
Curr Opin Immunol. Author manuscript; available in PMC 2010 Dec 1.
Published in final edited form as:
Curr Opin Immunol. 2009 Dec; 21(6): 612–618.
Published online 2009 Oct 23. doi: 10.1016/j.coi.2009.09.011
PMCID: PMC2787714
NIHMSID: NIHMS149540
PMID: 19854631
Regulatory T cells and inhibitory cytokines in autoimmunity
Maria Bettini and Dario A. A. Vignali
Author information Copyright and License information PMC Disclaimer
The publisher's final edited version of this article is available at Curr Opin Immunol
Summary of recent advances
Foxp3+ regulatory T cells (Tregs) contribute significantly to the maintenance of peripheral tolerance, but they ultimately fail in autoimmune diseases. The events that lead to Treg failure in controlling autoreactive effector T cells (Teffs) during autoimmunity are not completely understood. In this review, we discuss possible mechanisms for this subversion as they relate to type 1 diabetes (T1D) and multiple sclerosis (MS). Recent studies emphasize (i) the role of inflammatory cytokines, such as IL-6, in inhibiting or subverting Treg function, (ii) the issue of Treg plasticity, (iii) the possible resistance of autoimmune T cells to Treg-mediated control, and (iv) Treg-associated inhibitory cytokines TGFβ, IL-10 and IL-35 in facilitating Treg suppressive activity and promoting Treg generation. These recent advances place a large emphasis on the local tissue specific inflammatory environment as it relates to Treg function and disease development.
Foxp3+ 조절 T 세포(Treg)는
말초 내성 유지에 크게 기여.
하지만 자가면역 질환에서는 결국 실패합니다.
자가면역 중
자가 반응성 이펙터 T 세포(Teff)를 제어하는데
Treg가 실패하게 되는 사건은 완전히 이해되지 않았습니다.
https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2019.01198/full
이 리뷰에서는
제1형 당뇨병(T1D)과 다발성 경화증(MS)과 관련하여
이러한 전복의 가능한 메커니즘에 대해 논의합니다.
최근 연구에서는
(i) IL-6와 같은 염증성 사이토카인이Treg 기능을 억제하거나 전복하는 역할,
(ii)Treg 가소성 문제,
(iii) 자가 면역 T 세포의 Treg 매개 제어에 대한 저항 가능성,
(iv)Treg 억제 활동을 촉진하고 Treg 생성을 촉진하는 Treg관련 억제 사이토카인 TGFβ, IL-10 및 IL-35를 강조하고 있습니다.
이러한 최근의 발전은Treg 기능 및 질병 발생과 관련된 국소 조직 특이적 염증 환경에 큰 중점을 두고 있습니다.

Introduction
Autoimmunity ensues when central and/or peripheral tolerance barriers are overcome, thereby allowing the activation of self-reactive T cells, which induce tissue destruction. Most self-reactive T cells are deleted in the thymus, a process referred to as central tolerance. The few remaining self-reactive T cells that enter the periphery are controlled by peripheral tolerance mechanisms, in which Foxp3+ Tregs have emerged as the primary mediators [1]. Autoimmune disease develops when this last tolerance barrier is compromised. It has been shown that many individuals with no autoimmune manifestations harbor self-reactive T cells [2].
Thus, the continual battle between self-reactive T cells and suppressive Tregs is critical in determining whether autoimmunity commences. Many autoimmune diseases manifest themselves in an oscillating fashion, which is likely due to the aforementioned struggle between the regulatory and inflammatory arms of the immune system, suggesting that Tregs are still actively involved in regulating this self-reactive response. There are three mechanisms that might contribute to a breakdown in Treg control: (a) Treg numbers are reduced and/or Tregs are dysfunctional due to inherent deficiencies in autoimmune susceptible individuals, (b) Treg suppressive function is inhibited, diverted or converted by the chronic inflammation that occurs in autoimmunity, and/or (c) self-reactive effector T cells (Teff) become unusually aggressive and are refractory to regulation by otherwise functional Tregs because they either overwhelm regulatory control or express molecules that render them resistant. Here we will discuss how these possibilities might contribute to this loss of Treg control, illustrated by two key autoimmune diseases Type 1 Diabetes (T1D) and Multiple Sclerosis (MS), with occasional reference to other autoimmune or inflammatory diseases.
Even though multiple mechanisms of Treg-mediated suppression have been identified, the relative importance of each mechanism in vivo remains unclear [3]. Current dogma, based mostly on in vitro experiments, suggests that Treg function is contact dependent. However, a recent study illustrates that this may be inaccurate as Tregs that are optimally stimulated by contact with Teff can mediate potent third party suppression via soluble factors in vitro [4]. These data, together with significant support from in vivo observations, suggest that inhibitory cytokines such as TGFβ, IL-10, and IL-35 are major contributors to Treg function [3,5]. Consequently, this review will also focus on the contribution of these three inhibitory cytokines in mediating (or failing to mediate) Treg function in autoimmune disease.
중추 및/또는 말초 내성 장벽이 극복되면
자가 면역이 발생하여
조직 파괴를 유도하는
자가 반응성 T 세포가 활성화됩니다.
대부분의 자가 반응성 T 세포는
흉선에서 삭제되는데,
이 과정을 중추 관용이라고 합니다.
말초로 유입되는 소수의 남은 자가 반응성 T 세포는
말초 관용 메커니즘에 의해 제어되며,
여기서 Foxp3+ Tregs가 주요 매개체로 등장했습니다 [1].
자가면역질환은
이 마지막 내성 장벽이 손상될 때 발생합니다.
자가면역 증상이 없는 많은 사람들도
자기 반응성 T 세포를 보유하고 있는 것으로 나타났습니다[2].
따라서
자가 반응성 T세포와
억제성 Treg 사이의 지속적인 싸움은
자가 면역이 시작되는지 여부를 결정하는 데 매우 중요합니다.
https://www.nature.com/articles/s41392-023-01471-y

많은 자가면역 질환이 진동하는 방식으로 나타나는데,
이는 앞서 언급한 면역 체계의 조절 부문과
염증 부문 간의 투쟁 때문일 수 있으며,
이는 Tregs가 여전히 이러한 자가 반응 반응을 조절하는데
적극적으로 관여하고 있음을 시사합니다.
(a) 자가면역에 취약한 사람의 선천적 결핍으로 인해 Treg 수가 감소하거나 Treg가 기능 장애를 일으키는 경우,
(b) 자가면역에서 발생하는 만성 염증에 의해 Treg 억제 기능이 억제, diverted 또는 전환되는 경우,
(c) 자가 반응 효과 T 세포(Teff)가 비정상적으로 공격적이 되어 조절 기능을 압도하거나 저항력을 갖게 하는 분자를 발현하여 정상적으로 기능하는 Treg의 조절에 불응하는 경우 등 세 가지 메커니즘이 있을 수 있습니다.
