
Open AccessReview
B Cell Activation and Escape of Tolerance Checkpoints:
Recent Insights from Studying Autoreactive B Cells
by
Carlo G. Bonasia
Department of Rheumatology and Clinical Immunology, University Medical Center Groningen, University of Groningen, 9713 Groningen, GZ, The Netherlands
2
Department of Pathology and Medical Biology, University Medical Center Groningen, University of Groningen, 9713 Groningen, GZ, The Netherlands
*
Author to whom correspondence should be addressed.
Cells 2021, 10(5), 1190; https://doi.org/10.3390/cells10051190
Submission received: 15 April 2021 / Revised: 7 May 2021 / Accepted: 9 May 2021 / Published: 13 May 2021
(This article belongs to the Special Issue B Cell Signaling and Activation in Autoimmunity)
Downloadkeyboard_arrow_down
Abstract
Autoreactive B cells are key drivers of pathogenic processes in autoimmune diseases by the production of autoantibodies, secretion of cytokines, and presentation of autoantigens to T cells. However, the mechanisms that underlie the development of autoreactive B cells are not well understood. Here, we review recent studies leveraging novel techniques to identify and characterize (auto)antigen-specific B cells. The insights gained from such studies pertaining to the mechanisms involved in the escape of tolerance checkpoints and the activation of autoreactive B cells are discussed. In addition, we briefly highlight potential therapeutic strategies to target and eliminate autoreactive B cells in autoimmune diseases.
자가 반응성 B 세포는
자가 항체 생산,
사이토카인 분비,
자가 항원을 T 세포에 제시함으로써
자가 면역 질환에서 병원성 과정의 핵심 동인입니다.
https://www.nature.com/articles/nri.2017.19
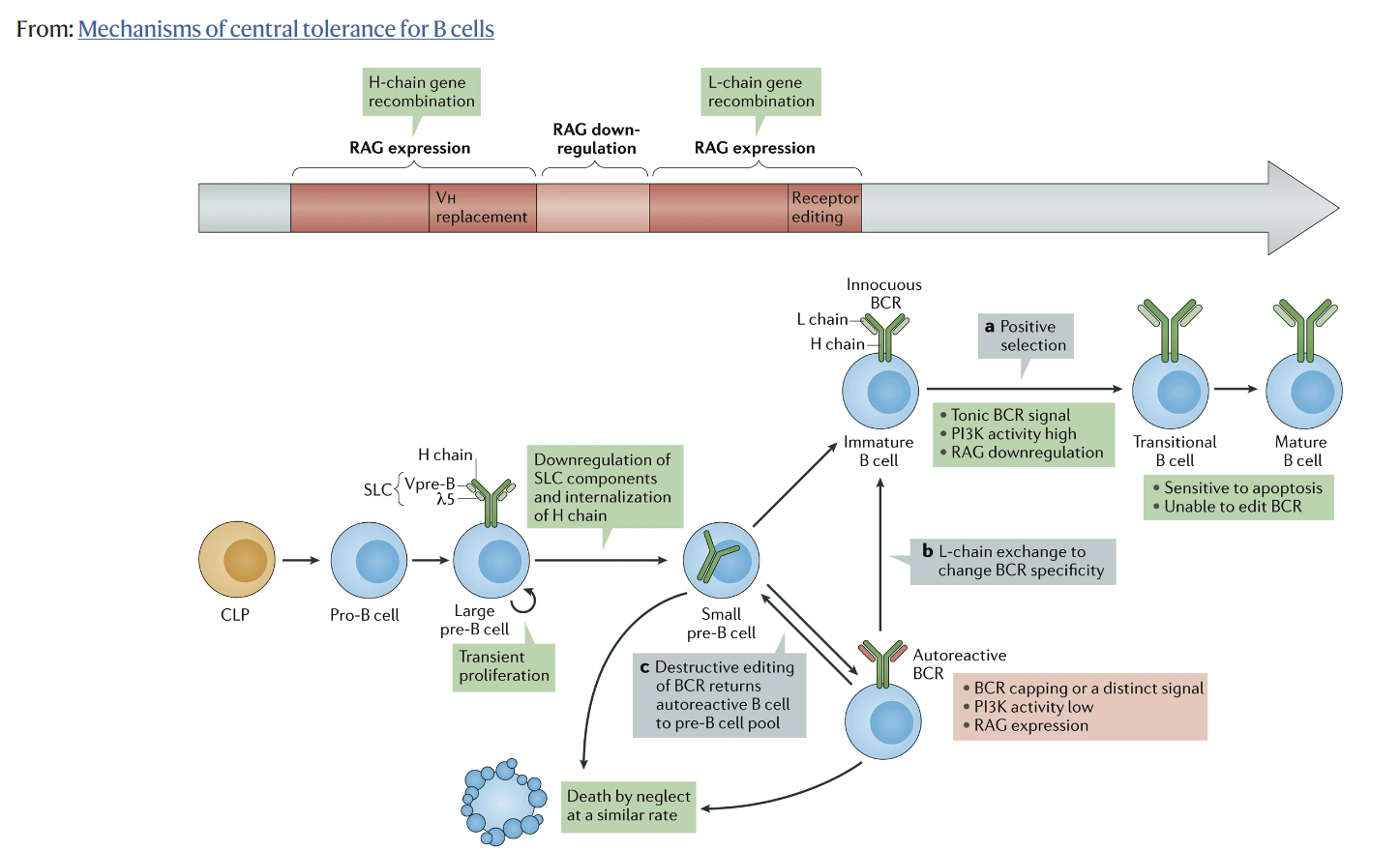
그러나
자가 반응성 B 세포의 발달의 기초가 되는
메커니즘은 잘 알려져 있지 않습니다.
여기에서는
(자가)항원 특이적 B 세포를 식별하고
특성화하기 위해 새로운 기술을 활용한 최근 연구를 검토합니다.
이러한 연구에서 얻은
관용 체크포인트 탈출과
자가 반응성 B 세포의 활성화에 관련된 메커니즘에 대한 통찰력을 논의합니다.
또한
자가면역질환에서
자가반응성 B 세포를 표적으로 삼아 제거하는
잠재적 치료 전략에 대해서도 간략히 소개합니다.
Keywords:
autoimmune diseases; B cells; autoreactive B cells; tolerance
1. Introduction
Autoimmune diseases are mostly chronic, complex, immune disorders that vary in severity from mild to lethal. It has been estimated that up to 9.4% of the world population is affected by an autoimmune disease [1]. In addition, the prevalence of autoimmune diseases is increasing, making autoimmune diseases a major disease burden globally [2]. Multiple factors are thought to be involved in the development of autoimmune diseases including genetic and environmental factors [3]. Autoimmune diseases are characterized by a loss of self-tolerance leading to an immune response against self-antigens. The autoimmune response can cause inflammation and damage to specific or multiple tissues and organs, depending on the target autoantigen.
B cells play a key role in the pathogenesis of many autoimmune diseases (Table 1). The presence of autoantibodies, and their described contribution to the pathogenesis of these B cell-mediated autoimmune diseases, is proof of B cell dependency. However, the most conclusive evidence that B cells act as key players in B cell-mediated autoimmune diseases is derived from the successful treatment of patients with therapies that specifically deplete B cells, mainly rituximab, a chimeric anti-CD20 monoclonal antibody that targets CD20+ B cells [4]. In addition to antibody production by terminally differentiated B cells (plasma cells), B cells can also contribute to disease development and progression by antibody-independent mechanisms. B cells are capable of producing cytokines which can affect the T cell response [5,6]. Moreover, B cells act as professional antigen-presenting cells (APCs) [6].
