host RNA. TLR9 recognizes viral CpG sequences and induces the induction of IFN-α 108, 109, 110. However, as membrane restriction prevents TLRs from sampling the cytosol where much of the viral lifecycle occurs, cytosolic PRRs provide comprehensive innate immune recognition. Thus, TLR3 and the adaptor TRIF are not required for viral induction of type I IFN in many tissues and cell types 111, although specialized innate antiviral cells, such as plasmacytoid DCs, rely more heavily on TLR mediated recognition of viral nucleic acids 112. Recognition of cytoplasmic dsDNA leading to NF-κB activation and type I interferon production has also been reported 113, 114. Although several candidates have been found, the relevant receptor has not yet been conclusively identified 115. This receptor(s) is predicted to be important for type I IFN production in response to viruses and intracellular pathogens, such as Listeria sp.
Although research on the recognition of PAMP/PRR pairs is an ongoing process of discovery, defective responses of MyD88-deficient cells indicate that many fungal and parasite species are capable of activating TLR pathways. TLR4 has been shown to recognize Aspergillus hyphae 116, and Cryptococcus neoformans capsular polysaccharide 117. TLR2 and TLR6 are required for recognition of yeast zymosan, while TLR4 is thought to recognize certain yeast mannans. The identification of parasite PAMPs has been more elusive, and their existence is somewhat controversial. Nevertheless, TLR2 heterodimers recognize various parasite GPI-anchored proteins and glycoinositolphospholipids from the parasitic protozoa Trypanosoma cruzi 118. TLR9 was initially reported to recognize the malarial pigment hemozoin, a byproduct of heme metabolism in infected erythrocytes 119, although subsequent studies suggest that DNA associated with hemozoin is the relevant TLR9 ligand 120. Instead, hemozoin may be recognized by the NLRP3 inflammasome 121. TLR11 recognizes a profilin-like protein that is conserved in apicomplexan parasites including Toxoplasma gondii 122. Recognition of helminths remains an area of controversy 123. In particular, it remains unclear how helminth PAMPs might preferentially induce TH2 responses that are characteristic of the immune responses to this class of pathogen. Direct skewing of adaptive responses by the mechanism of innate recognition is suggested to occur in other scenerios as well 124. For example, C-type lectins can directly affect TH activation and differentiantion 125, thus recognition of C. albicans α-mannans by dectin-2 induces a protective TH17-predominant response 126. Therefore, whether through cell type or PRR specificity, the recognition of specific PAMPS can shape the nature of the subsequent innate and adaptive immune responses.
Immediate anti-microbial responses
PAMP recognition initiates a complex series of events: the first is the mounting of immediate antimicrobial responses at the cellular level. This effective and evolutionarily conserved function of PRRs is dependent on activation of NF-κB gene programs. In particular, much of the early innate response has been demonstrated to depend on the canonical NF-κB pathway. Thus, rela−/−/ tnfr1−/− and ikbkb−/−/tnfr1−/− double-knockout mice have increased susceptibility to bacterial infection 57, 127, 128, 129. MEFs from Nemo-deficent mice do not exhibit NF-κB activation by LPS or IL-1 130. Therefore, activation of NF-κB responsive genes by the innate immune system depends on NEMO and likely progresses through the canonical NF-κB signaling pathway. Early anti-microbial effectors include NF-κB-dependent expression of defensins, which are cationic peptides that exert direct bactericidal activity by inducing membrane permeabilization. The classic mediators of defensin release are small intestinal Paneth cells, which secrete α-defensins into the intestinal lumen following exposure to bacterial PAMPs 131. The production of anti-microbial nitrogen and oxygen species, which are acutely toxic to a variety of microbes, augments the activity of anti-microbial peptides. Production of nitric oxide (NO) is mediated in part by inducible NO synthase (iNOS), which is coordinately regulated by NF-κB and STAT transcription factors 132.
Inflammation
Inflammation begins with epithelial or stromal cells of the infected tissue or tissue resident hematopoietic cells such as mast cells or DCs recognizing an inflammatory stimulus and propagating pro-inflammatory signals. These signals lead to the recruitment and activation of effector cells, initially neutrophils and later macrophages and other leukocytes, resulting in the tissue changes characteristic of inflammation – rubor, calor, dolor, and tumor (redness, heat, pain, and swelling, respectively). There is a vast amount of literature that correlates NF-κB activation with inflammation in a wide array of diseases and animal models. There are, likewise, numerous studies using gene targeting and inhibitors of NF-κB that have established the causative role of NF-κB in inflammatory processes.
NF-κB is responsible for the transcription of the genes encoding many pro-inflammatory cytokines and chemokines. The pathway from pathogen recognition to pro-inflammatory cytokine production demonstrates a particular reliance on NF-κB. The immediate targets of NF-κB-dependent pro-inflammatory cytokines, such as TNFα, tend to be receptors that, in turn, activate NF-κB. Therefore, NF-κB is crucial to the propagation and elaboration of cytokine responses. TNFα is particularly important for both local and systemic inflammation, and it is a potent and well-studied inducer of NF-κB. One important early target of these effectors is the vascular endothelium. Changes in vascular endothelial cells both recruit circulating leukocytes and provide them with a means of exiting the vasculature into the infected tissue. NF-κB regulates the expression of adhesion molecules, both on leukocytes and on endothelial cells, which allow the extravasation of leukocytes from the circulation to the site of infection 133. Thus, in the absence of p65, the recruitment of circulating leukocytes to sites of inflammation is impaired 127.
Recruited neutrophils are the key mediators of local inflammation and, as mentioned above, NF-κB is important for their survival and function under relatively toxic conditions 134. NF-κB is important for the production of the enzymes that generate prostaglandins and reactive oxygen species (e.g., iNOS and Cox, both NF-κB target genes) and may, furthermore, be involved in the signaling induced by prostaglandins 135, 136. NF-κB has also been implicated in the response to leukotrienes, which similar to prostaglandins are short-lived paracrine effectors. For example, leukotriene-induced IL-8 production appears to be dependent on rapid activation of NF-κB 137. Finally, matrix metalloporteinases are also crucial mediators of local inflammation and leukocyte chemotaxtis, and their expression is also regulated by NF-κB 138, 139, 140.
Resolution of inflammation and subsequent tissue repair is a crucial event, and its failure is a common source of pathology. Resolution of inflammation is an active process that is as complex as the inflammatory response itself, and involves numerous pathways that are not all directly relevant to NF-κB 141. Macrophages are key regulators of both inflammation and its resolution, and NF-κB plays a key role in the instruction of macrophage responses. Macrophage differentiation into pro-inflammatory (M1) or anti-inflammatory (M2) cells depends both on cell intrinsic NF-κB pathway activation as well as the local cytokine milieu, which is strongly influenced by NF-κB.
During acute inflammation, there are multiple negative feedback pathways that help to rein in inflammatory responses 3. It has long been known that cells, such as macrophages, become resistant to repeated pro-inflammatory stimuli 142. The IκB family protein Bcl-3 can function both as an inhibitor and mediator of NF-κB transcriptional programs 2. Following LPS stimulation, Bcl-3 is induced and binds p50 dimers, with which it can repress the expression of a subset of NF-κB target genes. In this manner, Bcl-3 mediates the selective inhibition of repeated LPS responses in macrophages 143. Although clearly not the only mechanism, induced transcriptional repression by Bcl-3/p50 likely contributes to LPS-tolerance. Furthermore by selectively affecting chromatin remodeling, Bcl-3 mediates repression of pro-inflammatory genes, and also facilitates the expression of the anti-inflammatory gene IL-10. Thus, it appears that Bcl-3 acts appropriately in the regulation of genes that are categorized as either “tolerizable or “non-tolerizable” 144. NF-κB p50 appears to be capable of mediating anti-inflammatory responses under several additional circumstances as well. For example, p50 negatively regulates IFNγ production by and proliferation of NK cells 145. Also, expression levels of p50 in M2-like tumor associated macrophages (TAMs) regulate the balance between anti- and pro-inflammatory functions. Thus, it was shown that TAMs overexpress p50 and that deletion of p50 renders these macrophages pro-inflammatory 146.
Inhibition of NF-κB during the resolution phase of inflammation can prolong the inflammatory process and prevent proper tissue repair 147. Furthermore, IKKα-deficient mice display increased inflammatory responses in models of local and systemic inflammation 148, and show increased production of pro-inflammatory chemokines and cytokines 148, 149. Rather unexpectedly, IKKα-deficient mice and embryonic-liver-derived macrophages from IKKα-deficient mice exhibit augmented inflammatory responses compared with wild-type mice 149. The mechanisms of IKKα-mediated repression of transcriptional responses seem to be through effects on the level of nuclear p65 and c-Rel 148. Although there are significant differences between these two reports with regard to the proposed mechanism of action, both, nevertheless, observe the same biological consequence of IKKα deficiency in macrophages. Furthermore, recent work has suggested that IKKβ also exhibits some anti-inflammatory capacity in macrophages. First, IKKβ suppresses the secretion of the potent pro-inflammatory cytokine IL-1β 150. Second, IKKβ is associated with the maintenance of an anti-inflammatory M2-like phenotype in TAMs 151. Third, IKKβ is thought to inhibit M1 macrophage function by interfering with the STAT pathway during infection 152.
Initiation of adaptive responses
The innate immune system invokes potent anti-microbial activities and maintains host protection under many settings. Nevertheless, initiating the adaptive immune response remains a crucial step for robust and durable pathogen clearance. The process of information transfer from the innate to adaptive immune system is mediated by activation and maturation of APCs. The nature of the PAMP and the signaling pathways activated by the cognate PRR, as well as the context within which pathogen recognition occurs, all influence the manner in which the APC instructs T and B cell responses.
DC maturation mediated by pathogen recognition is crucial for the initiation of the adaptive immune response. To activate naive T cells, DCs must alter their chemokine receptor expression to enable migration into lymphoid tissues. During activation DCs fine-tune their antigen processing machinery such that the presentation of pathogen epitopes is favored. Finally, the expression of co-stimulatory molecules including B7.1/B7.2 (or CD80/CD86) is upregulated. These co-stimulatory molecules ligate the co-stimulatory molecule CD28, thus providing the second signal necessary to induce full T-cell activation. The response to pathogens is tailored on the basis of the distribution of PRRs in different cell types, and the ability of different cell types to, in turn, interact with T cells in a biasing manner 153.
There continues to be debate surrounding how PRRs differentially induce TH1 versus TH2 responses. Maturation of DCs following viral infection depends on nucleic acid-binding PRRs 154, 155. Plasmacytoid DCs, which are robust inducers of interferon during viral infection, express viral PRRs including elevated expression of TLR7 and TLR9. Cytoplasmic nucleic acid-sensing PRRs, RLRs, are expressed more broadly, although they are further induced by type-I interferons, and they may, therefore, provide crucial amplification of antiviral responses following virus recognition by TLRs. While studies of anti-viral responses suggest that both cell type-specific distribution of PRRs, and selective activation of signaling pathways by certain PAMPs are key determinants of TH1 versus TH2 responses, the situation is less clear for other pathogens. It appears that multiple mechanisms converge in shaping how APCs interact with the adaptive immune system.
Role of NF-κB in the adaptive response
T-cell responses mediated by NF-κB
T-cell activation, differentiation, proliferation, and effector function are all influenced by gene programs regulated by NF-κB. The majority of effort directed at understanding the role of NF-κB in T cell activation has focused on CD4+ T cells (Figures 6 and and7).7). On the whole, the historical lack of CD8+ T-cell conditional knockouts and the selective loss of CD8 cells in the absence of functional NF-κB have impeded thorough characterization of the role of NF-κB in these cells. Nevertheless, there continues to be progress in this area and, in fact, there has been a resurgence of interest in recent years. CD4+ T cells have long been known to undergo differentiation into either TH1 or TH2 subsets depending on the cytokines present during activation 156. However, an appreciation of the ability of CD4+ helper T cells to differentiate into additional effector cell types, induced regulatory T cells, pro-inflammatory TH17 cells, TH22 cells TH9 cells, has prompted a fresh look at the contribution of NF-κB to T cell differentiation and effector function (Figure 7).
NF-κB in T cell activation. NF-κB participates in the maintenance, activation, differentiation and proliferation of naive T cells. Tonic TCR stimulation promotes T cell maintenance, of both memory and naive cells, through NF-κB activation. Activation occurs when the naive T cell recognizes its cognate antigen presented by an activated APC expressing both peptide:MHC and B7 family co-stimulatory molecules. NF-κB-dependent proliferation and differentiation ensue and are influenced by the local cytokine milieu. NF-κB supports proliferation, differentiation and survival as indicated (green arrows).
TH cell differentiation. NF-κB participates in differentiation of several TH cell types following activation of naive CD4+ TH cells. Differentiation pathways in which NF-κB is implicated are indicated with a green arrow. Key transcription factors in each differentiation pathway are indicated above each arrow, while cytokine responsible for skewing TH cells toward a given pathway are indicated below each arrow. Additional cytokines and transcription factors implicated in several of the pathways are not depicted.
Activation of naive T cells requires antigen-specific and co-stimulatory signaling provided by activated APCs. Binding of the TCR to its cognate peptide presented in the binding cleft of MHC supplies the antigen-specific signal, while co-stimulatory signaling results from ligation of CD28 by B7 molecules expressed on activated APCs. Activated naive T-cells proliferate rapidly while simultaneously differentiating into effector cells (Figure 6). In the case of CD4+ T cells, proliferation leads to differentiation into immature effector cells, TH0, which subsequently differentiate into TH1, TH2, TH9, TH17, TH22, or inducible Treg (iTreg) cells depending on the cytokine milieu.
Stimulation of T cells through TCR and CD28 results, initially, in activation of p65-containing NF-κB complexes followed by delayed and sustained activation of c-Rel complexes 157, 158, 159, 160. Rapidly proliferating activated T cells rely on NF-κB activity for protection from apoptosis as well as for the production of cytokines, especially IL-2, supporting proliferation and differentiation (Figure 6). As expected, inhibition of NF-κB in activated T cells facilitates progression toward activation-induced cell death (AICD) or apoptosis 161, 162, and stimulation of p65-deficient naive T cells induces cell death 163. In contrast, T cells lacking c-Rel do not undergo apoptosis, but nevertheless, fail to proliferate in response to typical mitogenic stimuli 164. T cells, in which p105 cannot undergo inducible processing to p50 also fail to proliferate normally 165. In vitro, c-Rel is needed for production of IL-2 and therefore, for proliferation, but this need is not recapitulated in vivo 166. Interestingly, c-Rel-deficient T cells appear to have a defect in TH1 proliferation and production of IFNγ, indicating a selective role for NF-κB family members in TH1/TH2 differentiation, independent of that mediated by the innate response.
Multiple transcriptional activators and repressors regulate expression of IL-2. Amongst these, members of the NF-κB family play multiple roles. In naive T cells, which do not express IL-2, repressive p50 homodimers are found associated with the IL-2 promoter 167. Failure of T-cell proliferative responses in c-Rel-knockout mice is attributable to a failure to produce IL-2 164. In naive T cells, c-Rel is responsible for mediating chromatin remodeling across the IL-2 locus following CD3/CD28 co-stimulation 158. Thus, naive T cells can be primed by activation of c-Rel by pro-inflammatory cytokines, such as those elicited following stimulation with TLR ligands 157. Priming, which requires NF-κB activation, results in a more robust response to CD3/CD28 co-stimulation 168. Conversely, overexpression of p65 with c-Jun can overcome the requirement for co-stimulation in naive T cells 169.
TH0 differentiation into TH1, TH2, TH9, TH17, Treg effector cells depends on the induction of specific transcription factors, namely T-bet, GATA3, PU.1, RORγt, and Foxp3, respectively (Figure 7). There is increasing evidence that NF-κB family members are key arbitrators of the decision. For example, mice lacking p50 are unable to mount an asthma-like, airway TH2 response 170. Indeed, p50-deficient T cells fail to induce GATA3 expression during T-cell stimulation under TH2 differentiating conditions. These results suggest a transcriptional role for p50, and it was subsequently found that Bcl-3-deficient T cells also fail to undergo TH2 differentiation, suggesting that p50/Bcl-3 complexes are crucial for TH2 differentiation 171. Conversely, the same authors found that RelB-deficient T cells are deficient in TH1 differentiation due to decreased expression of T-bet. RelB may mediate upregulation of STAT4, which is responsible for T-bet induction downstream of IFNγ. Therefore, it appears that NF-κB activation during TCR stimulation may render cells competent for both proliferative and differentiating stimuli. As a corollary, NF-κB transactivation of the IL-2 gene is repressed as TH cells differentiate into TH1 or TH2 and become dependent on TH1 and TH2 cytokines (e.g., IFNγ and IL-4). It is thought that direct binding of T-bet to p65 that is associated with the IL-2 gene enhancer, may mediate the repression of IL-2 production in TH1 cells 172. In TH2 cells the lack of IL-2 transcription may be because of the decreased levels of p65 activation 173. As discussed above, several groups recently implicated NF-κB, and specifically c-Rel, in the regulation of Foxp3 expression and Treg development 61, 62, 63, 174. The relative contribution of c-Rel to iTreg development still requires further clarification. While a recent report demonstrated that IκBζ cooperates with ROR and is required for TH17 differentiation 59, the more general contribution of NF-κB in this process, either in the thymus or periphery, remains unclear. While the role of NF-κB in TH9 cells has yet to be interrogated, there are interesting clues suggesting an important role for NF-κB. Recently, PU.1 was reported to be the transcription factor responsible for TH9 differentiation 175. In unrelated recent work on acute myelogenous leukemia, it was noted that NF-κB regulates the transcription of PU.1 176. Furthermore, it has been suggested that IL-9 expression in T cells is regulated by NF-κB 177. Thus, it would seem reasonable to speculate that NF-κB may play an important role in TH9 differentiation (Figure 7). The transcription factors responsible for the differentiation of TH22 cells have not yet been identified. However, the possible role of TNFα as a TH22 differentiating cytokine is suggestive that NF-κB may be an important mediator of this differentiation process as well. In summary, NF-κB participates in both the initial T cell responses by supporting proliferation and regulating apoptosis, and is increasingly being shown to have an important role in the regulation of TH differentiation.
B-cell responses mediated by NF-κB
There are many parallels in the functions of NF-κB in T and B cells. NF-κB acts to support proliferation, regulate apoptosis, and control the processes of differentiation and maturation in B cells (Figure 8). B-cell responses are classified into two groups: thymus-dependent (TD) or thymus-independent (TI). In TD response follicular B cells require co-stimulatory signaling from TH cells expressing CD40L and cytokines, such as IL-4. These initial steps trigger germinal center formation, in which somatic hypermutation, isotype switching, and plasma cell differentiation occur. Thus, in individuals with a mutation in CD40L B cells fail to undergo class switch recombination in response to T-dependent antigens 178. Signaling through CD40 activates both canonical and non-canonical NF-κB pathways, although several lines of evidence suggest that the non-canonical pathway is not required in the response to TD antigens. Thus, B cells lacking p52 are fully capable of appropriate TD antigen responses when transferred 21, and B cells from relB−/− mice, although crippled in their proliferative response, undergo normal IgM secretion and class switching 179. In contrast to these results, B cells lacking either functional NIK or IKKα, exhibit defective responses 180, 181. It remains unclear whether the role of NIK and IKKα in this context is to induce p100 processing (non-canonical pathway) or to contribute to activation of the canonical pathway. Although both pathways are triggered by CD40 ligation, the current data suggest that canonical pathway activation is of great importance.
NF-κB in B cell activation. NF-κB participates in the maintenance, activation, differentiation and proliferation of naive B cells. Tonic BCR and cytokine, BAFF, stimulation promotes naive B cell maintenance through NF-κB activation. Activation occurs when the naïve B cell recognizes its cognate antigen and receives co-stimulatory signaling (CD40L) from an activated TH cell within the germinal center. NF-κB-dependent proliferation and differentiation ensue and are coupled to BCR affinity maturation. NF-κB signaling in B cells expressing selected BCRs results in class switch recombination and differentiation into either memory B cells or plasma cells. NF-κB supports proliferation, differentiation and survival as indicated (green arrows).
Evidence also supports a role for the canonical pathway in class switch recombination. In contrast to B cells lacking RelB or p52, those from rela−/− mice exhibit markedly diminished class switching, despite a modest loss of lymphocyte proliferation following various stimuli 182. Likewise, c-Rel-deficient mice fail to generate protective humoral immune responses demonstrating the requirement for c-Rel in class switch recombination 164, 183, 184. B cells from nfbk1−/− mice exhibit decreased proliferation in response to mitogenic stimulation, and p50/p65 double-knockout B cells exhibit greater defects in proliferation and class switching 185, 186. Therefore, B cell proliferation and the TD B-cell maturation response are dependent on the canonical NF-κB pathway.
TI antigens are those that have an intrinsic ability to initiate B-cell responses in the absence of T-cell help. TI responses are carried out by marginal zone and CD5+ B cells. TI antigens function by acting as both antigen and B-cell mitogens, for example antigens that are PRR ligands or antigens with high avidity that are capable of crosslinking the BCR through repetitive structural features. In such cases it is expected that B-cell responses are more dependent on members of the canonical pathway that have well-documented roles in TLR and BCR signaling. For example, c-Rel-deficient B cells are highly sensitive to apoptosis following BCR cross-linking 187, 188, 189. As mentioned above, p50 and p50/RelA double-knockout B cells are deficient in responses to TI stimulation. Likewise, IKKβ-deficient B cells fail to mount TI or TD responses 190. These IKKβ-deficient B cells also exhibit increased spontaneous apoptosis, supporting that NF-κB is important in survival of B cells.
NF-κB and lymphocyte longevity
Lymphocyte homeostasis reflects balanced lymphocyte turnover and lymphopoiesis. Turnover of mature lymphocytes is complex as it depends upon both activation induced and homeostatic proliferation, and cell death. Lymphocyte survival is mediated through tonic stimulation provided by both the antigen receptor and certain cytokine receptors. As such, NF-κB can be assumed to play an important role in lymphocyte homeostasis and this assumption is supported by various genetic models.
T cells can be quite long-lived and naive T cells require continued contact with MHC:self-peptides for survival. These self-peptide complexes are most likely expressed on lymphoid DCs and generate a tonic low grade TCR signal. Survival of memory cells, on the other hand, is independent of continued contact with self-peptide:MHC complexes. B cells are formed at a far higher rate than T cells and therefore, B cells also undergo a significantly higher rate of turnover. Nonetheless, B cells too require maintenance signals to achieve peripheral homeostasis. The antigen receptor on B cells provides a basal level of signaling, albeit independent of the presence of antigen, which is required for maintenance of mature B cells. Thus, deletion of the BCR from mature B cells results in a complete loss of the peripheral B cell pool 191. The assumption is that BCR provides tonic activation of the NF-κB pathway resulting in the expression of anti-apoptotic proteins. Consistent with this assumption, loss of IKKβ, NEMO, or components of the CBM complex in mature B cells also results in complete loss of peripheral B cells 71, 190, 192. Furthermore, B cells from rela−/−, nfkb1−/−, nfkb2−/−, and c-rel−/− mice all have increased sensitivity to apoptosis and/or decreased survival ex vivo 67, 188, 193. In addition to contributing directly, it is thought that low level NF-κB activation in B cells contributes to survival through upregulation of p100, which is necessary for BAFFR-induced activation of the alternative pathway 67, 75, 166.
Several studies have implicated BAFFR in this aspect of the B lymphocyte survival 194, suggesting that the non-canonical NF-κB pathway is also relevant to B-cell survival. Consistent with this idea, constitutive activation of NIK or IKKα can circumvent the requirement for B cell BAFFR 74, 195. Conversely, B cells lacking IKKα exhibit defective survival 72, 196. The relevant targets of the non-canonical pathway downstream of BAFFR are thought to be Bcl-2 family members, e.g., the anti-apoptotic factor A1 187 or Bcl-XL, which are known to be regulated by p52/RelB-containing complexes, and are required for the maintenance of mature B cells. Indeed, Bcl-XL can complement B cells with a mutant BAFFR 197. Thus, tonic BCR stimulation, through upregulation of p100, may cooperate with BAFFR ligation and alternative pathway activation, to upregulate anti-apoptotic Bcl2 family members and mediate maintenance and survival of mature B cells.
Concluding remarks
The areas covered in the current review necessarily represent a subset of those in which NF-κB is a crucial regulator of immune responses. Recent work has highlighted the importance of NF-κB in the maintenance of epithelium barriers 198; as a regulator of inflammation associated with obesity and hyperlipidemia; and as a key factor in the development of inflammatory diseases and cancer. Our understanding of the immune system continues to improve, and coordinately our recognition of the many roles played by the NF-κB family of transcription factors also grows. As one example, the expanding universe of T-cell subsets opens up a new area for the analysis of the role of NF-κB in T-cell differentiation and effector function. It will be intriguing to see how NF-κB contributes to the development of TH9, TH17, TH22, and iTreg/TH3 cells. Deciphering the role of NF-κB in the effector function of these recently described cell types should also prove to be an area of great interest, particularly given the growing evidence for dysregulation of these cell types in autoimmune and inflammatory disease. There is much that remains unknown about the contributions of NF-κB in early and non-lymphoid hematopoiesis; in responses to danger signaling and allergens; in the coordination of pathogen-specific adaptive immune responses by the innate immune system; and in the differentiation and effector responses of both lymphoid and non-lymphoid innate immune cells. Thus moving forward, as in the past, much of the work done to understand the physiological and pathophysiological role of NF-κB will continue to focus on the role of this transcription factor family in Immunobiology.
Acknowledgments
Research in the authors' laboratory was supported by grants from the National Institutes of Health (to SG).