여기에서는
두 가지 주요 자가 면역 질환인
제1형 당뇨병(T1D)과 다발성 경화증(MS)을 예로 들어
이러한 가능성이 어떻게 Treg 조절 상실에 기여할 수 있는지에 대해 논의하고
다른 자가 면역 또는 염증성 질환에 대해서도 가끔씩 언급합니다.
여러 가지 Treg 매개 억제 메커니즘이 확인되었지만,
생체 내에서 각 메커니즘의 상대적 중요성은 아직 불분명합니다[3].
주로 시험관 내 실험에 근거한 현재의 정설은 Treg 기능이 접촉에 의존한다는 것입니다. 그러나 최근 연구에 따르면 Teff와의 접촉에 의해 최적의 자극을 받은 Treg가 체외에서 수용성 인자를 통해 강력한 제3자 억제를 매개할 수 있기 때문에 이는 부정확할 수 있습니다 [4].
이러한 데이터는 생체 내 관찰에서 얻은 상당한 지원과 함께
TGFβ, IL-10, IL-35와 같은 억제 사이토카인이
Treg 기능의 주요 원인이라는 것을 시사합니다[3,5].
따라서
이 리뷰에서는
자가면역질환에서 Treg 기능을 매개하거나 매개하지 못하는
이 세 가지 억제 사이토카인의 기여에 초점을 맞출 것입니다.
Why do Tregs fail to suppress during autoimmune disease?
Under normal conditions, Tregs effectively inhibit excessive inflammation and autoimmune manifestations [6]. However, in autoimmune diseases Tregs fail to control the initial inflammatory insult and subsequent disease progression due to cell-intrinsic and/or cell-extrinsic factors. Studies performed in a variety of autoimmune models suggest that Tregs are still actively and continuously involved in inflammatory regulation after disease onset [7,8], although this needs to be more definitively assessed. When naturally occurring Tregs are absent in Foxp3-deficient islet antigen-specific BDC2.5 TCR transgenic NOD mice, disease onset is significantly earlier [7]. Interestingly, the rate of islet infiltration in BDC2.5 mice is similar in the presence or absence of Tregs, however, the infiltrating cells cause immediate damage in their absence [7]. In experimental autoimmune encephalomyelitis (EAE), an induced murine model of MS, Foxp3+ Tregs accumulate in the central nervous system (CNS) as the disease progresses [9] and in some instances aid in natural recovery from EAE [8]. Depletion of CD25+ T cells prior to disease induction reduces recovery rate while their depletion after recovery obliterates their protection against secondary autoimmune disease induction [10]. Although Tregs have some control over autoimmune inflammation, there may be several reasons why they ultimately fail to prevent disease progression.
정상적인 조건에서 Tregs는
과도한 염증과
자가면역 증상을 효과적으로 억제합니다[6].
그러나
자가면역 질환에서
Tregs는
세포 내재적 및/또는 세포 외재적 요인으로 인해
초기 염증 유발 및 후속 질병 진행을 제어하지 못합니다.
다양한 자가면역 모델에서 수행된 연구에 따르면
Tregs는 질병 발병 후에도 여전히 활발하고
이에 대한 보다 명확한 평가가 필요합니다.
Foxp3 결핍 섬 항원 특이적 BDC2.5 TCR 형질전환 NOD 마우스에서 자연적으로 발생하는 Tregs가 없는 경우, 질병 발병이 훨씬 더 일찍 시작됩니다 [7]. 흥미롭게도 BDC2.5 마우스의 섬 침윤 속도는 Tregs의 존재 유무에 관계없이 비슷하지만, 침윤 세포는 부재 시 즉각적인 손상을 일으킵니다 [7]. 다발성 경화증의 유도 쥐 모델인 실험적 자가면역성 뇌척수염(EAE)에서는 질병이 진행됨에 따라 Foxp3+ Tregs가 중추신경계(CNS)에 축적되고[9] 일부 경우 EAE로부터 자연 회복을 돕기도 합니다[8]. 질병이 유발되기 전에 CD25+ T 세포가 고갈되면 회복 속도가 감소하고 회복 후 고갈되면 이차 자가면역 질환 유발에 대한 보호 기능이 소멸됩니다 [10].
Tregs가
자가면역 염증을 어느 정도 제어할 수 있지만,
궁극적으로 질병 진행을 막지 못하는 데에는
몇 가지 이유가 있을 수 있습니다.
Inherent defect in Treg number or function
A possible explanation for Treg failure is due to a reduction in their number and/or function in autoimmune predisposed individuals. This may not necessarily manifest systemically, and may be restricted to the site of autoimmune insult. Some studies have reported an age-related defect in NOD Treg function [11], while others were unable to detect any major deficiency [12]. In both studies Tregs were identified and isolated based on CD25 expression, which may have resulted in contamination with activated Teff thereby altering data interpretation. A more recent study took advantage of NOD Foxp3-GFP reporter mice to cleanly separate the two populations and detected no intrinsic defects in NOD Treg numbers or suppressive capabilities [13]. There is some evidence that Treg function in human patients with an early onset of diabetes might be compromised [14]. However, human studies have to rely on CD25 as a marker for Treg separation, and thus are not entirely conclusive. More recent studies were able to exclude contaminating Teff from Treg analysis based on CD127 (IL7Rα) expression that has restricted expression on activated T cells and is absent from Tregs [15,16]. Although these studies are preliminary, at least in the early diagnosed T1D patients Treg numbers appear to be decreased [16].
자가면역 질환이 있는 사람의 경우
Treg의 수 및/또는
기능이 감소하기 때문일 수 있습니다.
이는 반드시 전신적으로 나타나는 것은 아니며
자가면역 질환 부위에 국한되어 나타날 수도 있습니다.
일부 연구에서는 나이와 관련된 NOD Treg 기능의 결함이 보고된 반면[11], 다른 연구에서는 주요 결함을 발견하지 못했습니다[12]. 두 연구 모두 CD25 발현을 기준으로 Treg를 식별하고 분리했는데, 이로 인해 활성화된 Teff에 오염되어 데이터 해석이 달라졌을 수 있습니다. 보다 최근의 연구에서는 NOD Foxp3-GFP 리포터 마우스를 활용하여 두 집단을 깨끗하게 분리한 결과, NOD Treg 수나 억제 능력에 본질적인 결함이 발견되지 않았습니다 [13]. 당뇨병이 조기에 발병한 인간 환자의 Treg 기능이 손상될 수 있다는 일부 증거가 있습니다[14]. 그러나 인간을 대상으로 한 연구는 Treg 분리를 위한 마커로 CD25에 의존해야 하므로 완전히 결정적인 것은 아닙니다.
최근 연구에서는
활성화된 T세포에서 발현이 제한되고
Treg에는 없는 CD127(IL7Rα) 발현을 기반으로
Treg 분석에서 오염된 Teff를 배제할 수 있었습니다 [15,16].