자가면역질환은
대부분 만성적이고 복잡한 면역 질환으로,
경증부터 치명적인 질환까지 그 심각도가 다양합니다.
전 세계 인구의 최대 9.4%가
자가면역 질환의 영향을 받는 것으로 추정됩니다[1].
또한
자가면역질환의 유병률이 증가하고 있어
자가면역질환은 전 세계적으로 주요 질병 부담이 되고 있습니다[2].
자가면역질환의 발병에는
유전적 요인과 환경적 요인을 포함한 여러 요인이 관여하는 것으로 알려져 있습니다[3].
자가면역질환은
자기 항원에 대한 면역 반응으로 이어지는
자기 내성의 상실을 특징으로 합니다.
자가 면역 반응은
표적 자가 항원에 따라 특정 또는 여러 조직과 장기에
염증과 손상을 일으킬 수 있습니다.
B 세포는
많은 자가 면역 질환의 발병에 핵심적인 역할을 합니다(표 1).
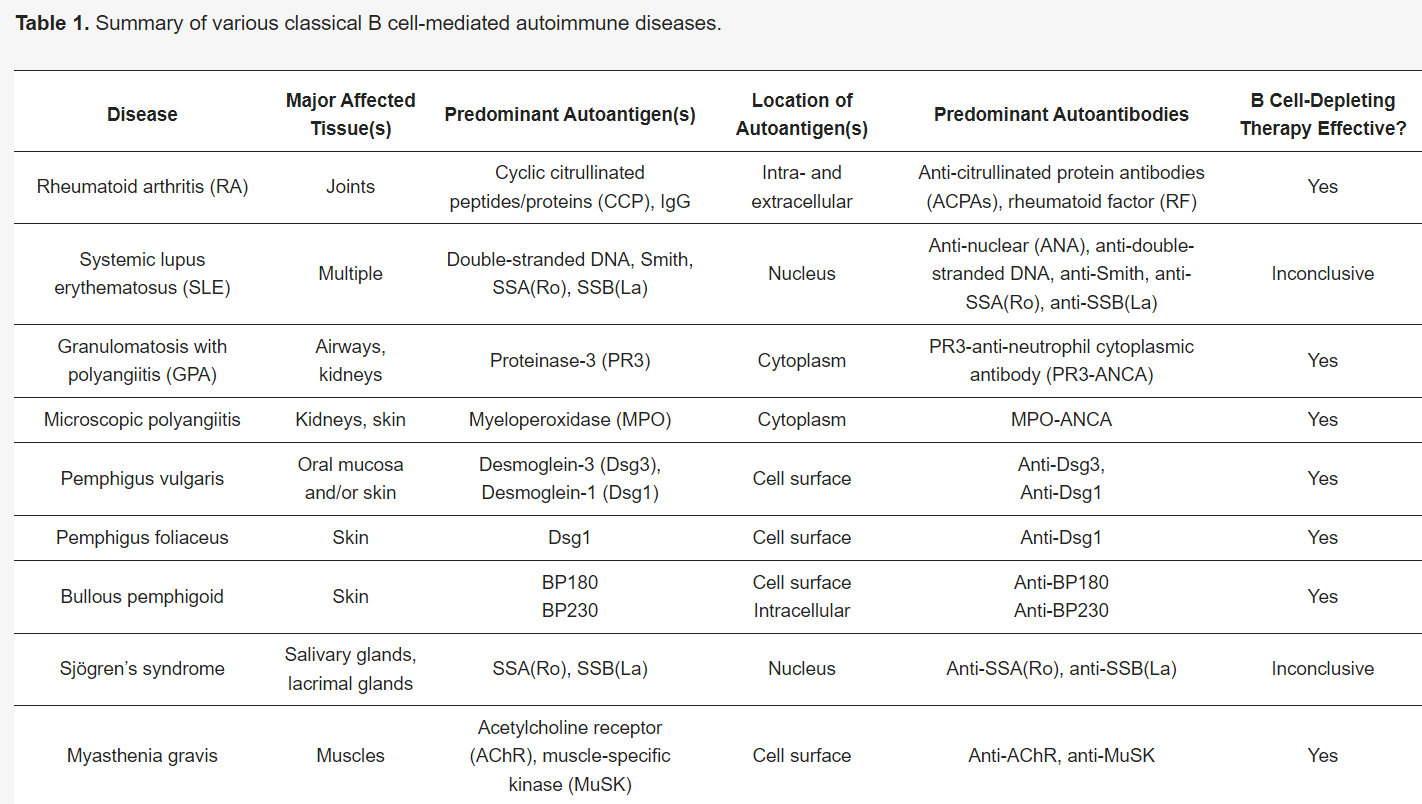
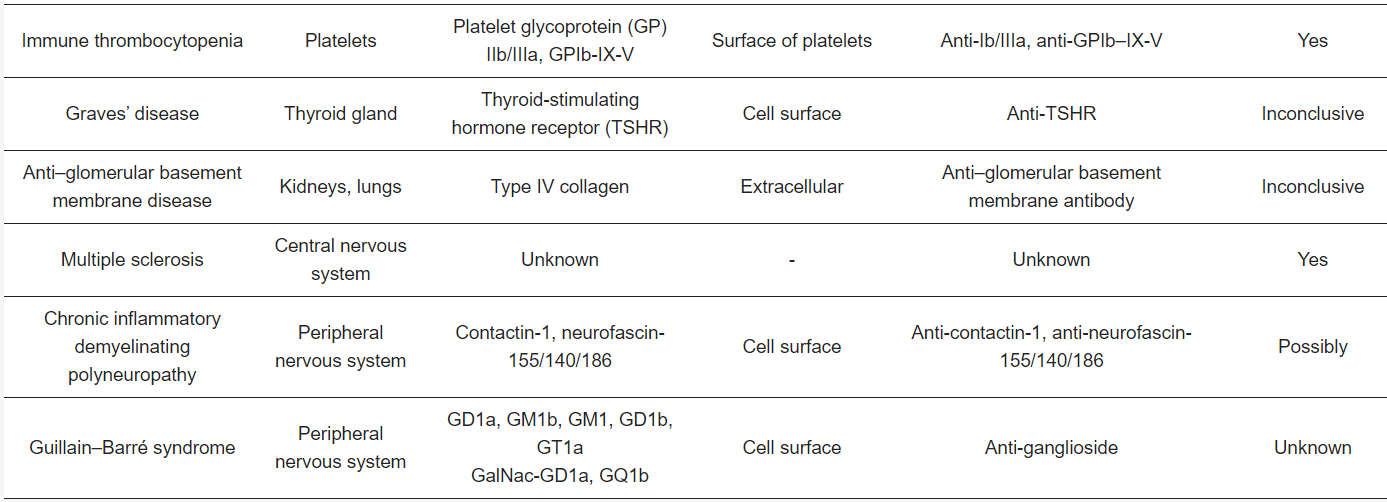
자가 항체의 존재와
이러한 B 세포 매개 자가 면역 질환의 발병 기전에 대한
자가 항체의 기여는 B 세포 의존성의 증거입니다.