References
- Hayden MS, Ghosh S. Signaling to NF-kappaB. Genes Dev. 2004;18:2195–2224. [PubMed] [Google Scholar]
- Hayden MS, West AP, Ghosh S. NF-kappaB and the immune response. Oncogene. 2006;25:6758–6780. [PubMed] [Google Scholar]
- Ghosh S, Hayden MS. New regulators of NF-kappaB in inflammation. Nat Rev Immunol. 2008;8:837–848. [PubMed] [Google Scholar]
- Burkly L, Hession C, Ogata L, et al. Expression of relB is required for the development of thymic medulla and dendritic cells. Nature. 1995;373:531–536. [PubMed] [Google Scholar]
- Irla M, Hugues S, Gill J, et al. Autoantigen-specific interactions with CD4+ thymocytes control mature medullary thymic epithelial cell cellularity. Immunity. 2008;29:451–463. [PubMed] [Google Scholar]
- Hikosaka Y, Nitta T, Ohigashi I, et al. The cytokine RANKL produced by positively selected thymocytes fosters medullary thymic epithelial cells that express autoimmune regulator. Immunity. 2008;29:438–450. [PubMed] [Google Scholar]
- Akiyama T, Shimo Y, Yanai H, et al. The tumor necrosis factor family receptors RANK and CD40 cooperatively establish the thymic medullary microenvironment and self-tolerance. Immunity. 2008;29:423–437. [PubMed] [Google Scholar]
- Anderson G, Jenkinson EJ, Rodewald HR. A roadmap for thymic epithelial cell development. Eur J Immunol. 2009;39:1694–1699. [PubMed] [Google Scholar]
- Ruddle NH, Akirav EM. Secondary lymphoid organs: responding to genetic and environmental cues in ontogeny and the immune response. J Immunol. 2009;183:2205–2212. [PMC free article] [PubMed] [Google Scholar]
- van de Pavert SA, Mebius RE. New insights into the development of lymphoid tissues. Nat Rev Immunol. 2010;10:664–674. [PubMed] [Google Scholar]
- Alcamo E, Hacohen N, Schulte LC, Rennert PD, Hynes RO, Baltimore D. Requirement for the NF-kappaB family member RelA in the development of secondary lymphoid organs. J Exp Med. 2002;195:233–244. [PMC free article] [PubMed] [Google Scholar]
- Miyawaki S, Nakamura Y, Suzuka H, et al. A new mutation, aly, that induces a generalized lack of lymph nodes accompanied by immunodeficiency in mice. Eur J Immunol. 1994;24:429–434. [PubMed] [Google Scholar]
- Koike R, Nishimura T, Yasumizu R, et al. The splenic marginal zone is absent in alymphoplastic aly mutant mice. Eur J Immunol. 1996;26:669–675. [PubMed] [Google Scholar]
- Shinkura R, Kitada K, Matsuda F, et al. Alymphoplasia is caused by a point mutation in the mouse gene encoding Nf-kappa b-inducing kinase. Nat Genet. 1999;22:74–77. [PubMed] [Google Scholar]
- Mebius RE. Organogenesis of lymphoid tissues. Nat Rev Immunol. 2003;3:292–303. [PubMed] [Google Scholar]
- Bonizzi G, Karin M. The two NF-kappaB activation pathways and their role in innate and adaptive immunity. Trends Immunol. 2004;25:280–288. [PubMed] [Google Scholar]
- Dougall WC, Glaccum M, Charrier K, et al. RANK is essential for osteoclast and lymph node development. Genes Dev. 1999;13:2412–2424. [PMC free article] [PubMed] [Google Scholar]
- Yilmaz ZB, Weih DS, Sivakumar V, Weih F. RelB is required for Peyer's patch development: differential regulation of p52-RelB by lymphotoxin and TNF. EMBO J. 2003;22:121–130. [PMC free article] [PubMed] [Google Scholar]
- Paxian S, Merkle H, Riemann M, et al. Abnormal organogenesis of Peyer's patches in mice deficient for NF-kappaB1, NF-kappaB2, and Bcl-3. Gastroenterology. 2002;122:1853–1868. [PubMed] [Google Scholar]
- Caamano JH, Rizzo CA, Durham SK, et al. Nuclear factor (NF)-kappa B2 (p100/p52) is required for normal splenic microarchitecture and B cell-mediated immune responses. J Exp Med. 1998;187:185–196. [PMC free article] [PubMed] [Google Scholar]
Cell Res. 2011 Feb; 21(2): 223–244.
Published online 2011 Jan 18. doi: 10.1038/cr.2011.13
PMCID: PMC3193440
PMID: 21243012
NF-κB in immunobiology
Matthew S Hayden1 and Sankar Ghosh1,*
Author information Copyright and License information PMC Disclaimer
Abstract
NF-κB was first discovered and characterized 25 years ago as a key regulator of inducible gene expression in the immune system. Thus, it is not surprising that the clearest biological role of NF-κB is in the development and function of the immune system. Both innate and adaptive immune responses as well as the development and maintenance of the cells and tissues that comprise the immune system are, at multiple steps, under the control of the NF-κB family of transcription factors. Although this is a well-studied area of NF-κB research, new and significant findings continue to accumulate. This review will focus on these areas of recent progress while also providing a broad overview of the roles of NF-κB in mammalian immunobiology.
Keywords: NF-κB, TLR, TCR, BCR, lymphoid organogenesis, inflammation, hematopoiesis
Introduction
As discussed in the introduction to the January special issue of Cell Research, the discovery and characterization of the NF-κB family of transcription factors resulted from studies in two major areas of research: immunology and cancer biology. Although the role of NF-κB in cancer biology is becoming progressively better established, historically much of our current knowledge of NF-κB resulted from efforts directed at understanding its role in the regulation and function of the immune response. This review will attempt to provide a comprehensive discussion of the diverse functions of NF-κB in immunobiology and update our previous efforts in reviewing the far-reaching role of NF-κB in this area of biology 1, 2, 3.
The mammalian immune response comprises various mechanisms by which the physiological and functional integrity of the host is maintained in the face of microbial insult. The bone marrow-derived cells of the immune system are chiefly, though by no means solely, responsible for performing these functions. Thus, we begin by reviewing the role of NF-κB in the development of these hematopoietic cells that mediate both innate and adaptive immune responses. With the relevant cell types and tissues in place, the immune response is triggered by host recognition of foreign pathogens. Our knowledge in this area of the immune response has expanded rapidly in the past decade. Pathogen recognition is followed by pathogen clearance, a highly variable response that may draw upon coordinated responses at the level of cell, tissue, and the organism. Finally, the immune response must be resolved, damage must be repaired and, whenever possible, the triggering insult remembered for future immunity.
In mammals, the NF-κB family is composed of five related transcription factors: p50, p52, p65 (also RelA), c-Rel, and RelB (Figure 1). These transcription factors share an N-terminal DNA-binding/dimerization domain, known as the Rel homology domain, through which they can form homo- and heterodimers. NF-κB dimers can bind to a variety of related target DNA sequences called κB sites to modulate gene expression. RelB, c-Rel, and p65 contain C-terminal transcription activation domains (TADs) that enable co-activator recruitment and target gene expression. As they lack TADs, p50 and p52 can activate transcription by forming heterodimers with p65, c-Rel, or RelB, or by recruiting other TAD-containing proteins. However, as homodimers lacking the ability to drive transcription, they can repress transcription on binding to DNA.
The mammalian NF-κB, IκB and IKK protein families. Relevant domains typifying each protein family are indicated and alternative nomenclatures are provided in parenthesis. The precursor proteins p100 and p105 function as both IκB and, when processed by the proteasome, NF-κB family members. Highly simplified schematics of canonical and non-canonical NF-κB pathways are shown. In the canonical pathway NEMO-containing IKK complexes are activated and induce phosphorylation and degradation of IκBα leading to the release of NF-κB dimers including p65:p50 dimers. In the non-canonical pathway, NEMO-independent activation of IKKα is mediated by the upstream kinase NIK. IKKα, with NIK, induces phosphorylation and processing of p100 to p52 resulting in the activation of predominantly p52:RelB complexes. Although they are illustrated separately here, as discussed in the text, signaling through these two pathways often is initiated by the same receptor – e.g., LTβR or CD40. (ANK, ankyrin-repeat domain; DD, death domain; RHD, REL homology domain; TAD, transactivation domain; LZ, leucine-zipper domain; GRR, glycine-rich region; HLH, helix-loop-helix domain; Z, zinc-finger domain; CC, coiled-coil domain; NBD, NEMO-binding domain; MOD/UBD, minimal oligomerization domain/ubiquitin-binding domain; PEST, proline, glutamic acid, serine, and threonine rich.)
In most cells, NF-κB complexes are inactive, residing predominantly in the cytoplasm in a complex with inhibitory IκB proteins (IκBα, IκBβ, IκBɛ, IκBζ, p100, p105, Bcl3, IκBns; Figure 1). When signaling pathways are activated, the IκB protein is degraded and NF-κB dimers enter the nucleus to modulate target gene expression. In almost all cases, the common step in this process is mediated by the IκB kinase (IKK) complex, which phosphorylates IκB and targets it for proteasomal degradation (see Liu and Chen, Cell Res 2011; 21:6–21). The IKK complex consists of two catalytically active kinases, IKKα (IKK1) and IKKβ (IKK2) and a regulatory scaffolding protein, NEMO (IKKγ Figure 1). These are the basic players in the NF-κB pathway; however, there is considerable complexity in how the individual proteins function and how they coordinate a nuanced NF-κB response (see Shih et al., Cell Res 2011; 21:86–102). The details of individual signaling pathways, the mechanism of action of upstream signaling intermediates, the role of various post-translational modifications, and cross-talk between NF-κB and other signaling pathways are covered in depth elsewhere in this special issue. Here, we focus on the function of NF-κB and the signaling pathways that regulate it in the mammalian immune response.
Development of the immune system
The mammalian immune system consists of a functionally linked group of anatomically disparate tissues and cell types. The dispersed cellular components of the immune system that arise from the bone marrow receive much of the attention in immunology and the study of NF-κB has likewise focused on these cells. However, lymphoid organs that facilitate coordination and dissemination of immune responses carried out by immune cells are also key sites of NF-κB function. Therefore, while this section is largely concerned with the role of NF-κB in hematopoiesis, the role of NF-κB in lymphoid organogenesis is also discussed briefly.
NF-κB and lymphoid organogenesis
NF-κB plays an important role in the development and function of primary (bone marrow, thymus) and secondary (lymph nodes, Peyer's patches, mucosal-associated lymphoid tissue, and the spleen) lymphoid tissues. There is clearly a role for NF-κB in the development and regulation of bone, and we refer the reader to the excellent review on the subject in this special issue (see Novack, Cell Res 2011; 21:169–182). The role of NF-κB in thymic architecture, originally thought to be limited to the important role of RelB in thymic architecture 4, has become clearer in terms of the development of medullary thymic epithelial cells 5, 6, 7, but still remains to be more fully elucidated 8. The secondary lymphoid tissues facilitate maintenance and activation of mature lymphocytes by providing an environment within which the interaction of lymphocytes and other leukocytes can be carefully orchestrated 9. The role of NF-κB in this process is now well appreciated. Multiple NF-κB knockouts exhibit defects in some aspect of secondary lymphoid organ development.
The initial events of lymphoid organogenesis involve the association of lymphotoxin (LT)α1β2-expressing hematopoietic cells and vascular cell adhesion molecule-1 (VCAM-1)-expressing stromal cells 10. This interaction initiates a positive feedback loop through NF-κB (Figure 2). LTα1β2, receptor activator of NF-κB ligand (RANKL), and TNFα are known to activate NF-κB, and figure prominently in lymphorganogenesis 10 (Figure 2). Also, mediators of lymphoid organogenesis and homeostasis, such as adhesion molecules (e.g., intercellular adhesion molecule, VCAM, peripheral node addressin, glycosylation-dependent cell adhesion molecule-1, and mucosal addressin cellular adhesion molecule (MadCAM)), cytokines (e.g., TNFα and LTα1β2) and organogenic chemokines (e.g., CXCL12 (GRO/MIP-2), CXCL13 (BLC), CCL19 (ELC), and CCL21 (SLC)), are regulated by NF-κB.
NF-κB in lymphoid organogenesis. NF-κB regulates key components of a positive feedback loop between hematopoietic and stromal cells. LTα1β2-expressing hematopoietic cells induce production of VCAM-1 through the canonical NF-κB pathway and chemokines through the non-canonical pathway in LTβR-expressing stromal cells. Stromal expression of chemokines induces the upregulation of integrins (α4β1) on hematopoietic cells resulting in increased recruitment of LTα1β2-expressing cells, and increased signaling through stromal LTβR. RANKL signaling through RANK on the hematopoietic cell leads to activation of NF-κB, and further upregulation of LTα1β2.
Signaling through TNFR1, LTβR, and RANK activates p65-containing complexes and, hence, it is not surprising that rela−/−/tnfr1−/− double-knockout mice lack Peyer's patches and lymph nodes, and exhibit a disorganized spleen 11. The requirement for p65 in the development of these tissues is attributable to its function in stromal cells, and is likely because of a combination of effects: regulation of apoptosis (e.g., that induced by TNF); regulation of expression of organogenic factors, such as VCAM and LTα1β2; and enhancement of the non-canonical p52/RelB NF-κB pathway. The non-canonical NF-κB pathway has a well-established role in secondary lymphoid organ development. Deletion of components of the non-canonical NF-κB signaling pathway, including NIK, IKKα, LT, and RANK, all lead to severe defects in secondary lymphoid organogenesis 12, 13, 14, 15, 16, 17. The target of the non-canonical pathway, the p52/RelB dimer, is the primary transcriptional regulator of key organogenic factors including CXCL12, CXCL13, CCL19, CCL21, and MadCAM-1 18. Thus, the p52 single knockout lacks normal B-cell follicles, germinal centers, and Peyer's patches 19, 20, 21, while RelB knockouts lack Peyer's patches and exhibit compromised lymphnode development 18.
Splenic architecture is crucial for B-cell development as well as for the initiation and maturation of B-cell responses. The spleen is divided functionally and histologically into white and red pulp zones. Macrophages in the red pulp are responsible for destroying damaged erythrocytes, while the white pulp is populated by splenic lymphocytes organized into B-cell follicles and T-cell zones. Yet splenic architecture is also dynamic, as exemplified by the formation of germinal centers during the initiation and maturation of B-cell responses. As with other secondary lymphoid organs, NF-κB figures prominently in the development and maintenance of splenic architecture. Mice in which p65 has been deleted exhibit defects in both static and dynamic splenic architecture 11. Deletion of RelB, NIK, or IKKα leads to defects in splenic architecture, similar to those of ltbr−/− spleens 12, 13. Similar to p65/TNFR1 knockouts, mice with a defective non-canonical pathway fail to segregate B cell/T cell zones and they fail to form germinal centers following immunization. Marginal zone macrophages, which line the border between red and white pulp areas, are also absent or disorganized in RelB, p52, NIK, or IKKα knockouts 21, 22. Recent work has highlighted the role of LTα1β2 in the development and maintenance of the marginal sinus architecture, suggesting that the role of NF-κB in splenic architecture should perhaps be revisited with an eye toward an analysis of endothelial function. Some splenic defects are also attributable to effects on hematopoietic cells, most notably of the myeloid lineages.
In summary, both the canonical and non-canonical NF-κB pathways are required for the development of most secondary lymphoid organs (Figure 2). However, the non-canonical pathway, as assessed by examining mice deficient for IKKα, p52, NIK, or RelB, is especially important both during organogenesis and for maintenance of splenic architecture. Recent advances suggest that NF-κB signaling in endothelial cells, which are crucial regulators of cellular movement into and out of lymphoid organs, should be better characterized. The functional consequences of defects in these processes are severe and have direct ramifications for the ability of host to mount a robust immune response. As such, the function of NF-κB in the development and regulation of these organs represents an important facet of the role of NF-κB in immunobiology.
NF-κB and hematopoiesis
The immune system includes cells of the lymphoid and myeloid lineages: T cells, B cells, monocytes, macrophages, dendritic cells (DCs), natural killer (NK) cells, basophils, eosinophils, neutrophils, and mast cells (Figure 3). These bone marrow-derived cells are the core constituents of both the innate and adaptive immune responses. Proliferation, differentiation, and apoptosis are the defining characteristic of hematopoiesis, and NF-κB participates in the regulation of each of these processes. While the study of the role of NF-κB in development and homeostasis of hematopoetic cells has focused largely on B cell and T lymphocytes, it is likely that the NF-κB pathway is also important for the development of NK cells, DCs, monocytes, granulocytes, and other cellular components of the immune system. In general, an anti-apoptotic and pro-proliferative role for NF-κB is invoked in the hematopoietic system (Figure 3). Indeed, deletion of the kinase TAK1, an upstream component of both NF-κB and AP1 pathways, results in decreased antiapoptotic gene expression, hematopoietic stem cell apoptosis, and failure of hemtopoiesis 23. However, in actuality, the situation is much more complex. For example, ikba−/−ikbe−/− cells, which have elevated NF-κB activation are broadly defective in myelopoiesis and exhibit defects in NK cells and lymphoid lineages 24, 25. Thus, NF-κB function must be interrogated at each point of cellular differentiation in order to fully appreciate its contribution to hematopoiesis. Greater insight into the majority of events that lead to the development of immune cells, awaits both further characterization of the developmental processes and generation of better genetic tools to examine the consequences of perturbing NF-κB function.
NF-κB in hematopoiesis. Red arrows indicate stages in which NF-κB activation is thought to contribute negatively and green arrows indicate a positive function in the development of the indicated lineages. Curved arrows indicate examples in which NF-κB contributes to the survival of cell population, either in the resting state or during immune responses. Gray arrows indicate developmental events for which NF-κB plays no role or for which the role of NF-κB has not been clearly demonstrated. (HSC, hematopoietic stem cell; CMP, common myeloid progenitor; MLP, myeloid/lymphoid progenitor; MEP, megakaryocyte erythrocyte progenitor; GMP, granulocyte monocyte progenitor; MDP, macrophage dendritic cell progenitor; CDP, common dendritic cell progenitor; CLP, common lymphoid progenitor; ETP, early thymic precursor; and B/NP, B-cell natural killer cell progenitor).
NF-κB in non-lymphocyte hematopoiesis
Although most, if not all cells of the body can exhibit some innate immune function, the ability to recognize microbes and initiate an antimicrobial response, we focus here only on the development of certain hematopoietic, non-lymphoid components of the innate immune system. In general, our understanding of the developmental origin of these innate cells lags far behind that of lymphocytes. Yet recent progress in characterizing the developmental process for several myeloid lineages should provide a better framework within which the contribution of NF-κB can be better assessed in the future.
Significant progress has been made recently in understanding DC development, including the characterization of a common DC progenitor 26, 27, 28, yet the contribution of NF-κB to early steps of DC lineage commitment remains poorly understood. The most notable contribution of NF-κB in this area is the requirement for RelB in development of the CD8α− DCs 4, 29, 30, 31. Although single knockouts of other NF-κB genes do not result in deficiencies in DC development, deletion of both p65 and p50 has a profound effect 32, 33. Again, however, the limitations of the experimental systems used to examine the contribution of NF-κB, particularly with regard to the need to either delete TNFR1 or utilize fetal liver transfer experiments, should be underscored. In the periphery, NF-κB is clearly required for DC maturation. Loss of IKKβ or inhibition of the IKK complex using a cell permeable peptide prevents DC maturation and antigen-presenting cell (APC) function 34, 35. In addition, peripheral differentiation and function of myeloid DCs, e.g., monocyte-derived DCs, is significantly impaired by NF-κB inhibition 36. DCs in the periphery survive for a short period of time following activation, but their survival can be prolonged by CD40L expression on T cells. CD40L activates both the canonical and non-canonical NF-κB pathways, and hence DCs deficient in both p50 and c-Rel, or DCs overexpressing a mutant super-repressor form of IκBα, demonstrate significantly decreased survival 32, 37. To date, the specific targets of NF-κB in DC development and, indeed, the role of NF-κB at individual stages of DC development, remain quite poorly characterized.
Genetic models of NF-κB deficiency and activation have revealed other roles for NF-κB in the myeloid lineage. Of the non-lymphocyte lineages, neutrophil development is perhaps best characterized in terms of the role of NF-κB. There remains, however, significant gaps in both our understanding of neutrophil development, and more so the role of NF-κB in this process. IκBα-knockout mice, which have elevated classical NF-κB activity, display robust granulocytosis 38. RelB-deficient mice exhibit an inflammatory phenotype with a marked increase in peripheral neutrophil numbers. Mice lacking c-Rel and p50, and heterozygous for p65, also exhibit neutrophilia 39. This increase in peripheral neutrophil numbers resulting from deletion of NF-κB appears to be related to both increased development and increased egress from the bone marrow 39. In mature neutrophils, the anti-apoptotic signal provided by NF-κB is crucial. In sharp contrast to lymphocytes, which are relatively long-lived in the absence of activation, neutrophils normally exhibit a very short lifespan but require protection from apoptosis during inflammation when they provide effector function. Neutrophils can respond by activating NF-κB in response to numerous pro-inflammatory stimuli, and the activated NF-κB can increase neutrophil survival 40. Although neutrophils are capable of activating NF-κB in response to many pro-inflammatory stimuli 41 they lack p52 and RelB 42, which are crucial for the maintenance of long-lived lymphocytes. Thus, in neutrophils the role of NF-κB is selective and cell type specific.
NF-κB in lymphopoiesis
Development of T and B cells has, historically, been the subject of much greater scrutiny than the development of cells of the myeloid lineages. NF-κB has been examined in many aspects of lymphopoiesis and found to be vital for the development and function of adaptive immune cells 43 (Figure 4). Despite their potential longevity in the periphery, lymphocyte development is characterized by abundant apoptosis. As a consequence, the anti-apoptotic properties of NF-κB play a key role in lymphopoeisis. Indeed, in many instances the requirement for NF-κB can be overcome by transgenic expression of the anti-apoptotic factor Bcl-2 44. Indeed, the role of NF-κB in B cell development was recently re-examined 45. Although NF-κB was expendable in cells undergoing rearrangement of the eponymous Igκ loci, NF-κB was required to protect the resulting cells from apoptosis, and for these cells to progress to rearrangement of the Igλ locus. This deficit in NF-κB function could be circumvented through overexpression of Bcl-2 45. The necessity of NF-κB for lymphopoesis is strikingly illustrated in human genetic diseases in which the gene encoding NEMO is inactivated by mutation. Because the NEMO gene is located on the X chromosome, it is usually subject to random inactivation in individual cells in females. However, in female patients who are heterozygous for a mutant version of NEMO, all peripheral lymphocytes possess an intact NEMO gene, rather than the 50% predicted by random inactivation, suggesting that in the absence of NEMO-dependent NF-κB signaling, B and T cells fail to develop.
NF-κB in lymphopoiesis. NF-κB plays a pro-survival role in common lymphoid precursor (CLP) cells which give rise to B- and T-cell lineages. B-cell development occurs in the bone marrow, where NF-κB protects pre-B cells from pro-apoptotic stimuli including TNFα. Signaling to NF-κB through the pre-B cell receptor mediates survival of Pre-B cells, which then undergo light chain recombination to produce a functional B cell receptor. NF-κB provides a necessary pro-survival signal during Igλ but not Igκ rearrangement. Expression of BCR leads to NF-κB-dependent differentiation into immature B cells. High levels of BCR signaling, i.e., through recognition of self-antigen, results in negative selection through the loss of NF-κB activity. Transitional B cells exit the bone marrow and migrate to the spleen, where they mature and differentiate, a process that also requires NF-κB. T-cell development occurs following migration of precursor cells into the thymus. Stimulation of NF-κB through pre-TCRα provides a pro-survival signal allowing recombination of the TCR α chain and maturation to the double-positive (DP) stage. Optimal signaling through the TCRα/β complex induces NF-κB-dependent survival pathways, while a failure to signal or high level signaling results in death by neglect or negative selection, respectively. Intermediate high NF-κB activation facilitates intrathymic regulatory T cell (Treg) development. NF-κB activity is required for the maintenance of long-lived B and T cells. (CLP, common lymphoid progenitor; ETP, early thymic progenitor; DN, double negative – CD4−CD8−; DP, double positive – CD4+CD8+; SP, single positive – either CD4+CD8− or CD4−CD8+)
The effects of NEMO inactivation in both mice and humans solidify the role of NF-κB in lymphopoiesis, although the details by which NF-κB functions in this process remain obscure. NF-κB plays diverse roles in lymphocyte development that can be grouped according to timing – that is, before, during, or after pre-antigen receptor signaling. Although no single NF-κB-subunit-knockout mouse has as severe a phenotype as NEMO knockouts with regard to the generation of mature lymphocytes, double knockouts of Rel proteins confirm the essential anti-apoptotic function of NF-κB. For example, loss of both p50 and p65, or both p65 and c-Rel, terminates lymphopoiesis before expression of the pre-antigen receptors 46, 47, suggesting that NF-κB regulates anti-apoptotic factors required for early lymphoid cell survival in response to pro-apoptotic stimuli (Figure 4). In fact, hematopoietic stem cells can activate NF-κB in response to TNFα, and in these cells NF-κB acts as a pro-survival factor 48. The role of NF-κB in early lymphocyte development seems clear – expression of either pre-T-cell receptor (pre-TCR) 49 or pre-BCR (pre-B-cell receptor) coincides with increasing NF-κB activity and induction of anti-apoptotic signals through NF-κB 50, 51. However, it remains unclear how NF-κB is activated downstream of the pre-AgR as the nature of the signaling pathway at play remains almost entirely uncharacterized. Whether NF-κB contributes to lineage choice made at these early stages or merely promotes survival is also unknown.
The extent of NF-κB activation serves as a rheostat in the selection of DP (double positive; CD4+CD8+) thymocytes (Figure 4). TCR-mediated NF-κB activation follows binding to peptide:major histocompatibility complex (MHC). A thymocyte that expresses a TCR that cannot bind MHC succumbs to “death by neglect”, whereas those that bind peptide:MHC are either positively or negatively selected depending on the strength of signaling. Thymocytes that bind self-peptide:MHC with very high affinity, are likely to be self-reactive, and hence are deleted through negative selection. Thus, only DP thymocytes that recognize self-peptide:MHC and signal within a defined range are positively selected and become single-positive (SP) T cells. During negative selection, NF-κB facilitates the induction of apoptosis following high affinity TCR ligation 52, perhaps by facilitating expression of pro-apoptotic genes and consequent sensitization to pro-apoptotic signals 53. The role of NF-κB in positive selection of thymocytes is more in keeping with the better-established role of NF-κB as an inducer of anti-apoptotic genes. Unlike in thymocytes, however, NF-κB functions as a pro-survival factor during negative selection of B cells. Immature B cells display constitutive NF-κB activity that is downregulated following BCR ligation 54 (Figure 4). Decreased NF-κB activity might then sensitize these cells to pro-apoptotic signals. Interestingly, some signaling components required for NF-κB activation in mature B and T cells can be genetically disrupted without affecting their development, suggesting that pathways leading to activation of NF-κB in developing B or T cells differ significantly from the pathways engaged following AgR ligation in mature lymphocytes.
Following positive and negative selection, DP thymocytes must make a lineage commitment as SP thymocytes (CD4+CD8− or CD4−CD8+) and thereafter emigrate from the thymus. This process requires NF-κB as deletion of NEMO in cd4-Cre mice results in loss of mature peripheral T cells 55. Equivalent deletion of the upstream kinase TAK1 has a similar outcome 56. The exact nature of NF-κB pathway requirement is somewhat unclear, as ikkb−/− chimeras, or cd4-Cre IKKβ conditional knockouts, are not defective in the production of naive T cells 55, 57. Further, the contribution of NF-κB to CD8 and CD4 lineages is not equivalent. CD8 SP cells have significantly higher levels of NF-κB activity than CD4 SP thymocytes 49, 58, yet the anti-apoptotic factor Bcl-2 is more highly expressed in CD4 than CD8 cells. Therefore, CD8 SP thymocytes are dependent on NF-κB for survival, while CD4 SP thymocytes are not 58. NF-κB is, however, clearly important in CD4 SP cell development, and forced activation of NF-κB in CD4 SP cells results in negative selection 58.
In addition to peripheral maturation and differentiation of T cells into various subsets, thymic differentiation into both TH17 cells and regulatory T cells (Tregs) is known to occur. The transcriptional control of TH17 cell differentiation and the role of thymic TH17 remain relatively obscure. One recent report has described an important role for IκBζ in TH17 differentiation 59. In this capacity, IκBζ acts with the nuclear orphan receptors RORγ and RORα to promote IL17A gene expression. Beyond this tangential role in TH17 development, a TH17-intrinsic role for NF-κB has been minimally interrogated. Instead the role of NF-κB in Treg development 60 is somewhat better understood. The differentiation of thymocytes into Foxp3/CD25-positive Tregs occurs in the thymus and depends on NF-κB. It is thought that NF-κB acts as a pioneer transcription factor enhancing accessibility of the foxp3 locus 61, 62, 63, 64.
