
- nature
- cell research
- review
- article
Crosstalk of reactive oxygen species and NF-κB signaling
- Review
- Published: 28 December 2010
Crosstalk of reactive oxygen species and NF-κB signaling
Cell Research volume 21, pages103–115 (2011)Cite this article
34k Accesses
2084 Citations
13 Altmetric
Abstract
NF-κB proteins are a family of transcription factors that are of central importance in inflammation and immunity. NF-κB also plays important roles in other processes, including development, cell growth and survival, and proliferation, and is involved in many pathological conditions. Reactive Oxygen Species (ROS) are created by a variety of cellular processes as part of cellular signaling events. While certain NF-κB-regulated genes play a major role in regulating the amount of ROS in the cell, ROS have various inhibitory or stimulatory roles in NF-κB signaling. Here we review the regulation of ROS levels by NF-κB targets and various ways in which ROS have been proposed to impact NF-κB signaling pathways.
NF-κB 단백질은
염증과 면역에 중요한 역할을 하는
전사인자 계열의 단백질입니다.
NF-κB는 또한
발달, 세포 성장 및 생존, 증식 등 다른 과정에서도
중요한 역할을 하며
많은 병리학적 상태에 관여합니다.
활성 산소종(ROS)은
세포 신호 전달 과정의 일부로
다양한 세포 프로세스에서 생성됩니다.
특정 NF-κB 조절 유전자가
세포 내 ROS의 양을 조절하는 데 중요한 역할을 하지만,
ROS는
NF-κB 신호에서 다양한 억제 또는 자극 역할을 합니다.
여기에서는
NF-κB 표적에 의한
ROS 수준의 조절과
ROS가
NF-κB 신호 경로에 영향을 미치는 다양한 방법을 검토합니다.
Similar content being viewed by others
Death by TNF: a road to inflammation
Article 15 November 2022

Innate immune and proinflammatory signals activate the Hippo pathway via a Tak1-STRIPAK-Tao axis
Article Open access02 January 2024
Article Open access30 July 2019
NF-κB
NF-κB proteins are a family of transcription factors and are of central importance in inflammation and immunity 1, 2. NF-κB transcription factors also regulate the expression of hundreds of genes that are involved in regulating cell growth, differentiation, development, and apoptosis. The mammalian NF-κB proteins consist of five different related family members that bind as homodimers or heterodimers to 10-base pair κB sites. All of these family members have a Rel-homology (RHD) domain that is essential for DNA binding and dimerization. The three Rel members of the family, RelA (also known as p65), RelB, and cRel, have a C-terminal transcription activation domain (TAD) that serves to positively regulate gene expression. The two other mammalian NF-κB proteins are synthesized as larger p105 and p100 precursor proteins, which have C-terminal ankyrin repeats that inhibit DNA binding until partially processed by proteasome to the smaller p50 and p52 products, respectively 1, 2, 3. In addition, these proteins lack a TAD and therefore do not generally activate transcription unless paired as a heterodimer with one of the Rel proteins. All NF-κB proteins are capable of homodimerization or heterodimerization with the other NF-κB proteins with the exception of RelB, which can only form heterodimers. Though most NF-κB dimer combinations result in the regulation of similar sets of genes, the ability to interact in various homo- and hetero- dimer configurations contributes to their ability to bind with varying affinities to κB sites in distinct DNA sequences, and they thus regulate unique, as well as overlapping, gene sets.
Given this diversity, it is therefore not surprising that a substantial variety of mechanisms have arisen to regulate the DNA binding activity of various NF-κB homo- and hetero- dimers. In general, NF-κB activity is principally regulated by the IκB proteins, which, like p100 and p105, possess ankyrin repeats and are generally inhibitory of DNA binding. Three of these proteins are considered “typical” IκBs, namely IκBα, IκBβ, and IκBɛ. These proteins bind to NF-κB proteins and mask their DNA binding domains. They also possess potent nuclear export signals (NES) and generally remove NF-κB proteins from the nucleus, and are thus inhibitory in multiple ways. Two other IκBs, IκBζ and Bcl-3, are considered “atypical”. They are found in the nucleus, are inducibly expressed, bind only to p50 and p52 homodimers, and under certain circumstances may act to repress or to activate these homodimers. The activity of the typical IκBs is controlled through phosphorylation by upstream IκB kinases (IKKs).
Although there are many ways of activating NF-κB, two main signaling pathways have been described that lead to the activation of NF-κB target genes. These are referred to as the canonical (or classical) and noncanonical (or alternative) pathways 1, 2, 4, 5. These two pathways may usually be distinguished by whether the p50 product of p105 (canonical) or p52 product of p100 (noncanonical) is involved. Since p50 is frequently associated with RelA and p52 is frequently associated with RelB, the regulation of these two NF-κB heterodimers has been the most studied and they are considered the prototypical heterodimers for the canonical and noncanonical pathways, respectively.
The canonical NF-κB pathway is activated mostly by the stimulation of proinflammatory receptors, such as the TNF Receptor superfamily, the Toll-Like receptor family (TLRs), and by cytokine receptors for the Interleukins. It is also activated by genotoxic agents as well. Upstream receptors that activate the canonical signaling pathway typically activate an IKK complex consisting of IKKα and IKKβ, which are the catalytic kinases, and IKKγ (also known as NEMO), which acts as a regulatory subunit. Recruitment and activation of this IKK complex is usually dependent on various TNF Receptor Associated Factor (TRAF) family proteins and sometimes on RIP kinases. The p105 (nfκb1 gene product) is constitutively processed by the proteosome into p50, which is held inactive as a heterodimer with RelA (or c-Rel) by its interaction with the inhibitory IκB proteins. (IκBα has been the most studied). Phosphorylation of IκBα on serines 32 and 36 by the IKK complex (primarily IKKβ) targets it for ubiquitination. Subsequently the ubiqutinated IκBα is degraded by the proteosome and this unmasks the DNA binding activity of the p50/RelA heterodimer and also allows it to translocate to the nucleus where it can bind to κB sites and activate gene transcription 1, 2, 4, 5.