그러나
B세포가
B세포 매개 자가면역질환에서
핵심적인 역할을 한다는 가장 결정적인 증거는
주로 CD20+ B세포를 표적으로 하는
키메라 항-CD20 단일클론 항체인 리툭시맙[4] 등
B세포를 특이적으로 고갈시키는 치료제로
환자를 성공적으로 치료한 데서 찾을 수 있습니다.

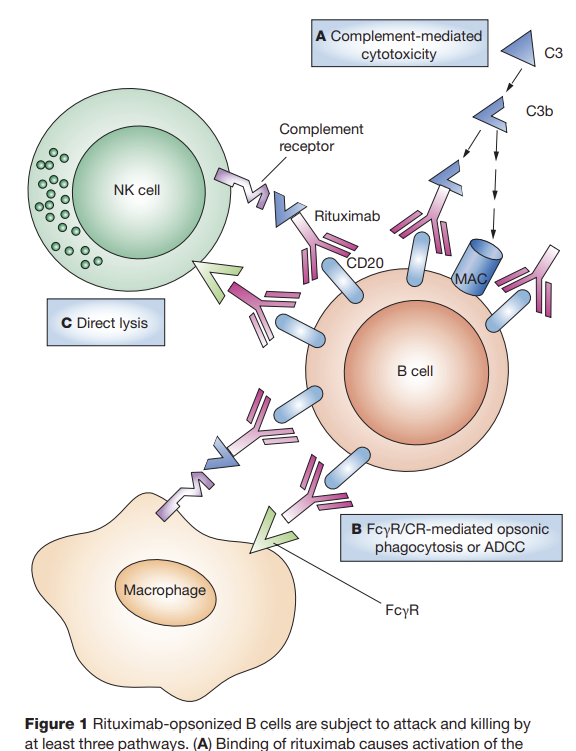
말기 분화된 B 세포(형질 세포)에 의한
항체 생산 외에도
B 세포는 항체 독립적인 메커니즘을 통해
질병 발생 및 진행에 기여할 수 있습니다.
B 세포는
T 세포 반응에 영향을 줄 수 있는
또한
B 세포는
전문 항원 제시 세포(APC)로도 작용합니다[6].
Table 1. Summary of various classical B cell-mediated autoimmune diseases.
Since B cells play a major role in autoimmune diseases, it is of high interest to understand the development of autoreactive B cells. However, our knowledge regarding autoreactive B cell development is rudimentary. During B cell development, immature B cells are generated in the bone marrow that express unique randomly assembled B cell receptors (BCRs). B cells mature in the periphery and B cell activation is initiated upon antigen binding. Activated B cells differentiate into memory and plasma cells, which provide protective immunity. As a consequence of the random process of assembling BCRs, a large proportion of the immature B cells generated in the bone marrow is autoreactive. To prevent autoimmunity, B cells are subjected to various self-tolerance checkpoints from the immature B cell stage until the plasma cell stage. Central self-tolerance checkpoint mechanisms in the bone marrow, in conjunction with self-tolerance checkpoint mechanisms in the periphery, normally prevent the occurrence of pathogenic autoreactive B cells. In autoimmune diseases, these self-tolerance checkpoints are breached; however, which exact checkpoints fail, and how this occurs, is still elusive. Moreover, the precursor cells from which pathogenic autoreactive B cells derive remain unknown. Furthermore, how autoreactive B cells are activated in disease is incompletely understood.
Research into autoreactive B cell development in humans has been hampered because techniques to detect, isolate, and characterize these low-frequency cells were lacking. In recent years, novel sophisticated and sensitive techniques have been developed that facilitate the study of low-frequency (auto)antigen-specific B cells [25].
In this review, we summarize our current knowledge pertaining to the development of autoreactive B cells with a main focus on the autoimmune diseases rheumatoid arthritis (RA), systemic lupus erythematosus (SLE), granulomatosis with polyangiitis (GPA), and pemphigus (Table 1). Following a brief overview of general B cell development and B cell tolerance, we discuss recent insights gained from studies focusing on the autoreactive B cells themselves and highlight their relevance for our understanding of disease pathogenesis and the design of novel therapeutic approaches.
B 세포는
자가 면역 질환에서 중요한 역할을 하기 때문에
자가 반응성 B 세포의 발달을 이해하는 것은 큰 관심사입니다.
그러나
자가 반응성 B 세포 발달에 관한 우리의 지식은 초보적인 수준입니다.
B 세포가 발달하는 동안
골수에서 무작위로 조립된 고유한 B 세포 수용체(BCR)를 발현하는
미성숙 B 세포가 생성됩니다.
B 세포는
말초에서 성숙하고 항원 결합에 따라
B 세포 활성화가 시작됩니다.
활성화된 B 세포는
기억 세포와 형질 세포로 분화하여
방어 면역을 제공합니다.
BCR이
무작위로 조립되는 과정의 결과로
골수에서 생성되는 미성숙 B 세포의 상당수가
자가 반응합니다.
자가 면역을 방지하기 위해
B 세포는 미성숙 B 세포 단계부터
혈장 세포 단계까지
다양한 자가 내성 체크포인트를 거칩니다.
골수의 중추 자가 관용 체크포인트 메커니즘은
말초의 자가 관용 체크포인트 메커니즘과 함께
병원성 자가 반응성 B 세포의 발생을
정상적으로 방지합니다.
자가면역질환에서는
이러한 자가관용 체크포인트에 문제가 발생하지만,
정확히 어떤 체크포인트가 어떻게 실패하는지는 아직 밝혀지지 않았습니다.
게다가
병원성 자가 반응성 B 세포가 유래하는 전구 세포는
아직 밝혀지지 않았습니다.
또한
질병에서
자가 반응성 B 세포가 어떻게 활성화되는지도
불완전하게 이해되고 있습니다.
이러한
저주파 세포를 검출, 분리, 특성화하는 기술이 부족하여 인간의 자가 반응성 B 세포 발달에 대한 연구는 난항을 겪어왔습니다. 최근에는 저주파 (자가)항원 특이적 B 세포의 연구를 용이하게 하는 정교하고 민감한 새로운 기술이 개발되었습니다 [25].
이 리뷰에서는
자가면역질환인 류마티스 관절염(RA),
전신 홍반성 루푸스(SLE),
다발성 혈관염을 동반한 육아종증(GPA),
천포창에 초점을 맞추어
자가 반응성 B 세포의 발달과 관련된 현재 지식을 요약합니다(표 1).
일반적인 B 세포 발달과
B 세포 내성에 대한 간략한 개요를 살펴본 다음,
자가 반응성 B 세포 자체에 초점을 맞춘 연구에서 얻은
최근 인사이트를 논의하고
질병 발병 기전에 대한 이해와 새로운 치료 접근법의 설계에 대한 관련성을 강조합니다.