Immature B cells complete development into either follicular or marginal zone B cells after exiting the bone marrow as transitional B cells. NF-κB-regulated expression of pro-survival factors is important to these final steps of B-cell development 65. Signaling through the TNFR superfamily member BAFFR is crucial for transitional B-cell survival through induction of anti-apoptotic Bcl-2 family members 66, 67, 68. Thus, BAFF-knockout mice exhibit a complete failure of transitional B-cell maturation, which mirrors that seen in Bcl-XL-knockout mice 66, 69, 70. BAFF activates both non-canonical and canonical NF-κB pathways, though the former is principally responsible for the anti-apoptotic function in transitional B cells. Nevertheless, deficiency in NEMO, IKKα, or IKKβ decreases the numbers of mature B cells 57, 71, 72. Likewise, p50/p52 or p65/c-Rel double-knockout progenitor cells are defective in their ability to mature beyond the transitional B-cell stage 47, 73. A requirement for both the canonical and non-canonical NF-κB pathways may explain why deletion of p50 and p52 produces a more complete block in B-cell development than loss of p65 and c-Rel. Thus, only those knockouts that target both the canonical and non-canonical NF-κB pathways have an effect that approximates the phenotype seen in BAFF or Bcl-XL deficiency. Constitutive hyperactivation of the canonical pathway can circumvent BAFF-R deletion 74, yet it remains unclear whether the canonical pathway merely supports the non-canonical pathway 75, or has an independent function.
NF-κB in innate immunity
Pattern recognition receptors
Recognizing pathogens is an absolute requirement for the initiation of effector functions. Mammals express several diverse classes of pattern recognition receptors (PRRs) that are dedicated to this purpose. These PPRs include transmembrane Toll-like receptors (TLRs), cytoplasmic Nod-like receptors (NLR) and RIG-I like receptors (RLR), scavenger receptors, C-type lectins, and the complement system. Although epithelial cells are frequently the first to encounter pathogens, they are also constantly exposed to non-pathogenic microbes. Therefore, while a variety of PRRs are differentially expressed in epidermis, gut, pulmonary, urinary, and reproductive epithelium, their expression and responsiveness is tightly controlled. Instead sentinel cells of the innate immune system, particularly tissue resident DCs and macrophages, express a more complete complement of PRRs, and exhibit a higher propensity to signal on exposure to pathogens.
TLRs are evolutionarily conserved pattern recognition receptors that recognize unique, essential molecules characteristic of various classes of microbes 76. The 11 characterized mammalian TLRs have varied tissue distribution and serve as recognition receptors for pathogen-associated molecular patterns (PAMPs) present on bacteria, viruses, fungi, and parasites. Perhaps because of the modular nature of the TLR extracellular domain, which consists of multiple leucine-rich repeats (LRRs), these receptors are capable of recognizing more than one microbial molecule (Figure 5). Heterodimerization of TLRs further expands the repertoire of recognized molecules. In general, the ability of TLRs to distinguish between pathogen types is translated into appropriate innate and adaptive responses through the selective activation of NF-κB, IRFs, and other inducible transcription factors. TLR signaling pathways are complex and have been extensively reviewed elsewhere 2, 77, 78. In general, TLR signaling is often divided into MyD88-dependent and TRIF-dependent pathways: MyD88 signaling predominantly leads to the activation of NF-κB, while TRIF signaling leads to both IRF3 and, to a lesser extent, NF-κB activation.
Pattern recognition receptors and their cognate ligands. TLRs 3, 7, 8, 9 and 11 have been reported to exhibit endosomal or intracellular localization while NOD1, NOD2, RIG-I, MDA-5, NALP1, NALP3, NLRC4, and the intracellular DNA sensor (ISD) function in the cytoplasm. Only a partial list of ligands or classes of ligands for each receptor is given.
In recent years, there has been significant activity in characterization of cytoplasmic PRRs, particularly those recognizing viral nucleic acids. These efforts followed the initial description of nucleotide oligomerization domain proteins (NOD), NOD1 and NOD2. NLRs, of which NOD1 and NOD2 are the best-known examples, are characterized by LRRs and NODs. NOD1, NOD2, and IPAF have CARD domains and can signal to NF-κB (see Blonska and Lin, Cell Res 2011; 21:55-70). NOD1 recognizes a peptidoglycan containing meso-diaminopimelic acid and induces NF-κB through a canonical pathway that includes activation of IKKβ. NOD2 recognizes muramyl dipeptide (MDP), a ubiquitous component of nearly all bacterial cell walls. The CARD-containing kinase RIP2 is required for NF-κB activation. The function of RIP2 tyrosine kinase activity is unclear, although it was recently shown that kinase inhibitors could prevent MDP-induced cytokine responses 79. Although RIP2 binds to NEMO, it is not thought that RIP2 tyrosine kinase activity mediates IKK phosphorylation directly. Instead it has been proposed that RIP2 directly mediates activation of the IKK complex through NEMO binding and induced proximity 80.
Retinoic acid inducible gene I (RIG-I) and melanoma differentiation-associated gene 5 (MDA5) are DExD/H-box RNA helicase-containing cytoplasmic proteins that bind directly to dsRNA 81, 82, 83. The details of RNA binding to RIG-I and MDA5 continue to be the subject of intense research. Thus, it was thought that RIG-I recognizes dsRNA replicative intermediates of negative stranded single-stranded RNA (ssRNA) viruses, while MDA5 responds to long dsRNA replicative intermediates of plus strand ssRNA viruses 84, 85, 86, 87. Indeed, the structure of RIG-I bound to short dsRNA products has recently been solved 88, 89. RIG-I has also recently been implicated in the recognition of DNA viruses by binding short dsRNAs generated by RNA polIII 90. However, it has been shown that RIG-I can directly recognize ssRNA genomes containing 5′ triphosphates, and suggested that for negative stranded RNA viruses, this may be the relevant PAMP 91. On binding to either the viral genome, viral replication intermediate, or RNA product, RIG-I, and MDA5 induce the activation of IRF3 and NF-κB. The CARD-containing protein MAVS (also known as CARDIF, IPS1, and VISA) mediates signaling from RIG-I 92, 93, 94, 95. RIG-I and MDA5 differentially recognize various groups of RNA viruses and are independently required for robust antiviral responses 86. Therefore, together with TLRs, RLRs provide an additional mechanism of sensing viral infection and mediating type-I IFN (IFN-α/β) responses.
Pathogen recognition in innate immunity
Pathogens recognized by PRRs can be categorized as bacterial, viral, or eukaryotic, and each of these categories has several well-described PAMPs 96. Both in terms of accessibility and uniqueness to prokaryotes, the bacterial cell well is the most abundant source of PAMPs for TLRs and other PRRs. Lipoproteins, glycolipids, and protein components of bacterial cell wells have all been shown to function as PAMPs. The TLR4 ligand LPS, a glycolipid component of the outer membrane of Gram-negative bacteria, is the most thoroughly studied PAMP and the most potent TLR ligand known. Trace amounts of LPS activates the innate immune system via TLR4, leading to the production of numerous proinflammatory mediators, such as TNFα, IL-1, and IL-6. TLR4-mediated responses to LPS require CD14 and MD-2.
Conserved differences in bacterial nucleic acid structures can also be recognized by the innate immune system. TLR9 recognizes bacterial DNA containing unmethylated CpG motifs, and TLR9-deficient mice are not responsive to CpG DNA challenge 97. The low frequency and high rate of methylation of CpG motifs prevent recognition of mammalian DNA by TLR9 under physiological circumstances. A recent report indicated that the intracellular, endosomal restriction of TLR9 is critical for discriminating between self and non-self DNA, as host DNA, unlike microbial DNA, does not usually enter the endosomal compartment 98.
Nucleic acids are also key viral PAMPs, and are recognized by TLRs 3, 7, 8, and 9, as well as by RLRs. Signaling from viral PAMPs results in the activation of NF-κB and IRFs, which cooperatively mediate the production of IFN-α/β 99, 100. TLR3 recognizes dsRNA, a common viral replicative intermediate 101. TLR7 and TLR8, initially found to recognize synthetic antiviral compounds 102, 103, recognize guanosine- or uridine-rich ssRNA derived from RNA viruses 104, 105, 106. Interestingly, mammalian RNA is significantly less stimulatory than bacterial RNA, suggesting that nucleoside modifications facilitate discrimination between endogenous and pathogen RNA 107. In addition, subcellular localization of TLR7 and TLR8 to endocytic compartments, and limited constitutive cell type expression, may also facilitate self/non-self discrimination by minimizing exposure to host RNA. TLR9 recognizes viral CpG sequences and induces the induction of IFN-α 108, 109, 110. However, as membrane restriction prevents TLRs from sampling the cytosol where much of the viral lifecycle occurs, cytosolic PRRs provide comprehensive innate immune recognition. Thus, TLR3 and the adaptor TRIF are not required for viral induction of type I IFN in many tissues and cell types 111, although specialized innate antiviral cells, such as plasmacytoid DCs, rely more heavily on TLR mediated recognition of viral nucleic acids 112. Recognition of cytoplasmic dsDNA leading to NF-κB activation and type I interferon production has also been reported 113, 114. Although several candidates have been found, the relevant receptor has not yet been conclusively identified 115. This receptor(s) is predicted to be important for type I IFN production in response to viruses and intracellular pathogens, such as Listeria sp.
Although research on the recognition of PAMP/PRR pairs is an ongoing process of discovery, defective responses of MyD88-deficient cells indicate that many fungal and parasite species are capable of activating TLR pathways. TLR4 has been shown to recognize Aspergillus hyphae 116, and Cryptococcus neoformans capsular polysaccharide 117. TLR2 and TLR6 are required for recognition of yeast zymosan, while TLR4 is thought to recognize certain yeast mannans. The identification of parasite PAMPs has been more elusive, and their existence is somewhat controversial. Nevertheless, TLR2 heterodimers recognize various parasite GPI-anchored proteins and glycoinositolphospholipids from the parasitic protozoa Trypanosoma cruzi 118. TLR9 was initially reported to recognize the malarial pigment hemozoin, a byproduct of heme metabolism in infected erythrocytes 119, although subsequent studies suggest that DNA associated with hemozoin is the relevant TLR9 ligand 120. Instead, hemozoin may be recognized by the NLRP3 inflammasome 121. TLR11 recognizes a profilin-like protein that is conserved in apicomplexan parasites including Toxoplasma gondii 122. Recognition of helminths remains an area of controversy 123. In particular, it remains unclear how helminth PAMPs might preferentially induce TH2 responses that are characteristic of the immune responses to this class of pathogen. Direct skewing of adaptive responses by the mechanism of innate recognition is suggested to occur in other scenerios as well 124. For example, C-type lectins can directly affect TH activation and differentiantion 125, thus recognition of C. albicans α-mannans by dectin-2 induces a protective TH17-predominant response 126. Therefore, whether through cell type or PRR specificity, the recognition of specific PAMPS can shape the nature of the subsequent innate and adaptive immune responses.
Immediate anti-microbial responses
PAMP recognition initiates a complex series of events: the first is the mounting of immediate antimicrobial responses at the cellular level. This effective and evolutionarily conserved function of PRRs is dependent on activation of NF-κB gene programs. In particular, much of the early innate response has been demonstrated to depend on the canonical NF-κB pathway. Thus, rela−/−/ tnfr1−/− and ikbkb−/−/tnfr1−/− double-knockout mice have increased susceptibility to bacterial infection 57, 127, 128, 129. MEFs from Nemo-deficent mice do not exhibit NF-κB activation by LPS or IL-1 130. Therefore, activation of NF-κB responsive genes by the innate immune system depends on NEMO and likely progresses through the canonical NF-κB signaling pathway. Early anti-microbial effectors include NF-κB-dependent expression of defensins, which are cationic peptides that exert direct bactericidal activity by inducing membrane permeabilization. The classic mediators of defensin release are small intestinal Paneth cells, which secrete α-defensins into the intestinal lumen following exposure to bacterial PAMPs 131. The production of anti-microbial nitrogen and oxygen species, which are acutely toxic to a variety of microbes, augments the activity of anti-microbial peptides. Production of nitric oxide (NO) is mediated in part by inducible NO synthase (iNOS), which is coordinately regulated by NF-κB and STAT transcription factors 132.
Inflammation
Inflammation begins with epithelial or stromal cells of the infected tissue or tissue resident hematopoietic cells such as mast cells or DCs recognizing an inflammatory stimulus and propagating pro-inflammatory signals. These signals lead to the recruitment and activation of effector cells, initially neutrophils and later macrophages and other leukocytes, resulting in the tissue changes characteristic of inflammation – rubor, calor, dolor, and tumor (redness, heat, pain, and swelling, respectively). There is a vast amount of literature that correlates NF-κB activation with inflammation in a wide array of diseases and animal models. There are, likewise, numerous studies using gene targeting and inhibitors of NF-κB that have established the causative role of NF-κB in inflammatory processes.
NF-κB is responsible for the transcription of the genes encoding many pro-inflammatory cytokines and chemokines. The pathway from pathogen recognition to pro-inflammatory cytokine production demonstrates a particular reliance on NF-κB. The immediate targets of NF-κB-dependent pro-inflammatory cytokines, such as TNFα, tend to be receptors that, in turn, activate NF-κB. Therefore, NF-κB is crucial to the propagation and elaboration of cytokine responses. TNFα is particularly important for both local and systemic inflammation, and it is a potent and well-studied inducer of NF-κB. One important early target of these effectors is the vascular endothelium. Changes in vascular endothelial cells both recruit circulating leukocytes and provide them with a means of exiting the vasculature into the infected tissue. NF-κB regulates the expression of adhesion molecules, both on leukocytes and on endothelial cells, which allow the extravasation of leukocytes from the circulation to the site of infection 133. Thus, in the absence of p65, the recruitment of circulating leukocytes to sites of inflammation is impaired 127.
Recruited neutrophils are the key mediators of local inflammation and, as mentioned above, NF-κB is important for their survival and function under relatively toxic conditions 134. NF-κB is important for the production of the enzymes that generate prostaglandins and reactive oxygen species (e.g., iNOS and Cox, both NF-κB target genes) and may, furthermore, be involved in the signaling induced by prostaglandins 135, 136. NF-κB has also been implicated in the response to leukotrienes, which similar to prostaglandins are short-lived paracrine effectors. For example, leukotriene-induced IL-8 production appears to be dependent on rapid activation of NF-κB 137. Finally, matrix metalloporteinases are also crucial mediators of local inflammation and leukocyte chemotaxtis, and their expression is also regulated by NF-κB 138, 139, 140.
Resolution of inflammation and subsequent tissue repair is a crucial event, and its failure is a common source of pathology. Resolution of inflammation is an active process that is as complex as the inflammatory response itself, and involves numerous pathways that are not all directly relevant to NF-κB 141. Macrophages are key regulators of both inflammation and its resolution, and NF-κB plays a key role in the instruction of macrophage responses. Macrophage differentiation into pro-inflammatory (M1) or anti-inflammatory (M2) cells depends both on cell intrinsic NF-κB pathway activation as well as the local cytokine milieu, which is strongly influenced by NF-κB.
During acute inflammation, there are multiple negative feedback pathways that help to rein in inflammatory responses 3. It has long been known that cells, such as macrophages, become resistant to repeated pro-inflammatory stimuli 142. The IκB family protein Bcl-3 can function both as an inhibitor and mediator of NF-κB transcriptional programs 2. Following LPS stimulation, Bcl-3 is induced and binds p50 dimers, with which it can repress the expression of a subset of NF-κB target genes. In this manner, Bcl-3 mediates the selective inhibition of repeated LPS responses in macrophages 143. Although clearly not the only mechanism, induced transcriptional repression by Bcl-3/p50 likely contributes to LPS-tolerance. Furthermore by selectively affecting chromatin remodeling, Bcl-3 mediates repression of pro-inflammatory genes, and also facilitates the expression of the anti-inflammatory gene IL-10. Thus, it appears that Bcl-3 acts appropriately in the regulation of genes that are categorized as either “tolerizable or “non-tolerizable” 144. NF-κB p50 appears to be capable of mediating anti-inflammatory responses under several additional circumstances as well. For example, p50 negatively regulates IFNγ production by and proliferation of NK cells 145. Also, expression levels of p50 in M2-like tumor associated macrophages (TAMs) regulate the balance between anti- and pro-inflammatory functions. Thus, it was shown that TAMs overexpress p50 and that deletion of p50 renders these macrophages pro-inflammatory 146.
Inhibition of NF-κB during the resolution phase of inflammation can prolong the inflammatory process and prevent proper tissue repair 147. Furthermore, IKKα-deficient mice display increased inflammatory responses in models of local and systemic inflammation 148, and show increased production of pro-inflammatory chemokines and cytokines 148, 149. Rather unexpectedly, IKKα-deficient mice and embryonic-liver-derived macrophages from IKKα-deficient mice exhibit augmented inflammatory responses compared with wild-type mice 149. The mechanisms of IKKα-mediated repression of transcriptional responses seem to be through effects on the level of nuclear p65 and c-Rel 148. Although there are significant differences between these two reports with regard to the proposed mechanism of action, both, nevertheless, observe the same biological consequence of IKKα deficiency in macrophages. Furthermore, recent work has suggested that IKKβ also exhibits some anti-inflammatory capacity in macrophages. First, IKKβ suppresses the secretion of the potent pro-inflammatory cytokine IL-1β 150. Second, IKKβ is associated with the maintenance of an anti-inflammatory M2-like phenotype in TAMs 151. Third, IKKβ is thought to inhibit M1 macrophage function by interfering with the STAT pathway during infection 152.
Initiation of adaptive responses
The innate immune system invokes potent anti-microbial activities and maintains host protection under many settings. Nevertheless, initiating the adaptive immune response remains a crucial step for robust and durable pathogen clearance. The process of information transfer from the innate to adaptive immune system is mediated by activation and maturation of APCs. The nature of the PAMP and the signaling pathways activated by the cognate PRR, as well as the context within which pathogen recognition occurs, all influence the manner in which the APC instructs T and B cell responses.
DC maturation mediated by pathogen recognition is crucial for the initiation of the adaptive immune response. To activate naive T cells, DCs must alter their chemokine receptor expression to enable migration into lymphoid tissues. During activation DCs fine-tune their antigen processing machinery such that the presentation of pathogen epitopes is favored. Finally, the expression of co-stimulatory molecules including B7.1/B7.2 (or CD80/CD86) is upregulated. These co-stimulatory molecules ligate the co-stimulatory molecule CD28, thus providing the second signal necessary to induce full T-cell activation. The response to pathogens is tailored on the basis of the distribution of PRRs in different cell types, and the ability of different cell types to, in turn, interact with T cells in a biasing manner 153.
There continues to be debate surrounding how PRRs differentially induce TH1 versus TH2 responses. Maturation of DCs following viral infection depends on nucleic acid-binding PRRs 154, 155. Plasmacytoid DCs, which are robust inducers of interferon during viral infection, express viral PRRs including elevated expression of TLR7 and TLR9. Cytoplasmic nucleic acid-sensing PRRs, RLRs, are expressed more broadly, although they are further induced by type-I interferons, and they may, therefore, provide crucial amplification of antiviral responses following virus recognition by TLRs. While studies of anti-viral responses suggest that both cell type-specific distribution of PRRs, and selective activation of signaling pathways by certain PAMPs are key determinants of TH1 versus TH2 responses, the situation is less clear for other pathogens. It appears that multiple mechanisms converge in shaping how APCs interact with the adaptive immune system.
Role of NF-κB in the adaptive response
T-cell responses mediated by NF-κB
T-cell activation, differentiation, proliferation, and effector function are all influenced by gene programs regulated by NF-κB. The majority of effort directed at understanding the role of NF-κB in T cell activation has focused on CD4+ T cells (Figures 6 and and7).7). On the whole, the historical lack of CD8+ T-cell conditional knockouts and the selective loss of CD8 cells in the absence of functional NF-κB have impeded thorough characterization of the role of NF-κB in these cells. Nevertheless, there continues to be progress in this area and, in fact, there has been a resurgence of interest in recent years. CD4+ T cells have long been known to undergo differentiation into either TH1 or TH2 subsets depending on the cytokines present during activation 156. However, an appreciation of the ability of CD4+ helper T cells to differentiate into additional effector cell types, induced regulatory T cells, pro-inflammatory TH17 cells, TH22 cells TH9 cells, has prompted a fresh look at the contribution of NF-κB to T cell differentiation and effector function (Figure 7).
NF-κB in T cell activation. NF-κB participates in the maintenance, activation, differentiation and proliferation of naive T cells. Tonic TCR stimulation promotes T cell maintenance, of both memory and naive cells, through NF-κB activation. Activation occurs when the naive T cell recognizes its cognate antigen presented by an activated APC expressing both peptide:MHC and B7 family co-stimulatory molecules. NF-κB-dependent proliferation and differentiation ensue and are influenced by the local cytokine milieu. NF-κB supports proliferation, differentiation and survival as indicated (green arrows).
TH cell differentiation. NF-κB participates in differentiation of several TH cell types following activation of naive CD4+ TH cells. Differentiation pathways in which NF-κB is implicated are indicated with a green arrow. Key transcription factors in each differentiation pathway are indicated above each arrow, while cytokine responsible for skewing TH cells toward a given pathway are indicated below each arrow. Additional cytokines and transcription factors implicated in several of the pathways are not depicted.
Activation of naive T cells requires antigen-specific and co-stimulatory signaling provided by activated APCs. Binding of the TCR to its cognate peptide presented in the binding cleft of MHC supplies the antigen-specific signal, while co-stimulatory signaling results from ligation of CD28 by B7 molecules expressed on activated APCs. Activated naive T-cells proliferate rapidly while simultaneously differentiating into effector cells (Figure 6). In the case of CD4+ T cells, proliferation leads to differentiation into immature effector cells, TH0, which subsequently differentiate into TH1, TH2, TH9, TH17, TH22, or inducible Treg (iTreg) cells depending on the cytokine milieu.
Stimulation of T cells through TCR and CD28 results, initially, in activation of p65-containing NF-κB complexes followed by delayed and sustained activation of c-Rel complexes 157, 158, 159, 160. Rapidly proliferating activated T cells rely on NF-κB activity for protection from apoptosis as well as for the production of cytokines, especially IL-2, supporting proliferation and differentiation (Figure 6). As expected, inhibition of NF-κB in activated T cells facilitates progression toward activation-induced cell death (AICD) or apoptosis 161, 162, and stimulation of p65-deficient naive T cells induces cell death 163. In contrast, T cells lacking c-Rel do not undergo apoptosis, but nevertheless, fail to proliferate in response to typical mitogenic stimuli 164. T cells, in which p105 cannot undergo inducible processing to p50 also fail to proliferate normally 165. In vitro, c-Rel is needed for production of IL-2 and therefore, for proliferation, but this need is not recapitulated in vivo 166. Interestingly, c-Rel-deficient T cells appear to have a defect in TH1 proliferation and production of IFNγ, indicating a selective role for NF-κB family members in TH1/TH2 differentiation, independent of that mediated by the innate response.
Multiple transcriptional activators and repressors regulate expression of IL-2. Amongst these, members of the NF-κB family play multiple roles. In naive T cells, which do not express IL-2, repressive p50 homodimers are found associated with the IL-2 promoter 167. Failure of T-cell proliferative responses in c-Rel-knockout mice is attributable to a failure to produce IL-2 164. In naive T cells, c-Rel is responsible for mediating chromatin remodeling across the IL-2 locus following CD3/CD28 co-stimulation 158. Thus, naive T cells can be primed by activation of c-Rel by pro-inflammatory cytokines, such as those elicited following stimulation with TLR ligands 157. Priming, which requires NF-κB activation, results in a more robust response to CD3/CD28 co-stimulation 168. Conversely, overexpression of p65 with c-Jun can overcome the requirement for co-stimulation in naive T cells 169.
TH0 differentiation into TH1, TH2, TH9, TH17, Treg effector cells depends on the induction of specific transcription factors, namely T-bet, GATA3, PU.1, RORγt, and Foxp3, respectively (Figure 7). There is increasing evidence that NF-κB family members are key arbitrators of the decision. For example, mice lacking p50 are unable to mount an asthma-like, airway TH2 response 170. Indeed, p50-deficient T cells fail to induce GATA3 expression during T-cell stimulation under TH2 differentiating conditions. These results suggest a transcriptional role for p50, and it was subsequently found that Bcl-3-deficient T cells also fail to undergo TH2 differentiation, suggesting that p50/Bcl-3 complexes are crucial for TH2 differentiation 171. Conversely, the same authors found that RelB-deficient T cells are deficient in TH1 differentiation due to decreased expression of T-bet. RelB may mediate upregulation of STAT4, which is responsible for T-bet induction downstream of IFNγ. Therefore, it appears that NF-κB activation during TCR stimulation may render cells competent for both proliferative and differentiating stimuli. As a corollary, NF-κB transactivation of the IL-2 gene is repressed as TH cells differentiate into TH1 or TH2 and become dependent on TH1 and TH2 cytokines (e.g., IFNγ and IL-4). It is thought that direct binding of T-bet to p65 that is associated with the IL-2 gene enhancer, may mediate the repression of IL-2 production in TH1 cells 172. In TH2 cells the lack of IL-2 transcription may be because of the decreased levels of p65 activation 173. As discussed above, several groups recently implicated NF-κB, and specifically c-Rel, in the regulation of Foxp3 expression and Treg development 61, 62, 63, 174. The relative contribution of c-Rel to iTreg development still requires further clarification. While a recent report demonstrated that IκBζ cooperates with ROR and is required for TH17 differentiation 59, the more general contribution of NF-κB in this process, either in the thymus or periphery, remains unclear. While the role of NF-κB in TH9 cells has yet to be interrogated, there are interesting clues suggesting an important role for NF-κB. Recently, PU.1 was reported to be the transcription factor responsible for TH9 differentiation 175. In unrelated recent work on acute myelogenous leukemia, it was noted that NF-κB regulates the transcription of PU.1 176. Furthermore, it has been suggested that IL-9 expression in T cells is regulated by NF-κB 177. Thus, it would seem reasonable to speculate that NF-κB may play an important role in TH9 differentiation (Figure 7). The transcription factors responsible for the differentiation of TH22 cells have not yet been identified. However, the possible role of TNFα as a TH22 differentiating cytokine is suggestive that NF-κB may be an important mediator of this differentiation process as well. In summary, NF-κB participates in both the initial T cell responses by supporting proliferation and regulating apoptosis, and is increasingly being shown to have an important role in the regulation of TH differentiation.
B-cell responses mediated by NF-κB
There are many parallels in the functions of NF-κB in T and B cells. NF-κB acts to support proliferation, regulate apoptosis, and control the processes of differentiation and maturation in B cells (Figure 8). B-cell responses are classified into two groups: thymus-dependent (TD) or thymus-independent (TI). In TD response follicular B cells require co-stimulatory signaling from TH cells expressing CD40L and cytokines, such as IL-4. These initial steps trigger germinal center formation, in which somatic hypermutation, isotype switching, and plasma cell differentiation occur. Thus, in individuals with a mutation in CD40L B cells fail to undergo class switch recombination in response to T-dependent antigens 178. Signaling through CD40 activates both canonical and non-canonical NF-κB pathways, although several lines of evidence suggest that the non-canonical pathway is not required in the response to TD antigens. Thus, B cells lacking p52 are fully capable of appropriate TD antigen responses when transferred 21, and B cells from relB−/− mice, although crippled in their proliferative response, undergo normal IgM secretion and class switching 179. In contrast to these results, B cells lacking either functional NIK or IKKα, exhibit defective responses 180, 181. It remains unclear whether the role of NIK and IKKα in this context is to induce p100 processing (non-canonical pathway) or to contribute to activation of the canonical pathway. Although both pathways are triggered by CD40 ligation, the current data suggest that canonical pathway activation is of great importance.