Noncanonical NF-κB activation is stimulated by specific TNF receptor family members that signal through the recruitment of TRAF2 and TRAF3. These include LTβR, CD40, CD27, CD30, BAFF-R, RANK, and others 6, 7, 8. The upstream kinase in the noncanonical pathway is the NF-κB-inducing kinase (NIK). Continual degradation of NIK in resting cells prevents constitutive activation of the noncanonical NF-κB pathway 9. Degradation of NIK is mediated by a complex between TRAF3, TRAF2, and cIAPs 1 and 2, which ubiquitinates NIK, targeting it for proteosomal degradation, and the degradation of TRAF2 or TRAF3 by receptor-stimulated processes prevents NIK degradation by this complex and results in NIK stability 10, 11. Stabilization of NIK results in the stimulation of downstream noncanonical signaling events.
In contrast to the canonical pathway, p100 is processed to p52 only after the noncanonical pathway is stimulated. The ankyrin-repeat inhibitory portion of unprocessed p100 therefore acts like a negative regulator by inhibiting DNA binding and nuclear localization of the NF-κB heterodimer. Thus p100 processing is a critical step in the noncanonical NF-κB signal pathway. Processing of p100 is triggered by its phosphorylation, which, unlike in canonical signaling, is dependent on an IKK complex made of homodimeric IKKα. IKKα is in turn activated by its phosphorylation by the recently stabilized NIK. The phosphorylation of p100 by IKKα on its C-terminus targets it for ubiquitination and partial degradation by the proteosome 6, 7, 12, thus freeing p52 and its hetero- or homo-dimeric partner to bind to DNA in the nucleus and affect transcription.
Reactive oxygen species (ROS)
There are many cellular sources of reactive oxygen species (ROS) within a cell (Figure 1). These sources can be broadly divided into two main categories. Firstly, there are those biological processes that release ROS as a byproduct, or a waste product, of various other necessary reactions and secondly, there are those processes that generate ROS intentionally, either in molecular synthesis or breakdown, as part of a signal transduction pathway, or as part of a cell defense mechanism.
NF-κB 단백질은 전사인자의 일종으로 염증과 면역에서 매우 중요한 역할을 합니다1, 2. 또한 NF-κB 전사인자는 세포 성장, 분화, 발달 및 세포 사멸을 조절하는 데 관여하는 수백 개의 유전자의 발현을 조절합니다. 포유류의 NF-κB 단백질은 10염기쌍 κB 부위에 호모다이머 또는 헤테로다이머로 결합하는 5개의 서로 다른 관련 계열로 구성됩니다. 이 모든 패밀리 멤버는 DNA 결합 및 이량체화에 필수적인 Rel 상동성(RHD) 도메인을 가지고 있습니다. RelA(p65라고도 함), RelB, cRel의 세 가지 Rel 계열은 유전자 발현을 긍정적으로 조절하는 역할을 하는 C-말단 전사 활성화 도메인(TAD)을 가지고 있습니다. 다른 두 포유류 NF-κB 단백질은 더 큰 p105 및 p100 전구체 단백질로 합성되며, 프로테아좀에 의해 부분적으로 처리될 때까지 DNA 결합을 억제하는 C-말단 안키린 반복을 가진 작은 p50 및 p52 생성물(각각 1, 2, 3)로 합성됩니다. 또한 이러한 단백질은 TAD가 없기 때문에 일반적으로 Rel 단백질 중 하나와 헤테로다이머로 짝을 이루지 않는 한 전사를 활성화하지 않습니다. 이종이합체만 형성할 수 있는 RelB를 제외한 모든 NF-κB 단백질은 다른 NF-κB 단백질과 동이합체화 또는 이종이합체화할 수 있습니다. 대부분의 NF-κB 이합체 조합은 유사한 유전자 집합을 조절하지만, 다양한 동이합체 및 이합체 구성으로 상호 작용하는 능력은 서로 다른 DNA 서열의 κB 부위에 다양한 친화력으로 결합하는 능력에 기여하므로 중복되는 유전자 집합뿐만 아니라 고유한 유전자 집합을 조절할 수 있습니다.
이러한 다양성을 고려할 때, 다양한 NF-κB 동형 및 이종 이합체의 DNA 결합 활성을 조절하기 위해 상당히 다양한 메커니즘이 생겨난 것은 놀라운 일이 아닙니다. 일반적으로 NF-κB 활성은 주로 p100 및 p105와 같이 안키린 반복을 가지고 있으며 일반적으로 DNA 결합을 억제하는 IκB 단백질에 의해 조절됩니다. 이 중 세 가지 단백질, 즉 IκBα, IκBβ, IκBɛ가 "전형적인" IκB로 간주됩니다. 이 단백질들은 NF-κB 단백질과 결합하여 DNA 결합 도메인을 가립니다. 또한 강력한 핵 내보내기 신호(NES)를 가지고 있으며 일반적으로 핵에서 NF-κB 단백질을 제거하므로 다양한 방식으로 억제 작용을 합니다. 다른 두 개의 IκB인 IκBζ와 Bcl-3는 "비정형"으로 간주됩니다. 이들은 핵에서 발견되고 유도적으로 발현되며 p50 및 p52 호모다이머에만 결합하며 특정 상황에서 이러한 호모다이머를 억제하거나 활성화하는 작용을 할 수 있습니다. 전형적인 IκB의 활성은 업스트림 IκB 키나아제(IKK)에 의한 인산화를 통해 조절됩니다.