이러한 연구는 예비적이지만,
적어도
초기 진단된 T1D 환자에서
Treg 수치가 감소하는 것으로 보입니다 [16].
Treg defects at the site of inflammation
Another possibility for Treg failure is that the chronic inflammation and consequent molecular milieu at the site of autoimmune destruction could have a negative effect on Treg function. This could be manifested as a reduction in Treg inhibitory function, apoptosis, or conversion into a pro-inflammatory T cell lineage. Recently, the fate of Foxp3+ Tregs over the course of diabetes development in NOD mice was followed. Treg function at the site of inflammation was found to be diminished due to decreased levels of IL-2, a critical Treg growth factor [17]. These tissue-infiltrating Tregs were more prone to apoptosis and thus failed to control autoimmune responses. This study underlines the dependence of Treg function on local IL-2 production and the inflammatory milieu.
Treg stability and plasticity during autoimmune response has recently become a hot issue, especially since administration of in vitro differentiated Tregs is being considered for human therapy. Recent studies addressing the stability of Foxp3+ T cells have shown that both lymphopenic and inflammatory environments can result in the loss of Foxp3 expression in Tregs [18–20]. The apparent plasticity of Treg lineage might result in the instability of this population during chronic autoinflammatory responses and lead to loss of Treg function in an autoimmune environment.
염증 부위의 Treg 결함
Treg 기능 부전의 또 다른 가능성은
자가면역 파괴 부위의 만성 염증과
그에 따른 분자 환경이 Treg 기능에 부정적인 영향을 미칠 수 있다는 것입니다.
이는
Treg 억제 기능의 감소,
세포 자멸사 또는
염증성 T 세포 계통으로의 전환으로 나타날 수 있습니다.
최근에는 NOD 마우스에서 당뇨병이 발생하는 동안 Foxp3+ Treg의 운명을 추적했습니다.
염증 부위에서
Treg 기능은 중요한 트레그 성장 인자인
IL-2의 수치 감소로 인해 감소하는 것으로 밝혀졌습니다 [17].
이렇게 조직에 침윤한 Treg는 세포 사멸에 더 취약하여 자가 면역 반응을 제어하지 못했습니다. 이 연구는 국소 IL-2 생산과 염증 환경에 대한 Treg 기능의 의존성을 강조합니다.
자가면역 반응 중
Treg의 안정성과 가소성 Treg stability and plasticity 은
최근 뜨거운 이슈가 되고 있으며,
특히 체외 분화 Treg의 투여가 인간 치료로 고려되고 있습니다. 최근 Foxp3+ T 세포의 안정성에 관한 연구에 따르면 림프구 감소 및 염증 환경 모두 Treg에서 Foxp3 발현이 손실될 수 있습니다 [18-20].
Treg 계통의 명백한 가소성은
만성 자가 염증 반응 중에 이 집단의 불안정성을 초래하고
자가 면역 환경에서
Treg 기능의 상실로 이어질 수 있습니다.
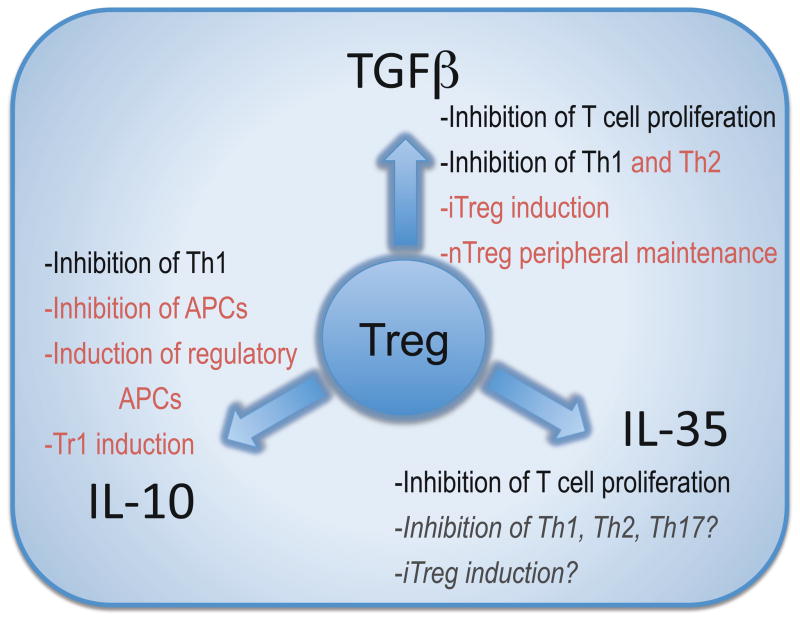
Clearly, Tregs possess other modes of action in addition to the three inhibitory cytokines discussed here [3,21]. There is still some controversy over the relative importance of soluble factors versus cell contact-dependent inhibition in mediating Treg function. Substantial in vivo data, together with a recent study indicate that cytokines contribute significantly to Treg function when cell contact is absent [4]. However, to unequivocally show the importance of cytokines in mediating Treg function one would have to assess inhibition in their absence. The extent of redundancy within these three inhibitory cytokines remains unclear. At first glance, it seems that there is significant overlap between the functions of the three Treg inhibitory cytokines (Figure 1). Both IL-10 and TGFβ can inhibit Th1 type responses, both are able to convert T cells into a regulatory population, albeit with quite distinct phenotypes, and all three cytokines seem to be play a role in the gut homeostasis. However, recent studies suggest that different transcriptional ‘programs’ are utilized by Tregs depending on the Th subset they are attempting to suppress which may likewise lead to the utilization of distinct suppressive functions [22,23]. Interestingly, Tregs appear to utilize similar transcription factors as the T helper cells they are suppressing, raising the possibility of a co-evolutionary progression. IRF4 deficiency in Tregs resulted in selective ablation of IL-10 transcription, partial decrease in EBI3 expression, and no affect on TGFβ. These findings suggest that Tregs may respond with a distinct inhibitory cytokine profile depending on the inflammatory milieu [22]. Thus, it is possible that further examples of Treg functional plasticity may emerge. Therefore, it is possible that although all three cytokines are utilized by Tregs in the same tissue they may control different cellular populations or that different cytokines may be more important in different organs, in a manner that cannot be fully gleaned from the studies conducted thus far. Given the brevity of cellular targets and unique microenvironments within which Tregs have to operate, it would not be surprising if there is limited functional redundancy for these cytokines, as has been suggested for IL-10 [24]. It will also be important to assess the effect of losing one inhibitory mechanism on the remaining functional landscape as it is conceivable that Tregs may make adjustments to compensate for such a loss given the functional plasticity already described.
분명히 Treg는
여기에서 설명한 세 가지 억제 사이토카인 외에도
Treg 기능을 매개하는 데 있어
수용성 인자와 세포 접촉 의존적 억제의 상대적 중요성에 대해서는
여전히 논란이 있습니다.