2. General B Cell Development and B Cell Tolerance
2.1. B Cell Generation and Central Tolerance
B cell generation starts in the bone marrow, in which hematopoietic stem cells transit into immature B cells through a series of developmental steps. Immature B cells are the first cells to express a complete BCR during B cell development. Moreover, immature B cells express unique BCRs, and therefore each cell has a unique antigen specificity, as a result of the random recombination of variable (V), diversity (D), and joining (J) immunoglobulin gene segments [26]. Since the process of V(D)J recombination is random, a portion of immature B cells is autoreactive. In fact, it has been estimated that 75% of the immature B cells express BCRs that bind self-antigens [27]. Autoreactive immature B cells are subjected to central tolerance mechanisms in order to eliminate these cells from the B cell pool (Figure 1). These mechanisms include: (i) clonal deletion, i.e., removal of autoreactive B cells by the induction of apoptosis; (ii) anergy, i.e., a state of B cells characterized by unresponsiveness to antigens, downregulation of BCR expression, and a short life-span; and (iii) receptor editing, i.e., a mechanism that replaces the light chain of the BCR with a newly recombined light chain, resulting in a BCR with a different antigen specificity [28,29,30,31]. The fate of immature B cells is controlled by BCR signaling. Immature B cells with functionally unligated BCRs exhibit tonic BCR signaling, a constitutive BCR signal that is important for B cell survival and development. Moreover, whether immature B cells are subjected to central tolerance mechanisms is dependent on the strength of the interaction between the BCR and self-antigens. A moderate avidity BCR–self-antigen interaction stimulates cell survival and the continuation of development. Contrarily, a high avidity BCR–self-antigen interaction causes BCR signaling above tonic levels, resulting in developmental arrest and the subjection of cells to central tolerance mechanisms [28]. Of the total immature B cell population, around 40% are still autoreactive after the central tolerance checkpoint [27]. This relatively large remaining proportion of autoreactive immature B cells is probably because immature B cells solely encounter the self-antigens present in the microenvironment of the bone marrow. Furthermore, most autoreactivity at this stage is considered to be due to the presence of B cells that express BCRs with a low affinity for self-antigens.
2.1. B Cell Generation and Central Tolerance
B 세포 생성은
골수에서 시작되며,
조혈 줄기 세포는 일련의 발달 단계를 거쳐
미성숙 B 세포로 전환됩니다.
미성숙 B 세포는
B 세포 발달 과정에서
완전한 BCR을 발현하는 최초의 세포입니다.
또한 미성숙 B 세포는
가변성(V), 다양성(D), 결합성(J) 면역글로불린 유전자 세그먼트의
무작위 재조합의 결과로
각 세포는 고유한 BCR을 발현하므로
고유한 항원 특이성을 갖습니다 [26].
V(D)J 재조합 과정은
무작위로 이루어지기 때문에
미성숙 B 세포의 일부는 자가 반응합니다.
실제로 미성숙 B 세포의 75%는
자가 항원과 결합하는
BCR을 발현하는 것으로 추정됩니다 [27].
자가 반응하는 미성숙 B 세포는
B 세포 풀에서 이러한 세포를 제거하기 위해
중앙 관용 메커니즘을 거치게 됩니다(그림 1).
이러한 메커니즘에는 다음이 포함됩니다:
(i) 클론 결실, 즉 세포사멸 유도에 의한 자가 반응성 B 세포 제거,
(ii) 항원에 대한 무반응성, BCR 발현의 하향 조절, 짧은 수명을 특징으로 하는 B 세포의 상태,
(iii) 수용체 편집, 즉 BCR의 경쇄를 새로 재조합된 경쇄로 대체하여 다른 항원 특이성을 가진 BCR을 생성하는 메커니즘 [28,29,30,31] 등이 있습니다.
The fate of immature B cells is controlled by BCR signaling. Immature B cells with functionally unligated BCRs exhibit tonic BCR signaling, a constitutive BCR signal that is important for B cell survival and development. Moreover, whether immature B cells are subjected to central tolerance mechanisms is dependent on the strength of the interaction between the BCR and self-antigens. A moderate avidity BCR–self-antigen interaction stimulates cell survival and the continuation of development. Contrarily, a high avidity BCR–self-antigen interaction causes BCR signaling above tonic levels, resulting in developmental arrest and the subjection of cells to central tolerance mechanisms
미성숙 B 세포의 운명은
BCR 신호에 의해 제어됩니다.
기능적으로 결합이 해제된 BCR을 가진 미성숙 B 세포는
B 세포 생존과 발달에 중요한 구성적 constitutive BCR 신호인 tonic BCR 신호를 나타냅니다.
또한 미성숙 B 세포가
중추 내성 메커니즘에 노출되는지 여부는
BCR과 자가 항원 간의 상호작용 강도에 따라 달라집니다.
strength of the interaction between the BCR and self-antigens
중간 정도의 BCR-자가 항원 상호작용은
세포 생존과 발달의 지속을 자극합니다.
moderate avidity BCR–self-antigen interaction
반대로,
BCR-자가 항원 상호작용이 tonic level 수준 이상으로 높으면
BCR 신호가 발생하여
발달이 정지되고 세포가 중앙 내성 메커니즘에 복종하게 됩니다[28].
전체 미성숙 B 세포 집단 중 약 40%는
중앙 내성 체크포인트 이후에도
여전히 자가 반응합니다[27].
자가 반응성 미성숙 B 세포의 상대적으로 큰 비율은
아마도 미성숙 B 세포가 골수의 미세 환경에 존재하는
자기 항원만을 만나기 때문일 것입니다.
또한, 이 단계에서
대부분의 자가 반응성은 자가 항원에 대한 친화도가 낮은
BCR을 발현하는 B 세포의 존재로 인한 것으로 간주됩니다.
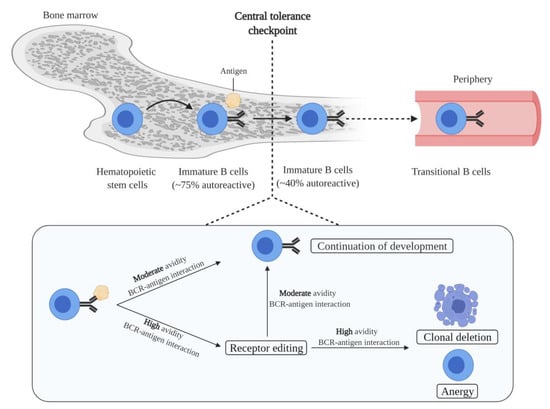
Figure 1. Elimination of autoreactive B cells by central tolerance mechanisms. The development of immature B cells from hematopoietic stem cells is accompanied by the generation of autoreactivity as the result of random variable (V), diversity (D), and joining (J) recombination. In the bone marrow, a significant proportion of autoreactive immature B cells is reduced at the central tolerance checkpoint. Central tolerance mechanisms include clonal deletion, anergy, and receptor editing. Central tolerance mechanisms are induced dependent on the binding strength between the BCR of immature B cells and self-antigens present in the bone marrow. Immature B cells that have escaped central tolerance mechanisms migrate into the periphery as transitional B cells. Figure was created with BioRender.
중앙 관용 메커니즘에 의한 자가 반응성 B 세포의 제거.
조혈 줄기세포에서 미성숙 B 세포가 발달하면
무작위 변수(V), 다양성(D), 결합(J) 재조합의 결과로
자가 반응성이 생성됩니다.
골수에서는 중앙 관용 체크포인트에서
자가 반응성 미성숙 B 세포의
상당 비율이 감소합니다.
중앙관용 메커니즘에는
클론 결실, 알레르기 및 수용체 편집이 포함됩니다.