NF-κB를 활성화하는 방법은 여러 가지가 있지만, NF-κB 표적 유전자의 활성화로 이어지는 두 가지 주요 신호 경로가 설명되어 있습니다. 이를 정식(또는 고전적) 및 비정식(또는 대체) 경로 1, 2, 4, 5라고 합니다. 이 두 경로는 일반적으로 p105의 p50 생성물(정식 경로) 또는 p100의 p52 생성물(비정식 경로)이 관여하는지 여부에 따라 구분할 수 있습니다. p50은 RelA와 자주 연관되고 p52는 RelB와 자주 연관되기 때문에 이 두 NF-κB 이합체의 조절이 가장 많이 연구되어 왔으며 각각 정식 경로와 비정식 경로의 대표적인 이합체로 간주됩니다.
정식 NF-κB 경로는 주로 TNF 수용체 수퍼패밀리, 톨-유사 수용체 패밀리(TLR), 인터루킨의 사이토카인 수용체와 같은 전 염증성 수용체의 자극에 의해 활성화됩니다. 또한 유전 독성 물질에 의해서도 활성화됩니다. 정식 신호 경로를 활성화하는 상류 수용체는 일반적으로 촉매 키나아제인 IKKα 및 IKKβ와 조절 서브유닛으로 작용하는 IKKγ(NEMO라고도 함)로 구성된 IKK 복합체를 활성화합니다. 이 IKK 복합체의 모집과 활성화는 일반적으로 다양한 TNF 수용체 관련 인자(TRAF) 계열 단백질과 때로는 RIP 키나제에 의존합니다. p105(nfκb1 유전자 산물)는 프로테오좀에 의해 p50으로 구성적으로 처리되며, 이는 억제성 IκB 단백질과의 상호 작용에 의해 RelA(또는 c-Rel)와 헤테로다이머로 비활성 상태로 유지됩니다. (IκBα가 가장 많이 연구되었습니다). 세린 32와 36에 있는 IκBα의 인산화는 IKK 복합체(주로 IKKβ)에 의해 유비퀴틴화를 표적으로 삼습니다. 이후 유비퀴틴화된 IκBα는 프로테오좀에 의해 분해되고 이는 p50/RelA 이합체의 DNA 결합 활성을 제거하고 핵으로 이동하여 κB 부위에 결합하여 유전자 전사 1, 2, 4, 5를 활성화할 수 있게 해줍니다.
비규범적 NF-κB 활성화는 TRAF2 및 TRAF3의 모집을 통해 신호를 보내는 특정 TNF 수용체 계열에 의해 자극됩니다. 여기에는 LTβR, CD40, CD27, CD30, BAFF-R, RANK 및 기타 6, 7, 8이 포함됩니다. 비규범적 경로의 업스트림 키나아제는 NF-κB 유도 키나아제(NIK)입니다. 휴면 세포에서 NIK가 지속적으로 분해되면 비규범적 NF-κB 경로의 구성적 활성화가 방지됩니다 9. NIK의 분해는 NIK를 유비퀴틴화하여 프로테오솜 분해를 표적으로 하는 TRAF3, TRAF2 및 cIAP 1과 2 사이의 복합체에 의해 매개되며, 수용체 자극 과정에 의한 TRAF2 또는 TRAF3의 분해는 이 복합체에 의한 NIK 분해를 방지하고 NIK 안정성을 초래합니다 10, 11. NIK의 안정화는 다운스트림 비정규 신호 이벤트의 자극을 초래합니다.
정식 경로와 달리, 비정식 경로가 자극을 받은 후에야 p100이 p52로 처리됩니다. 따라서 처리되지 않은 p100의 안키린 반복 억제 부분은 NF-κB 헤테로다이머의 DNA 결합과 핵 국소화를 억제하여 음성 조절자처럼 작용합니다. 따라서 p100 처리는 비규범적 NF-κB 신호 경로에서 중요한 단계입니다. p100의 처리는 인산화에 의해 촉발되며, 이는 정식 신호와 달리 동이합체 IKKα로 구성된 IKK 복합체에 의존합니다. IKKα는 최근 안정화된 NIK에 의한 인산화에 의해 활성화됩니다. C-말단에 있는 IKKα에 의한 p100의 인산화는 프로테오좀 6, 7, 12에 의한 유비퀴틴화 및 부분 분해를 표적으로 하여 p52와 그 헤테로 또는 호모 이합체 파트너가 핵의 DNA에 결합하여 전사에 영향을 미칠 수 있도록 합니다.
활성 산소 종(ROS)
세포 내에는 많은 세포 내 활성 산소 종(ROS)의 공급원이 있습니다(그림 1). 이러한 활성산소는 크게 두 가지 범주로 나눌 수 있습니다. 첫째, 다양한 기타 필수 반응의 부산물 또는 노폐물로 ROS를 방출하는 생물학적 과정과 둘째, 분자 합성 또는 분해 과정에서 신호 전달 경로의 일부 또는 세포 방어 메커니즘의 일부로 의도적으로 ROS를 생성하는 과정이 있습니다.
Figure 1
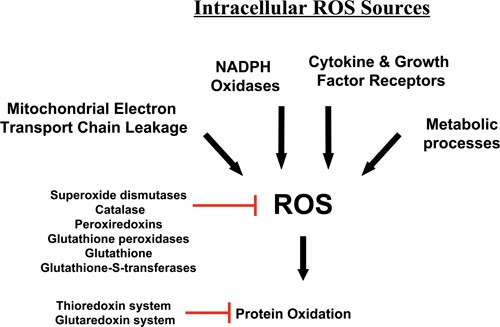
Intracellular Sources of ROS. The mitochondria are a major source of ROS, especially through electron leakage from Complexes I and III. ROS are also produced by NAD(P)H oxidases, sometimes in response to cytokines and other growth factor receptors, which may also utilize other pathways to produce ROS for use in their signaling pathways. Lastly, metabolic enzymes often create ROS as side products or through nonspecific reactions.