NF-κB in B cell activation. NF-κB participates in the maintenance, activation, differentiation and proliferation of naive B cells. Tonic BCR and cytokine, BAFF, stimulation promotes naive B cell maintenance through NF-κB activation. Activation occurs when the naïve B cell recognizes its cognate antigen and receives co-stimulatory signaling (CD40L) from an activated TH cell within the germinal center. NF-κB-dependent proliferation and differentiation ensue and are coupled to BCR affinity maturation. NF-κB signaling in B cells expressing selected BCRs results in class switch recombination and differentiation into either memory B cells or plasma cells. NF-κB supports proliferation, differentiation and survival as indicated (green arrows).
Evidence also supports a role for the canonical pathway in class switch recombination. In contrast to B cells lacking RelB or p52, those from rela−/− mice exhibit markedly diminished class switching, despite a modest loss of lymphocyte proliferation following various stimuli 182. Likewise, c-Rel-deficient mice fail to generate protective humoral immune responses demonstrating the requirement for c-Rel in class switch recombination 164, 183, 184. B cells from nfbk1−/− mice exhibit decreased proliferation in response to mitogenic stimulation, and p50/p65 double-knockout B cells exhibit greater defects in proliferation and class switching 185, 186. Therefore, B cell proliferation and the TD B-cell maturation response are dependent on the canonical NF-κB pathway.
TI antigens are those that have an intrinsic ability to initiate B-cell responses in the absence of T-cell help. TI responses are carried out by marginal zone and CD5+ B cells. TI antigens function by acting as both antigen and B-cell mitogens, for example antigens that are PRR ligands or antigens with high avidity that are capable of crosslinking the BCR through repetitive structural features. In such cases it is expected that B-cell responses are more dependent on members of the canonical pathway that have well-documented roles in TLR and BCR signaling. For example, c-Rel-deficient B cells are highly sensitive to apoptosis following BCR cross-linking 187, 188, 189. As mentioned above, p50 and p50/RelA double-knockout B cells are deficient in responses to TI stimulation. Likewise, IKKβ-deficient B cells fail to mount TI or TD responses 190. These IKKβ-deficient B cells also exhibit increased spontaneous apoptosis, supporting that NF-κB is important in survival of B cells.
NF-κB and lymphocyte longevity
Lymphocyte homeostasis reflects balanced lymphocyte turnover and lymphopoiesis. Turnover of mature lymphocytes is complex as it depends upon both activation induced and homeostatic proliferation, and cell death. Lymphocyte survival is mediated through tonic stimulation provided by both the antigen receptor and certain cytokine receptors. As such, NF-κB can be assumed to play an important role in lymphocyte homeostasis and this assumption is supported by various genetic models.
T cells can be quite long-lived and naive T cells require continued contact with MHC:self-peptides for survival. These self-peptide complexes are most likely expressed on lymphoid DCs and generate a tonic low grade TCR signal. Survival of memory cells, on the other hand, is independent of continued contact with self-peptide:MHC complexes. B cells are formed at a far higher rate than T cells and therefore, B cells also undergo a significantly higher rate of turnover. Nonetheless, B cells too require maintenance signals to achieve peripheral homeostasis. The antigen receptor on B cells provides a basal level of signaling, albeit independent of the presence of antigen, which is required for maintenance of mature B cells. Thus, deletion of the BCR from mature B cells results in a complete loss of the peripheral B cell pool 191. The assumption is that BCR provides tonic activation of the NF-κB pathway resulting in the expression of anti-apoptotic proteins. Consistent with this assumption, loss of IKKβ, NEMO, or components of the CBM complex in mature B cells also results in complete loss of peripheral B cells 71, 190, 192. Furthermore, B cells from rela−/−, nfkb1−/−, nfkb2−/−, and c-rel−/− mice all have increased sensitivity to apoptosis and/or decreased survival ex vivo 67, 188, 193. In addition to contributing directly, it is thought that low level NF-κB activation in B cells contributes to survival through upregulation of p100, which is necessary for BAFFR-induced activation of the alternative pathway 67, 75, 166.
Several studies have implicated BAFFR in this aspect of the B lymphocyte survival 194, suggesting that the non-canonical NF-κB pathway is also relevant to B-cell survival. Consistent with this idea, constitutive activation of NIK or IKKα can circumvent the requirement for B cell BAFFR 74, 195. Conversely, B cells lacking IKKα exhibit defective survival 72, 196. The relevant targets of the non-canonical pathway downstream of BAFFR are thought to be Bcl-2 family members, e.g., the anti-apoptotic factor A1 187 or Bcl-XL, which are known to be regulated by p52/RelB-containing complexes, and are required for the maintenance of mature B cells. Indeed, Bcl-XL can complement B cells with a mutant BAFFR 197. Thus, tonic BCR stimulation, through upregulation of p100, may cooperate with BAFFR ligation and alternative pathway activation, to upregulate anti-apoptotic Bcl2 family members and mediate maintenance and survival of mature B cells.
Concluding remarks
The areas covered in the current review necessarily represent a subset of those in which NF-κB is a crucial regulator of immune responses. Recent work has highlighted the importance of NF-κB in the maintenance of epithelium barriers 198; as a regulator of inflammation associated with obesity and hyperlipidemia; and as a key factor in the development of inflammatory diseases and cancer. Our understanding of the immune system continues to improve, and coordinately our recognition of the many roles played by the NF-κB family of transcription factors also grows. As one example, the expanding universe of T-cell subsets opens up a new area for the analysis of the role of NF-κB in T-cell differentiation and effector function. It will be intriguing to see how NF-κB contributes to the development of TH9, TH17, TH22, and iTreg/TH3 cells. Deciphering the role of NF-κB in the effector function of these recently described cell types should also prove to be an area of great interest, particularly given the growing evidence for dysregulation of these cell types in autoimmune and inflammatory disease. There is much that remains unknown about the contributions of NF-κB in early and non-lymphoid hematopoiesis; in responses to danger signaling and allergens; in the coordination of pathogen-specific adaptive immune responses by the innate immune system; and in the differentiation and effector responses of both lymphoid and non-lymphoid innate immune cells. Thus moving forward, as in the past, much of the work done to understand the physiological and pathophysiological role of NF-κB will continue to focus on the role of this transcription factor family in Immunobiology.
Acknowledgments
Research in the authors' laboratory was supported by grants from the National Institutes of Health (to SG).
References
- Hayden MS, Ghosh S. Signaling to NF-kappaB. Genes Dev. 2004;18:2195–2224. [PubMed] [Google Scholar]
- Hayden MS, West AP, Ghosh S. NF-kappaB and the immune response. Oncogene. 2006;25:6758–6780. [PubMed] [Google Scholar]
- Ghosh S, Hayden MS. New regulators of NF-kappaB in inflammation. Nat Rev Immunol. 2008;8:837–848. [PubMed] [Google Scholar]
- Burkly L, Hession C, Ogata L, et al. Expression of relB is required for the development of thymic medulla and dendritic cells. Nature. 1995;373:531–536. [PubMed] [Google Scholar]
- Irla M, Hugues S, Gill J, et al. Autoantigen-specific interactions with CD4+ thymocytes control mature medullary thymic epithelial cell cellularity. Immunity. 2008;29:451–463. [PubMed] [Google Scholar]
- Hikosaka Y, Nitta T, Ohigashi I, et al. The cytokine RANKL produced by positively selected thymocytes fosters medullary thymic epithelial cells that express autoimmune regulator. Immunity. 2008;29:438–450. [PubMed] [Google Scholar]
- Akiyama T, Shimo Y, Yanai H, et al. The tumor necrosis factor family receptors RANK and CD40 cooperatively establish the thymic medullary microenvironment and self-tolerance. Immunity. 2008;29:423–437. [PubMed] [Google Scholar]
- Anderson G, Jenkinson EJ, Rodewald HR. A roadmap for thymic epithelial cell development. Eur J Immunol. 2009;39:1694–1699. [PubMed] [Google Scholar]
- Ruddle NH, Akirav EM. Secondary lymphoid organs: responding to genetic and environmental cues in ontogeny and the immune response. J Immunol. 2009;183:2205–2212. [PMC free article] [PubMed] [Google Scholar]
- van de Pavert SA, Mebius RE. New insights into the development of lymphoid tissues. Nat Rev Immunol. 2010;10:664–674. [PubMed] [Google Scholar]
- Alcamo E, Hacohen N, Schulte LC, Rennert PD, Hynes RO, Baltimore D. Requirement for the NF-kappaB family member RelA in the development of secondary lymphoid organs. J Exp Med. 2002;195:233–244. [PMC free article] [PubMed] [Google Scholar]
- Miyawaki S, Nakamura Y, Suzuka H, et al. A new mutation, aly, that induces a generalized lack of lymph nodes accompanied by immunodeficiency in mice. Eur J Immunol. 1994;24:429–434. [PubMed] [Google Scholar]
- Koike R, Nishimura T, Yasumizu R, et al. The splenic marginal zone is absent in alymphoplastic aly mutant mice. Eur J Immunol. 1996;26:669–675. [PubMed] [Google Scholar]
- Shinkura R, Kitada K, Matsuda F, et al. Alymphoplasia is caused by a point mutation in the mouse gene encoding Nf-kappa b-inducing kinase. Nat Genet. 1999;22:74–77. [PubMed] [Google Scholar]
- Mebius RE. Organogenesis of lymphoid tissues. Nat Rev Immunol. 2003;3:292–303. [PubMed] [Google Scholar]
- Bonizzi G, Karin M. The two NF-kappaB activation pathways and their role in innate and adaptive immunity. Trends Immunol. 2004;25:280–288. [PubMed] [Google Scholar]
- Dougall WC, Glaccum M, Charrier K, et al. RANK is essential for osteoclast and lymph node development. Genes Dev. 1999;13:2412–2424. [PMC free article] [PubMed] [Google Scholar]
- Yilmaz ZB, Weih DS, Sivakumar V, Weih F. RelB is required for Peyer's patch development: differential regulation of p52-RelB by lymphotoxin and TNF. EMBO J. 2003;22:121–130. [PMC free article] [PubMed] [Google Scholar]
- Paxian S, Merkle H, Riemann M, et al. Abnormal organogenesis of Peyer's patches in mice deficient for NF-kappaB1, NF-kappaB2, and Bcl-3. Gastroenterology. 2002;122:1853–1868. [PubMed] [Google Scholar]
- Caamano JH, Rizzo CA, Durham SK, et al. Nuclear factor (NF)-kappa B2 (p100/p52) is required for normal splenic microarchitecture and B cell-mediated immune responses. J Exp Med. 1998;187:185–196. [PMC free article] [PubMed] [Google Scholar]
Cell Res. 2011 Feb; 21(2): 223–244.
Published online 2011 Jan 18. doi: 10.1038/cr.2011.13
PMCID: PMC3193440
PMID: 21243012
NF-κB in immunobiology
Matthew S Hayden1 and Sankar Ghosh1,*
Author information Copyright and License information PMC Disclaimer
Abstract
NF-κB was first discovered and characterized 25 years ago as a key regulator of inducible gene expression in the immune system. Thus, it is not surprising that the clearest biological role of NF-κB is in the development and function of the immune system. Both innate and adaptive immune responses as well as the development and maintenance of the cells and tissues that comprise the immune system are, at multiple steps, under the control of the NF-κB family of transcription factors. Although this is a well-studied area of NF-κB research, new and significant findings continue to accumulate. This review will focus on these areas of recent progress while also providing a broad overview of the roles of NF-κB in mammalian immunobiology.
Keywords: NF-κB, TLR, TCR, BCR, lymphoid organogenesis, inflammation, hematopoiesis
Introduction
As discussed in the introduction to the January special issue of Cell Research, the discovery and characterization of the NF-κB family of transcription factors resulted from studies in two major areas of research: immunology and cancer biology. Although the role of NF-κB in cancer biology is becoming progressively better established, historically much of our current knowledge of NF-κB resulted from efforts directed at understanding its role in the regulation and function of the immune response. This review will attempt to provide a comprehensive discussion of the diverse functions of NF-κB in immunobiology and update our previous efforts in reviewing the far-reaching role of NF-κB in this area of biology 1, 2, 3.
The mammalian immune response comprises various mechanisms by which the physiological and functional integrity of the host is maintained in the face of microbial insult. The bone marrow-derived cells of the immune system are chiefly, though by no means solely, responsible for performing these functions. Thus, we begin by reviewing the role of NF-κB in the development of these hematopoietic cells that mediate both innate and adaptive immune responses. With the relevant cell types and tissues in place, the immune response is triggered by host recognition of foreign pathogens. Our knowledge in this area of the immune response has expanded rapidly in the past decade. Pathogen recognition is followed by pathogen clearance, a highly variable response that may draw upon coordinated responses at the level of cell, tissue, and the organism. Finally, the immune response must be resolved, damage must be repaired and, whenever possible, the triggering insult remembered for future immunity.
In mammals, the NF-κB family is composed of five related transcription factors: p50, p52, p65 (also RelA), c-Rel, and RelB (Figure 1). These transcription factors share an N-terminal DNA-binding/dimerization domain, known as the Rel homology domain, through which they can form homo- and heterodimers. NF-κB dimers can bind to a variety of related target DNA sequences called κB sites to modulate gene expression. RelB, c-Rel, and p65 contain C-terminal transcription activation domains (TADs) that enable co-activator recruitment and target gene expression. As they lack TADs, p50 and p52 can activate transcription by forming heterodimers with p65, c-Rel, or RelB, or by recruiting other TAD-containing proteins. However, as homodimers lacking the ability to drive transcription, they can repress transcription on binding to DNA.
The mammalian NF-κB, IκB and IKK protein families. Relevant domains typifying each protein family are indicated and alternative nomenclatures are provided in parenthesis. The precursor proteins p100 and p105 function as both IκB and, when processed by the proteasome, NF-κB family members. Highly simplified schematics of canonical and non-canonical NF-κB pathways are shown. In the canonical pathway NEMO-containing IKK complexes are activated and induce phosphorylation and degradation of IκBα leading to the release of NF-κB dimers including p65:p50 dimers. In the non-canonical pathway, NEMO-independent activation of IKKα is mediated by the upstream kinase NIK. IKKα, with NIK, induces phosphorylation and processing of p100 to p52 resulting in the activation of predominantly p52:RelB complexes. Although they are illustrated separately here, as discussed in the text, signaling through these two pathways often is initiated by the same receptor – e.g., LTβR or CD40. (ANK, ankyrin-repeat domain; DD, death domain; RHD, REL homology domain; TAD, transactivation domain; LZ, leucine-zipper domain; GRR, glycine-rich region; HLH, helix-loop-helix domain; Z, zinc-finger domain; CC, coiled-coil domain; NBD, NEMO-binding domain; MOD/UBD, minimal oligomerization domain/ubiquitin-binding domain; PEST, proline, glutamic acid, serine, and threonine rich.)
In most cells, NF-κB complexes are inactive, residing predominantly in the cytoplasm in a complex with inhibitory IκB proteins (IκBα, IκBβ, IκBɛ, IκBζ, p100, p105, Bcl3, IκBns; Figure 1). When signaling pathways are activated, the IκB protein is degraded and NF-κB dimers enter the nucleus to modulate target gene expression. In almost all cases, the common step in this process is mediated by the IκB kinase (IKK) complex, which phosphorylates IκB and targets it for proteasomal degradation (see Liu and Chen, Cell Res 2011; 21:6–21). The IKK complex consists of two catalytically active kinases, IKKα (IKK1) and IKKβ (IKK2) and a regulatory scaffolding protein, NEMO (IKKγ Figure 1). These are the basic players in the NF-κB pathway; however, there is considerable complexity in how the individual proteins function and how they coordinate a nuanced NF-κB response (see Shih et al., Cell Res 2011; 21:86–102). The details of individual signaling pathways, the mechanism of action of upstream signaling intermediates, the role of various post-translational modifications, and cross-talk between NF-κB and other signaling pathways are covered in depth elsewhere in this special issue. Here, we focus on the function of NF-κB and the signaling pathways that regulate it in the mammalian immune response.
Development of the immune system
The mammalian immune system consists of a functionally linked group of anatomically disparate tissues and cell types. The dispersed cellular components of the immune system that arise from the bone marrow receive much of the attention in immunology and the study of NF-κB has likewise focused on these cells. However, lymphoid organs that facilitate coordination and dissemination of immune responses carried out by immune cells are also key sites of NF-κB function. Therefore, while this section is largely concerned with the role of NF-κB in hematopoiesis, the role of NF-κB in lymphoid organogenesis is also discussed briefly.
NF-κB and lymphoid organogenesis
NF-κB plays an important role in the development and function of primary (bone marrow, thymus) and secondary (lymph nodes, Peyer's patches, mucosal-associated lymphoid tissue, and the spleen) lymphoid tissues. There is clearly a role for NF-κB in the development and regulation of bone, and we refer the reader to the excellent review on the subject in this special issue (see Novack, Cell Res 2011; 21:169–182). The role of NF-κB in thymic architecture, originally thought to be limited to the important role of RelB in thymic architecture 4, has become clearer in terms of the development of medullary thymic epithelial cells 5, 6, 7, but still remains to be more fully elucidated 8. The secondary lymphoid tissues facilitate maintenance and activation of mature lymphocytes by providing an environment within which the interaction of lymphocytes and other leukocytes can be carefully orchestrated 9. The role of NF-κB in this process is now well appreciated. Multiple NF-κB knockouts exhibit defects in some aspect of secondary lymphoid organ development.
The initial events of lymphoid organogenesis involve the association of lymphotoxin (LT)α1β2-expressing hematopoietic cells and vascular cell adhesion molecule-1 (VCAM-1)-expressing stromal cells 10. This interaction initiates a positive feedback loop through NF-κB (Figure 2). LTα1β2, receptor activator of NF-κB ligand (RANKL), and TNFα are known to activate NF-κB, and figure prominently in lymphorganogenesis 10 (Figure 2). Also, mediators of lymphoid organogenesis and homeostasis, such as adhesion molecules (e.g., intercellular adhesion molecule, VCAM, peripheral node addressin, glycosylation-dependent cell adhesion molecule-1, and mucosal addressin cellular adhesion molecule (MadCAM)), cytokines (e.g., TNFα and LTα1β2) and organogenic chemokines (e.g., CXCL12 (GRO/MIP-2), CXCL13 (BLC), CCL19 (ELC), and CCL21 (SLC)), are regulated by NF-κB.
NF-κB in lymphoid organogenesis. NF-κB regulates key components of a positive feedback loop between hematopoietic and stromal cells. LTα1β2-expressing hematopoietic cells induce production of VCAM-1 through the canonical NF-κB pathway and chemokines through the non-canonical pathway in LTβR-expressing stromal cells. Stromal expression of chemokines induces the upregulation of integrins (α4β1) on hematopoietic cells resulting in increased recruitment of LTα1β2-expressing cells, and increased signaling through stromal LTβR. RANKL signaling through RANK on the hematopoietic cell leads to activation of NF-κB, and further upregulation of LTα1β2.
Signaling through TNFR1, LTβR, and RANK activates p65-containing complexes and, hence, it is not surprising that rela−/−/tnfr1−/− double-knockout mice lack Peyer's patches and lymph nodes, and exhibit a disorganized spleen 11. The requirement for p65 in the development of these tissues is attributable to its function in stromal cells, and is likely because of a combination of effects: regulation of apoptosis (e.g., that induced by TNF); regulation of expression of organogenic factors, such as VCAM and LTα1β2; and enhancement of the non-canonical p52/RelB NF-κB pathway. The non-canonical NF-κB pathway has a well-established role in secondary lymphoid organ development. Deletion of components of the non-canonical NF-κB signaling pathway, including NIK, IKKα, LT, and RANK, all lead to severe defects in secondary lymphoid organogenesis 12, 13, 14, 15, 16, 17. The target of the non-canonical pathway, the p52/RelB dimer, is the primary transcriptional regulator of key organogenic factors including CXCL12, CXCL13, CCL19, CCL21, and MadCAM-1 18. Thus, the p52 single knockout lacks normal B-cell follicles, germinal centers, and Peyer's patches 19, 20, 21, while RelB knockouts lack Peyer's patches and exhibit compromised lymphnode development 18.
Splenic architecture is crucial for B-cell development as well as for the initiation and maturation of B-cell responses. The spleen is divided functionally and histologically into white and red pulp zones. Macrophages in the red pulp are responsible for destroying damaged erythrocytes, while the white pulp is populated by splenic lymphocytes organized into B-cell follicles and T-cell zones. Yet splenic architecture is also dynamic, as exemplified by the formation of germinal centers during the initiation and maturation of B-cell responses. As with other secondary lymphoid organs, NF-κB figures prominently in the development and maintenance of splenic architecture. Mice in which p65 has been deleted exhibit defects in both static and dynamic splenic architecture 11. Deletion of RelB, NIK, or IKKα leads to defects in splenic architecture, similar to those of ltbr−/− spleens 12, 13. Similar to p65/TNFR1 knockouts, mice with a defective non-canonical pathway fail to segregate B cell/T cell zones and they fail to form germinal centers following immunization. Marginal zone macrophages, which line the border between red and white pulp areas, are also absent or disorganized in RelB, p52, NIK, or IKKα knockouts 21, 22. Recent work has highlighted the role of LTα1β2 in the development and maintenance of the marginal sinus architecture, suggesting that the role of NF-κB in splenic architecture should perhaps be revisited with an eye toward an analysis of endothelial function. Some splenic defects are also attributable to effects on hematopoietic cells, most notably of the myeloid lineages.
In summary, both the canonical and non-canonical NF-κB pathways are required for the development of most secondary lymphoid organs (Figure 2). However, the non-canonical pathway, as assessed by examining mice deficient for IKKα, p52, NIK, or RelB, is especially important both during organogenesis and for maintenance of splenic architecture. Recent advances suggest that NF-κB signaling in endothelial cells, which are crucial regulators of cellular movement into and out of lymphoid organs, should be better characterized. The functional consequences of defects in these processes are severe and have direct ramifications for the ability of host to mount a robust immune response. As such, the function of NF-κB in the development and regulation of these organs represents an important facet of the role of NF-κB in immunobiology.
NF-κB and hematopoiesis
The immune system includes cells of the lymphoid and myeloid lineages: T cells, B cells, monocytes, macrophages, dendritic cells (DCs), natural killer (NK) cells, basophils, eosinophils, neutrophils, and mast cells (Figure 3). These bone marrow-derived cells are the core constituents of both the innate and adaptive immune responses. Proliferation, differentiation, and apoptosis are the defining characteristic of hematopoiesis, and NF-κB participates in the regulation of each of these processes. While the study of the role of NF-κB in development and homeostasis of hematopoetic cells has focused largely on B cell and T lymphocytes, it is likely that the NF-κB pathway is also important for the development of NK cells, DCs, monocytes, granulocytes, and other cellular components of the immune system. In general, an anti-apoptotic and pro-proliferative role for NF-κB is invoked in the hematopoietic system (Figure 3). Indeed, deletion of the kinase TAK1, an upstream component of both NF-κB and AP1 pathways, results in decreased antiapoptotic gene expression, hematopoietic stem cell apoptosis, and failure of hemtopoiesis 23. However, in actuality, the situation is much more complex. For example, ikba−/−ikbe−/− cells, which have elevated NF-κB activation are broadly defective in myelopoiesis and exhibit defects in NK cells and lymphoid lineages 24, 25. Thus, NF-κB function must be interrogated at each point of cellular differentiation in order to fully appreciate its contribution to hematopoiesis. Greater insight into the majority of events that lead to the development of immune cells, awaits both further characterization of the developmental processes and generation of better genetic tools to examine the consequences of perturbing NF-κB function.
NF-κB in hematopoiesis. Red arrows indicate stages in which NF-κB activation is thought to contribute negatively and green arrows indicate a positive function in the development of the indicated lineages. Curved arrows indicate examples in which NF-κB contributes to the survival of cell population, either in the resting state or during immune responses. Gray arrows indicate developmental events for which NF-κB plays no role or for which the role of NF-κB has not been clearly demonstrated. (HSC, hematopoietic stem cell; CMP, common myeloid progenitor; MLP, myeloid/lymphoid progenitor; MEP, megakaryocyte erythrocyte progenitor; GMP, granulocyte monocyte progenitor; MDP, macrophage dendritic cell progenitor; CDP, common dendritic cell progenitor; CLP, common lymphoid progenitor; ETP, early thymic precursor; and B/NP, B-cell natural killer cell progenitor).
NF-κB in non-lymphocyte hematopoiesis
Although most, if not all cells of the body can exhibit some innate immune function, the ability to recognize microbes and initiate an antimicrobial response, we focus here only on the development of certain hematopoietic, non-lymphoid components of the innate immune system. In general, our understanding of the developmental origin of these innate cells lags far behind that of lymphocytes. Yet recent progress in characterizing the developmental process for several myeloid lineages should provide a better framework within which the contribution of NF-κB can be better assessed in the future.
Significant progress has been made recently in understanding DC development, including the characterization of a common DC progenitor 26, 27, 28, yet the contribution of NF-κB to early steps of DC lineage commitment remains poorly understood. The most notable contribution of NF-κB in this area is the requirement for RelB in development of the CD8α− DCs 4, 29, 30, 31. Although single knockouts of other NF-κB genes do not result in deficiencies in DC development, deletion of both p65 and p50 has a profound effect 32, 33. Again, however, the limitations of the experimental systems used to examine the contribution of NF-κB, particularly with regard to the need to either delete TNFR1 or utilize fetal liver transfer experiments, should be underscored. In the periphery, NF-κB is clearly required for DC maturation. Loss of IKKβ or inhibition of the IKK complex using a cell permeable peptide prevents DC maturation and antigen-presenting cell (APC) function 34, 35. In addition, peripheral differentiation and function of myeloid DCs, e.g., monocyte-derived DCs, is significantly impaired by NF-κB inhibition 36. DCs in the periphery survive for a short period of time following activation, but their survival can be prolonged by CD40L expression on T cells. CD40L activates both the canonical and non-canonical NF-κB pathways, and hence DCs deficient in both p50 and c-Rel, or DCs overexpressing a mutant super-repressor form of IκBα, demonstrate significantly decreased survival 32, 37. To date, the specific targets of NF-κB in DC development and, indeed, the role of NF-κB at individual stages of DC development, remain quite poorly characterized.
Genetic models of NF-κB deficiency and activation have revealed other roles for NF-κB in the myeloid lineage. Of the non-lymphocyte lineages, neutrophil development is perhaps best characterized in terms of the role of NF-κB. There remains, however, significant gaps in both our understanding of neutrophil development, and more so the role of NF-κB in this process. IκBα-knockout mice, which have elevated classical NF-κB activity, display robust granulocytosis 38. RelB-deficient mice exhibit an inflammatory phenotype with a marked increase in peripheral neutrophil numbers. Mice lacking c-Rel and p50, and heterozygous for p65, also exhibit neutrophilia 39. This increase in peripheral neutrophil numbers resulting from deletion of NF-κB appears to be related to both increased development and increased egress from the bone marrow 39. In mature neutrophils, the anti-apoptotic signal provided by NF-κB is crucial. In sharp contrast to lymphocytes, which are relatively long-lived in the absence of activation, neutrophils normally exhibit a very short lifespan but require protection from apoptosis during inflammation when they provide effector function. Neutrophils can respond by activating NF-κB in response to numerous pro-inflammatory stimuli, and the activated NF-κB can increase neutrophil survival 40. Although neutrophils are capable of activating NF-κB in response to many pro-inflammatory stimuli 41 they lack p52 and RelB 42, which are crucial for the maintenance of long-lived lymphocytes. Thus, in neutrophils the role of NF-κB is selective and cell type specific.