중추관용 메커니즘은
미성숙 B 세포의 BCR과 골수에 존재하는
자가 항원 사이의 결합 강도에 따라 유도됩니다.
중추 내성 메커니즘을 벗어난
미성숙 B 세포는
과도기적 B 세포로 말초로 이동합니다.
그림은 BioRender로 생성되었습니다.
2.2. Peripheral B Cell Maturation and Tolerance
Immature B cells emerge from the bone marrow into the periphery as transitional B cells which home to the spleen. In the spleen, transitional B cells acquire IgD expression in addition to IgM and develop into mature naive B cells. As part of peripheral tolerance, transitional B cells undergo clonal deletion or attain an anergic state in response to high avidity BCR–self-antigen interactions (Figure 2A) [6]. Since other self-antigens are expressed in the spleen compared to the bone marrow, the frequency of autoreactive B cells further reduces.
미성숙 B 세포는
골수에서 비장으로 이동하는 전이 B 세포로 말초로 나옵니다.
비장에서 전이 B 세포는
IgM과 더불어 IgD 발현을 획득하고
성숙한 순진한 B 세포로 발전합니다.
말초 관용의 일환으로 과도기 B 세포는 클론 결실을 겪거나 높은 열성 BCR-자가 항원 상호 작용에 반응하여 과민성 상태에 도달합니다(그림 2A) [6]. 골수에 비해 비장에서 다른 자가 항원이 발현되기 때문에 자가 반응성 B 세포의 빈도는 더욱 감소합니다.
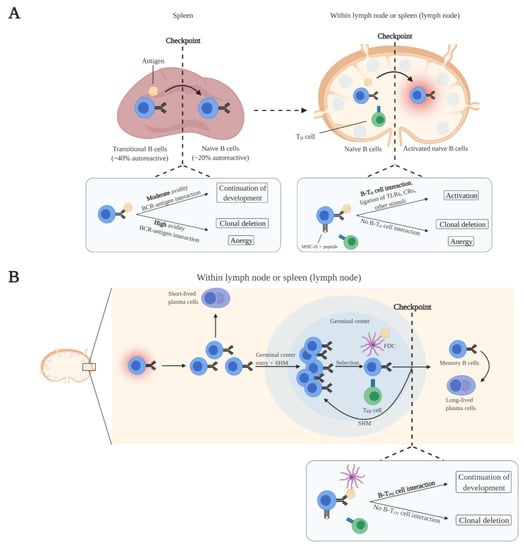
Figure 2. Elimination of autoreactive B cells by peripheral tolerance mechanisms at various checkpoints. (A) Maturation of transitional B cells takes place in the spleen. Transitional B cells that strongly bind self-antigens present in the spleen undergo clonal deletion or anergy, which reduces the frequency of autoreactive B cells. The transitional B cells that moderately bind self-antigens mature into naive B cells. Naive B cells predominantly encounter antigens within lymph nodes and the spleen. Activation of naive B cells is dependent on the binding of antigens and interaction with CD4+ T helper (TH) cells with the same antigen specificity in which B cells receive costimulatory signals. Naive B cells that do not have interaction with TH cells undergo clonal deletion or anergy, which further reduces the frequency of autoreactive B cells. (B) Activated B cells enter germinal centers within lymph nodes and the spleen, undergo somatic hypermution (SHM) and isotype switching, and ultimately mature into memory B cells and long-lived plasma cells. These maturation processes are dependent on costimulatory signals from CD4+ follicular T helper (TFH) cells with the same antigen specificity. Clonal deletion is induced in B cells that do not receive costimulatory signals from TFH cells, resulting in the removal of autoreactive B cells from the B cell pool. Figure was created with BioRender.
다양한 체크포인트에서 말초 내성 메커니즘에 의한 자가 반응성 B 세포의 제거.
(A) 비장에서 전이 B 세포의 성숙이 일어납니다. 비장에 존재하는 자가 항원에 강하게 결합하는 과도기적 B 세포는 클론 결실 또는 항원을 겪으며 자가 반응성 B 세포의 빈도를 감소시킵니다. 자기 항원과 적당히 결합하는 과도기적 B 세포는 순진한 B 세포로 성숙합니다. 순진한 B 세포는 주로 림프절과 비장에서 항원을 만나게 됩니다. 순진한 B 세포의 활성화는 항원의 결합과 B 세포가 비용 자극 신호를 수신하는 동일한 항원 특이성을 가진 CD4+ T 헬퍼(TH) 세포와의 상호작용에 따라 달라집니다. TH 세포와 상호 작용하지 않는 순진한 B 세포는 클론 결실 또는 알레르기를 겪으며, 이는 자가 반응성 B 세포의 빈도를 더욱 감소시킵니다.
(B) 활성화된 B 세포는 림프절과 비장 내 생식 센터로 들어가 체세포 과분화(SHM) 및 동종 전환을 거쳐 궁극적으로 기억 B 세포와 장수 혈장 세포로 성숙합니다. 이러한 성숙 과정은 동일한 항원 특이성을 가진 CD4+ 여포성 T 헬퍼(TFH) 세포의 비용 자극 신호에 따라 달라집니다. TFH 세포로부터 비용 자극 신호를 받지 않는 B 세포에서 클론 결실이 유도되어 B 세포 풀에서 자가 반응성 B 세포가 제거됩니다. 그림은 BioRender로 생성되었습니다.
Naive B cells continuously circulate through the blood and lymphatic system and primarily populate follicular sites within lymph nodes and the spleen. Naive B cell activation is initiated upon antigen binding. Naive B cells take up the antigen and present peptides from the antigen via MHC-II to CD4+ T helper (TH) cells with the same antigen specificity. Additionally, naive B cells express B7 upon antigen binding. B7 binds to CD28 of MHC-II-peptide-bound antigen-specific TH cells, leading to their activation. Subsequently, B7–CD28 interaction induces the expression of CD40L in TH cells. Complete activation of naive B cells is achieved by CD40L costimulation from activated antigen-specific TH cells [32,33]. In addition, other signals can aid in overcoming the BCR signaling threshold for complete naive B cell activation. These include the activation of Toll-like receptors (TLRs) and complement receptors (CRs) on naive B cells [34,35]. Concerning the elimination of autoreactive naive B cells, clonal deletion or anergy is induced in antigen-activated naive B cells when costimulation by TH cells is absent (Figure 2A).
순진한 B 세포는
혈액과 림프계를 지속적으로 순환하며
주로 림프절과 비장 내의 여포 부위에 서식합니다.
항원 결합 시 순진한 B 세포 활성화가 시작됩니다.
순진한 B 세포는
항원을 받아들여 MHC-II를 통해
항원의 펩타이드를 동일한 항원 특이성을 가진
CD4+ T 헬퍼(TH) 세포에 제시합니다.
또한, 순진한 B 세포는
항원 결합 시 B7을 발현합니다.
B7은
MHC-II-펩타이드 결합 항원 특이적 TH 세포의
CD28에 결합하여 활성화로 이어집니다.