In the first category, the mitochondria are in a large measure the greatest source of ROS, since the reactions that occur during oxidative phosphorylation processes frequently lose electrons during their transfer between electron transport chain complexes. These electrons react with molecular oxygen to produce ROS. In consequence of this, the toxic buildup of ROS and cellular oxidation is usually alleviated by enzymes such as the superoxide dismutases, catalase, and peroxiredoxins, as well as systems of antioxidants and their associated enzymes, such as the thioredoxin and glutathione systems (Figure 1) 13, 14, 15. These systems not only serve to repair oxidative damage, but also contribute to the overall response of the cell to ROS by acting as oxidative sensors in signal transduction pathways. For instance, thioredoxin-1 oxidation has been proposed to serve in translating information on the redox state of the cell into ASK1 kinase activity through various mechanisms 16, 17, 18, 19, 20.
In the second category of ROS sources are many enzymes that generate ROS for diverse purposes, although none more robustly than the phagocytic NADPH oxidase, NOX2 (gp91), which uses NADPH to reduce molecular oxygen, thus producing superoxide 21, 22. This superoxide is typically used as a defense tool against infectious pathogens, and is converted in phagosomal compartments by superoxide dismutase and myeloperoxidase to hypochlorous acid (HOCl), which is a potent microbicidal compound 23. However, during this process, some leakage of ROS from the phagosome occurs; and they enter the cytosol, contributing to the oxidative stress of the cell. In addition to NOX2, other NADPH oxidases of this family have been characterized in many cell types 21, 22, though the family members typically produce ROS less robustly. In addition to their cell defense function, local recruitment of NADPH oxidases, including non-phagocytic oxidases, has more recently been implicated in the production of ROS by growth factors and cytokines, though some of these may produce ROS by other means 24. Consequently, these ROS are generated intentionally by the cell to function specifically within signaling pathways. Likewise, other ROS-generating enzymes, such as lipoxygenases and cyclooxygenases. create ROS to function within specific catabolic or anabolic processes, and typically produce substantially less ROS than NADPH oxidases. Thus, there are many different potential intracellular sources of ROS (Figure 1), much of which is capable of influencing, or being influenced by, NF-κB activity.
NF-κB: Protecting from ROS
As mentioned previously, ROS are toxic in cells at certain levels, due to the oxidative stress they exert by their reaction with proteins, lipids, and nucleic acids. The correct cellular response to ROS production is consequently critical in order to prevent further oxidative damage, and to maintain cell survival. However, when too much cellular damage has occurred, it is to the advantage of a multicellular organism to remove the cell for the benefit of the surrounding cells. Reactive oxygen species can therefore trigger both apoptotic and necrotic cell death depending on the severity of the oxidative stress 25, 26, 27. Although there are a few exceptions where NF-κB contributes to cell death 28, in most cases the expression of NF-κB target genes typically promotes cellular survival. Therefore it is not surprising that ROS would modulate an NF-κB response and that NF-κB target genes would attenuate ROS to promote survival. One of the main signaling pathways that intersects with NF-κB with regard to ROS and cell death is the crosstalk that occurs between NF-κB and JNK. Crosstalk from NF-κB to JNK is known to prevent sustained JNK activation and thus prevents cell death through both apoptosis and necrosis 29, 30, 31. This extensive crosstalk occurs in multiple ways, and has been reviewed elsewhere 32, 33, 34, 35, 36.
Antioxidant NF-κB targets
One of the most important ways in which NF-κB activity influences ROS levels is via increased expression of antioxidant proteins (Figure 2). Here we discuss a few of the known as well as proposed NF-κB targets that may contribute to protection from ROS.
Figure 2
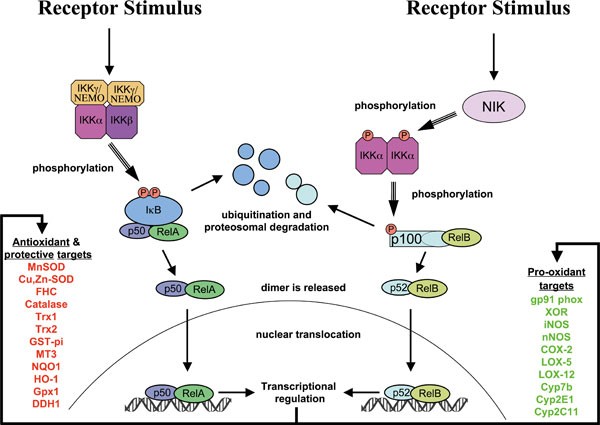
Activation of NF-κB and regulation of downstream transcriptional antioxidant and pro-oxidant targets. NF-κB is activated primarily by two pathways: the canonical pathway (shown on left) and the noncanonical pathway (shown on right). Downstream binding of the NF-κB proteins to DNA regulates downstream transcriptional targets. Shown are many potential antioxidant and pro-oxidant targets that have been proposed in the literature.
Manganese Superoxide Dismutase (MnSOD, or SOD2) is perhaps the most famous of NF-κB targets with antioxidant activity, due to numerous studies 37, 38, 39, 40. MnSOD is a mitochondrial enzyme that protects cells from oxidative stress by converting .O2− into H2O2. Mice lacking MnSOD die perinatally after birth due to massive oxidative stress 41. It is down-regulated in many oxidative diseases 42, and may be up-regulated in some cancers 43.