NF-κB in lymphopoiesis
Development of T and B cells has, historically, been the subject of much greater scrutiny than the development of cells of the myeloid lineages. NF-κB has been examined in many aspects of lymphopoiesis and found to be vital for the development and function of adaptive immune cells 43 (Figure 4). Despite their potential longevity in the periphery, lymphocyte development is characterized by abundant apoptosis. As a consequence, the anti-apoptotic properties of NF-κB play a key role in lymphopoeisis. Indeed, in many instances the requirement for NF-κB can be overcome by transgenic expression of the anti-apoptotic factor Bcl-2 44. Indeed, the role of NF-κB in B cell development was recently re-examined 45. Although NF-κB was expendable in cells undergoing rearrangement of the eponymous Igκ loci, NF-κB was required to protect the resulting cells from apoptosis, and for these cells to progress to rearrangement of the Igλ locus. This deficit in NF-κB function could be circumvented through overexpression of Bcl-2 45. The necessity of NF-κB for lymphopoesis is strikingly illustrated in human genetic diseases in which the gene encoding NEMO is inactivated by mutation. Because the NEMO gene is located on the X chromosome, it is usually subject to random inactivation in individual cells in females. However, in female patients who are heterozygous for a mutant version of NEMO, all peripheral lymphocytes possess an intact NEMO gene, rather than the 50% predicted by random inactivation, suggesting that in the absence of NEMO-dependent NF-κB signaling, B and T cells fail to develop.
NF-κB in lymphopoiesis. NF-κB plays a pro-survival role in common lymphoid precursor (CLP) cells which give rise to B- and T-cell lineages. B-cell development occurs in the bone marrow, where NF-κB protects pre-B cells from pro-apoptotic stimuli including TNFα. Signaling to NF-κB through the pre-B cell receptor mediates survival of Pre-B cells, which then undergo light chain recombination to produce a functional B cell receptor. NF-κB provides a necessary pro-survival signal during Igλ but not Igκ rearrangement. Expression of BCR leads to NF-κB-dependent differentiation into immature B cells. High levels of BCR signaling, i.e., through recognition of self-antigen, results in negative selection through the loss of NF-κB activity. Transitional B cells exit the bone marrow and migrate to the spleen, where they mature and differentiate, a process that also requires NF-κB. T-cell development occurs following migration of precursor cells into the thymus. Stimulation of NF-κB through pre-TCRα provides a pro-survival signal allowing recombination of the TCR α chain and maturation to the double-positive (DP) stage. Optimal signaling through the TCRα/β complex induces NF-κB-dependent survival pathways, while a failure to signal or high level signaling results in death by neglect or negative selection, respectively. Intermediate high NF-κB activation facilitates intrathymic regulatory T cell (Treg) development. NF-κB activity is required for the maintenance of long-lived B and T cells. (CLP, common lymphoid progenitor; ETP, early thymic progenitor; DN, double negative – CD4−CD8−; DP, double positive – CD4+CD8+; SP, single positive – either CD4+CD8− or CD4−CD8+)
The effects of NEMO inactivation in both mice and humans solidify the role of NF-κB in lymphopoiesis, although the details by which NF-κB functions in this process remain obscure. NF-κB plays diverse roles in lymphocyte development that can be grouped according to timing – that is, before, during, or after pre-antigen receptor signaling. Although no single NF-κB-subunit-knockout mouse has as severe a phenotype as NEMO knockouts with regard to the generation of mature lymphocytes, double knockouts of Rel proteins confirm the essential anti-apoptotic function of NF-κB. For example, loss of both p50 and p65, or both p65 and c-Rel, terminates lymphopoiesis before expression of the pre-antigen receptors 46, 47, suggesting that NF-κB regulates anti-apoptotic factors required for early lymphoid cell survival in response to pro-apoptotic stimuli (Figure 4). In fact, hematopoietic stem cells can activate NF-κB in response to TNFα, and in these cells NF-κB acts as a pro-survival factor 48. The role of NF-κB in early lymphocyte development seems clear – expression of either pre-T-cell receptor (pre-TCR) 49 or pre-BCR (pre-B-cell receptor) coincides with increasing NF-κB activity and induction of anti-apoptotic signals through NF-κB 50, 51. However, it remains unclear how NF-κB is activated downstream of the pre-AgR as the nature of the signaling pathway at play remains almost entirely uncharacterized. Whether NF-κB contributes to lineage choice made at these early stages or merely promotes survival is also unknown.
The extent of NF-κB activation serves as a rheostat in the selection of DP (double positive; CD4+CD8+) thymocytes (Figure 4). TCR-mediated NF-κB activation follows binding to peptide:major histocompatibility complex (MHC). A thymocyte that expresses a TCR that cannot bind MHC succumbs to “death by neglect”, whereas those that bind peptide:MHC are either positively or negatively selected depending on the strength of signaling. Thymocytes that bind self-peptide:MHC with very high affinity, are likely to be self-reactive, and hence are deleted through negative selection. Thus, only DP thymocytes that recognize self-peptide:MHC and signal within a defined range are positively selected and become single-positive (SP) T cells. During negative selection, NF-κB facilitates the induction of apoptosis following high affinity TCR ligation 52, perhaps by facilitating expression of pro-apoptotic genes and consequent sensitization to pro-apoptotic signals 53. The role of NF-κB in positive selection of thymocytes is more in keeping with the better-established role of NF-κB as an inducer of anti-apoptotic genes. Unlike in thymocytes, however, NF-κB functions as a pro-survival factor during negative selection of B cells. Immature B cells display constitutive NF-κB activity that is downregulated following BCR ligation 54 (Figure 4). Decreased NF-κB activity might then sensitize these cells to pro-apoptotic signals. Interestingly, some signaling components required for NF-κB activation in mature B and T cells can be genetically disrupted without affecting their development, suggesting that pathways leading to activation of NF-κB in developing B or T cells differ significantly from the pathways engaged following AgR ligation in mature lymphocytes.
Following positive and negative selection, DP thymocytes must make a lineage commitment as SP thymocytes (CD4+CD8− or CD4−CD8+) and thereafter emigrate from the thymus. This process requires NF-κB as deletion of NEMO in cd4-Cre mice results in loss of mature peripheral T cells 55. Equivalent deletion of the upstream kinase TAK1 has a similar outcome 56. The exact nature of NF-κB pathway requirement is somewhat unclear, as ikkb−/− chimeras, or cd4-Cre IKKβ conditional knockouts, are not defective in the production of naive T cells 55, 57. Further, the contribution of NF-κB to CD8 and CD4 lineages is not equivalent. CD8 SP cells have significantly higher levels of NF-κB activity than CD4 SP thymocytes 49, 58, yet the anti-apoptotic factor Bcl-2 is more highly expressed in CD4 than CD8 cells. Therefore, CD8 SP thymocytes are dependent on NF-κB for survival, while CD4 SP thymocytes are not 58. NF-κB is, however, clearly important in CD4 SP cell development, and forced activation of NF-κB in CD4 SP cells results in negative selection 58.
In addition to peripheral maturation and differentiation of T cells into various subsets, thymic differentiation into both TH17 cells and regulatory T cells (Tregs) is known to occur. The transcriptional control of TH17 cell differentiation and the role of thymic TH17 remain relatively obscure. One recent report has described an important role for IκBζ in TH17 differentiation 59. In this capacity, IκBζ acts with the nuclear orphan receptors RORγ and RORα to promote IL17A gene expression. Beyond this tangential role in TH17 development, a TH17-intrinsic role for NF-κB has been minimally interrogated. Instead the role of NF-κB in Treg development 60 is somewhat better understood. The differentiation of thymocytes into Foxp3/CD25-positive Tregs occurs in the thymus and depends on NF-κB. It is thought that NF-κB acts as a pioneer transcription factor enhancing accessibility of the foxp3 locus 61, 62, 63, 64.
Immature B cells complete development into either follicular or marginal zone B cells after exiting the bone marrow as transitional B cells. NF-κB-regulated expression of pro-survival factors is important to these final steps of B-cell development 65. Signaling through the TNFR superfamily member BAFFR is crucial for transitional B-cell survival through induction of anti-apoptotic Bcl-2 family members 66, 67, 68. Thus, BAFF-knockout mice exhibit a complete failure of transitional B-cell maturation, which mirrors that seen in Bcl-XL-knockout mice 66, 69, 70. BAFF activates both non-canonical and canonical NF-κB pathways, though the former is principally responsible for the anti-apoptotic function in transitional B cells. Nevertheless, deficiency in NEMO, IKKα, or IKKβ decreases the numbers of mature B cells 57, 71, 72. Likewise, p50/p52 or p65/c-Rel double-knockout progenitor cells are defective in their ability to mature beyond the transitional B-cell stage 47, 73. A requirement for both the canonical and non-canonical NF-κB pathways may explain why deletion of p50 and p52 produces a more complete block in B-cell development than loss of p65 and c-Rel. Thus, only those knockouts that target both the canonical and non-canonical NF-κB pathways have an effect that approximates the phenotype seen in BAFF or Bcl-XL deficiency. Constitutive hyperactivation of the canonical pathway can circumvent BAFF-R deletion 74, yet it remains unclear whether the canonical pathway merely supports the non-canonical pathway 75, or has an independent function.
NF-κB in innate immunity
Pattern recognition receptors
Recognizing pathogens is an absolute requirement for the initiation of effector functions. Mammals express several diverse classes of pattern recognition receptors (PRRs) that are dedicated to this purpose. These PPRs include transmembrane Toll-like receptors (TLRs), cytoplasmic Nod-like receptors (NLR) and RIG-I like receptors (RLR), scavenger receptors, C-type lectins, and the complement system. Although epithelial cells are frequently the first to encounter pathogens, they are also constantly exposed to non-pathogenic microbes. Therefore, while a variety of PRRs are differentially expressed in epidermis, gut, pulmonary, urinary, and reproductive epithelium, their expression and responsiveness is tightly controlled. Instead sentinel cells of the innate immune system, particularly tissue resident DCs and macrophages, express a more complete complement of PRRs, and exhibit a higher propensity to signal on exposure to pathogens.
TLRs are evolutionarily conserved pattern recognition receptors that recognize unique, essential molecules characteristic of various classes of microbes 76. The 11 characterized mammalian TLRs have varied tissue distribution and serve as recognition receptors for pathogen-associated molecular patterns (PAMPs) present on bacteria, viruses, fungi, and parasites. Perhaps because of the modular nature of the TLR extracellular domain, which consists of multiple leucine-rich repeats (LRRs), these receptors are capable of recognizing more than one microbial molecule (Figure 5). Heterodimerization of TLRs further expands the repertoire of recognized molecules. In general, the ability of TLRs to distinguish between pathogen types is translated into appropriate innate and adaptive responses through the selective activation of NF-κB, IRFs, and other inducible transcription factors. TLR signaling pathways are complex and have been extensively reviewed elsewhere 2, 77, 78. In general, TLR signaling is often divided into MyD88-dependent and TRIF-dependent pathways: MyD88 signaling predominantly leads to the activation of NF-κB, while TRIF signaling leads to both IRF3 and, to a lesser extent, NF-κB activation.
Pattern recognition receptors and their cognate ligands. TLRs 3, 7, 8, 9 and 11 have been reported to exhibit endosomal or intracellular localization while NOD1, NOD2, RIG-I, MDA-5, NALP1, NALP3, NLRC4, and the intracellular DNA sensor (ISD) function in the cytoplasm. Only a partial list of ligands or classes of ligands for each receptor is given.
In recent years, there has been significant activity in characterization of cytoplasmic PRRs, particularly those recognizing viral nucleic acids. These efforts followed the initial description of nucleotide oligomerization domain proteins (NOD), NOD1 and NOD2. NLRs, of which NOD1 and NOD2 are the best-known examples, are characterized by LRRs and NODs. NOD1, NOD2, and IPAF have CARD domains and can signal to NF-κB (see Blonska and Lin, Cell Res 2011; 21:55-70). NOD1 recognizes a peptidoglycan containing meso-diaminopimelic acid and induces NF-κB through a canonical pathway that includes activation of IKKβ. NOD2 recognizes muramyl dipeptide (MDP), a ubiquitous component of nearly all bacterial cell walls. The CARD-containing kinase RIP2 is required for NF-κB activation. The function of RIP2 tyrosine kinase activity is unclear, although it was recently shown that kinase inhibitors could prevent MDP-induced cytokine responses 79. Although RIP2 binds to NEMO, it is not thought that RIP2 tyrosine kinase activity mediates IKK phosphorylation directly. Instead it has been proposed that RIP2 directly mediates activation of the IKK complex through NEMO binding and induced proximity 80.
Retinoic acid inducible gene I (RIG-I) and melanoma differentiation-associated gene 5 (MDA5) are DExD/H-box RNA helicase-containing cytoplasmic proteins that bind directly to dsRNA 81, 82, 83. The details of RNA binding to RIG-I and MDA5 continue to be the subject of intense research. Thus, it was thought that RIG-I recognizes dsRNA replicative intermediates of negative stranded single-stranded RNA (ssRNA) viruses, while MDA5 responds to long dsRNA replicative intermediates of plus strand ssRNA viruses 84, 85, 86, 87. Indeed, the structure of RIG-I bound to short dsRNA products has recently been solved 88, 89. RIG-I has also recently been implicated in the recognition of DNA viruses by binding short dsRNAs generated by RNA polIII 90. However, it has been shown that RIG-I can directly recognize ssRNA genomes containing 5′ triphosphates, and suggested that for negative stranded RNA viruses, this may be the relevant PAMP 91. On binding to either the viral genome, viral replication intermediate, or RNA product, RIG-I, and MDA5 induce the activation of IRF3 and NF-κB. The CARD-containing protein MAVS (also known as CARDIF, IPS1, and VISA) mediates signaling from RIG-I 92, 93, 94, 95. RIG-I and MDA5 differentially recognize various groups of RNA viruses and are independently required for robust antiviral responses 86. Therefore, together with TLRs, RLRs provide an additional mechanism of sensing viral infection and mediating type-I IFN (IFN-α/β) responses.
Pathogen recognition in innate immunity
Pathogens recognized by PRRs can be categorized as bacterial, viral, or eukaryotic, and each of these categories has several well-described PAMPs 96. Both in terms of accessibility and uniqueness to prokaryotes, the bacterial cell well is the most abundant source of PAMPs for TLRs and other PRRs. Lipoproteins, glycolipids, and protein components of bacterial cell wells have all been shown to function as PAMPs. The TLR4 ligand LPS, a glycolipid component of the outer membrane of Gram-negative bacteria, is the most thoroughly studied PAMP and the most potent TLR ligand known. Trace amounts of LPS activates the innate immune system via TLR4, leading to the production of numerous proinflammatory mediators, such as TNFα, IL-1, and IL-6. TLR4-mediated responses to LPS require CD14 and MD-2.
Conserved differences in bacterial nucleic acid structures can also be recognized by the innate immune system. TLR9 recognizes bacterial DNA containing unmethylated CpG motifs, and TLR9-deficient mice are not responsive to CpG DNA challenge 97. The low frequency and high rate of methylation of CpG motifs prevent recognition of mammalian DNA by TLR9 under physiological circumstances. A recent report indicated that the intracellular, endosomal restriction of TLR9 is critical for discriminating between self and non-self DNA, as host DNA, unlike microbial DNA, does not usually enter the endosomal compartment 98.
Nucleic acids are also key viral PAMPs, and are recognized by TLRs 3, 7, 8, and 9, as well as by RLRs. Signaling from viral PAMPs results in the activation of NF-κB and IRFs, which cooperatively mediate the production of IFN-α/β 99, 100. TLR3 recognizes dsRNA, a common viral replicative intermediate 101. TLR7 and TLR8, initially found to recognize synthetic antiviral compounds 102, 103, recognize guanosine- or uridine-rich ssRNA derived from RNA viruses 104, 105, 106. Interestingly, mammalian RNA is significantly less stimulatory than bacterial RNA, suggesting that nucleoside modifications facilitate discrimination between endogenous and pathogen RNA 107. In addition, subcellular localization of TLR7 and TLR8 to endocytic compartments, and limited constitutive cell type expression, may also facilitate self/non-self discrimination by minimizing exposure to host RNA. TLR9 recognizes viral CpG sequences and induces the induction of IFN-α 108, 109, 110. However, as membrane restriction prevents TLRs from sampling the cytosol where much of the viral lifecycle occurs, cytosolic PRRs provide comprehensive innate immune recognition. Thus, TLR3 and the adaptor TRIF are not required for viral induction of type I IFN in many tissues and cell types 111, although specialized innate antiviral cells, such as plasmacytoid DCs, rely more heavily on TLR mediated recognition of viral nucleic acids 112. Recognition of cytoplasmic dsDNA leading to NF-κB activation and type I interferon production has also been reported 113, 114. Although several candidates have been found, the relevant receptor has not yet been conclusively identified 115. This receptor(s) is predicted to be important for type I IFN production in response to viruses and intracellular pathogens, such as Listeria sp.
Although research on the recognition of PAMP/PRR pairs is an ongoing process of discovery, defective responses of MyD88-deficient cells indicate that many fungal and parasite species are capable of activating TLR pathways. TLR4 has been shown to recognize Aspergillus hyphae 116, and Cryptococcus neoformans capsular polysaccharide 117. TLR2 and TLR6 are required for recognition of yeast zymosan, while TLR4 is thought to recognize certain yeast mannans. The identification of parasite PAMPs has been more elusive, and their existence is somewhat controversial. Nevertheless, TLR2 heterodimers recognize various parasite GPI-anchored proteins and glycoinositolphospholipids from the parasitic protozoa Trypanosoma cruzi 118. TLR9 was initially reported to recognize the malarial pigment hemozoin, a byproduct of heme metabolism in infected erythrocytes 119, although subsequent studies suggest that DNA associated with hemozoin is the relevant TLR9 ligand 120. Instead, hemozoin may be recognized by the NLRP3 inflammasome 121. TLR11 recognizes a profilin-like protein that is conserved in apicomplexan parasites including Toxoplasma gondii 122. Recognition of helminths remains an area of controversy 123. In particular, it remains unclear how helminth PAMPs might preferentially induce TH2 responses that are characteristic of the immune responses to this class of pathogen. Direct skewing of adaptive responses by the mechanism of innate recognition is suggested to occur in other scenerios as well 124. For example, C-type lectins can directly affect TH activation and differentiantion 125, thus recognition of C. albicans α-mannans by dectin-2 induces a protective TH17-predominant response 126. Therefore, whether through cell type or PRR specificity, the recognition of specific PAMPS can shape the nature of the subsequent innate and adaptive immune responses.
Immediate anti-microbial responses
PAMP recognition initiates a complex series of events: the first is the mounting of immediate antimicrobial responses at the cellular level. This effective and evolutionarily conserved function of PRRs is dependent on activation of NF-κB gene programs. In particular, much of the early innate response has been demonstrated to depend on the canonical NF-κB pathway. Thus, rela−/−/ tnfr1−/− and ikbkb−/−/tnfr1−/− double-knockout mice have increased susceptibility to bacterial infection 57, 127, 128, 129. MEFs from Nemo-deficent mice do not exhibit NF-κB activation by LPS or IL-1 130. Therefore, activation of NF-κB responsive genes by the innate immune system depends on NEMO and likely progresses through the canonical NF-κB signaling pathway. Early anti-microbial effectors include NF-κB-dependent expression of defensins, which are cationic peptides that exert direct bactericidal activity by inducing membrane permeabilization. The classic mediators of defensin release are small intestinal Paneth cells, which secrete α-defensins into the intestinal lumen following exposure to bacterial PAMPs 131. The production of anti-microbial nitrogen and oxygen species, which are acutely toxic to a variety of microbes, augments the activity of anti-microbial peptides. Production of nitric oxide (NO) is mediated in part by inducible NO synthase (iNOS), which is coordinately regulated by NF-κB and STAT transcription factors 132.
Inflammation
Inflammation begins with epithelial or stromal cells of the infected tissue or tissue resident hematopoietic cells such as mast cells or DCs recognizing an inflammatory stimulus and propagating pro-inflammatory signals. These signals lead to the recruitment and activation of effector cells, initially neutrophils and later macrophages and other leukocytes, resulting in the tissue changes characteristic of inflammation – rubor, calor, dolor, and tumor (redness, heat, pain, and swelling, respectively). There is a vast amount of literature that correlates NF-κB activation with inflammation in a wide array of diseases and animal models. There are, likewise, numerous studies using gene targeting and inhibitors of NF-κB that have established the causative role of NF-κB in inflammatory processes.
NF-κB is responsible for the transcription of the genes encoding many pro-inflammatory cytokines and chemokines. The pathway from pathogen recognition to pro-inflammatory cytokine production demonstrates a particular reliance on NF-κB. The immediate targets of NF-κB-dependent pro-inflammatory cytokines, such as TNFα, tend to be receptors that, in turn, activate NF-κB. Therefore, NF-κB is crucial to the propagation and elaboration of cytokine responses. TNFα is particularly important for both local and systemic inflammation, and it is a potent and well-studied inducer of NF-κB. One important early target of these effectors is the vascular endothelium. Changes in vascular endothelial cells both recruit circulating leukocytes and provide them with a means of exiting the vasculature into the infected tissue. NF-κB regulates the expression of adhesion molecules, both on leukocytes and on endothelial cells, which allow the extravasation of leukocytes from the circulation to the site of infection 133. Thus, in the absence of p65, the recruitment of circulating leukocytes to sites of inflammation is impaired 127.
Recruited neutrophils are the key mediators of local inflammation and, as mentioned above, NF-κB is important for their survival and function under relatively toxic conditions 134. NF-κB is important for the production of the enzymes that generate prostaglandins and reactive oxygen species (e.g., iNOS and Cox, both NF-κB target genes) and may, furthermore, be involved in the signaling induced by prostaglandins 135, 136. NF-κB has also been implicated in the response to leukotrienes, which similar to prostaglandins are short-lived paracrine effectors. For example, leukotriene-induced IL-8 production appears to be dependent on rapid activation of NF-κB 137. Finally, matrix metalloporteinases are also crucial mediators of local inflammation and leukocyte chemotaxtis, and their expression is also regulated by NF-κB 138, 139, 140.
Resolution of inflammation and subsequent tissue repair is a crucial event, and its failure is a common source of pathology. Resolution of inflammation is an active process that is as complex as the inflammatory response itself, and involves numerous pathways that are not all directly relevant to NF-κB 141. Macrophages are key regulators of both inflammation and its resolution, and NF-κB plays a key role in the instruction of macrophage responses. Macrophage differentiation into pro-inflammatory (M1) or anti-inflammatory (M2) cells depends both on cell intrinsic NF-κB pathway activation as well as the local cytokine milieu, which is strongly influenced by NF-κB.
During acute inflammation, there are multiple negative feedback pathways that help to rein in inflammatory responses 3. It has long been known that cells, such as macrophages, become resistant to repeated pro-inflammatory stimuli 142. The IκB family protein Bcl-3 can function both as an inhibitor and mediator of NF-κB transcriptional programs 2. Following LPS stimulation, Bcl-3 is induced and binds p50 dimers, with which it can repress the expression of a subset of NF-κB target genes. In this manner, Bcl-3 mediates the selective inhibition of repeated LPS responses in macrophages 143. Although clearly not the only mechanism, induced transcriptional repression by Bcl-3/p50 likely contributes to LPS-tolerance. Furthermore by selectively affecting chromatin remodeling, Bcl-3 mediates repression of pro-inflammatory genes, and also facilitates the expression of the anti-inflammatory gene IL-10. Thus, it appears that Bcl-3 acts appropriately in the regulation of genes that are categorized as either “tolerizable or “non-tolerizable” 144. NF-κB p50 appears to be capable of mediating anti-inflammatory responses under several additional circumstances as well. For example, p50 negatively regulates IFNγ production by and proliferation of NK cells 145. Also, expression levels of p50 in M2-like tumor associated macrophages (TAMs) regulate the balance between anti- and pro-inflammatory functions. Thus, it was shown that TAMs overexpress p50 and that deletion of p50 renders these macrophages pro-inflammatory 146.
Inhibition of NF-κB during the resolution phase of inflammation can prolong the inflammatory process and prevent proper tissue repair 147. Furthermore, IKKα-deficient mice display increased inflammatory responses in models of local and systemic inflammation 148, and show increased production of pro-inflammatory chemokines and cytokines 148, 149. Rather unexpectedly, IKKα-deficient mice and embryonic-liver-derived macrophages from IKKα-deficient mice exhibit augmented inflammatory responses compared with wild-type mice 149. The mechanisms of IKKα-mediated repression of transcriptional responses seem to be through effects on the level of nuclear p65 and c-Rel 148. Although there are significant differences between these two reports with regard to the proposed mechanism of action, both, nevertheless, observe the same biological consequence of IKKα deficiency in macrophages. Furthermore, recent work has suggested that IKKβ also exhibits some anti-inflammatory capacity in macrophages. First, IKKβ suppresses the secretion of the potent pro-inflammatory cytokine IL-1β 150. Second, IKKβ is associated with the maintenance of an anti-inflammatory M2-like phenotype in TAMs 151. Third, IKKβ is thought to inhibit M1 macrophage function by interfering with the STAT pathway during infection 152.
Initiation of adaptive responses
The innate immune system invokes potent anti-microbial activities and maintains host protection under many settings. Nevertheless, initiating the adaptive immune response remains a crucial step for robust and durable pathogen clearance. The process of information transfer from the innate to adaptive immune system is mediated by activation and maturation of APCs. The nature of the PAMP and the signaling pathways activated by the cognate PRR, as well as the context within which pathogen recognition occurs, all influence the manner in which the APC instructs T and B cell responses.
DC maturation mediated by pathogen recognition is crucial for the initiation of the adaptive immune response. To activate naive T cells, DCs must alter their chemokine receptor expression to enable migration into lymphoid tissues. During activation DCs fine-tune their antigen processing machinery such that the presentation of pathogen epitopes is favored. Finally, the expression of co-stimulatory molecules including B7.1/B7.2 (or CD80/CD86) is upregulated. These co-stimulatory molecules ligate the co-stimulatory molecule CD28, thus providing the second signal necessary to induce full T-cell activation. The response to pathogens is tailored on the basis of the distribution of PRRs in different cell types, and the ability of different cell types to, in turn, interact with T cells in a biasing manner 153.
There continues to be debate surrounding how PRRs differentially induce TH1 versus TH2 responses. Maturation of DCs following viral infection depends on nucleic acid-binding PRRs 154, 155. Plasmacytoid DCs, which are robust inducers of interferon during viral infection, express viral PRRs including elevated expression of TLR7 and TLR9. Cytoplasmic nucleic acid-sensing PRRs, RLRs, are expressed more broadly, although they are further induced by type-I interferons, and they may, therefore, provide crucial amplification of antiviral responses following virus recognition by TLRs. While studies of anti-viral responses suggest that both cell type-specific distribution of PRRs, and selective activation of signaling pathways by certain PAMPs are key determinants of TH1 versus TH2 responses, the situation is less clear for other pathogens. It appears that multiple mechanisms converge in shaping how APCs interact with the adaptive immune system.