이후 B7-CD28 상호 작용은 TH 세포에서 CD40L의 발현을 유도합니다. 순진한 B 세포의 완전한 활성화는 활성화된 항원 특이적 TH 세포의 CD40L 비용 자극에 의해 이루어집니다 [32,33]. 또한 다른 신호가 완전한 순진한 B 세포 활성화를 위한 BCR 신호 역치를 극복하는 데 도움이 될 수 있습니다. 여기에는 순진한 B 세포에서 톨 유사 수용체(TLR)와 보체 수용체(CR)의 활성화가 포함됩니다 [34,35].
자가 반응성 순진한 B 세포의 제거와 관련하여,
TH 세포에 의한 비용 자극이 없을 때
항원 활성화된 순진한 B 세포에서 클론 결실 또는
알레르기가 유도됩니다(그림 2A).
Costimulation is absent as a result of the elimination of autoantigen-specific T cells by central tolerance mechanisms in the thymus and peripheral tolerance mechanisms. Ultimately, the autoreactive B cell pool is greatly reduced. It has been demonstrated that approximately 20% of the peripheral naive B cells are autoreactive [27]. However, this presumably represents a mix of low-affinity autoreactive naive B cells together with autoreactive naive B cells undergoing anergy or clonal deletion.
When naive B cells are fully activated, they proliferate and differentiate into short-lived plasma cells at extrafollicular sites within the lymph node or the spleen. Short-lived plasma cells predominantly express low-affinity IgM antibodies. Besides migration and development at extrafollicular sites, activated naive B cells migrate into germinal centers. There, the B cells proliferate and undergo somatic hypermutation (SHM) of their immunoglobulin V genes, which affects BCR affinity. B cells that have the highest affinity for antigen retained on follicular dendritic cells (FDCs), and that present antigen to CD4+ follicular T helper (TFH) cells with the same antigen specificity, are positively selected. These positively selected B cells receive costimulatory signals (CD40L) from TFH cells to undergo repetitive rounds of proliferation, SHM, and selection [36,37]. Ultimately, positively selected high-affinity B cells undergo class switching and will mature into memory B cells and long-lived plasma cells [37,38]. Regarding autoreactivity and tolerance, SHM may lead to the generation of autoreactive B cells because this is a random process. The self-tolerance checkpoints after B cell activation are not well-defined. It has been described that B cells that do not receive costimulatory signals from TFH cells are removed by clonal deletion (Figure 2B) [39]. The absence of costimulatory signals is the result of the purge of autoantigen-specific TFH cells by T cell tolerance mechanisms during T cell development. Furthermore, as a byproduct of SHM, SHM-mediated receptor editing of autoreactive B cells can decrease the affinity for autoantigens, thereby removing autoreactivity [39].
While the majority of autoreactive B cells are deleted during B cell maturation, a small fraction of the normal B cell repertoire in healthy individuals is autoreactive. These non-pathogenic natural autoreactive B cells, which do not enter germinal centers, predominantly produce low-affinity polyreactive IgM antibodies and to a lesser extent low-affinity polyreactive IgG and IgA antibodies [40,41]. Various functions have been ascribed to natural autoreactive B cells, including clearance of cellular debris from apoptotic cells and inhibition of autoimmune inflammatory processes by masking self-antigens [40,41].
Overall, several self-tolerance checkpoints are present during normal B cell development, at various (micro)anatomical sites, and during different developmental stages, to prevent the occurrence of pathogenic autoreactive B cells.
흉선의 중추 내성 메커니즘과 말초 내성 메커니즘에 의한
자가 항원 특이적 T 세포의 제거로 인해
비용 자극이 부재합니다.
궁극적으로
자가 반응성 B 세포 풀이 크게 감소합니다.
말초 순진한 B 세포의
약 20%가 자가 반응성이 있다는 것이 입증되었습니다 [27].
그러나 이는 아마도
저친화성 자가반응성 순진한 B 세포와
알레르기 또는 클론 결실을 겪는
자가반응성 순진한 B 세포가 혼합된 것으로 추정됩니다.
순진한 B 세포가 완전히 활성화되면
림프절 또는 비장 내 여포 외 부위에서
단수명 형질 세포로 증식 및 분화합니다.
단수명 형질세포는 주로
저친화성 IgM 항체를 발현합니다.
여포 외 부위에서의 이동 및 발달 외에도 활성화된 순진한 B 세포는 생식 센터로 이동합니다. 그곳에서 B세포는 증식하고 면역글로불린 V 유전자의 체세포 과돌연변이(SHM)를 겪으며, 이는 BCR 친화성에 영향을 미칩니다. 여포 수지상 세포(FDC)에 보유된 항원에 대한 친화력이 가장 높고 동일한 항원 특이성을 가진 CD4+ 여포 T 헬퍼(TFH) 세포에 항원을 제시하는 B 세포가 양성으로 선택됩니다. 이렇게 양성으로 선택된 B 세포는 TFH 세포로부터 비용 자극 신호(CD40L)를 받아 증식, SHM, 선택 과정을 반복적으로 거치게 됩니다[36,37]. 궁극적으로 양성으로 선택된 고친화성 B 세포는 클래스 전환을 거쳐 기억 B 세포와 수명이 긴 형질 세포로 성숙하게 됩니다[37,38]. 자가 반응성 및 내성과 관련하여 SHM은 무작위 과정이기 때문에 자가 반응성 B 세포의 생성으로 이어질 수 있습니다. B 세포 활성화 후 자가 내성 체크포인트는 잘 정의되어 있지 않습니다. TFH 세포로부터 비용 자극 신호를 받지 않는 B 세포는 클론 결실에 의해 제거된다고 설명되어 있습니다(그림 2B) [39]. 비용 자극 신호의 부재는 T 세포 발달 과정에서 T 세포 관용 메커니즘에 의해 자가 항원 특이적 TFH 세포가 제거된 결과입니다. 또한, SHM의 부산물로서 자가 반응성 B 세포의 SHM 매개 수용체 편집은 자가 항원에 대한 친화력을 감소시켜 자가 반응성을 제거할 수 있습니다 [39].
자가 반응성 B 세포의 대부분은 B 세포 성숙 과정에서 삭제되지만, 건강한 개인의 정상 B 세포 레퍼토리 중 일부는 자가 반응성을 보입니다. 이러한 비병원성 자연 자가 반응성 B 세포는 생식 센터에 들어가지 않으며, 주로 저친화성 다중 반응성 IgM 항체를 생산하고 저친화성 다중 반응성 IgG 및 IgA 항체를 덜 생산합니다[40,41]. 자연적인 자가 반응 B 세포에는 세포 사멸 세포에서 세포 파편 제거, 자가 항원을 마스킹하여 자가 면역 염증 과정의 억제 등 다양한 기능이 있습니다[40,41].
전반적으로, 병원성 자가 반응 B 세포의 발생을 방지하기 위해 정상적인 B 세포 발달 과정, 다양한 (미세) 해부학적 부위 및 다양한 발달 단계에 여러 자기 내성 체크포인트가 존재합니다.
3. Autoreactive B Cell Development in Autoimmune Diseases
3.1. Compromised Self-Tolerance Checkpoints and Precursor Cells of Autoreactive B Cells
Two issues in the field of autoimmunity that have remained unresolved thus far are the identification of the precise self-tolerance checkpoints that are breached and the characterization of the precursors from which autoreactive B cells derive. Recently, multiple studies have focused on the B cell subset distribution and phenotypical characterization of autoreactive B cells in autoimmune diseases and health, providing novel insights into the mechanisms underlying the development of autoreactive B cells.