Likewise, its cytoplasmic relative, Copper-Zinc Superoxide Dismutase (Cu,Zn-SOD, or SOD1) has been shown to be an NF-κB target in at least one study 44. It catalyzes a similar reaction, causing the dismutation of .O2− into H2O2. SOD1-deficient mice have a shortened life span, have persistent oxidative damage and develop hepatocellular carcinoma 45.
Ferritin Heavy Chain (FHC) is the second-most well-known NF-κB target that protects from oxidative damage 46. An iron storage protein, FHC does not directly scavenge ROS, but protects the cell from oxidative damage by preventing iron-mediated generation of highly reactive .OH radicals from H2O2 (Fenton reaction). Thus FHC may synergize with MnSOD to rid the cell of ROS by preventing the generation of more highly reactive species (.O2− and .OH) and promoting the breakdown of H2O2 into water by peroxidases and catalases 47.
Little evidence exists for the regulation of Catalase by NF-κB. However, one report suggests that this is the case 48, and one other study suggests that catalase could be the target of inhibitory p50 homodimers, since its promoter is bound by p50 in unstimulated cells and catalase is down-regulated when canonical NF-κB activation occurs 49.
While the evidence is not yet overwhelming, both thioredoxins, Thioredoxin-1 (Trx1) and Thioredoxin-2 (Trx2), two of the most important cellular antioxidants in the cell, have been reported to be regulated by NF-κB 38, 39. Thioredoxins protect from oxidative stress by means of their 2-cysteine active site that reacts with ROS and is also able to reduce oxidized proteins. They also serve as hydrogen donors to the thioredoxin-dependent peroxide reductases. Trx1 is expressed in the cytoplasm and nucleus. Inactivation of Trx1 in mice also results in early embryonic lethality 50. Trx2 is localized within the mitochondria and is also indispensable for cell survival 51, 52. Deletion of Trx2 causes massive apoptosis due to the accumulation of intracellular ROS, resulting in early embryonic lethality in homozygous mice 52.
Glutathione S-transferase pi (GST-pi) is up-regulated by oxidative stress through NF-κB 53. GST-pi is a phase II enzyme that catalyzes the reaction of the GSH thiolate to toxic electrophilic compounds, thus allowing highly reactive carcinogens or radicals to be eliminated by excretion machinery 54, 55. It also is proposed to contribute to the repair of damage from oxidative stress. Disruption of the gene encoding GST-Pi in HCT116 cells showed that GSTP1 protects HCT116 cells from oxidative stress and resultant apoptosis under growth-limiting conditions 56.
Metallothionein-3 (MT3) has been shown to be an NF-κB target in keratinocytes and fibroblasts 57. Metallothioneins are low-molecular-weight, cysteine-rich proteins which bind to many different metals 58. In addition to regulating metal toxicity, the cysteine residues in metallothioneins can scavenge .O2− and .OH radicals 59.
NAD(P)H dehydrogenase [quinone] 1 (NQO1) is an NF-κB target that is activated in response to the DNA crosslinking agent mitomycin C 60. This FAD-binding protein is a cytoplasmic 2-electron reductase that reduces quinones to hydroquinones. Since it is a 2-electron reductase, its enzymatic activity prevents the one electron reduction of quinones that produces radical species 61. Interestingly, NQO1 deletion also prevents the activation of NF-κB 62.
HO-1 is a heme oxygenase that is up-regulated by NF-κB 63, 64, 65 and other transcription factors in response to oxidative stress and hypoxia. Heme oxygenase-1 catalyzes heme degradation, resulting in the formation of carbon monoxide and biliverdin, which is subsequently reduced to bilirubin by biliverdin reductase 66. Since bilirubin is a potent antioxidant, it is thought that HO-1 is therefore protective from oxidative stress.
Glutathione peroxidase-1 (Gpx1) is an abundant cytoplasmic enzyme that catalyzes the conversion of H2O2 into water using glutathione as a substrate 67. It is one of the most important members of antioxidant proteins. Although it prefers H2O2 as a substrate, it also can reduce lipid peroxides 67, as well as peroxynitrite 68. In skeletal muscles cells, glutathione peroxidase is up-regulated by NF-κB in response to oxidative stress. The glutathione peroxidase promoter is bound by all five of NF-κB subunits in U937 cells in response to LPS, signifiying an important NF-κB target 49.
Dihydrodiol dehydrogenase (DDH1 or AKR1C1) is one of many dehydrogenase enzymes regulated by NF-κB 69. DDH1 is a phase-2 aldoketo reductase and oxidizes transdihydrodiols of polycyclic aromatic hydrocarbons. Like many phase-2 enzymes that activate toxic compounds to eliminate them from the body, the reactive products downstream of its reaction have been associated with induction of ROS 70. However, ectopic expression of DDH1 has been shown to lower the basal levels of ROS in some cell types, suggesting that DDH1 can act as a protective enzyme 71.
Pro-ROS NF-κB targets
Since NF-κB is important in inflammation, some enzymes that promote the production of ROS are also regulated as its targets, especially in cells of the immune system. Below, we discuss a few involved in the generation of ROS.
During the inflammatory process, expression of the phagocytic NADPH oxidase NOX2 (gp91 phox) is dependent on, and induced by, NF-κB 72. As mentioned previously, NADPH oxidase enzymes are specifically devoted to the production of ROS. NADPH oxidases use NADPH to produce superoxide, which is used in immune defenses, and also is used for cell signaling.