최근의 연구와 함께
상당한 생체 내 데이터에 따르면
세포 접촉이 없을 때 사이토카인이 Treg 기능에 크게 기여하는 것으로 나타났습니다 [4].
그러나
Treg 기능을 매개하는 사이토카인의 중요성을 명확하게 보여주기 위해서는
사이토카인이 없을 때의 억제를 평가해야 합니다.
이 세 가지 억제 사이토카인의 중복 정도는 아직 불분명합니다.
언뜻 보기에는
세 가지 Treg 억제 사이토카인의 기능 사이에
상당한 중복이 있는 것으로 보입니다(그림 1).
IL-10과 TGFβ는
모두 Th1 유형 반응을 억제할 수 있고,
표현형은 매우 다르지만 모두 T세포를 조절 집단으로 전환할 수 있으며,
세 사이토카인 모두 장내 항상성에 중요한 역할을 하는 것으로 보입니다.
그러나 최근 연구에 따르면
Treg는 억제하려는 Th 하위 집합에 따라
서로 다른 전사 '프로그램'을 활용하며,
이는 마찬가지로 뚜렷한 억제 기능의 활용으로 이어질 수 있습니다 [22,23].
흥미롭게도 Treg는
그들이 억제하는 T 헬퍼 세포와 유사한 전사인자를 활용하는 것으로 보이며,
이는 공진화 가능성을 제기합니다.
Tregs에서
IRF4 결핍은
IL-10 전사의 선택적 제거,
EBI3 발현의 부분적 감소를 가져왔으며
TGFβ에는 영향을 미치지 않았습니다.
이러한 결과는
Tregs가 염증 환경에 따라
뚜렷한 억제 사이토카인 프로파일로 반응할 수 있음을 시사합니다[22].
따라서
Treg의 기능적 가소성에 대한 추가적인 예가 나타날 수 있습니다.
따라서
세 가지 사이토카인이
모두 같은 조직에서
Treg에 의해 활용되더라도
지금까지 수행된 연구에서 완전히 파악할 수 없는 방식으로
서로 다른 세포 집단을 제어하거나
다른 기관에서 서로 다른 사이토카인이 더 중요할 수 있습니다.
세포 표적의 간결성과 Tregs가 작동해야 하는
고유한 미세 환경을 고려할 때
IL-10에서 제안된 것처럼
이러한 사이토카인에 대한 기능적 중복성이 제한적이라고 해도 놀랄 일이 아닙니다 [24].
또한 이미 설명한 기능적 가소성을 고려할 때
Tregs가 이러한 손실을 보상하기 위해 조정할 수 있기 때문에
하나의 억제 메커니즘을 잃는 것이
나머지 기능적 환경에 미치는 영향을 평가하는 것도 중요합니다.
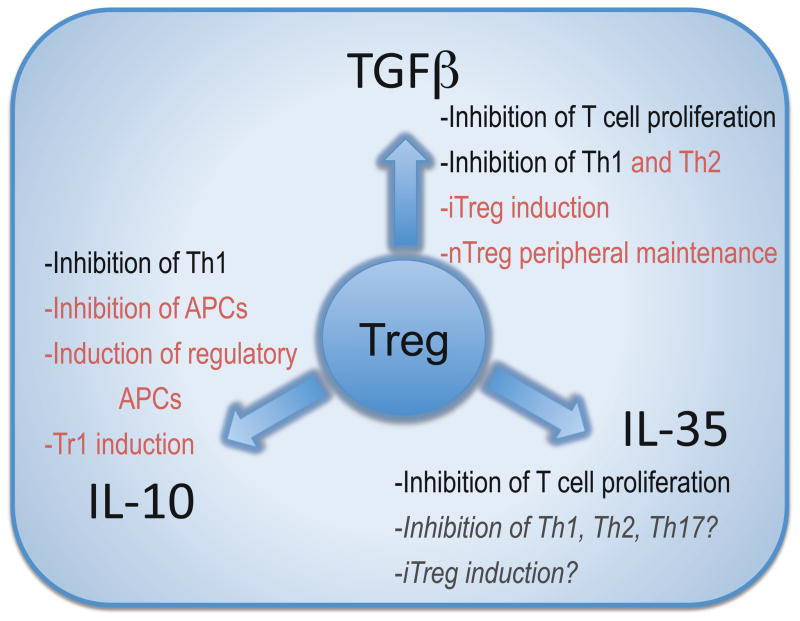
Redundancy and specificity of Treg inhibitory cytokine function.
Teff resistance to inhibition
A third possibility is that highly activated self-reactive Teffs become resistant to Treg-mediated control. Studies performed in both spontaneous T1D and induced EAE models report normal Treg function during disease, but show that the Teffs are hyper activated and do not capitulate to suppression by Tregs [7,9,13,25,26]. NOD Treg function, as measured by in vitro assays, was reported to be fairly comparable to B6 Tregs. In contrast, NOD Teffs exhibited an increased ability to proliferate and were unresponsive to Treg-mediated suppression [13]. Similarly, Teffs from human T1D patients are more refractory to inhibition by Tregs than Teffs from healthy controls [27].
Mounting evidence shows that inhibitory actions of Treg-associated cytokines can be subverted in highly inflammatory environments. In particular, IL-6 has been shown to have a negative effect on Treg function [28], and in the CNS of EAE mice a typically inhibitory cytokine TGFβ synergizes with IL-6 to induce pro-inflammatory Th17 cells [29,30]. Moreover, when directly compared to Th1 and Th2 cells, Th17 cells were more likely to overcome the inhibition by Tregs [31]. These data have raised the possibility that Th17 cells are more resistant to regulatory control than other T helper subsets. The molecular basis for this resistance remains obscure.
Thus it is possible that Treg function is normal during autoimmune disease, but the chronic inflammatory environment at the tissue site of autoimmune damage counteracts and overwhelms the suppressive Treg activity. While the issue of Treg fitness during autoimmunity remains a hot topic of discussion, it is possible that multiple pathways lead to the breakdown of peripheral tolerance. It is clear, however, that Tregs function has to be considered and assessed in a context of the inflammatory environment where they operate rather than in the periphery.
억제에 대한 Teff 내성
세 번째 가능성은
고도로 활성화된 자가 반응성 Teff가
Treg 매개 제어에 내성을 갖게 되는 것입니다.
자발적 T1D 및 유도된 EAE 모델 모두에서 수행된 연구에 따르면
질병 동안 정상적인 Treg 기능이 보고되었지만,
Teff가 과도하게 활성화되어
Treg에 의한 억제에 항복하지 않는 것으로 나타났습니다 [7,9,13,25,26].