Within the circulating pool of autoreactive B cells, an enrichment of memory/class-switched memory B cells has been demonstrated in RA, GPA, and pemphigus vulgaris patients when compared to the total B cell compartment of patients and/or the (natural) autoreactive B cell compartment of healthy individuals [42,43,44]. These observations imply a breach of self-tolerance within germinal centers in these diseases. Normally, a self-tolerance checkpoint exists within germinal centers in which autoreactive B cells are unable to develop in germinal centers due to the absence of TFH cell support (Figure 2B). Hence, this would also suggest that the self-tolerance of TFH cells is defective, or that other mechanisms are present that circumvent TFH dependency. Moreover, these observations indicate that autoreactive B cells in RA, GPA, and pemphigus vulgaris derive from germinal centers since only memory/class-switched memory B cells were enriched and memory B cell generation and class switching mostly take place at these sites. Furthermore, autoreactive B cells may be generated from non-autoreactive B cells or non-pathogenic autoreactive B cells with a very low affinity for the autoantigen, as recently proposed by Cho et al. [45]. In their study, single-cell repertoire analysis of circulating Dsg3-specific memory B cells from pemphigus vulgaris patients revealed that these cells were mostly class-switched and affinity-matured. Interestingly, when monoclonal Dsg3-specific autoantibodies derived from patients were germline-reverted, they could no longer bind Dsg3, implying that these cells have acquired pathogenic autoreactive properties through SHM. Similarly, another study found that germline-reverted ACPAs lost or reduced reactivity to citrullinated proteins, suggesting a similar mechanism in RA [46]. The observation that germinal center-derived circulating autoreactive class-switched memory B cells, which are most likely affinity-matured cells, are enriched in GPA suggests also a similar mechanism in this disease [43].
A different mechanism for the development of autoreactive B cells has been proposed for SLE. Analysis of the circulating nuclear antigen-specific B cell frequencies, within the different B cell subsets, demonstrated that the nuclear antigen-specific B cells in SLE patients are eliminated to the same extent as the nuclear antigen-specific B cells in healthy individuals, suggesting properly functioning central and peripheral tolerance checkpoints in SLE [47]. However, anergy induction of circulating naive nuclear antigen-specific B cells in SLE patients appeared to be defective since the frequency of circulating anergic cells (IgMlow) within the naive nuclear antigen-specific B cell compartment was found to be significantly lower compared to healthy individuals [47]. Moreover, de la Varga-Martiınez and colleagues observed a higher frequency of CD69+ activated B cells within the circulating nuclear antigen-specific B cell compartment of SLE patients compared to the total B cell compartment of the same patients [48]. Intriguingly, the activated nuclear antigen-specific B cells were mostly of the naive subset. These studies indicate that a fraction of naive nuclear antigen-specific B cells in SLE possibly escape early tolerance checkpoints due to failure of anergy induction and can be activated. This suggests that activated naive autoreactive B cells may be the precursor cells of autoreactive B cells in SLE. However, tolerance could also be breached in germinal centers, and autoreactive B cells can be generated from non-autoreactive B cells/non-pathogenic autoreactive B cells with a low affinity for autoantigens, since germline-reverted autoantibodies of SLE patients were demonstrated to be unreactive to SLE-related autoantigens [49,50].
Together, a breach of tolerance checkpoints in germinal centers appears to be a common mechanism that permits autoreactive B cells development across different autoimmune diseases. Moreover, current evidence regarding the precursors of autoreactive B cells, in diseases with high-affinity autoantibodies, points to non-autoreactive B cells/non-pathogenic autoreactive B cells with low affinity for the autoantigen that are further selected for high affinity in germinal centers after SHM. In SLE, early checkpoints may be additionally breached, and autoreactive B cells may also derive from activated naive autoreactive B cells, in which anergy induction is impaired.
3.1. 손상된 자가 내성 체크포인트와 자가 반응성 B 세포의 전구 세포
자가면역 분야에서 지금까지 해결되지 않은 두 가지 문제는 자가면역 체크포인트를 정확히 파악하는 것과 자가반응성 B 세포가 유래하는 전구세포의 특성을 규명하는 것입니다. 최근에는 자가면역 질환과 건강에서 자가반응성 B 세포의 B 세포 하위 집합 분포와 표현형 특성 분석에 초점을 맞춘 여러 연구가 진행되어 자가반응성 B 세포의 발생 메커니즘에 대한 새로운 통찰력을 제공하고 있습니다.
자가 반응성 B 세포의 순환 풀 내에서 환자의 전체 B 세포 구획 및/또는 건강한 개인의 (자연) 자가 반응성 B 세포 구획과 비교할 때 RA, GPA 및 천포창 환자에서 기억/클래스 전환 기억 B 세포의 농축이 입증되었습니다 [42,43,44]. 이러한 관찰은 이러한 질환의 생식세포 내 자가 내성이 위반되었음을 의미합니다. 일반적으로 자가관용 체크포인트는 배아 중심 내에 존재하며, TFH 세포의 지원이 없기 때문에 자가반응성 B 세포가 배아 중심 내에서 발달할 수 없습니다(그림 2B). 따라서 이는 TFH 세포의 자가 내성에 결함이 있거나 TFH 의존성을 우회하는 다른 메커니즘이 존재한다는 것을 시사합니다. 또한, 이러한 관찰은 RA, GPA 및 천포창의 자가 반응성 B 세포가 배아 중심부에서 유래한다는 것을 나타내는데, 이는 기억/클래스 전환 기억 B 세포만 농축되었고 기억 B 세포 생성 및 클래스 전환이 대부분 이 부위에서 일어나기 때문입니다. 또한, 자가 반응성 B 세포는 비자가 반응성 B 세포 또는 자가 항원에 대한 친화력이 매우 낮은 비병원성 자가 반응성 B 세포에서 생성될 수 있으며, 최근 Cho 등이 제안한 것처럼 [45]. 이들의 연구에서 천포창 환자의 순환하는 Dsg3 특이적 기억 B 세포의 단일 세포 레퍼토리를 분석한 결과, 이 세포는 대부분 클래스 전환 및 친화성 성숙 세포로 밝혀졌습니다. 흥미롭게도 환자에서 추출한 단일 클론 Dsg3 특이적 자가항체를 생식세포로 되돌린 경우 더 이상 Dsg3에 결합할 수 없었으며, 이는 이 세포가 SHM을 통해 병원성 자가반응 특성을 획득했음을 시사합니다. 마찬가지로, 또 다른 연구에서는 생식세포로 되돌아간 ACPA가 시트룰린화 단백질에 대한 반응성을 잃거나 감소시키는 것으로 나타나 RA에서도 유사한 메커니즘이 있음을 시사했습니다 [46]. 친화성 성숙 세포일 가능성이 가장 높은 배아 중심 유래 순환 자가 반응성 클래스 전환 기억 B 세포가 GPA에 풍부하다는 관찰도 이 질환에서 유사한 메커니즘을 시사합니다 [43].