Role of NF-κB in the adaptive response
T-cell responses mediated by NF-κB
T-cell activation, differentiation, proliferation, and effector function are all influenced by gene programs regulated by NF-κB. The majority of effort directed at understanding the role of NF-κB in T cell activation has focused on CD4+ T cells (Figures 6 and and7).7). On the whole, the historical lack of CD8+ T-cell conditional knockouts and the selective loss of CD8 cells in the absence of functional NF-κB have impeded thorough characterization of the role of NF-κB in these cells. Nevertheless, there continues to be progress in this area and, in fact, there has been a resurgence of interest in recent years. CD4+ T cells have long been known to undergo differentiation into either TH1 or TH2 subsets depending on the cytokines present during activation 156. However, an appreciation of the ability of CD4+ helper T cells to differentiate into additional effector cell types, induced regulatory T cells, pro-inflammatory TH17 cells, TH22 cells TH9 cells, has prompted a fresh look at the contribution of NF-κB to T cell differentiation and effector function (Figure 7).
NF-κB in T cell activation. NF-κB participates in the maintenance, activation, differentiation and proliferation of naive T cells. Tonic TCR stimulation promotes T cell maintenance, of both memory and naive cells, through NF-κB activation. Activation occurs when the naive T cell recognizes its cognate antigen presented by an activated APC expressing both peptide:MHC and B7 family co-stimulatory molecules. NF-κB-dependent proliferation and differentiation ensue and are influenced by the local cytokine milieu. NF-κB supports proliferation, differentiation and survival as indicated (green arrows).
TH cell differentiation. NF-κB participates in differentiation of several TH cell types following activation of naive CD4+ TH cells. Differentiation pathways in which NF-κB is implicated are indicated with a green arrow. Key transcription factors in each differentiation pathway are indicated above each arrow, while cytokine responsible for skewing TH cells toward a given pathway are indicated below each arrow. Additional cytokines and transcription factors implicated in several of the pathways are not depicted.
Activation of naive T cells requires antigen-specific and co-stimulatory signaling provided by activated APCs. Binding of the TCR to its cognate peptide presented in the binding cleft of MHC supplies the antigen-specific signal, while co-stimulatory signaling results from ligation of CD28 by B7 molecules expressed on activated APCs. Activated naive T-cells proliferate rapidly while simultaneously differentiating into effector cells (Figure 6). In the case of CD4+ T cells, proliferation leads to differentiation into immature effector cells, TH0, which subsequently differentiate into TH1, TH2, TH9, TH17, TH22, or inducible Treg (iTreg) cells depending on the cytokine milieu.
Stimulation of T cells through TCR and CD28 results, initially, in activation of p65-containing NF-κB complexes followed by delayed and sustained activation of c-Rel complexes 157, 158, 159, 160. Rapidly proliferating activated T cells rely on NF-κB activity for protection from apoptosis as well as for the production of cytokines, especially IL-2, supporting proliferation and differentiation (Figure 6). As expected, inhibition of NF-κB in activated T cells facilitates progression toward activation-induced cell death (AICD) or apoptosis 161, 162, and stimulation of p65-deficient naive T cells induces cell death 163. In contrast, T cells lacking c-Rel do not undergo apoptosis, but nevertheless, fail to proliferate in response to typical mitogenic stimuli 164. T cells, in which p105 cannot undergo inducible processing to p50 also fail to proliferate normally 165. In vitro, c-Rel is needed for production of IL-2 and therefore, for proliferation, but this need is not recapitulated in vivo 166. Interestingly, c-Rel-deficient T cells appear to have a defect in TH1 proliferation and production of IFNγ, indicating a selective role for NF-κB family members in TH1/TH2 differentiation, independent of that mediated by the innate response.
Multiple transcriptional activators and repressors regulate expression of IL-2. Amongst these, members of the NF-κB family play multiple roles. In naive T cells, which do not express IL-2, repressive p50 homodimers are found associated with the IL-2 promoter 167. Failure of T-cell proliferative responses in c-Rel-knockout mice is attributable to a failure to produce IL-2 164. In naive T cells, c-Rel is responsible for mediating chromatin remodeling across the IL-2 locus following CD3/CD28 co-stimulation 158. Thus, naive T cells can be primed by activation of c-Rel by pro-inflammatory cytokines, such as those elicited following stimulation with TLR ligands 157. Priming, which requires NF-κB activation, results in a more robust response to CD3/CD28 co-stimulation 168. Conversely, overexpression of p65 with c-Jun can overcome the requirement for co-stimulation in naive T cells 169.
TH0 differentiation into TH1, TH2, TH9, TH17, Treg effector cells depends on the induction of specific transcription factors, namely T-bet, GATA3, PU.1, RORγt, and Foxp3, respectively (Figure 7). There is increasing evidence that NF-κB family members are key arbitrators of the decision. For example, mice lacking p50 are unable to mount an asthma-like, airway TH2 response 170. Indeed, p50-deficient T cells fail to induce GATA3 expression during T-cell stimulation under TH2 differentiating conditions. These results suggest a transcriptional role for p50, and it was subsequently found that Bcl-3-deficient T cells also fail to undergo TH2 differentiation, suggesting that p50/Bcl-3 complexes are crucial for TH2 differentiation 171. Conversely, the same authors found that RelB-deficient T cells are deficient in TH1 differentiation due to decreased expression of T-bet. RelB may mediate upregulation of STAT4, which is responsible for T-bet induction downstream of IFNγ. Therefore, it appears that NF-κB activation during TCR stimulation may render cells competent for both proliferative and differentiating stimuli. As a corollary, NF-κB transactivation of the IL-2 gene is repressed as TH cells differentiate into TH1 or TH2 and become dependent on TH1 and TH2 cytokines (e.g., IFNγ and IL-4). It is thought that direct binding of T-bet to p65 that is associated with the IL-2 gene enhancer, may mediate the repression of IL-2 production in TH1 cells 172. In TH2 cells the lack of IL-2 transcription may be because of the decreased levels of p65 activation 173. As discussed above, several groups recently implicated NF-κB, and specifically c-Rel, in the regulation of Foxp3 expression and Treg development 61, 62, 63, 174. The relative contribution of c-Rel to iTreg development still requires further clarification. While a recent report demonstrated that IκBζ cooperates with ROR and is required for TH17 differentiation 59, the more general contribution of NF-κB in this process, either in the thymus or periphery, remains unclear. While the role of NF-κB in TH9 cells has yet to be interrogated, there are interesting clues suggesting an important role for NF-κB. Recently, PU.1 was reported to be the transcription factor responsible for TH9 differentiation 175. In unrelated recent work on acute myelogenous leukemia, it was noted that NF-κB regulates the transcription of PU.1 176. Furthermore, it has been suggested that IL-9 expression in T cells is regulated by NF-κB 177. Thus, it would seem reasonable to speculate that NF-κB may play an important role in TH9 differentiation (Figure 7). The transcription factors responsible for the differentiation of TH22 cells have not yet been identified. However, the possible role of TNFα as a TH22 differentiating cytokine is suggestive that NF-κB may be an important mediator of this differentiation process as well. In summary, NF-κB participates in both the initial T cell responses by supporting proliferation and regulating apoptosis, and is increasingly being shown to have an important role in the regulation of TH differentiation.
B-cell responses mediated by NF-κB
There are many parallels in the functions of NF-κB in T and B cells. NF-κB acts to support proliferation, regulate apoptosis, and control the processes of differentiation and maturation in B cells (Figure 8). B-cell responses are classified into two groups: thymus-dependent (TD) or thymus-independent (TI). In TD response follicular B cells require co-stimulatory signaling from TH cells expressing CD40L and cytokines, such as IL-4. These initial steps trigger germinal center formation, in which somatic hypermutation, isotype switching, and plasma cell differentiation occur. Thus, in individuals with a mutation in CD40L B cells fail to undergo class switch recombination in response to T-dependent antigens 178. Signaling through CD40 activates both canonical and non-canonical NF-κB pathways, although several lines of evidence suggest that the non-canonical pathway is not required in the response to TD antigens. Thus, B cells lacking p52 are fully capable of appropriate TD antigen responses when transferred 21, and B cells from relB−/− mice, although crippled in their proliferative response, undergo normal IgM secretion and class switching 179. In contrast to these results, B cells lacking either functional NIK or IKKα, exhibit defective responses 180, 181. It remains unclear whether the role of NIK and IKKα in this context is to induce p100 processing (non-canonical pathway) or to contribute to activation of the canonical pathway. Although both pathways are triggered by CD40 ligation, the current data suggest that canonical pathway activation is of great importance.
NF-κB in B cell activation. NF-κB participates in the maintenance, activation, differentiation and proliferation of naive B cells. Tonic BCR and cytokine, BAFF, stimulation promotes naive B cell maintenance through NF-κB activation. Activation occurs when the naïve B cell recognizes its cognate antigen and receives co-stimulatory signaling (CD40L) from an activated TH cell within the germinal center. NF-κB-dependent proliferation and differentiation ensue and are coupled to BCR affinity maturation. NF-κB signaling in B cells expressing selected BCRs results in class switch recombination and differentiation into either memory B cells or plasma cells. NF-κB supports proliferation, differentiation and survival as indicated (green arrows).
Evidence also supports a role for the canonical pathway in class switch recombination. In contrast to B cells lacking RelB or p52, those from rela−/− mice exhibit markedly diminished class switching, despite a modest loss of lymphocyte proliferation following various stimuli 182. Likewise, c-Rel-deficient mice fail to generate protective humoral immune responses demonstrating the requirement for c-Rel in class switch recombination 164, 183, 184. B cells from nfbk1−/− mice exhibit decreased proliferation in response to mitogenic stimulation, and p50/p65 double-knockout B cells exhibit greater defects in proliferation and class switching 185, 186. Therefore, B cell proliferation and the TD B-cell maturation response are dependent on the canonical NF-κB pathway.
TI antigens are those that have an intrinsic ability to initiate B-cell responses in the absence of T-cell help. TI responses are carried out by marginal zone and CD5+ B cells. TI antigens function by acting as both antigen and B-cell mitogens, for example antigens that are PRR ligands or antigens with high avidity that are capable of crosslinking the BCR through repetitive structural features. In such cases it is expected that B-cell responses are more dependent on members of the canonical pathway that have well-documented roles in TLR and BCR signaling. For example, c-Rel-deficient B cells are highly sensitive to apoptosis following BCR cross-linking 187, 188, 189. As mentioned above, p50 and p50/RelA double-knockout B cells are deficient in responses to TI stimulation. Likewise, IKKβ-deficient B cells fail to mount TI or TD responses 190. These IKKβ-deficient B cells also exhibit increased spontaneous apoptosis, supporting that NF-κB is important in survival of B cells.
NF-κB and lymphocyte longevity
Lymphocyte homeostasis reflects balanced lymphocyte turnover and lymphopoiesis. Turnover of mature lymphocytes is complex as it depends upon both activation induced and homeostatic proliferation, and cell death. Lymphocyte survival is mediated through tonic stimulation provided by both the antigen receptor and certain cytokine receptors. As such, NF-κB can be assumed to play an important role in lymphocyte homeostasis and this assumption is supported by various genetic models.
T cells can be quite long-lived and naive T cells require continued contact with MHC:self-peptides for survival. These self-peptide complexes are most likely expressed on lymphoid DCs and generate a tonic low grade TCR signal. Survival of memory cells, on the other hand, is independent of continued contact with self-peptide:MHC complexes. B cells are formed at a far higher rate than T cells and therefore, B cells also undergo a significantly higher rate of turnover. Nonetheless, B cells too require maintenance signals to achieve peripheral homeostasis. The antigen receptor on B cells provides a basal level of signaling, albeit independent of the presence of antigen, which is required for maintenance of mature B cells. Thus, deletion of the BCR from mature B cells results in a complete loss of the peripheral B cell pool 191. The assumption is that BCR provides tonic activation of the NF-κB pathway resulting in the expression of anti-apoptotic proteins. Consistent with this assumption, loss of IKKβ, NEMO, or components of the CBM complex in mature B cells also results in complete loss of peripheral B cells 71, 190, 192. Furthermore, B cells from rela−/−, nfkb1−/−, nfkb2−/−, and c-rel−/− mice all have increased sensitivity to apoptosis and/or decreased survival ex vivo 67, 188, 193. In addition to contributing directly, it is thought that low level NF-κB activation in B cells contributes to survival through upregulation of p100, which is necessary for BAFFR-induced activation of the alternative pathway 67, 75, 166.
Several studies have implicated BAFFR in this aspect of the B lymphocyte survival 194, suggesting that the non-canonical NF-κB pathway is also relevant to B-cell survival. Consistent with this idea, constitutive activation of NIK or IKKα can circumvent the requirement for B cell BAFFR 74, 195. Conversely, B cells lacking IKKα exhibit defective survival 72, 196. The relevant targets of the non-canonical pathway downstream of BAFFR are thought to be Bcl-2 family members, e.g., the anti-apoptotic factor A1 187 or Bcl-XL, which are known to be regulated by p52/RelB-containing complexes, and are required for the maintenance of mature B cells. Indeed, Bcl-XL can complement B cells with a mutant BAFFR 197. Thus, tonic BCR stimulation, through upregulation of p100, may cooperate with BAFFR ligation and alternative pathway activation, to upregulate anti-apoptotic Bcl2 family members and mediate maintenance and survival of mature B cells.
Concluding remarks
The areas covered in the current review necessarily represent a subset of those in which NF-κB is a crucial regulator of immune responses. Recent work has highlighted the importance of NF-κB in the maintenance of epithelium barriers 198; as a regulator of inflammation associated with obesity and hyperlipidemia; and as a key factor in the development of inflammatory diseases and cancer. Our understanding of the immune system continues to improve, and coordinately our recognition of the many roles played by the NF-κB family of transcription factors also grows. As one example, the expanding universe of T-cell subsets opens up a new area for the analysis of the role of NF-κB in T-cell differentiation and effector function. It will be intriguing to see how NF-κB contributes to the development of TH9, TH17, TH22, and iTreg/TH3 cells. Deciphering the role of NF-κB in the effector function of these recently described cell types should also prove to be an area of great interest, particularly given the growing evidence for dysregulation of these cell types in autoimmune and inflammatory disease. There is much that remains unknown about the contributions of NF-κB in early and non-lymphoid hematopoiesis; in responses to danger signaling and allergens; in the coordination of pathogen-specific adaptive immune responses by the innate immune system; and in the differentiation and effector responses of both lymphoid and non-lymphoid innate immune cells. Thus moving forward, as in the past, much of the work done to understand the physiological and pathophysiological role of NF-κB will continue to focus on the role of this transcription factor family in Immunobiology.
Acknowledgments
Research in the authors' laboratory was supported by grants from the National Institutes of Health (to SG).
References
- Hayden MS, Ghosh S. Signaling to NF-kappaB. Genes Dev. 2004;18:2195–2224. [PubMed] [Google Scholar]
- Hayden MS, West AP, Ghosh S. NF-kappaB and the immune response. Oncogene. 2006;25:6758–6780. [PubMed] [Google Scholar]
- Ghosh S, Hayden MS. New regulators of NF-kappaB in inflammation. Nat Rev Immunol. 2008;8:837–848. [PubMed] [Google Scholar]
- Burkly L, Hession C, Ogata L, et al. Expression of relB is required for the development of thymic medulla and dendritic cells. Nature. 1995;373:531–536. [PubMed] [Google Scholar]
- Irla M, Hugues S, Gill J, et al. Autoantigen-specific interactions with CD4+ thymocytes control mature medullary thymic epithelial cell cellularity. Immunity. 2008;29:451–463. [PubMed] [Google Scholar]
- Hikosaka Y, Nitta T, Ohigashi I, et al. The cytokine RANKL produced by positively selected thymocytes fosters medullary thymic epithelial cells that express autoimmune regulator. Immunity. 2008;29:438–450. [PubMed] [Google Scholar]
- Akiyama T, Shimo Y, Yanai H, et al. The tumor necrosis factor family receptors RANK and CD40 cooperatively establish the thymic medullary microenvironment and self-tolerance. Immunity. 2008;29:423–437. [PubMed] [Google Scholar]
- Anderson G, Jenkinson EJ, Rodewald HR. A roadmap for thymic epithelial cell development. Eur J Immunol. 2009;39:1694–1699. [PubMed] [Google Scholar]
- Ruddle NH, Akirav EM. Secondary lymphoid organs: responding to genetic and environmental cues in ontogeny and the immune response. J Immunol. 2009;183:2205–2212. [PMC free article] [PubMed] [Google Scholar]
- van de Pavert SA, Mebius RE. New insights into the development of lymphoid tissues. Nat Rev Immunol. 2010;10:664–674. [PubMed] [Google Scholar]
- Alcamo E, Hacohen N, Schulte LC, Rennert PD, Hynes RO, Baltimore D. Requirement for the NF-kappaB family member RelA in the development of secondary lymphoid organs. J Exp Med. 2002;195:233–244. [PMC free article] [PubMed] [Google Scholar]
- Miyawaki S, Nakamura Y, Suzuka H, et al. A new mutation, aly, that induces a generalized lack of lymph nodes accompanied by immunodeficiency in mice. Eur J Immunol. 1994;24:429–434. [PubMed] [Google Scholar]
- Koike R, Nishimura T, Yasumizu R, et al. The splenic marginal zone is absent in alymphoplastic aly mutant mice. Eur J Immunol. 1996;26:669–675. [PubMed] [Google Scholar]
- Shinkura R, Kitada K, Matsuda F, et al. Alymphoplasia is caused by a point mutation in the mouse gene encoding Nf-kappa b-inducing kinase. Nat Genet. 1999;22:74–77. [PubMed] [Google Scholar]
- Mebius RE. Organogenesis of lymphoid tissues. Nat Rev Immunol. 2003;3:292–303. [PubMed] [Google Scholar]
- Bonizzi G, Karin M. The two NF-kappaB activation pathways and their role in innate and adaptive immunity. Trends Immunol. 2004;25:280–288. [PubMed] [Google Scholar]
- Dougall WC, Glaccum M, Charrier K, et al. RANK is essential for osteoclast and lymph node development. Genes Dev. 1999;13:2412–2424. [PMC free article] [PubMed] [Google Scholar]
- Yilmaz ZB, Weih DS, Sivakumar V, Weih F. RelB is required for Peyer's patch development: differential regulation of p52-RelB by lymphotoxin and TNF. EMBO J. 2003;22:121–130. [PMC free article] [PubMed] [Google Scholar]
- Paxian S, Merkle H, Riemann M, et al. Abnormal organogenesis of Peyer's patches in mice deficient for NF-kappaB1, NF-kappaB2, and Bcl-3. Gastroenterology. 2002;122:1853–1868. [PubMed] [Google Scholar]
- Caamano JH, Rizzo CA, Durham SK, et al. Nuclear factor (NF)-kappa B2 (p100/p52) is required for normal splenic microarchitecture and B cell-mediated immune responses. J Exp Med. 1998;187:185–196. [PMC free article] [PubMed] [Google Scholar]
Cell Res. 2011 Feb; 21(2): 223–244.
Published online 2011 Jan 18. doi: 10.1038/cr.2011.13
PMCID: PMC3193440
PMID: 21243012
NF-κB in immunobiology
Matthew S Hayden1 and Sankar Ghosh1,*
Author information Copyright and License information PMC Disclaimer
Abstract
NF-κB was first discovered and characterized 25 years ago as a key regulator of inducible gene expression in the immune system. Thus, it is not surprising that the clearest biological role of NF-κB is in the development and function of the immune system. Both innate and adaptive immune responses as well as the development and maintenance of the cells and tissues that comprise the immune system are, at multiple steps, under the control of the NF-κB family of transcription factors. Although this is a well-studied area of NF-κB research, new and significant findings continue to accumulate. This review will focus on these areas of recent progress while also providing a broad overview of the roles of NF-κB in mammalian immunobiology.
Keywords: NF-κB, TLR, TCR, BCR, lymphoid organogenesis, inflammation, hematopoiesis
Introduction
As discussed in the introduction to the January special issue of Cell Research, the discovery and characterization of the NF-κB family of transcription factors resulted from studies in two major areas of research: immunology and cancer biology. Although the role of NF-κB in cancer biology is becoming progressively better established, historically much of our current knowledge of NF-κB resulted from efforts directed at understanding its role in the regulation and function of the immune response. This review will attempt to provide a comprehensive discussion of the diverse functions of NF-κB in immunobiology and update our previous efforts in reviewing the far-reaching role of NF-κB in this area of biology 1, 2, 3.
The mammalian immune response comprises various mechanisms by which the physiological and functional integrity of the host is maintained in the face of microbial insult. The bone marrow-derived cells of the immune system are chiefly, though by no means solely, responsible for performing these functions. Thus, we begin by reviewing the role of NF-κB in the development of these hematopoietic cells that mediate both innate and adaptive immune responses. With the relevant cell types and tissues in place, the immune response is triggered by host recognition of foreign pathogens. Our knowledge in this area of the immune response has expanded rapidly in the past decade. Pathogen recognition is followed by pathogen clearance, a highly variable response that may draw upon coordinated responses at the level of cell, tissue, and the organism. Finally, the immune response must be resolved, damage must be repaired and, whenever possible, the triggering insult remembered for future immunity.
In mammals, the NF-κB family is composed of five related transcription factors: p50, p52, p65 (also RelA), c-Rel, and RelB (Figure 1). These transcription factors share an N-terminal DNA-binding/dimerization domain, known as the Rel homology domain, through which they can form homo- and heterodimers. NF-κB dimers can bind to a variety of related target DNA sequences called κB sites to modulate gene expression. RelB, c-Rel, and p65 contain C-terminal transcription activation domains (TADs) that enable co-activator recruitment and target gene expression. As they lack TADs, p50 and p52 can activate transcription by forming heterodimers with p65, c-Rel, or RelB, or by recruiting other TAD-containing proteins. However, as homodimers lacking the ability to drive transcription, they can repress transcription on binding to DNA.
The mammalian NF-κB, IκB and IKK protein families. Relevant domains typifying each protein family are indicated and alternative nomenclatures are provided in parenthesis. The precursor proteins p100 and p105 function as both IκB and, when processed by the proteasome, NF-κB family members. Highly simplified schematics of canonical and non-canonical NF-κB pathways are shown. In the canonical pathway NEMO-containing IKK complexes are activated and induce phosphorylation and degradation of IκBα leading to the release of NF-κB dimers including p65:p50 dimers. In the non-canonical pathway, NEMO-independent activation of IKKα is mediated by the upstream kinase NIK. IKKα, with NIK, induces phosphorylation and processing of p100 to p52 resulting in the activation of predominantly p52:RelB complexes. Although they are illustrated separately here, as discussed in the text, signaling through these two pathways often is initiated by the same receptor – e.g., LTβR or CD40. (ANK, ankyrin-repeat domain; DD, death domain; RHD, REL homology domain; TAD, transactivation domain; LZ, leucine-zipper domain; GRR, glycine-rich region; HLH, helix-loop-helix domain; Z, zinc-finger domain; CC, coiled-coil domain; NBD, NEMO-binding domain; MOD/UBD, minimal oligomerization domain/ubiquitin-binding domain; PEST, proline, glutamic acid, serine, and threonine rich.)
In most cells, NF-κB complexes are inactive, residing predominantly in the cytoplasm in a complex with inhibitory IκB proteins (IκBα, IκBβ, IκBɛ, IκBζ, p100, p105, Bcl3, IκBns; Figure 1). When signaling pathways are activated, the IκB protein is degraded and NF-κB dimers enter the nucleus to modulate target gene expression. In almost all cases, the common step in this process is mediated by the IκB kinase (IKK) complex, which phosphorylates IκB and targets it for proteasomal degradation (see Liu and Chen, Cell Res 2011; 21:6–21). The IKK complex consists of two catalytically active kinases, IKKα (IKK1) and IKKβ (IKK2) and a regulatory scaffolding protein, NEMO (IKKγ Figure 1). These are the basic players in the NF-κB pathway; however, there is considerable complexity in how the individual proteins function and how they coordinate a nuanced NF-κB response (see Shih et al., Cell Res 2011; 21:86–102). The details of individual signaling pathways, the mechanism of action of upstream signaling intermediates, the role of various post-translational modifications, and cross-talk between NF-κB and other signaling pathways are covered in depth elsewhere in this special issue. Here, we focus on the function of NF-κB and the signaling pathways that regulate it in the mammalian immune response.
Development of the immune system
The mammalian immune system consists of a functionally linked group of anatomically disparate tissues and cell types. The dispersed cellular components of the immune system that arise from the bone marrow receive much of the attention in immunology and the study of NF-κB has likewise focused on these cells. However, lymphoid organs that facilitate coordination and dissemination of immune responses carried out by immune cells are also key sites of NF-κB function. Therefore, while this section is largely concerned with the role of NF-κB in hematopoiesis, the role of NF-κB in lymphoid organogenesis is also discussed briefly.
NF-κB and lymphoid organogenesis
NF-κB plays an important role in the development and function of primary (bone marrow, thymus) and secondary (lymph nodes, Peyer's patches, mucosal-associated lymphoid tissue, and the spleen) lymphoid tissues. There is clearly a role for NF-κB in the development and regulation of bone, and we refer the reader to the excellent review on the subject in this special issue (see Novack, Cell Res 2011; 21:169–182). The role of NF-κB in thymic architecture, originally thought to be limited to the important role of RelB in thymic architecture 4, has become clearer in terms of the development of medullary thymic epithelial cells 5, 6, 7, but still remains to be more fully elucidated 8. The secondary lymphoid tissues facilitate maintenance and activation of mature lymphocytes by providing an environment within which the interaction of lymphocytes and other leukocytes can be carefully orchestrated 9. The role of NF-κB in this process is now well appreciated. Multiple NF-κB knockouts exhibit defects in some aspect of secondary lymphoid organ development.
The initial events of lymphoid organogenesis involve the association of lymphotoxin (LT)α1β2-expressing hematopoietic cells and vascular cell adhesion molecule-1 (VCAM-1)-expressing stromal cells 10. This interaction initiates a positive feedback loop through NF-κB (Figure 2). LTα1β2, receptor activator of NF-κB ligand (RANKL), and TNFα are known to activate NF-κB, and figure prominently in lymphorganogenesis 10 (Figure 2). Also, mediators of lymphoid organogenesis and homeostasis, such as adhesion molecules (e.g., intercellular adhesion molecule, VCAM, peripheral node addressin, glycosylation-dependent cell adhesion molecule-1, and mucosal addressin cellular adhesion molecule (MadCAM)), cytokines (e.g., TNFα and LTα1β2) and organogenic chemokines (e.g., CXCL12 (GRO/MIP-2), CXCL13 (BLC), CCL19 (ELC), and CCL21 (SLC)), are regulated by NF-κB.
NF-κB in lymphoid organogenesis. NF-κB regulates key components of a positive feedback loop between hematopoietic and stromal cells. LTα1β2-expressing hematopoietic cells induce production of VCAM-1 through the canonical NF-κB pathway and chemokines through the non-canonical pathway in LTβR-expressing stromal cells. Stromal expression of chemokines induces the upregulation of integrins (α4β1) on hematopoietic cells resulting in increased recruitment of LTα1β2-expressing cells, and increased signaling through stromal LTβR. RANKL signaling through RANK on the hematopoietic cell leads to activation of NF-κB, and further upregulation of LTα1β2.