시험관 내 분석으로 측정한 NOD Treg 기능은 B6 Treg와 상당히 유사한 것으로 보고되었습니다. 이와 대조적으로, NOD Teff는 증식 능력이 증가했으며 Treg 매개 억제에 반응하지 않는 것으로 나타났습니다 [13]. 마찬가지로, 인간 T1D 환자의 Teff는 건강한 대조군의 Teff보다 Treg에 의한 억제에 더 잘 저항합니다 [27].
증가하는 증거에 따르면 염증도가 높은 환경에서는 Treg 관련 사이토카인의 억제 작용이 무력화될 수 있습니다. 특히 IL-6는 Treg 기능에 부정적인 영향을 미치는 것으로 나타났으며[28], EAE 마우스의 CNS에서는 일반적으로 억제성 사이토카인 TGFβ가 IL-6와 시너지 효과를 발휘하여 전염증성 Th17 세포를 유도합니다[29,30]. 또한 Th1 및 Th2 세포와 직접 비교했을 때 Th17 세포는 Tregs에 의한 억제를 극복할 가능성이 더 높았습니다 [31]. 이러한 데이터는 Th17 세포가 다른 T 헬퍼 하위 집합보다 조절 제어에 더 저항성이 있을 가능성을 제기했습니다. 이러한 저항성의 분자적 근거는 아직 불분명합니다.
따라서
자가면역 질환 중에는
Treg 기능이 정상이지만
자가면역 손상 조직 부위의 만성 염증 환경이
억제된 Treg 활동을 상쇄하고 압도할 가능성이 있습니다.
자가 면역 중 Treg 적합성 문제는
여전히 뜨거운 논의의 주제이지만,
여러 경로를 통해 말초 내성이 무너질 수 있습니다.
그러나 분명한 것은
말초가 아닌 염증 환경의 맥락에서
Treg 기능을 고려하고 평가해야 한다는 것입니다.
Inhibitory cytokines facilitate Treg-mediated control of Teffs and promote the induction of induced Tregs
As discussed in the previous section, the local inflammatory milieu at the site of autoimmune destruction can have a significant effect on Treg function. Cytokines secreted by Tregs, Teffs and other cellular infiltrates contribute to the generation of this environment. Inhibitory cytokines, such as TGFβ and IL-10, can directly inhibit cells that participate in this autoimmune destruction and/or can mediate the generation of induced T regulatory cells (iTregs) [3]. More recently IL-35 has also been shown to directly inhibit T cell proliferation [4,32], although its ability to affect other cell populations or its capacity to mediate/influence the induction of iTregs has yet to be established.
억제 사이토카인은
Treg를 매개로 한 Teff의 조절을 촉진하고
유도된 Treg의 유도를 촉진합니다.
이전 섹션에서 설명한 것처럼
자가 면역 파괴 부위의 국소 염증 환경은
Treg 기능에 상당한 영향을 미칠 수 있습니다.
Treg, Teff 및
기타 세포 침윤에 의해 분비되는 사이토카인이
이러한 환경의 생성에 기여합니다.
TGFβ 및 IL-10과 같은 억제 사이토카인은
이러한 자가 면역 파괴에 참여하는 세포를
직접 억제하거나
유도된 T 조절 세포(iTregs)의 생성을 매개할 수 있습니다[3].
최근에는
IL-35도 T세포 증식을 직접 억제하는 것으로 밝혀졌지만[4,32],
다른 세포 집단에 영향을 미치는 능력이나 iTreg의 유도를 매개/영향하는 능력은 아직 확립되지 않았습니다.
Inhibitory cytokines regulate Teffs
The in vivo role of Treg-derived cytokines in modulating autoimmune diseases has been clearly established. Mice deficient in TGFβ succumb to spontaneous autoimmune disease at 4–5 weeks of age [33,34], while the IL-10 knock out animals are more susceptible to induced autoimmune disease [35]. In TGFβ deficient mice, the dysregulated inflammatory response is alleviated after the depletion of CD4+ and CD8+ T cells [36,37]. Although, Tregs are not the only source of inhibitory cytokines, it has become more evident that Treg secreted cytokines contribute significantly to Treg function in vivo.
TGFβ has been implicated in the function of Tregs in T1D and inflammatory bowel disease (IBD) mouse models [38,39]. Depletion of TGFβ with neutralizing antibodies results in a decrease in both mouse and human Treg function [39–41]. T cell transfer and bone marrow chimeric studies revealed that TGFβ has both cell-intrinsic and cell-extrinsic regulatory effects [42,43]. These data show that TGFβ helps to preserve peripheral tolerance by induction of iTregs and maintenance of nTregs in the periphery.
IL-10 is a homodimeric cytokine with a wide range of inhibitory actions. IL-10 is produced by a variety of cells, including monocytes and T cells, and can exert its effects on both myeloid and lymphoid cells [35]. Its regulatory activity is mediated largely through its effects on APCs [44]. Interestingly, IL-10 and IL-10 receptor-deficient mice, unlike their TGFβ counterparts, do not develop spontaneous autoimmune symptoms which only becomes apparent following inflammatory insult. In the absence of IL-10, mice develop colitis in response to some intestinal microorganisms [45,46]. In other murine autoimmune models, such as EAE, mice lacking IL-10 exhibit an exacerbated form of the disease [35]. An increase in IL-10 production by Foxp3+ Tregs is associated with the recovery phase of EAE [8], and transfer of in vitro generated iTregs or exogenously purified natural Tregs was shown to prevent the induction of EAE through the production of IL-10 [47]. IL-10 plays a significant role in intestinal homeostasis. Although, IL-10-deficient Tregs are not devoid of regulatory activity, their function is particularly affected in controlling already established gut inflammation [48], and mice with Treg-specific deletion of IL-10 are susceptible to colitis induced by commensal flora [24]. Similarly to TGFβ, IL-10 can also mediate the generation of an induced regulatory T population, Tr1, that is characterized by secretion of IL-10 [49].
IL-35 is a recently discovered heterodimeric cytokine that is produced by Foxp3+ Tregs and contributes to their suppressive function [32]. IL-35 is composed of two chains, IL-12α (p35) and EBI3 that it shares with two other heterodimeric cytokines IL-12 and IL-27, respectively. IL-35 is required for maximal regulatory function in vivo as Tregs deficient in either chain are unable to control homeostatic T cell expansion and IBD [32]. Given that IL-35 emerged more recently, we still have a limited understanding of its biological activity which has also been complicated by the lack of appropriate tools for its functional dissection. Recombinant IL-35 has proven particularly challenging to generate and purify, compared with IL-12 and IL-27. It is tempting to speculate that the apparent poor stability of IL-35 might underlie important physiological features of this cytokine, such as limited potency over short-range. Alternatively, DCs that secrete IL-12 or IL-27 may be precluded from generating IL-35, due to preferential pairing of IL-12 and IL-27.