An enzyme that exists in two intraconverible forms that catalyze either reduction or oxidation reactions, Xanthine Oxidase/Dehydrogenase (XOR, or Xanthine Oxidoreductase) is regulated by NF-κB 73. The dehydrogenase form is the most dominant form in vivo, however it may be converted to the oxidase form through the oxidation of its protein sulphydryl groups 74. XOR typically catalyzes the interconversion of Xanthine and Urate with NAD+ and water as cofactors. However, the enzyme has very low specificity, and transfer of electrons to O2 instead of NAD+ results in the generation of superoxide and hydrogen peroxide 75. ROS production from XOR is implicated in several pathological conditions, including heart failure 75.
The Inducible Nitric Oxide Synthase, or iNOS (NOS2) is heavily upregulated by NF-κB 76, 77, 78, 79 and Neuronal Nitric Oxide Synthase, or nNOS (NOS1) is also an NF-κB target 80, 81. While technically nitric oxide synthases actually produce a reactive nitrogen species (i.e., nitric oxide, or NO) and not reactive oxygen species, we mention them here because NO is often produced where it can react with superoxide leading to formation of the highly reactive peroxynitrite. While peroxynitrite itself is highly reactive as both an oxidant and nitrating agent, it also reacts with CO2 to form Nitrosoperoxycarbonate (ONOOCO2-), which then homolyzes to form carbonate (CO3.-) and nitrogen dioxide radicals (NO2.). Peroxynitrite can cause various kinds of cellular damage, including damage to DNA, and can activate cell death pathways 82. At low levels, peroxynitrite, and its resultant radicals may participate in signal transduction pathways, in a large part by tyrosine nitration 83. Thus the expression of nitric oxide synthases can potentiate ROS damage as well as signaling.
Cyclooxygenase-2 (COX-2, also known as Prostaglandin G/H synthase 2) is a well-known NF-κB target involved in inflammation 84, 85 that converts arachidonic acid into prostaglandin H2 (PGH2) by a free radical mechanism involving a protein tyrosyl radical generated by cooperation from a heme prosthethic group 86. During the second step of the reaction that produces PGH2, superoxide is also generated 86. Thus, superoxide is a side product of this reaction, and may contribute to oxidative stress as well as signaling.
Other enzymes, in addition to COX-2, that generate ROS during arachidonic acid metabolism have also been reported to be NF-κB targets. Among these are arachidonate 12-lipoxygenase (LOX-12, or ALOX12) 87 and arachidonate 5-lipoxygenase (LOX-5, or ALOX5) 88. As with COX-2-mediated reactions, oxidized metabolites and byproducts of these enzymatic reactions contribute to ROS within the cell 89, 90. In addition, the metabolic products of LOX-12 and LOX-5 , 12(S)- hydroxyeicosatetranoic acid and leukotriene B4, respectively, have been shown to activate and induce NADPH oxidases 91.
Cytochrome p450 enzymes, which are phase I enzymes that detoxify toxic compounds, have long been known to produce ROS when uncoupled, particularly H2O2 and hydroxyl radicals 92, 93, 94, 95. Cyp2E1, Cyp2C11, and Cyp7b are all known to have NF-κB promoter elements. Cyp2E1 and Cyp2C11 are both down-regulated by pro-inflammatory cytokines 96, 97, suggesting a negative regulation by NF-κB, while Cyp7b is up-regulated 98. Both Cyp2E1 and Cyp2C11 are known to be able to produce ROS through uncoupled reactions 95, 99, 100, 101.
Influence of ROS on NF-κB activation
Having examined some of the transcriptional targets of NF-κB that affect ROS amounts within the cells, let us now turn to the ways in which ROS affect the activity of NF-κB (Figure 3). A difficulty in defining ROS contributions to signaling is that ROS can often function in multiple places (i.e. upstream or downstream) within a given pathway, and sometimes in opposing ways (i.e. inhibitory or stimulatory). Such seems to be the case with regards to ROS functions in the NF-κB pathway. For instance ROS often stimulates the NF-κB pathway in the cytoplasm, but inhibits NF-κB activity in the nucleus 102. Thus, overexpression of the antioxidant protein TRX1 was shown to diminish NF-κB activation by inhibiting IκB degradation 103, but others have shown that TRX1 translocation to the nucleus during TNF or PMA stimulation serves to enhance NF-κB DNA binding 104, 105. ROS have been reported to both activate and to repress NF-κB signaling. While many of the differences noted in the literature are probably due to the use of different methodology, many of the differences are also attributed to the study of different upstream pathways and cell-specific differences.
Figure 3
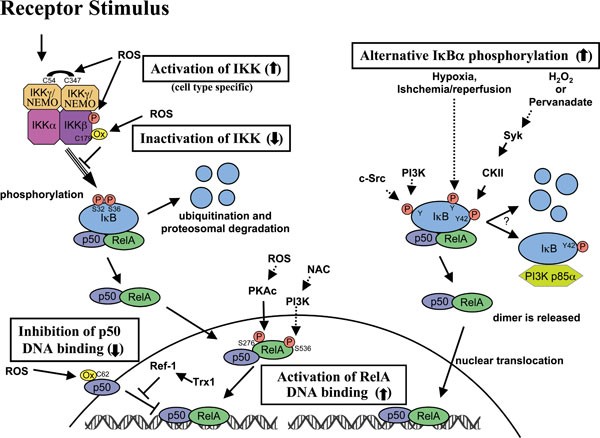
Crosstalk of ROS with NF-κB signaling pathways. ROS interacts with NF-κB at various places within the signaling pathway. Many of these interactions occur in a cell type-specific manner. ROS has been proposed to both activate and inactivate the IKK complex leading to an effect on the downstream targets. Often ROS has been shown to activate NF-κB through alternative IκBα phosphorylation, which may or may not result in the degradation of IκBα. Lastly, ROS may influence the DNA binding properties of the NF-κB proteins themselves. Oxidation of p50 on its DNA binding domain has been shown to prevent its DNA binding, and must be reversed in the nucleus by a Trx1-dependent process involving Ref-1. On the other hand, the phosphorylation of RelA that is influenced by ROS-dependent processes leads to greater NF-κB activation.