Signaling through TNFR1, LTβR, and RANK activates p65-containing complexes and, hence, it is not surprising that rela−/−/tnfr1−/− double-knockout mice lack Peyer's patches and lymph nodes, and exhibit a disorganized spleen 11. The requirement for p65 in the development of these tissues is attributable to its function in stromal cells, and is likely because of a combination of effects: regulation of apoptosis (e.g., that induced by TNF); regulation of expression of organogenic factors, such as VCAM and LTα1β2; and enhancement of the non-canonical p52/RelB NF-κB pathway. The non-canonical NF-κB pathway has a well-established role in secondary lymphoid organ development. Deletion of components of the non-canonical NF-κB signaling pathway, including NIK, IKKα, LT, and RANK, all lead to severe defects in secondary lymphoid organogenesis 12, 13, 14, 15, 16, 17. The target of the non-canonical pathway, the p52/RelB dimer, is the primary transcriptional regulator of key organogenic factors including CXCL12, CXCL13, CCL19, CCL21, and MadCAM-1 18. Thus, the p52 single knockout lacks normal B-cell follicles, germinal centers, and Peyer's patches 19, 20, 21, while RelB knockouts lack Peyer's patches and exhibit compromised lymphnode development 18.
Splenic architecture is crucial for B-cell development as well as for the initiation and maturation of B-cell responses. The spleen is divided functionally and histologically into white and red pulp zones. Macrophages in the red pulp are responsible for destroying damaged erythrocytes, while the white pulp is populated by splenic lymphocytes organized into B-cell follicles and T-cell zones. Yet splenic architecture is also dynamic, as exemplified by the formation of germinal centers during the initiation and maturation of B-cell responses. As with other secondary lymphoid organs, NF-κB figures prominently in the development and maintenance of splenic architecture. Mice in which p65 has been deleted exhibit defects in both static and dynamic splenic architecture 11. Deletion of RelB, NIK, or IKKα leads to defects in splenic architecture, similar to those of ltbr−/− spleens 12, 13. Similar to p65/TNFR1 knockouts, mice with a defective non-canonical pathway fail to segregate B cell/T cell zones and they fail to form germinal centers following immunization. Marginal zone macrophages, which line the border between red and white pulp areas, are also absent or disorganized in RelB, p52, NIK, or IKKα knockouts 21, 22. Recent work has highlighted the role of LTα1β2 in the development and maintenance of the marginal sinus architecture, suggesting that the role of NF-κB in splenic architecture should perhaps be revisited with an eye toward an analysis of endothelial function. Some splenic defects are also attributable to effects on hematopoietic cells, most notably of the myeloid lineages.
In summary, both the canonical and non-canonical NF-κB pathways are required for the development of most secondary lymphoid organs (Figure 2). However, the non-canonical pathway, as assessed by examining mice deficient for IKKα, p52, NIK, or RelB, is especially important both during organogenesis and for maintenance of splenic architecture. Recent advances suggest that NF-κB signaling in endothelial cells, which are crucial regulators of cellular movement into and out of lymphoid organs, should be better characterized. The functional consequences of defects in these processes are severe and have direct ramifications for the ability of host to mount a robust immune response. As such, the function of NF-κB in the development and regulation of these organs represents an important facet of the role of NF-κB in immunobiology.
NF-κB and hematopoiesis
The immune system includes cells of the lymphoid and myeloid lineages: T cells, B cells, monocytes, macrophages, dendritic cells (DCs), natural killer (NK) cells, basophils, eosinophils, neutrophils, and mast cells (Figure 3). These bone marrow-derived cells are the core constituents of both the innate and adaptive immune responses. Proliferation, differentiation, and apoptosis are the defining characteristic of hematopoiesis, and NF-κB participates in the regulation of each of these processes. While the study of the role of NF-κB in development and homeostasis of hematopoetic cells has focused largely on B cell and T lymphocytes, it is likely that the NF-κB pathway is also important for the development of NK cells, DCs, monocytes, granulocytes, and other cellular components of the immune system. In general, an anti-apoptotic and pro-proliferative role for NF-κB is invoked in the hematopoietic system (Figure 3). Indeed, deletion of the kinase TAK1, an upstream component of both NF-κB and AP1 pathways, results in decreased antiapoptotic gene expression, hematopoietic stem cell apoptosis, and failure of hemtopoiesis 23. However, in actuality, the situation is much more complex. For example, ikba−/−ikbe−/− cells, which have elevated NF-κB activation are broadly defective in myelopoiesis and exhibit defects in NK cells and lymphoid lineages 24, 25. Thus, NF-κB function must be interrogated at each point of cellular differentiation in order to fully appreciate its contribution to hematopoiesis. Greater insight into the majority of events that lead to the development of immune cells, awaits both further characterization of the developmental processes and generation of better genetic tools to examine the consequences of perturbing NF-κB function.
NF-κB in hematopoiesis. Red arrows indicate stages in which NF-κB activation is thought to contribute negatively and green arrows indicate a positive function in the development of the indicated lineages. Curved arrows indicate examples in which NF-κB contributes to the survival of cell population, either in the resting state or during immune responses. Gray arrows indicate developmental events for which NF-κB plays no role or for which the role of NF-κB has not been clearly demonstrated. (HSC, hematopoietic stem cell; CMP, common myeloid progenitor; MLP, myeloid/lymphoid progenitor; MEP, megakaryocyte erythrocyte progenitor; GMP, granulocyte monocyte progenitor; MDP, macrophage dendritic cell progenitor; CDP, common dendritic cell progenitor; CLP, common lymphoid progenitor; ETP, early thymic precursor; and B/NP, B-cell natural killer cell progenitor).
NF-κB in non-lymphocyte hematopoiesis
Although most, if not all cells of the body can exhibit some innate immune function, the ability to recognize microbes and initiate an antimicrobial response, we focus here only on the development of certain hematopoietic, non-lymphoid components of the innate immune system. In general, our understanding of the developmental origin of these innate cells lags far behind that of lymphocytes. Yet recent progress in characterizing the developmental process for several myeloid lineages should provide a better framework within which the contribution of NF-κB can be better assessed in the future.
Significant progress has been made recently in understanding DC development, including the characterization of a common DC progenitor 26, 27, 28, yet the contribution of NF-κB to early steps of DC lineage commitment remains poorly understood. The most notable contribution of NF-κB in this area is the requirement for RelB in development of the CD8α− DCs 4, 29, 30, 31. Although single knockouts of other NF-κB genes do not result in deficiencies in DC development, deletion of both p65 and p50 has a profound effect 32, 33. Again, however, the limitations of the experimental systems used to examine the contribution of NF-κB, particularly with regard to the need to either delete TNFR1 or utilize fetal liver transfer experiments, should be underscored. In the periphery, NF-κB is clearly required for DC maturation. Loss of IKKβ or inhibition of the IKK complex using a cell permeable peptide prevents DC maturation and antigen-presenting cell (APC) function 34, 35. In addition, peripheral differentiation and function of myeloid DCs, e.g., monocyte-derived DCs, is significantly impaired by NF-κB inhibition 36. DCs in the periphery survive for a short period of time following activation, but their survival can be prolonged by CD40L expression on T cells. CD40L activates both the canonical and non-canonical NF-κB pathways, and hence DCs deficient in both p50 and c-Rel, or DCs overexpressing a mutant super-repressor form of IκBα, demonstrate significantly decreased survival 32, 37. To date, the specific targets of NF-κB in DC development and, indeed, the role of NF-κB at individual stages of DC development, remain quite poorly characterized.
Genetic models of NF-κB deficiency and activation have revealed other roles for NF-κB in the myeloid lineage. Of the non-lymphocyte lineages, neutrophil development is perhaps best characterized in terms of the role of NF-κB. There remains, however, significant gaps in both our understanding of neutrophil development, and more so the role of NF-κB in this process. IκBα-knockout mice, which have elevated classical NF-κB activity, display robust granulocytosis 38. RelB-deficient mice exhibit an inflammatory phenotype with a marked increase in peripheral neutrophil numbers. Mice lacking c-Rel and p50, and heterozygous for p65, also exhibit neutrophilia 39. This increase in peripheral neutrophil numbers resulting from deletion of NF-κB appears to be related to both increased development and increased egress from the bone marrow 39. In mature neutrophils, the anti-apoptotic signal provided by NF-κB is crucial. In sharp contrast to lymphocytes, which are relatively long-lived in the absence of activation, neutrophils normally exhibit a very short lifespan but require protection from apoptosis during inflammation when they provide effector function. Neutrophils can respond by activating NF-κB in response to numerous pro-inflammatory stimuli, and the activated NF-κB can increase neutrophil survival 40. Although neutrophils are capable of activating NF-κB in response to many pro-inflammatory stimuli 41 they lack p52 and RelB 42, which are crucial for the maintenance of long-lived lymphocytes. Thus, in neutrophils the role of NF-κB is selective and cell type specific.
NF-κB in lymphopoiesis
Development of T and B cells has, historically, been the subject of much greater scrutiny than the development of cells of the myeloid lineages. NF-κB has been examined in many aspects of lymphopoiesis and found to be vital for the development and function of adaptive immune cells 43 (Figure 4). Despite their potential longevity in the periphery, lymphocyte development is characterized by abundant apoptosis. As a consequence, the anti-apoptotic properties of NF-κB play a key role in lymphopoeisis. Indeed, in many instances the requirement for NF-κB can be overcome by transgenic expression of the anti-apoptotic factor Bcl-2 44. Indeed, the role of NF-κB in B cell development was recently re-examined 45. Although NF-κB was expendable in cells undergoing rearrangement of the eponymous Igκ loci, NF-κB was required to protect the resulting cells from apoptosis, and for these cells to progress to rearrangement of the Igλ locus. This deficit in NF-κB function could be circumvented through overexpression of Bcl-2 45. The necessity of NF-κB for lymphopoesis is strikingly illustrated in human genetic diseases in which the gene encoding NEMO is inactivated by mutation. Because the NEMO gene is located on the X chromosome, it is usually subject to random inactivation in individual cells in females. However, in female patients who are heterozygous for a mutant version of NEMO, all peripheral lymphocytes possess an intact NEMO gene, rather than the 50% predicted by random inactivation, suggesting that in the absence of NEMO-dependent NF-κB signaling, B and T cells fail to develop.
NF-κB in lymphopoiesis. NF-κB plays a pro-survival role in common lymphoid precursor (CLP) cells which give rise to B- and T-cell lineages. B-cell development occurs in the bone marrow, where NF-κB protects pre-B cells from pro-apoptotic stimuli including TNFα. Signaling to NF-κB through the pre-B cell receptor mediates survival of Pre-B cells, which then undergo light chain recombination to produce a functional B cell receptor. NF-κB provides a necessary pro-survival signal during Igλ but not Igκ rearrangement. Expression of BCR leads to NF-κB-dependent differentiation into immature B cells. High levels of BCR signaling, i.e., through recognition of self-antigen, results in negative selection through the loss of NF-κB activity. Transitional B cells exit the bone marrow and migrate to the spleen, where they mature and differentiate, a process that also requires NF-κB. T-cell development occurs following migration of precursor cells into the thymus. Stimulation of NF-κB through pre-TCRα provides a pro-survival signal allowing recombination of the TCR α chain and maturation to the double-positive (DP) stage. Optimal signaling through the TCRα/β complex induces NF-κB-dependent survival pathways, while a failure to signal or high level signaling results in death by neglect or negative selection, respectively. Intermediate high NF-κB activation facilitates intrathymic regulatory T cell (Treg) development. NF-κB activity is required for the maintenance of long-lived B and T cells. (CLP, common lymphoid progenitor; ETP, early thymic progenitor; DN, double negative – CD4−CD8−; DP, double positive – CD4+CD8+; SP, single positive – either CD4+CD8− or CD4−CD8+)
The effects of NEMO inactivation in both mice and humans solidify the role of NF-κB in lymphopoiesis, although the details by which NF-κB functions in this process remain obscure. NF-κB plays diverse roles in lymphocyte development that can be grouped according to timing – that is, before, during, or after pre-antigen receptor signaling. Although no single NF-κB-subunit-knockout mouse has as severe a phenotype as NEMO knockouts with regard to the generation of mature lymphocytes, double knockouts of Rel proteins confirm the essential anti-apoptotic function of NF-κB. For example, loss of both p50 and p65, or both p65 and c-Rel, terminates lymphopoiesis before expression of the pre-antigen receptors 46, 47, suggesting that NF-κB regulates anti-apoptotic factors required for early lymphoid cell survival in response to pro-apoptotic stimuli (Figure 4). In fact, hematopoietic stem cells can activate NF-κB in response to TNFα, and in these cells NF-κB acts as a pro-survival factor 48. The role of NF-κB in early lymphocyte development seems clear – expression of either pre-T-cell receptor (pre-TCR) 49 or pre-BCR (pre-B-cell receptor) coincides with increasing NF-κB activity and induction of anti-apoptotic signals through NF-κB 50, 51. However, it remains unclear how NF-κB is activated downstream of the pre-AgR as the nature of the signaling pathway at play remains almost entirely uncharacterized. Whether NF-κB contributes to lineage choice made at these early stages or merely promotes survival is also unknown.
The extent of NF-κB activation serves as a rheostat in the selection of DP (double positive; CD4+CD8+) thymocytes (Figure 4). TCR-mediated NF-κB activation follows binding to peptide:major histocompatibility complex (MHC). A thymocyte that expresses a TCR that cannot bind MHC succumbs to “death by neglect”, whereas those that bind peptide:MHC are either positively or negatively selected depending on the strength of signaling. Thymocytes that bind self-peptide:MHC with very high affinity, are likely to be self-reactive, and hence are deleted through negative selection. Thus, only DP thymocytes that recognize self-peptide:MHC and signal within a defined range are positively selected and become single-positive (SP) T cells. During negative selection, NF-κB facilitates the induction of apoptosis following high affinity TCR ligation 52, perhaps by facilitating expression of pro-apoptotic genes and consequent sensitization to pro-apoptotic signals 53. The role of NF-κB in positive selection of thymocytes is more in keeping with the better-established role of NF-κB as an inducer of anti-apoptotic genes. Unlike in thymocytes, however, NF-κB functions as a pro-survival factor during negative selection of B cells. Immature B cells display constitutive NF-κB activity that is downregulated following BCR ligation 54 (Figure 4). Decreased NF-κB activity might then sensitize these cells to pro-apoptotic signals. Interestingly, some signaling components required for NF-κB activation in mature B and T cells can be genetically disrupted without affecting their development, suggesting that pathways leading to activation of NF-κB in developing B or T cells differ significantly from the pathways engaged following AgR ligation in mature lymphocytes.
Following positive and negative selection, DP thymocytes must make a lineage commitment as SP thymocytes (CD4+CD8− or CD4−CD8+) and thereafter emigrate from the thymus. This process requires NF-κB as deletion of NEMO in cd4-Cre mice results in loss of mature peripheral T cells 55. Equivalent deletion of the upstream kinase TAK1 has a similar outcome 56. The exact nature of NF-κB pathway requirement is somewhat unclear, as ikkb−/− chimeras, or cd4-Cre IKKβ conditional knockouts, are not defective in the production of naive T cells 55, 57. Further, the contribution of NF-κB to CD8 and CD4 lineages is not equivalent. CD8 SP cells have significantly higher levels of NF-κB activity than CD4 SP thymocytes 49, 58, yet the anti-apoptotic factor Bcl-2 is more highly expressed in CD4 than CD8 cells. Therefore, CD8 SP thymocytes are dependent on NF-κB for survival, while CD4 SP thymocytes are not 58. NF-κB is, however, clearly important in CD4 SP cell development, and forced activation of NF-κB in CD4 SP cells results in negative selection 58.
In addition to peripheral maturation and differentiation of T cells into various subsets, thymic differentiation into both TH17 cells and regulatory T cells (Tregs) is known to occur. The transcriptional control of TH17 cell differentiation and the role of thymic TH17 remain relatively obscure. One recent report has described an important role for IκBζ in TH17 differentiation 59. In this capacity, IκBζ acts with the nuclear orphan receptors RORγ and RORα to promote IL17A gene expression. Beyond this tangential role in TH17 development, a TH17-intrinsic role for NF-κB has been minimally interrogated. Instead the role of NF-κB in Treg development 60 is somewhat better understood. The differentiation of thymocytes into Foxp3/CD25-positive Tregs occurs in the thymus and depends on NF-κB. It is thought that NF-κB acts as a pioneer transcription factor enhancing accessibility of the foxp3 locus 61, 62, 63, 64.
Immature B cells complete development into either follicular or marginal zone B cells after exiting the bone marrow as transitional B cells. NF-κB-regulated expression of pro-survival factors is important to these final steps of B-cell development 65. Signaling through the TNFR superfamily member BAFFR is crucial for transitional B-cell survival through induction of anti-apoptotic Bcl-2 family members 66, 67, 68. Thus, BAFF-knockout mice exhibit a complete failure of transitional B-cell maturation, which mirrors that seen in Bcl-XL-knockout mice 66, 69, 70. BAFF activates both non-canonical and canonical NF-κB pathways, though the former is principally responsible for the anti-apoptotic function in transitional B cells. Nevertheless, deficiency in NEMO, IKKα, or IKKβ decreases the numbers of mature B cells 57, 71, 72. Likewise, p50/p52 or p65/c-Rel double-knockout progenitor cells are defective in their ability to mature beyond the transitional B-cell stage 47, 73. A requirement for both the canonical and non-canonical NF-κB pathways may explain why deletion of p50 and p52 produces a more complete block in B-cell development than loss of p65 and c-Rel. Thus, only those knockouts that target both the canonical and non-canonical NF-κB pathways have an effect that approximates the phenotype seen in BAFF or Bcl-XL deficiency. Constitutive hyperactivation of the canonical pathway can circumvent BAFF-R deletion 74, yet it remains unclear whether the canonical pathway merely supports the non-canonical pathway 75, or has an independent function.
NF-κB in innate immunity
Pattern recognition receptors
Recognizing pathogens is an absolute requirement for the initiation of effector functions. Mammals express several diverse classes of pattern recognition receptors (PRRs) that are dedicated to this purpose. These PPRs include transmembrane Toll-like receptors (TLRs), cytoplasmic Nod-like receptors (NLR) and RIG-I like receptors (RLR), scavenger receptors, C-type lectins, and the complement system. Although epithelial cells are frequently the first to encounter pathogens, they are also constantly exposed to non-pathogenic microbes. Therefore, while a variety of PRRs are differentially expressed in epidermis, gut, pulmonary, urinary, and reproductive epithelium, their expression and responsiveness is tightly controlled. Instead sentinel cells of the innate immune system, particularly tissue resident DCs and macrophages, express a more complete complement of PRRs, and exhibit a higher propensity to signal on exposure to pathogens.
TLRs are evolutionarily conserved pattern recognition receptors that recognize unique, essential molecules characteristic of various classes of microbes 76. The 11 characterized mammalian TLRs have varied tissue distribution and serve as recognition receptors for pathogen-associated molecular patterns (PAMPs) present on bacteria, viruses, fungi, and parasites. Perhaps because of the modular nature of the TLR extracellular domain, which consists of multiple leucine-rich repeats (LRRs), these receptors are capable of recognizing more than one microbial molecule (Figure 5). Heterodimerization of TLRs further expands the repertoire of recognized molecules. In general, the ability of TLRs to distinguish between pathogen types is translated into appropriate innate and adaptive responses through the selective activation of NF-κB, IRFs, and other inducible transcription factors. TLR signaling pathways are complex and have been extensively reviewed elsewhere 2, 77, 78. In general, TLR signaling is often divided into MyD88-dependent and TRIF-dependent pathways: MyD88 signaling predominantly leads to the activation of NF-κB, while TRIF signaling leads to both IRF3 and, to a lesser extent, NF-κB activation.
Pattern recognition receptors and their cognate ligands. TLRs 3, 7, 8, 9 and 11 have been reported to exhibit endosomal or intracellular localization while NOD1, NOD2, RIG-I, MDA-5, NALP1, NALP3, NLRC4, and the intracellular DNA sensor (ISD) function in the cytoplasm. Only a partial list of ligands or classes of ligands for each receptor is given.
In recent years, there has been significant activity in characterization of cytoplasmic PRRs, particularly those recognizing viral nucleic acids. These efforts followed the initial description of nucleotide oligomerization domain proteins (NOD), NOD1 and NOD2. NLRs, of which NOD1 and NOD2 are the best-known examples, are characterized by LRRs and NODs. NOD1, NOD2, and IPAF have CARD domains and can signal to NF-κB (see Blonska and Lin, Cell Res 2011; 21:55-70). NOD1 recognizes a peptidoglycan containing meso-diaminopimelic acid and induces NF-κB through a canonical pathway that includes activation of IKKβ. NOD2 recognizes muramyl dipeptide (MDP), a ubiquitous component of nearly all bacterial cell walls. The CARD-containing kinase RIP2 is required for NF-κB activation. The function of RIP2 tyrosine kinase activity is unclear, although it was recently shown that kinase inhibitors could prevent MDP-induced cytokine responses 79. Although RIP2 binds to NEMO, it is not thought that RIP2 tyrosine kinase activity mediates IKK phosphorylation directly. Instead it has been proposed that RIP2 directly mediates activation of the IKK complex through NEMO binding and induced proximity 80.
Retinoic acid inducible gene I (RIG-I) and melanoma differentiation-associated gene 5 (MDA5) are DExD/H-box RNA helicase-containing cytoplasmic proteins that bind directly to dsRNA 81, 82, 83. The details of RNA binding to RIG-I and MDA5 continue to be the subject of intense research. Thus, it was thought that RIG-I recognizes dsRNA replicative intermediates of negative stranded single-stranded RNA (ssRNA) viruses, while MDA5 responds to long dsRNA replicative intermediates of plus strand ssRNA viruses 84, 85, 86, 87. Indeed, the structure of RIG-I bound to short dsRNA products has recently been solved 88, 89. RIG-I has also recently been implicated in the recognition of DNA viruses by binding short dsRNAs generated by RNA polIII 90. However, it has been shown that RIG-I can directly recognize ssRNA genomes containing 5′ triphosphates, and suggested that for negative stranded RNA viruses, this may be the relevant PAMP 91. On binding to either the viral genome, viral replication intermediate, or RNA product, RIG-I, and MDA5 induce the activation of IRF3 and NF-κB. The CARD-containing protein MAVS (also known as CARDIF, IPS1, and VISA) mediates signaling from RIG-I 92, 93, 94, 95. RIG-I and MDA5 differentially recognize various groups of RNA viruses and are independently required for robust antiviral responses 86. Therefore, together with TLRs, RLRs provide an additional mechanism of sensing viral infection and mediating type-I IFN (IFN-α/β) responses.
Pathogen recognition in innate immunity
Pathogens recognized by PRRs can be categorized as bacterial, viral, or eukaryotic, and each of these categories has several well-described PAMPs 96. Both in terms of accessibility and uniqueness to prokaryotes, the bacterial cell well is the most abundant source of PAMPs for TLRs and other PRRs. Lipoproteins, glycolipids, and protein components of bacterial cell wells have all been shown to function as PAMPs. The TLR4 ligand LPS, a glycolipid component of the outer membrane of Gram-negative bacteria, is the most thoroughly studied PAMP and the most potent TLR ligand known. Trace amounts of LPS activates the innate immune system via TLR4, leading to the production of numerous proinflammatory mediators, such as TNFα, IL-1, and IL-6. TLR4-mediated responses to LPS require CD14 and MD-2.
Conserved differences in bacterial nucleic acid structures can also be recognized by the innate immune system. TLR9 recognizes bacterial DNA containing unmethylated CpG motifs, and TLR9-deficient mice are not responsive to CpG DNA challenge 97. The low frequency and high rate of methylation of CpG motifs prevent recognition of mammalian DNA by TLR9 under physiological circumstances. A recent report indicated that the intracellular, endosomal restriction of TLR9 is critical for discriminating between self and non-self DNA, as host DNA, unlike microbial DNA, does not usually enter the endosomal compartment 98.
Nucleic acids are also key viral PAMPs, and are recognized by TLRs 3, 7, 8, and 9, as well as by RLRs. Signaling from viral PAMPs results in the activation of NF-κB and IRFs, which cooperatively mediate the production of IFN-α/β 99, 100. TLR3 recognizes dsRNA, a common viral replicative intermediate 101. TLR7 and TLR8, initially found to recognize synthetic antiviral compounds 102, 103, recognize guanosine- or uridine-rich ssRNA derived from RNA viruses 104, 105, 106. Interestingly, mammalian RNA is significantly less stimulatory than bacterial RNA, suggesting that nucleoside modifications facilitate discrimination between endogenous and pathogen RNA 107. In addition, subcellular localization of TLR7 and TLR8 to endocytic compartments, and limited constitutive cell type expression, may also facilitate self/non-self discrimination by minimizing exposure to host RNA. TLR9 recognizes viral CpG sequences and induces the induction of IFN-α 108, 109, 110. However, as membrane restriction prevents TLRs from sampling the cytosol where much of the viral lifecycle occurs, cytosolic PRRs provide comprehensive innate immune recognition. Thus, TLR3 and the adaptor TRIF are not required for viral induction of type I IFN in many tissues and cell types 111, although specialized innate antiviral cells, such as plasmacytoid DCs, rely more heavily on TLR mediated recognition of viral nucleic acids 112. Recognition of cytoplasmic dsDNA leading to NF-κB activation and type I interferon production has also been reported 113, 114. Although several candidates have been found, the relevant receptor has not yet been conclusively identified 115. This receptor(s) is predicted to be important for type I IFN production in response to viruses and intracellular pathogens, such as Listeria sp.
Although research on the recognition of PAMP/PRR pairs is an ongoing process of discovery, defective responses of MyD88-deficient cells indicate that many fungal and parasite species are capable of activating TLR pathways. TLR4 has been shown to recognize Aspergillus hyphae 116, and Cryptococcus neoformans capsular polysaccharide 117. TLR2 and TLR6 are required for recognition of yeast zymosan, while TLR4 is thought to recognize certain yeast mannans. The identification of parasite PAMPs has been more elusive, and their existence is somewhat controversial. Nevertheless, TLR2 heterodimers recognize various parasite GPI-anchored proteins and glycoinositolphospholipids from the parasitic protozoa Trypanosoma cruzi 118. TLR9 was initially reported to recognize the malarial pigment hemozoin, a byproduct of heme metabolism in infected erythrocytes 119, although subsequent studies suggest that DNA associated with hemozoin is the relevant TLR9 ligand 120. Instead, hemozoin may be recognized by the NLRP3 inflammasome 121. TLR11 recognizes a profilin-like protein that is conserved in apicomplexan parasites including Toxoplasma gondii 122. Recognition of helminths remains an area of controversy 123. In particular, it remains unclear how helminth PAMPs might preferentially induce TH2 responses that are characteristic of the immune responses to this class of pathogen. Direct skewing of adaptive responses by the mechanism of innate recognition is suggested to occur in other scenerios as well 124. For example, C-type lectins can directly affect TH activation and differentiantion 125, thus recognition of C. albicans α-mannans by dectin-2 induces a protective TH17-predominant response 126. Therefore, whether through cell type or PRR specificity, the recognition of specific PAMPS can shape the nature of the subsequent innate and adaptive immune responses.