It is clearly important to determine the bioactivity of IL-35. Whether IL-35 can inhibit all T helper subsets and other cellular populations, such as B cells, macrophages or DCs, or has more selective targets remains to be determined. Significant insight is likely to be gained from the determination of the IL-35 receptor and its expression pattern. It is tempting to speculate that since IL-35 represents a novel pairing of the IL-12 cytokine chains, its receptor will also be a new combination of the known family receptor chains [50]. Alternatively, the receptor might be composed of novel subunits. If it is the former, a key question will be how cells can translate similar signaling pathways into completely different downstream functional consequences, i.e. stimulatory signaling induced by IL-12 and IL-27 vs inhibitory signal delivered by IL-35. Interestingly, precedence does exist for a similar signaling cascade mediating quite different functional outcomes, as exemplified by the IL-6 and IL-10 receptor which both use STAT3 [51]. Lastly, an open question is whether IL-35 can mediate the generation of an induced regulatory T cell population. This is clearly a feature of the other two inhibitory cytokines IL-10 and TGFβ and thus it remains plausible that IL-35 might have a similar capacity. TGFβ-inuced iTregs (Th3) and IL-10-induced Tr1 clearly have very distinct transcriptional and functional profiles [3,52] and so it is possible that any IL-35-induced regulatory populations may also be quite distinct.
억제 사이토카인이 Teff를 조절
자가면역 질환을 조절하는
Treg 유래 사이토카인의 생체 내 역할은
명확히 밝혀졌습니다.
TGFβ가 결핍된 마우스는
생후 4~5주에 자연적인 자가면역 질환에 걸리며[33,34],
IL-10이 결핍된 동물은
유도된 자가면역 질환에 더 취약합니다[35].
TGFβ 결핍 마우스에서는
CD4+ 및 CD8+ T 세포가 고갈된 후 조절 장애가 있는 염증 반응이 완화됩니다 [36,37].
비록 Treg가
억제 사이토카인의 유일한 공급원은 아니지만,
생체 내에서 Treg 분비 사이토카인이
Treg 기능에 크게 기여한다는 것이 더욱 분명해졌습니다.
TGFβ는 T1D 및 염증성 장 질환(IBD) 마우스 모델에서 Treg의 기능에 관여하는 것으로 밝혀졌습니다[38,39]. 중화 항체로 TGFβ를 고갈시키면 마우스와 인간의 Treg 기능이 모두 감소합니다 [39-41]. T 세포 전이 및 골수 키메라 연구에 따르면 TGFβ는 세포 내재적 및 세포 외재적 조절 효과를 모두 가지고 있는 것으로 나타났습니다 [42,43]. 이러한 데이터는 TGFβ가 말초에서 iTreg를 유도하고 nTreg를 유지함으로써 말초 내성을 보존하는 데 도움이 된다는 것을 보여줍니다.
IL-10은
광범위한 억제 작용을 하는
동이형 사이토카인입니다.
IL-10은
단핵구와 T세포를 포함한 다양한 세포에서 생성되며
골수 세포와 림프 세포 모두에 효과를 발휘할 수 있습니다[35].
IL-10의 조절 활동은
주로 APC에 미치는 영향을 통해 매개됩니다 [44].
흥미롭게도 IL-10 및 IL-10 수용체가 결핍된 마우스는 TGFβ가 결핍된 마우스와 달리 염증성 모욕 후에만 나타나는 자발적인 자가 면역 증상이 나타나지 않습니다. IL-10이 없는 생쥐는 일부 장내 미생물에 반응하여 대장염이 발생합니다[45,46]. EAE와 같은 다른 쥐 자가면역 모델에서 IL-10이 결핍된 마우스는 질병의 악화 형태를 보입니다 [35]. Foxp3+ Tregs에 의한 IL-10 생산의 증가는 EAE의 회복 단계와 관련이 있으며[8], 시험관 내에서 생성된 iTregs 또는 외인성 정제된 천연 Tregs의 전달은 IL-10 생산을 통한 EAE의 유도를 막는 것으로 나타났습니다[47]. IL-10은 장 항상성 유지에 중요한 역할을 합니다. IL-10이 결핍된 Treg는 조절 활동이 없는 것은 아니지만, 이미 발생한 장 염증을 조절하는 데 특히 영향을 받으며[48], Treg 특이적으로 IL-10이 결핍된 마우스는 공생 세균총에 의해 유발된 대장염에 취약합니다[24]. TGFβ와 유사하게 IL-10은 IL-10의 분비를 특징으로 하는 유도된 조절 T 집단인 Tr1의 생성을 매개할 수도 있습니다 [49].
IL-35는
최근에 발견된 이합체 사이토카인으로,
Foxp3+ Treg에 의해 생성되며 이들의 억제 기능에 기여합니다[32].
IL-35는 두 개의 사슬, 즉 IL-12α(p35)와 EBI3로 구성되어 있으며, 각각 다른 두 개의 이종이합체 사이토카인 IL-12 및 IL-27과 공유합니다. 두 사슬 중 하나라도 결핍된 Treg는 항상성 T 세포 확장 및 IBD를 제어할 수 없으므로 생체 내 조절 기능을 극대화하려면 IL-35가 필요합니다 [32]. IL-35가 최근에 등장한 점을 감안할 때, 우리는 여전히 그 생물학적 활성에 대한 이해가 제한적이며 기능적 해부를 위한 적절한 도구의 부족으로 인해 복잡해졌습니다. 재조합 IL-35는 IL-12 및 IL-27에 비해 생성 및 정제가 특히 까다로운 것으로 입증되었습니다. IL-35의 낮은 안정성이 단거리에서 제한된 효능과 같은 이 사이토카인의 중요한 생리적 특징의 기저에 있을 수 있다고 추측할 수 있습니다. 또는 IL-12 또는 IL-27을 분비하는 DC는 IL-12와 IL-27의 우선적 결합으로 인해 IL-35를 생성하지 못할 수도 있습니다.
IL-35의 생체 활성을 결정하는 것은 분명히 중요합니다. IL-35가 모든 T 헬퍼 하위 집합과 B세포, 대식세포 또는 DC와 같은 다른 세포 집단을 억제할 수 있는지 또는 더 선택적인 표적을 가지고 있는지 여부는 아직 밝혀지지 않았습니다. IL-35 수용체와 그 발현 패턴을 결정하면 상당한 통찰력을 얻을 수 있을 것으로 보입니다. IL-35는 IL-12 사이토카인 사슬의 새로운 쌍을 나타내므로, 그 수용체도 알려진 계열 수용체 사슬의 새로운 조합일 것이라고 추측할 수 있습니다 [50]. 또는 수용체가 새로운 서브유닛으로 구성될 수도 있습니다. 전자의 경우, 핵심 질문은 세포가 어떻게 유사한 신호 경로를 완전히 다른 다운스트림 기능적 결과, 즉 IL-12 및 IL-27에 의해 유도되는 자극 신호와 IL-35에 의해 전달되는 억제 신호로 변환할 수 있는지가 될 것입니다. 흥미롭게도, STAT3를 사용하는 IL-6 및 IL-10 수용체 [51]에서 예시된 것처럼 유사한 신호 캐스케이드가 상당히 다른 기능적 결과를 매개하는 우선순위가 존재합니다. 마지막으로, IL-35가 유도된 조절 T 세포 집단의 생성을 매개할 수 있는지에 대한 의문이 남아 있습니다. 이는 다른 두 가지 억제 사이토카인인 IL-10과 TGFβ의 특징이므로 IL-35도 비슷한 능력을 가질 수 있을 것으로 보입니다. TGFβ에 의해 유도된 iTregs(Th3)와 IL-10에 의해 유도된 Tr1은 매우 뚜렷한 전사 및 기능적 프로파일을 가지고 있으므로[3,52], IL-35에 의해 유도된 조절 집단도 매우 뚜렷할 수 있습니다.