What are the targets of ROS in the NF-κB pathway? The best characterized way in which ROS affects signaling is through its reaction with cysteine, especially at an enzyme's catalytic sites, where the cysteine has a low pKa and exists in the thiolate form 106. A primary example is that of protein tyrosine phosphatases, which have been shown to be inactivated by ROS through oxidation of catalytic cysteines 107, 108, 109. ROS has also been shown to inactivate dual specificity phosphatases 110, which dephosphorylate phospholipids, in addition to both phospho-tyrosine and phospho-serine/threonine residues. In the absence of phosphatase activity, prolonged phosphorylation stimulates the activity of kinases and other enzymes within the cell.
Initial oxidation of cysteines, resulting in sulfenic acid is usually reversible by the cellular antioxidant machinery, but further oxidation to sulfinic and then to sulfonic acids results in irreversible inactivation of the phosphatases by ROS 107, 111. Sulfenic acid is usually unstable and may react with cellular glutathione to form a disulfide bond resulting in an S-glutathionated protein. While a S-glutathionated enzyme is often still inactive, it may then be reduced to its normal state by glutaredoxin in the cytoplasm. If another cysteine instead of glutathione is in close proximity to a recently oxidized cysteine in the form of sulfenic acid, an intramolecular disulfide bond can form, possibly leading to a change in protein conformation and thus preventing or initiating protein activity.
Direct regulation of NF-κB heterodimers by ROS
Direct oxidation of NF-κB by ROS inhibits its DNA binding ability 112. A specific cysteine of p50 is especially sensitive to oxidation. This cysteine, Cys-62 is in the RHD and therefore its oxidation inhibits DNA binding 113, 114, 115. This oxidation is probably followed by an S-glutathionylation event since glutathionated NF-κB has been shown to have less transcriptional activity 116, 117. This oxidation may be selectively reduced and the p50 DNA binding restored by a nuclear enzyme that is associated with base excision repair called Ref-1 (also called APE1) 118, apparently in part through its direct interaction with TRX1 104, 119. This same cysteine may likewise be S-nitrosylated by NO 120, which is produced by the iNOS protein that is up-regulated as an NF-κB target, thus acting as a negative feedback loop 121.
There are other more indirect ways that ROS influences DNA binding of NF-κB proteins. The phosphorylation of RelA on Ser-276 is required for expression of a subset of NF-κB-dependent genes 122. Phosphorylated Ser-276 is necessary for the interaction of RelA with CBP/300 123, as well as the positive transcription elongation factor b 122. PKAc mediates phosphorylation of Ser-276 123, 124, and this event is thought to be dependent on ROS based on a variety of reasons 125, among which is the finding that anti-oxidant treatment inhibits Ser-276 phosphorylation and CBP/300 binding 126.
Aside from the regulation of Ser-276 phosphorylation by ROS, Ser-536 phosphorylation of RelA is induced by NAC through a PI3-kinase-mediated mechanism in a variety of cell lines and this event contributes to its DNA-binding activity 127.
ROS regulation of upstream NF-κB activating pathways.
Exogenously added H2O2 regulates NF-κB activation, and it does so in part through alternative phosphorylation of IκBα (Figure 3). While typically IκBα is usually phosphorylated on serines 32 and 36, which leads to its ubiquitination and degradation, H2O2 affects the phosphorylation of IκBα on Tyr42 or other tyrosine residues, and IκBα may or may not be degraded as part of the process 128, 129, 130, 131. Although IKK is phosphorylated, IKK is not required in this case, and IκBα phosphorylation may be mediated by casein kinase II, possibly downstream of Syk 129, 130. Although this event is observed in response to exogenous H2O2, there is reason to believe that alternative phosphorylation and inactivation of IκBα occurs under physiological conditions because it is also observed under conditions of pervanadate treatment, indicating a possible role for ROS in inhibiting phosphatases, and has also been observed during hypoxia, reoxygenation following hypoxia and in ischemia/reperfusion injury in vivo 132, 133, 134, 135, 136, 137, 138. Degradation of IκBα may not be necessary in this case because Tyr42-phosphorylated IκBα is bound by the SH2 domains of p85α regulatory subunit of PI3K, thus unmasking NF-κB and allowing it to translocate to the nucleus 136. PI3K as well as c-Src has been implicated in alternative tyrosine phosphorylation of IκBα 132, 134, 136.
ROS modification of IκBα has also been shown to lead to inhibition of NF-κB activation. Glutathionylation of IκBα has been detected at cysteine 189, thus preventing phosphorylation events and subsequent degradation 139. However, most of the inhibitory action of ROS on NF-κB with respect to IκBα has been tied to IκBα stability due to the inhibition of the proteasome 140.
IKK is another primary target for ROS in influencing NF-κB signaling. H2O2 has been shown in some cells to inactivate IKK 141, 142, 143, 144. An inhibitory effect may be mediated by ROS oxidation of IKKβ on cysteine 179, since it is found to be S-glutathionated upon exposure to ROS, thus inactivating its kinase activity 142 and leading to a reduction in NF-κB signaling. Deficiency of glutaredoxin 1, which repairs S-glutathionation, leads to reduced nuclear translocation of RelA and substantial loss of NF-κB binding 142. IKK inactivation through oxidation of Cys-179 of IKKβ has also been shown upon arsenite treament 145 and this cysteine residue is also a target of S-nitrosylation by nitric oxide 146. More importantly, Cys-179 is the oxidation target of anti-inflammatory cyclopentone prostaglandins, PGA and 15d-PGJ2 147, suggesting that oxidation of this residue regulates IKK in physiological settings.