Immediate anti-microbial responses
PAMP recognition initiates a complex series of events: the first is the mounting of immediate antimicrobial responses at the cellular level. This effective and evolutionarily conserved function of PRRs is dependent on activation of NF-κB gene programs. In particular, much of the early innate response has been demonstrated to depend on the canonical NF-κB pathway. Thus, rela−/−/ tnfr1−/− and ikbkb−/−/tnfr1−/− double-knockout mice have increased susceptibility to bacterial infection 57, 127, 128, 129. MEFs from Nemo-deficent mice do not exhibit NF-κB activation by LPS or IL-1 130. Therefore, activation of NF-κB responsive genes by the innate immune system depends on NEMO and likely progresses through the canonical NF-κB signaling pathway. Early anti-microbial effectors include NF-κB-dependent expression of defensins, which are cationic peptides that exert direct bactericidal activity by inducing membrane permeabilization. The classic mediators of defensin release are small intestinal Paneth cells, which secrete α-defensins into the intestinal lumen following exposure to bacterial PAMPs 131. The production of anti-microbial nitrogen and oxygen species, which are acutely toxic to a variety of microbes, augments the activity of anti-microbial peptides. Production of nitric oxide (NO) is mediated in part by inducible NO synthase (iNOS), which is coordinately regulated by NF-κB and STAT transcription factors 132.
Inflammation
Inflammation begins with epithelial or stromal cells of the infected tissue or tissue resident hematopoietic cells such as mast cells or DCs recognizing an inflammatory stimulus and propagating pro-inflammatory signals. These signals lead to the recruitment and activation of effector cells, initially neutrophils and later macrophages and other leukocytes, resulting in the tissue changes characteristic of inflammation – rubor, calor, dolor, and tumor (redness, heat, pain, and swelling, respectively). There is a vast amount of literature that correlates NF-κB activation with inflammation in a wide array of diseases and animal models. There are, likewise, numerous studies using gene targeting and inhibitors of NF-κB that have established the causative role of NF-κB in inflammatory processes.
NF-κB is responsible for the transcription of the genes encoding many pro-inflammatory cytokines and chemokines. The pathway from pathogen recognition to pro-inflammatory cytokine production demonstrates a particular reliance on NF-κB. The immediate targets of NF-κB-dependent pro-inflammatory cytokines, such as TNFα, tend to be receptors that, in turn, activate NF-κB. Therefore, NF-κB is crucial to the propagation and elaboration of cytokine responses. TNFα is particularly important for both local and systemic inflammation, and it is a potent and well-studied inducer of NF-κB. One important early target of these effectors is the vascular endothelium. Changes in vascular endothelial cells both recruit circulating leukocytes and provide them with a means of exiting the vasculature into the infected tissue. NF-κB regulates the expression of adhesion molecules, both on leukocytes and on endothelial cells, which allow the extravasation of leukocytes from the circulation to the site of infection 133. Thus, in the absence of p65, the recruitment of circulating leukocytes to sites of inflammation is impaired 127.
Recruited neutrophils are the key mediators of local inflammation and, as mentioned above, NF-κB is important for their survival and function under relatively toxic conditions 134. NF-κB is important for the production of the enzymes that generate prostaglandins and reactive oxygen species (e.g., iNOS and Cox, both NF-κB target genes) and may, furthermore, be involved in the signaling induced by prostaglandins 135, 136. NF-κB has also been implicated in the response to leukotrienes, which similar to prostaglandins are short-lived paracrine effectors. For example, leukotriene-induced IL-8 production appears to be dependent on rapid activation of NF-κB 137. Finally, matrix metalloporteinases are also crucial mediators of local inflammation and leukocyte chemotaxtis, and their expression is also regulated by NF-κB 138, 139, 140.
Resolution of inflammation and subsequent tissue repair is a crucial event, and its failure is a common source of pathology. Resolution of inflammation is an active process that is as complex as the inflammatory response itself, and involves numerous pathways that are not all directly relevant to NF-κB 141. Macrophages are key regulators of both inflammation and its resolution, and NF-κB plays a key role in the instruction of macrophage responses. Macrophage differentiation into pro-inflammatory (M1) or anti-inflammatory (M2) cells depends both on cell intrinsic NF-κB pathway activation as well as the local cytokine milieu, which is strongly influenced by NF-κB.
During acute inflammation, there are multiple negative feedback pathways that help to rein in inflammatory responses 3. It has long been known that cells, such as macrophages, become resistant to repeated pro-inflammatory stimuli 142. The IκB family protein Bcl-3 can function both as an inhibitor and mediator of NF-κB transcriptional programs 2. Following LPS stimulation, Bcl-3 is induced and binds p50 dimers, with which it can repress the expression of a subset of NF-κB target genes. In this manner, Bcl-3 mediates the selective inhibition of repeated LPS responses in macrophages 143. Although clearly not the only mechanism, induced transcriptional repression by Bcl-3/p50 likely contributes to LPS-tolerance. Furthermore by selectively affecting chromatin remodeling, Bcl-3 mediates repression of pro-inflammatory genes, and also facilitates the expression of the anti-inflammatory gene IL-10. Thus, it appears that Bcl-3 acts appropriately in the regulation of genes that are categorized as either “tolerizable or “non-tolerizable” 144. NF-κB p50 appears to be capable of mediating anti-inflammatory responses under several additional circumstances as well. For example, p50 negatively regulates IFNγ production by and proliferation of NK cells 145. Also, expression levels of p50 in M2-like tumor associated macrophages (TAMs) regulate the balance between anti- and pro-inflammatory functions. Thus, it was shown that TAMs overexpress p50 and that deletion of p50 renders these macrophages pro-inflammatory 146.
Inhibition of NF-κB during the resolution phase of inflammation can prolong the inflammatory process and prevent proper tissue repair 147. Furthermore, IKKα-deficient mice display increased inflammatory responses in models of local and systemic inflammation 148, and show increased production of pro-inflammatory chemokines and cytokines 148, 149. Rather unexpectedly, IKKα-deficient mice and embryonic-liver-derived macrophages from IKKα-deficient mice exhibit augmented inflammatory responses compared with wild-type mice 149. The mechanisms of IKKα-mediated repression of transcriptional responses seem to be through effects on the level of nuclear p65 and c-Rel 148. Although there are significant differences between these two reports with regard to the proposed mechanism of action, both, nevertheless, observe the same biological consequence of IKKα deficiency in macrophages. Furthermore, recent work has suggested that IKKβ also exhibits some anti-inflammatory capacity in macrophages. First, IKKβ suppresses the secretion of the potent pro-inflammatory cytokine IL-1β 150. Second, IKKβ is associated with the maintenance of an anti-inflammatory M2-like phenotype in TAMs 151. Third, IKKβ is thought to inhibit M1 macrophage function by interfering with the STAT pathway during infection 152.
Initiation of adaptive responses
The innate immune system invokes potent anti-microbial activities and maintains host protection under many settings. Nevertheless, initiating the adaptive immune response remains a crucial step for robust and durable pathogen clearance. The process of information transfer from the innate to adaptive immune system is mediated by activation and maturation of APCs. The nature of the PAMP and the signaling pathways activated by the cognate PRR, as well as the context within which pathogen recognition occurs, all influence the manner in which the APC instructs T and B cell responses.
DC maturation mediated by pathogen recognition is crucial for the initiation of the adaptive immune response. To activate naive T cells, DCs must alter their chemokine receptor expression to enable migration into lymphoid tissues. During activation DCs fine-tune their antigen processing machinery such that the presentation of pathogen epitopes is favored. Finally, the expression of co-stimulatory molecules including B7.1/B7.2 (or CD80/CD86) is upregulated. These co-stimulatory molecules ligate the co-stimulatory molecule CD28, thus providing the second signal necessary to induce full T-cell activation. The response to pathogens is tailored on the basis of the distribution of PRRs in different cell types, and the ability of different cell types to, in turn, interact with T cells in a biasing manner 153.
There continues to be debate surrounding how PRRs differentially induce TH1 versus TH2 responses. Maturation of DCs following viral infection depends on nucleic acid-binding PRRs 154, 155. Plasmacytoid DCs, which are robust inducers of interferon during viral infection, express viral PRRs including elevated expression of TLR7 and TLR9. Cytoplasmic nucleic acid-sensing PRRs, RLRs, are expressed more broadly, although they are further induced by type-I interferons, and they may, therefore, provide crucial amplification of antiviral responses following virus recognition by TLRs. While studies of anti-viral responses suggest that both cell type-specific distribution of PRRs, and selective activation of signaling pathways by certain PAMPs are key determinants of TH1 versus TH2 responses, the situation is less clear for other pathogens. It appears that multiple mechanisms converge in shaping how APCs interact with the adaptive immune system.
Role of NF-κB in the adaptive response
T-cell responses mediated by NF-κB
T-cell activation, differentiation, proliferation, and effector function are all influenced by gene programs regulated by NF-κB. The majority of effort directed at understanding the role of NF-κB in T cell activation has focused on CD4+ T cells (Figures 6 and and7).7). On the whole, the historical lack of CD8+ T-cell conditional knockouts and the selective loss of CD8 cells in the absence of functional NF-κB have impeded thorough characterization of the role of NF-κB in these cells. Nevertheless, there continues to be progress in this area and, in fact, there has been a resurgence of interest in recent years. CD4+ T cells have long been known to undergo differentiation into either TH1 or TH2 subsets depending on the cytokines present during activation 156. However, an appreciation of the ability of CD4+ helper T cells to differentiate into additional effector cell types, induced regulatory T cells, pro-inflammatory TH17 cells, TH22 cells TH9 cells, has prompted a fresh look at the contribution of NF-κB to T cell differentiation and effector function (Figure 7).
NF-κB in T cell activation. NF-κB participates in the maintenance, activation, differentiation and proliferation of naive T cells. Tonic TCR stimulation promotes T cell maintenance, of both memory and naive cells, through NF-κB activation. Activation occurs when the naive T cell recognizes its cognate antigen presented by an activated APC expressing both peptide:MHC and B7 family co-stimulatory molecules. NF-κB-dependent proliferation and differentiation ensue and are influenced by the local cytokine milieu. NF-κB supports proliferation, differentiation and survival as indicated (green arrows).
TH cell differentiation. NF-κB participates in differentiation of several TH cell types following activation of naive CD4+ TH cells. Differentiation pathways in which NF-κB is implicated are indicated with a green arrow. Key transcription factors in each differentiation pathway are indicated above each arrow, while cytokine responsible for skewing TH cells toward a given pathway are indicated below each arrow. Additional cytokines and transcription factors implicated in several of the pathways are not depicted.
Activation of naive T cells requires antigen-specific and co-stimulatory signaling provided by activated APCs. Binding of the TCR to its cognate peptide presented in the binding cleft of MHC supplies the antigen-specific signal, while co-stimulatory signaling results from ligation of CD28 by B7 molecules expressed on activated APCs. Activated naive T-cells proliferate rapidly while simultaneously differentiating into effector cells (Figure 6). In the case of CD4+ T cells, proliferation leads to differentiation into immature effector cells, TH0, which subsequently differentiate into TH1, TH2, TH9, TH17, TH22, or inducible Treg (iTreg) cells depending on the cytokine milieu.
Stimulation of T cells through TCR and CD28 results, initially, in activation of p65-containing NF-κB complexes followed by delayed and sustained activation of c-Rel complexes 157, 158, 159, 160. Rapidly proliferating activated T cells rely on NF-κB activity for protection from apoptosis as well as for the production of cytokines, especially IL-2, supporting proliferation and differentiation (Figure 6). As expected, inhibition of NF-κB in activated T cells facilitates progression toward activation-induced cell death (AICD) or apoptosis 161, 162, and stimulation of p65-deficient naive T cells induces cell death 163. In contrast, T cells lacking c-Rel do not undergo apoptosis, but nevertheless, fail to proliferate in response to typical mitogenic stimuli 164. T cells, in which p105 cannot undergo inducible processing to p50 also fail to proliferate normally 165. In vitro, c-Rel is needed for production of IL-2 and therefore, for proliferation, but this need is not recapitulated in vivo 166. Interestingly, c-Rel-deficient T cells appear to have a defect in TH1 proliferation and production of IFNγ, indicating a selective role for NF-κB family members in TH1/TH2 differentiation, independent of that mediated by the innate response.
Multiple transcriptional activators and repressors regulate expression of IL-2. Amongst these, members of the NF-κB family play multiple roles. In naive T cells, which do not express IL-2, repressive p50 homodimers are found associated with the IL-2 promoter 167. Failure of T-cell proliferative responses in c-Rel-knockout mice is attributable to a failure to produce IL-2 164. In naive T cells, c-Rel is responsible for mediating chromatin remodeling across the IL-2 locus following CD3/CD28 co-stimulation 158. Thus, naive T cells can be primed by activation of c-Rel by pro-inflammatory cytokines, such as those elicited following stimulation with TLR ligands 157. Priming, which requires NF-κB activation, results in a more robust response to CD3/CD28 co-stimulation 168. Conversely, overexpression of p65 with c-Jun can overcome the requirement for co-stimulation in naive T cells 169.
TH0 differentiation into TH1, TH2, TH9, TH17, Treg effector cells depends on the induction of specific transcription factors, namely T-bet, GATA3, PU.1, RORγt, and Foxp3, respectively (Figure 7). There is increasing evidence that NF-κB family members are key arbitrators of the decision. For example, mice lacking p50 are unable to mount an asthma-like, airway TH2 response 170. Indeed, p50-deficient T cells fail to induce GATA3 expression during T-cell stimulation under TH2 differentiating conditions. These results suggest a transcriptional role for p50, and it was subsequently found that Bcl-3-deficient T cells also fail to undergo TH2 differentiation, suggesting that p50/Bcl-3 complexes are crucial for TH2 differentiation 171. Conversely, the same authors found that RelB-deficient T cells are deficient in TH1 differentiation due to decreased expression of T-bet. RelB may mediate upregulation of STAT4, which is responsible for T-bet induction downstream of IFNγ. Therefore, it appears that NF-κB activation during TCR stimulation may render cells competent for both proliferative and differentiating stimuli. As a corollary, NF-κB transactivation of the IL-2 gene is repressed as TH cells differentiate into TH1 or TH2 and become dependent on TH1 and TH2 cytokines (e.g., IFNγ and IL-4). It is thought that direct binding of T-bet to p65 that is associated with the IL-2 gene enhancer, may mediate the repression of IL-2 production in TH1 cells 172. In TH2 cells the lack of IL-2 transcription may be because of the decreased levels of p65 activation 173. As discussed above, several groups recently implicated NF-κB, and specifically c-Rel, in the regulation of Foxp3 expression and Treg development 61, 62, 63, 174. The relative contribution of c-Rel to iTreg development still requires further clarification. While a recent report demonstrated that IκBζ cooperates with ROR and is required for TH17 differentiation 59, the more general contribution of NF-κB in this process, either in the thymus or periphery, remains unclear. While the role of NF-κB in TH9 cells has yet to be interrogated, there are interesting clues suggesting an important role for NF-κB. Recently, PU.1 was reported to be the transcription factor responsible for TH9 differentiation 175. In unrelated recent work on acute myelogenous leukemia, it was noted that NF-κB regulates the transcription of PU.1 176. Furthermore, it has been suggested that IL-9 expression in T cells is regulated by NF-κB 177. Thus, it would seem reasonable to speculate that NF-κB may play an important role in TH9 differentiation (Figure 7). The transcription factors responsible for the differentiation of TH22 cells have not yet been identified. However, the possible role of TNFα as a TH22 differentiating cytokine is suggestive that NF-κB may be an important mediator of this differentiation process as well. In summary, NF-κB participates in both the initial T cell responses by supporting proliferation and regulating apoptosis, and is increasingly being shown to have an important role in the regulation of TH differentiation.
B-cell responses mediated by NF-κB
There are many parallels in the functions of NF-κB in T and B cells. NF-κB acts to support proliferation, regulate apoptosis, and control the processes of differentiation and maturation in B cells (Figure 8). B-cell responses are classified into two groups: thymus-dependent (TD) or thymus-independent (TI). In TD response follicular B cells require co-stimulatory signaling from TH cells expressing CD40L and cytokines, such as IL-4. These initial steps trigger germinal center formation, in which somatic hypermutation, isotype switching, and plasma cell differentiation occur. Thus, in individuals with a mutation in CD40L B cells fail to undergo class switch recombination in response to T-dependent antigens 178. Signaling through CD40 activates both canonical and non-canonical NF-κB pathways, although several lines of evidence suggest that the non-canonical pathway is not required in the response to TD antigens. Thus, B cells lacking p52 are fully capable of appropriate TD antigen responses when transferred 21, and B cells from relB−/− mice, although crippled in their proliferative response, undergo normal IgM secretion and class switching 179. In contrast to these results, B cells lacking either functional NIK or IKKα, exhibit defective responses 180, 181. It remains unclear whether the role of NIK and IKKα in this context is to induce p100 processing (non-canonical pathway) or to contribute to activation of the canonical pathway. Although both pathways are triggered by CD40 ligation, the current data suggest that canonical pathway activation is of great importance.
NF-κB in B cell activation. NF-κB participates in the maintenance, activation, differentiation and proliferation of naive B cells. Tonic BCR and cytokine, BAFF, stimulation promotes naive B cell maintenance through NF-κB activation. Activation occurs when the naïve B cell recognizes its cognate antigen and receives co-stimulatory signaling (CD40L) from an activated TH cell within the germinal center. NF-κB-dependent proliferation and differentiation ensue and are coupled to BCR affinity maturation. NF-κB signaling in B cells expressing selected BCRs results in class switch recombination and differentiation into either memory B cells or plasma cells. NF-κB supports proliferation, differentiation and survival as indicated (green arrows).
Evidence also supports a role for the canonical pathway in class switch recombination. In contrast to B cells lacking RelB or p52, those from rela−/− mice exhibit markedly diminished class switching, despite a modest loss of lymphocyte proliferation following various stimuli 182. Likewise, c-Rel-deficient mice fail to generate protective humoral immune responses demonstrating the requirement for c-Rel in class switch recombination 164, 183, 184. B cells from nfbk1−/− mice exhibit decreased proliferation in response to mitogenic stimulation, and p50/p65 double-knockout B cells exhibit greater defects in proliferation and class switching 185, 186. Therefore, B cell proliferation and the TD B-cell maturation response are dependent on the canonical NF-κB pathway.
TI antigens are those that have an intrinsic ability to initiate B-cell responses in the absence of T-cell help. TI responses are carried out by marginal zone and CD5+ B cells. TI antigens function by acting as both antigen and B-cell mitogens, for example antigens that are PRR ligands or antigens with high avidity that are capable of crosslinking the BCR through repetitive structural features. In such cases it is expected that B-cell responses are more dependent on members of the canonical pathway that have well-documented roles in TLR and BCR signaling. For example, c-Rel-deficient B cells are highly sensitive to apoptosis following BCR cross-linking 187, 188, 189. As mentioned above, p50 and p50/RelA double-knockout B cells are deficient in responses to TI stimulation. Likewise, IKKβ-deficient B cells fail to mount TI or TD responses 190. These IKKβ-deficient B cells also exhibit increased spontaneous apoptosis, supporting that NF-κB is important in survival of B cells.
NF-κB and lymphocyte longevity
Lymphocyte homeostasis reflects balanced lymphocyte turnover and lymphopoiesis. Turnover of mature lymphocytes is complex as it depends upon both activation induced and homeostatic proliferation, and cell death. Lymphocyte survival is mediated through tonic stimulation provided by both the antigen receptor and certain cytokine receptors. As such, NF-κB can be assumed to play an important role in lymphocyte homeostasis and this assumption is supported by various genetic models.
T cells can be quite long-lived and naive T cells require continued contact with MHC:self-peptides for survival. These self-peptide complexes are most likely expressed on lymphoid DCs and generate a tonic low grade TCR signal. Survival of memory cells, on the other hand, is independent of continued contact with self-peptide:MHC complexes. B cells are formed at a far higher rate than T cells and therefore, B cells also undergo a significantly higher rate of turnover. Nonetheless, B cells too require maintenance signals to achieve peripheral homeostasis. The antigen receptor on B cells provides a basal level of signaling, albeit independent of the presence of antigen, which is required for maintenance of mature B cells. Thus, deletion of the BCR from mature B cells results in a complete loss of the peripheral B cell pool 191. The assumption is that BCR provides tonic activation of the NF-κB pathway resulting in the expression of anti-apoptotic proteins. Consistent with this assumption, loss of IKKβ, NEMO, or components of the CBM complex in mature B cells also results in complete loss of peripheral B cells 71, 190, 192. Furthermore, B cells from rela−/−, nfkb1−/−, nfkb2−/−, and c-rel−/− mice all have increased sensitivity to apoptosis and/or decreased survival ex vivo 67, 188, 193. In addition to contributing directly, it is thought that low level NF-κB activation in B cells contributes to survival through upregulation of p100, which is necessary for BAFFR-induced activation of the alternative pathway 67, 75, 166.
Several studies have implicated BAFFR in this aspect of the B lymphocyte survival 194, suggesting that the non-canonical NF-κB pathway is also relevant to B-cell survival. Consistent with this idea, constitutive activation of NIK or IKKα can circumvent the requirement for B cell BAFFR 74, 195. Conversely, B cells lacking IKKα exhibit defective survival 72, 196. The relevant targets of the non-canonical pathway downstream of BAFFR are thought to be Bcl-2 family members, e.g., the anti-apoptotic factor A1 187 or Bcl-XL, which are known to be regulated by p52/RelB-containing complexes, and are required for the maintenance of mature B cells. Indeed, Bcl-XL can complement B cells with a mutant BAFFR 197. Thus, tonic BCR stimulation, through upregulation of p100, may cooperate with BAFFR ligation and alternative pathway activation, to upregulate anti-apoptotic Bcl2 family members and mediate maintenance and survival of mature B cells.
Concluding remarks
The areas covered in the current review necessarily represent a subset of those in which NF-κB is a crucial regulator of immune responses. Recent work has highlighted the importance of NF-κB in the maintenance of epithelium barriers 198; as a regulator of inflammation associated with obesity and hyperlipidemia; and as a key factor in the development of inflammatory diseases and cancer. Our understanding of the immune system continues to improve, and coordinately our recognition of the many roles played by the NF-κB family of transcription factors also grows. As one example, the expanding universe of T-cell subsets opens up a new area for the analysis of the role of NF-κB in T-cell differentiation and effector function. It will be intriguing to see how NF-κB contributes to the development of TH9, TH17, TH22, and iTreg/TH3 cells. Deciphering the role of NF-κB in the effector function of these recently described cell types should also prove to be an area of great interest, particularly given the growing evidence for dysregulation of these cell types in autoimmune and inflammatory disease. There is much that remains unknown about the contributions of NF-κB in early and non-lymphoid hematopoiesis; in responses to danger signaling and allergens; in the coordination of pathogen-specific adaptive immune responses by the innate immune system; and in the differentiation and effector responses of both lymphoid and non-lymphoid innate immune cells. Thus moving forward, as in the past, much of the work done to understand the physiological and pathophysiological role of NF-κB will continue to focus on the role of this transcription factor family in Immunobiology.
Acknowledgments
Research in the authors' laboratory was supported by grants from the National Institutes of Health (to SG).
References
- Hayden MS, Ghosh S. Signaling to NF-kappaB. Genes Dev. 2004;18:2195–2224. [PubMed] [Google Scholar]
- Hayden MS, West AP, Ghosh S. NF-kappaB and the immune response. Oncogene. 2006;25:6758–6780. [PubMed] [Google Scholar]
- Ghosh S, Hayden MS. New regulators of NF-kappaB in inflammation. Nat Rev Immunol. 2008;8:837–848. [PubMed] [Google Scholar]
- Burkly L, Hession C, Ogata L, et al. Expression of relB is required for the development of thymic medulla and dendritic cells. Nature. 1995;373:531–536. [PubMed] [Google Scholar]
- Irla M, Hugues S, Gill J, et al. Autoantigen-specific interactions with CD4+ thymocytes control mature medullary thymic epithelial cell cellularity. Immunity. 2008;29:451–463. [PubMed] [Google Scholar]
- Hikosaka Y, Nitta T, Ohigashi I, et al. The cytokine RANKL produced by positively selected thymocytes fosters medullary thymic epithelial cells that express autoimmune regulator. Immunity. 2008;29:438–450. [PubMed] [Google Scholar]
- Akiyama T, Shimo Y, Yanai H, et al. The tumor necrosis factor family receptors RANK and CD40 cooperatively establish the thymic medullary microenvironment and self-tolerance. Immunity. 2008;29:423–437. [PubMed] [Google Scholar]
- Anderson G, Jenkinson EJ, Rodewald HR. A roadmap for thymic epithelial cell development. Eur J Immunol. 2009;39:1694–1699. [PubMed] [Google Scholar]
- Ruddle NH, Akirav EM. Secondary lymphoid organs: responding to genetic and environmental cues in ontogeny and the immune response. J Immunol. 2009;183:2205–2212. [PMC free article] [PubMed] [Google Scholar]
- van de Pavert SA, Mebius RE. New insights into the development of lymphoid tissues. Nat Rev Immunol. 2010;10:664–674. [PubMed] [Google Scholar]
- Alcamo E, Hacohen N, Schulte LC, Rennert PD, Hynes RO, Baltimore D. Requirement for the NF-kappaB family member RelA in the development of secondary lymphoid organs. J Exp Med. 2002;195:233–244. [PMC free article] [PubMed] [Google Scholar]
- Miyawaki S, Nakamura Y, Suzuka H, et al. A new mutation, aly, that induces a generalized lack of lymph nodes accompanied by immunodeficiency in mice. Eur J Immunol. 1994;24:429–434. [PubMed] [Google Scholar]
- Koike R, Nishimura T, Yasumizu R, et al. The splenic marginal zone is absent in alymphoplastic aly mutant mice. Eur J Immunol. 1996;26:669–675. [PubMed] [Google Scholar]
- Shinkura R, Kitada K, Matsuda F, et al. Alymphoplasia is caused by a point mutation in the mouse gene encoding Nf-kappa b-inducing kinase. Nat Genet. 1999;22:74–77. [PubMed] [Google Scholar]
- Mebius RE. Organogenesis of lymphoid tissues. Nat Rev Immunol. 2003;3:292–303. [PubMed] [Google Scholar]
- Bonizzi G, Karin M. The two NF-kappaB activation pathways and their role in innate and adaptive immunity. Trends Immunol. 2004;25:280–288. [PubMed] [Google Scholar]
- Dougall WC, Glaccum M, Charrier K, et al. RANK is essential for osteoclast and lymph node development. Genes Dev. 1999;13:2412–2424. [PMC free article] [PubMed] [Google Scholar]
- Yilmaz ZB, Weih DS, Sivakumar V, Weih F. RelB is required for Peyer's patch development: differential regulation of p52-RelB by lymphotoxin and TNF. EMBO J. 2003;22:121–130. [PMC free article] [PubMed] [Google Scholar]
- Paxian S, Merkle H, Riemann M, et al. Abnormal organogenesis of Peyer's patches in mice deficient for NF-kappaB1, NF-kappaB2, and Bcl-3. Gastroenterology. 2002;122:1853–1868. [PubMed] [Google Scholar]
- Caamano JH, Rizzo CA, Durham SK, et al. Nuclear factor (NF)-kappa B2 (p100/p52) is required for normal splenic microarchitecture and B cell-mediated immune responses. J Exp Med. 1998;187:185–196. [PMC free article] [PubMed] [Google Scholar]