참고) 운동을 하면 ....
https://www.nature.com/articles/s41574-022-00641-2

Inhibitory cytokines promote infectious tolerance
It is clear that Foxp3+ Tregs play a major role in most autoimmune diseases. What remains unknown is the relative contribution of natural, thymus-derived Tregs (nTregs) versus iTregs in mediating control of autoimmunity. Furthermore, recent studies suggest that the importance of iTregs in controlling autoimmunity may vary depending on the site of tissue inflammation [4,32].
Recent studies have shown that Tregs and Treg-derived cytokines have long lasting tolerance effects in vivo [4,32]. Even though diabetes onset can be prevented by Treg transfer into NOD mice, the endogenous Foxp3+ Treg population dominates at the site of the original autoimmune response in the pancreas while the transferred population is significantly reduced [38]. In a mouse model of IBD, while TGFβ is essential for downregulating the autoimmune response, Treg-derived TGFβ was not critical for protection [39,42]. This suggests that the main function of TGFβ may be to mediate infectious tolerance (ie. generation of iTregs) which may not necessarily be Treg-derived [53]. It is possible that in order to achieve infectious tolerance several simultaneous signals are required in addition to TGFβ. These can be other inhibitory cytokines, such as IL-10, or cell surface molecules that require cell contact for ligation [54].
Recent findings suggest that different organs or tissues are more suitable or permissive to Treg differentiation due to their cellular and/or molecular composition. Mucosal CD103+ dendritic cells are exceptionally adept at inducing Tregs through production of TGFβ and the Vitamin A metabolite retinoic acid (RA) [55,56]. RA counteracts the effects of inflammatory cytokines and aides in skewing T cells towards a regulatory phenotype [57–59]. CD8+CD205+ dendritic cells in the spleen can also mediate iTreg development in part through production of TGFβ [60]. The CNS, however, does not seem to favor the generation of Foxp3+ iTregs [9]. This is in part due to Teff-derived IL-6, which in combination with TGFβ induces Th17 cells rather than Foxp3+ iTregs [29]. DC phenotype is initially induced by microbial interactions leading to the induction of either inflammatory or inhibitory/toleragenic DCs [61]. It is conceivable that the complete Freund’s adjuvant used to induce EAE, which contains mycobacterial cell wall components, results in the generation of DCs that are suboptimal for iTreg generation. In support to this, mice immunized with myelin peptide in incomplete Freund’s adjuvant that lacks Mycobacterium tuberculosis exhibited a decrease in IL-6 production and an increase in antigen specific Foxp3+ T cells in the spleen. However, there was only a modest increase in iTregs and whether these were present in the CNS is unknown [29]. Taken together, these studies suggest that the stimulus received by DCs may be more critical than the particular site or organ that is affected in determining iTreg development. Thus, under alternate stimulatory conditions the CNS might be an acceptable location for iTreg differentiation.
억제 사이토카인은 감염성 내성을 촉진
대부분의 자가면역 질환에서
Foxp3+ Tregs가 중요한 역할을 한다는 것은 분명합니다.
자가면역 조절을 매개하는 데 있어
자연적인 흉선 유래 Treg(nTreg)와 iTreg의 상대적인 기여도는 아직 알려지지 않았습니다.
또한,
최근 연구에 따르면
자가면역 조절에 있어 iTreg의 중요성은
조직 염증 부위에 따라 달라질 수 있다고 합니다[4,32].
최근 연구에 따르면 Treg 및 Treg 유래 사이토카인은 생체 내에서 오래 지속되는 내성 효과가 있는 것으로 나타났습니다 [4,32]. NOD 마우스로 Treg를 이식하면 당뇨병 발병을 예방할 수 있지만, 췌장의 원래 자가 면역 반응 부위에서는 내인성 Foxp3+ Treg 집단이 우세한 반면, 이식된 집단은 현저히 감소합니다 [38]. IBD의 마우스 모델에서 TGFβ는 자가 면역 반응을 하향 조절하는 데 필수적이지만, Treg 유래 TGFβ는 보호에 중요하지 않았습니다 [39,42]. 이는 TGFβ의 주요 기능이 반드시 Treg 유래가 아닐 수도 있는 감염성 내성(즉, iTreg의 생성)을 매개하는 것일 수 있음을 시사합니다 [53]. 감염성 내성을 달성하기 위해서는 TGFβ 외에도 여러 가지 동시 신호가 필요할 수 있습니다. 이는 IL-10과 같은 다른 억제 사이토카인 또는 결합을 위해 세포 접촉이 필요한 세포 표면 분자일 수 있습니다 [54].
최근 연구 결과에 따르면 세포 및/또는 분자 구성에 따라 기관이나 조직에 따라 Treg 분화에 더 적합하거나 허용적인 것으로 나타났습니다. 점막 CD103+ 수지상 세포는 TGFβ와 비타민 A 대사산물인 레티노산(RA)의 생성을 통해 Treg를 유도하는 데 매우 능숙합니다[55,56]. RA는 염증성 사이토카인의 효과를 상쇄하고 T 세포를 조절 표현형으로 왜곡하는 데 도움을 줍니다[57-59]. 비장의 CD8+CD205+ 수지상 세포도 부분적으로 TGFβ 생성을 통해 iTreg 발달을 매개할 수 있습니다 [60]. 그러나 CNS는 Foxp3+ iTreg의 생성을 선호하지 않는 것으로 보입니다 [9]. 이는 부분적으로 Teff 유래 IL-6가 TGFβ와 결합하여 Foxp3+ iTreg가 아닌 Th17 세포를 유도하기 때문입니다 [29]. DC 표현형은 처음에 미생물 상호작용에 의해 유도되어 염증성 또는 억제성/관용성 DC를 유도합니다 [61]. 마이코박테리아 세포벽 성분을 포함하는 EAE를 유도하는 데 사용되는 완전한 Freund의 보조제는 iTreg 생성에 최적이 아닌 DC를 생성하는 결과를 초래할 수 있다고 생각할 수 있습니다. 이를 뒷받침하기 위해 결핵균이 결여된 불완전한 프룬트 보조제에 미엘린 펩타이드를 넣어 면역시킨 쥐는 비장에서 IL-6 생산이 감소하고 항원 특이적 Foxp3+ T 세포가 증가하는 것으로 나타났습니다. 그러나 iTregs는 약간만 증가했으며 이것이 중추신경계에 존재하는지 여부는 알려지지 않았습니다 [29]. 이러한 연구 결과를 종합해 보면 iTreg 발달을 결정하는 데 영향을 받는 특정 부위나 기관보다 DC가 받는 자극이 더 중요할 수 있음을 시사합니다. 따라서 대체 자극 조건 하에서 CNS는 iTreg 분화에 적합한 위치가 될 수 있습니다.