Conversely, some studies have shown that ROS, in particular, H2O2, can activate IKKs in some cell types. Dimerization of IKKγ/NEMO was potentiated by H2O2 through formation of disulfide bonds between Cys54 and Cys 347, thus implicating the NEMO subunit in positive regulation by ROS 148, though the other IKK subunits were likely inhibited. Another study reported activation of IKK by H2O2, but in this report NF-κB activation was shown to be inhibited due to lack of IκBα degradation despite its ubiquitination, suggesting that the proteasome was also inhibited 149. One study showed that phosphorylation of both catalytic IKK subunits was potentiated by H2O2 150, which may suggest inhibition of an IKK phosphatase.
Kinases upstream of IKK could be potentially regulated by ROS. MEKK1 was originally suggested to play a role in NF-κB activation. Since it is a redox-sensitive kinase that is inactivated by glutathionylation at C1238, this could potentially be a link to ROS inactivation of IKK in some cells 151. However, an essential role for MEKK1 in IKK activation has been dismissed based on findings in MEKK1-deficient cells 152, and MEKK3, which may be required instead of MEKK1 153, 154, lacks the cysteine that makes MEKK1 a redox-sensitive kinase 155. To our knowledge, TAK1, which has also been suggested to be required for canonical NF-κB activation 154, 156, 157, 158, 159, is not known to be redox-regulated.
NIK, the upstream kinase in the noncanonical pathway is believed to be activated by ROS through inhibition of phosphatases 160. NIK phosphorylation of IKKα is increased upon treatment of H2O2 within a narrow range. However, given that ROS can sometimes inactivate the proteasome, the increased activity of NIK may be in part due to its increased stability as well since it is constitutively degraded by the proteasome.
One potential redox-regulated kinase that affects IKK activation is Akt. Akt positively influences IKKβ-mediated NF-κB activation through the downstream activation of mTOR in association with Raptor 161, 162. Akt has a kinase domain that is subject to oxidation events that inactivate the kinase activity by forming a disulfide bond between Cys-297 and Cys-311 163, and thus ROS could prevent IKK activation by inhibiting Akt. However, not only is Akt itself regulated by ROS, but PTEN, an upstream inhibitor of Akt activation, has a catalytic cysteine that is oxidized by ROS, thus inactivating its phosphatase activity 164. Thus, Akt can be regulated both positively and negatively by ROS.
Conclusion
In summary, ROS interacts with NF-κB signaling pathways in many ways. The transcription of NF-κB-dependent genes influences the levels of ROS in the cell, and in turn, the levels of NF-κB activity are also regulated by the levels of ROS. Depending on the context, ROS can both activate and inhibit NF-κB signaling. A high degree of complexity characterizes ROS interactions with NF-κB pathways owing to the capability for ROS to act in many ways and at numerous places simultaneously. Another complication is that many ROS effects and interactions appear to be cell type-specific. Though we have learned much about the way that ROS influences signaling there is still doubtless a great deal that is yet to be elucidated.
References
Hayden MS, Ghosh S . Shared principles in NF-kappaB signaling. Cell 2008; 132:344–362.
Vallabhapurapu S, Karin M . Regulation and function of NF-kappaB transcription factors in the immune system. Annu Rev Immunol 2009; 27:693–733.
Hatada EN, Nieters A, Wulczyn FG, et al. The ankyrin repeat domains of the NF-kappa B precursor p105 and the protooncogene bcl-3 act as specific inhibitors of NF-kappa B DNA binding. Proc Natl Sci USA 1992; 89:2489–2493.
Bonizzi G, Karin M . The two NF-kappaB activation pathways and their role in innate and adaptive immunity. Trends Immunol 2004; 25:280–288.
Beinke S, Ley SC . Functions of NF-kappaB1 and NF-kappaB2 in immune cell biology. Biochem J 2004; 382(Pt 2):393–409.
Senftleben U, Cao Y, Xiao G, et al. Activation by IKKalpha of a second, evolutionary conserved, NF-kappa B signaling pathway. Science 2001; 293:1495–1499.
Xiao G, Harhaj EW, Sun SC . NF-kappaB-inducing kinase regulates the processing of NF-kappaB2 p100. Mol Cell 2001; 7:401–409.
Dejardin E, Droin NM, Delhase M, et al. The lymphotoxin-beta receptor induces different patterns of gene expression via two NF-kappaB pathways. Immunity 2002; 17:525–535.
Liao G, Zhang M, Harhaj EW, Sun SC . Regulation of the NF-kappaB-inducing kinase by tumor necrosis factor receptor-associated factor 3-induced degradation. J Biol Chem 2004; 279:26243–26250.
Zarnegar BJ, Wang Y, Mahoney DJ, et al. Noncanonical NF-kappaB activation requires coordinated assembly of a regulatory complex of the adaptors cIAP1, cIAP2, TRAF2 and TRAF3 and the kinase NIK. Nat Immunol 2008; 9:1371–1378.
Vallabhapurapu S, Matsuzawa A, Zhang W, et al. Nonredundant and complementary functions of TRAF2 and TRAF3 in a ubiquitination cascade that activates NIK-dependent alternative NF-kappaB signaling. Nat Immunol 2008; 9:1364–1370.
Xiao G, Fong A, Sun SC . Induction of p100 processing by NF-kappaB-inducing kinase involves docking IkappaB kinase alpha (IKKalpha) to p100 and IKKalpha-mediated phosphorylation. J Biol Chem 2004; 279:30099–30105.
Rhee SG, Yang KS, Kang SW, Woo HA, Chang TS . Controlled elimination of intracellular H(2)O(2): regulation of pero