Concluding remarks
Recent advances have placed significant emphasis on dissecting Treg function in autoimmunity and assessing their therapeutic utility. The local inflammatory environment, consisting of specialized APCs, metabolic components, growth factors and inhibitory and/or inflammatory cytokines, can significantly alter the efficacy and generation of Tregs. Given the complexity of Treg function in vivo, several key questions remain before we fully understand molecular and cellular defects that occur in autoimmunity, as they pertain to Tregs, and how this knowledge might be utilized in a clinical setting.
최근의 발전은
자가 면역에서 Treg 기능을 해부하고 치료적 유용성을 평가하는 데 상당한 중점을 두고 있습니다.
특수 APC, 대사 성분, 성장 인자, 억제 및/또는
염증성 사이토카인으로 구성된 국소 염증 환경은
Treg의 효능과 생성을 크게 변화시킬 수 있습니다.
생체 내 Treg 기능의 복잡성을 고려할 때,
자가 면역에서 발생하는 분자 및 세포 결함을 완전히 이해하기 전에
Treg와 관련된 몇 가지 핵심 질문과 이
러한 지식이 임상 환경에서 어떻게 활용될 수 있는지에 대한 몇 가지 핵심 질문이 남아 있습니다.
- 다양한 조직 부위의 염증이 Treg 가소성에 어떤 영향을 미치며, 염증성 사이토카인이 Treg 안정성에 미치는 부정적인 영향에 어떻게 대응할 수 있을까요? 우리는 시험관 내에서 iTreg를 유도하기 위한 최소한의 요건(TGFβ, IL-2, TCR 자극)을 알고 있지만, Foxp3 유전자좌의 안정화를 위해 어떤 신호가 필요한지는 아직 밝혀내지 못했습니다. 이는 Foxp3+ iTreg의 치료적 사용에 있어 핵심이 될 것으로 보입니다. 또한 어떤 분자적 사건이 nTreg 가소성을 매개하며 어떻게 이를 예방할 수 있는지도 아직 불분명합니다.
- 성공적인 Treg 치료를 위해 항원 특이성이 필요한 자가면역질환은 무엇이며, 그 항원 표적은 무엇이어야 할까요? 체외에서 유도된 조절 세포 집단, 즉 TGFβ 유도 iTreg와 IL-10 유도 Tr1 세포의 치료적 사용은 질병 부위에 효과적으로 서식하고 활성화될 수 있는 항원 특이적 조절 세포 집단을 생성할 수 있기 때문에 매력적인 가능성입니다 [49]. 예를 들어, 최근 T세포가 섬으로 이동하는 것은 세포 자율적 사건이라는 사실이 밝혀져, T1D의 핵심 개시 항원에 대한 논란이 여전히 존재하지만 섬 항원 특이적 iTreg가 내관 형성 치료의 효과적인 매개체가 될 수 있음을 시사했습니다 [62]. 경구 항원 투여는 항원 특이적 Tregs를 유도하며 잠재적으로 유망한 치료 접근법이지만, 치료의 성공 여부를 명확하게 평가하기 위해서는 추가적인 임상시험이 필요합니다 [49,63,64]. 그러나 많은 자가면역 질환의 경우 표적 항원의 선택이 모호하고 그 중요성이 제대로 정의되지 않은 채로 남아 있습니다.
- 사이토카인 치료의 효능과 안전성을 개선하고 효과적인 국소 전달을 달성하려면 어떻게 해야 할까요? 사이토카인 치료는 플레오트로픽 활성과 잠재적 독성으로 인해 투여하기 가장 어려운 치료법 중 하나일 수 있습니다. 따라서 향후 치료법은 복합적일 수 있으며, 특정 부위에 사이토카인을 표적으로 하는 변형 및/또는 생체 활성 또는 안정성을 향상시키는 변형이 포함될 수도 있습니다. 표적 사이토카인 전달은 어렵지만 치료 효과를 개선하고 부작용을 줄일 수 있습니다. 한 연구에서는 유전자 변형 락토코커스 락티스를 사용하여 IBD 환자의 위장관 내로 IL-10을 국소 투여했습니다[65]. 또는 IL-10 유전자와 융합된 젤라틴 나노 입자를 사용하여 IL-10의 국소 전달에 효과를 보았습니다 [66]. IL-35에 대해서는 아직 많은 것이 밝혀지지 않았지만, 지금까지는 활성화 또는 염증 활동이 없는 것처럼 보이는 기능적 단성인 것으로 보입니다 [32]. 이 기능이 확인된다면 생체 내 독성이 제한적일 수 있으므로 치료적으로 바람직할 것입니다.
Acknowledgments
DAAV is supported by the Juvenile Diabetes Research Foundation International (1-2004-141 [The Robert and Janice Compton Research Grant, In Honor of Elizabeth S. Compton] and 1-2006-847), the NIH (AI072239), the St Jude Cancer Center Support CORE grant (CA-21765) and the American Lebanese Syrian Associated Charities (ALSAC). MB is supported by a Juvenile Diabetes Research Foundation International post-doctoral fellowship (3-2009-594).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and recommended reading
Papers of particular interest, published within the past two years, have been highlighted as:
• of special interest
•• of outstanding interest
1. Sakaguchi S, Ono M, Setoguchi R, Yagi H, Hori S, Fehervari Z, Shimizu J, Takahashi T, Nomura T. Foxp3+ CD25+ CD4+ natural regulatory T cells in dominant self-tolerance and autoimmune disease. Immunol Rev. 2006;212:8–27. [PubMed] [Google Scholar]
2. Hellings N, Baree M, Verhoeven C, D'Hooghe MB, Medaer R, Bernard CC, Raus J, Stinissen P. T-cell reactivity to multiple myelin antigens in multiple sclerosis patients and healthy controls. J Neurosci Res. 2001;63:290–302. [PubMed] [Google Scholar]
3. Vignali DA, Collison LW, Workman CJ. How regulatory T cells work. Nat Rev Immunol. 2008;8:523–532. [PMC free article] [PubMed] [Google Scholar]