자가 반응성 B 세포의 발달에 대한 다른 메커니즘이 SLE에 대해 제안되었습니다. 다양한 B 세포 하위 집합 내에서 순환하는 핵 항원 특이 B 세포 주파수를 분석한 결과, SLE 환자의 핵 항원 특이 B 세포가 건강한 사람의 핵 항원 특이 B 세포와 동일한 정도로 제거되어 SLE에서 중추 및 말초 내성 체크포인트가 제대로 기능하는 것으로 나타났습니다 [47]. 그러나, 순진한 핵항원 특이 B 세포 구획 내에서 순진한 핵항원 특이 B 세포의 순환 알레르기 세포(IgMlow)의 빈도가 건강한 개인에 비해 현저히 낮은 것으로 밝혀졌기 때문에 SLE 환자에서 순환 순진한 핵항원 특이 B 세포의 알레르기 유발에 결함이 있는 것으로 나타났습니다 [47]. 또한 드 라 바르가-마르티네즈와 동료들은 같은 환자의 전체 B 세포 구획에 비해 SLE 환자의 순환 핵 항원 특이적 B 세포 구획 내에서 CD69+ 활성화된 B 세포의 빈도가 더 높다는 것을 관찰했습니다 [48]. 흥미롭게도 활성화된 핵 항원 특이적 B 세포는 대부분 순진한 하위 집합에 속했습니다. 이러한 연구는 SLE에서 순진한 핵 항원 특이 B 세포의 일부가 알레르기 유도의 실패로 인해 초기 내성 체크포인트에서 벗어나 활성화될 수 있음을 나타냅니다. 이는 활성화된 순진한 자가 반응성 B 세포가 SLE에서 자가 반응성 B 세포의 전구 세포일 수 있음을 시사합니다. 그러나 내성은 생식 센터에서도 위반될 수 있으며, 자가항원에 대한 친화력이 낮은 비자가 반응성 B 세포/비병원성 자가 반응성 B 세포에서 자가 반응성 B 세포가 생성될 수 있는데, 이는 SLE 환자의 생식선 회귀 자가항체가 SLE 관련 자가항원에 대해 반응하지 않는 것으로 입증되었기 때문입니다 [49,50].
종합하면,
생식 중추의 내성 체크포인트 위반은
다양한 자가면역 질환에서 자가반응성 B 세포의 발달을 허용하는
공통 메커니즘인 것으로 보입니다.
또한, 고친화성 자가항체가 있는 질환에서
자가반응성 B 세포의 전구체에 관한 현재의 증거는
자가항원에 대한 친화력이 낮은 비자가반응성 B 세포/비병원성 자가반응성 B 세포가
SHM 이후 생식 중추에서 높은 친화력을 위해 추가로 선택된다는 점을 지적합니다.
SLE에서는 초기 체크포인트가 추가로 위반될 수 있으며, 자가 반응성 B 세포는 알레르기 유도가 손상된 활성화된 순진한 자가 반응성 B 세포에서 유래할 수도 있습니다.
3.2. Activation of Autoreactive B Cells
T cell-dependent activation of B cells requires the availability of antigens that can bind to the BCR and are subsequently internalized, processed, and presented by MHC-II molecules on the cell surface to activate antigen-specific TH cells. Additionally, stimulation of innate immune receptors, such as TLRs and CRs, can help to promote the activation of B cells. Data on the activation of autoreactive B cells, and whether this deviates from normal B cell activation, are scarce since most studies have not specifically investigated activation of the autoreactive B cells themselves. Therefore, we summarize the activation mechanisms mainly from studies that have investigated these mechanisms indirectly.
B cells encounter extracellular antigens circulating through the secondary lymphoid organs or presented by specialized APCs that present antigens in their native state [51]. Autoantigens can be present at various intra- and extracellular locations depending on the disease type and target autoantigen(s) (Table 1). RA is an example in which autoantigens are persistently available since most citrullinated peptides, including citrullinated fibrinogen and type II collagen, are located extracellularly [52,53]. However, in many autoimmune diseases, the autoantigens are usually not located extracellularly and therefore not available to autoreactive B cells. Infection, injury, and inflammation are likely important factors for increasing the availability of such autoantigens. These factors cause cellular damage and the release of autoantigens. Importantly, disruptive cellular debris clearance, necrosis, and impaired degradation of neutrophil extracellular traps have been associated with increased susceptibility to autoimmune diseases due to an increased presence of autoantigens extracellularly [54]. In SLE in particular, it has been reported that apoptotic cell clearance is deficient, due to decreased phagocytosis, leading to secondary necrosis of apoptotic cells [55]. Subsequently, intracellular antigens are released. In addition, degradation of neutrophil extracellular traps is impaired in SLE causing intracellular antigens to be present longer extracellularly [55]. Moreover, intracellular autoantigens can be released by cells during inflammatory processes as part of their normal function. As an example, the autoantigen in GPA, proteinase 3 (PR3), is a protease involved in the degradation of extracellular matrix proteins, which is released from neutrophils upon activation during inflammation, thus exposing PR3-specific B cells to their autoantigen [56,57,58].
Since T cell-dependent activation of B cells requires T cell help for complete activation, T cells with the same antigen specificity as the autoreactive B cells should be present in autoimmune diseases to support autoreactive B cell activation. Autoantigen-specific T cells have been detected in several autoimmune diseases including RA, SLE, GPA, and pemphigus vulgaris [59,60,61,62]. In RA patients for example, T cells reactive against various RA-associated citrullinated peptides have been detected in the peripheral blood and lymphoid tissues [59]. For the detection, tetramers were used consisting of citrullinated peptides bound to HLA-DRB1*04:01, which is encoded by an HLA class II RA risk allele associated with binding and presenting citrullinated peptides. The binding of autoantigen-MHC-II to T cells suggests that T cells are present that recognize autoantigens displayed by MHC-II, thus having the same antigen specificity as autoreactive B cells. These studies suggest that, in these autoimmune diseases, in addition to a breach in B cell tolerance, T cell tolerance has failed as well.
B cells express various pattern recognition receptors including several TLRs, which act as co-receptors for the BCR. Among the TLRs expressed by B cells, activation of TLR7 and TLR9 has been associated with autoimmunity [63]. TLR7 and TLR9 are located in the endosomal compartment of cells and are activated upon binding of ssRNA and DNA with unmethylated CpGs, respectively [64]. Synergistic activation of the BCR and TLR7 or TLR9 with nuclear antigens has been shown to activate autoreactive rheumatoid factor (RF)-specific murine B cells [65,66,67]. Intriguingly, TLR7/TLR9 activation and BAFF overexpression have been associated with the activation of autoreactive B cells in SLE-like mouse models independent of T cells [68,69]. Since autoantigens in SLE are mostly ligands for both the BCR and TLRs, it can be hypothesized that stimulation by such antigens alone could be sufficient for the activation of autoreactive B cells independent of T cell signals [53]. These data implicate that extracellular nuclear antigens, when available, may contribute to autoreactive B cell activation by simultaneously activating TLRs.
Taken together, T cell-dependent activation of autoreactive B cells can be initiated, depending on the autoimmune disease, by autoantigens that are persistently expressed extracellularly. However, in most diseases, the autoantigens are not located extracellularly and factors such as infection and defective apoptotic cell clearance are responsible for autoantigens to be released and become accessible for autoreactive B cells. The observation that autoantigen-specific T cells are found in many autoimmune diseases suggests that in these diseases autoreactive B cell-autoantigen-specific T cell interaction can take place for the activation of autoreactive B cells. Furthermore, activation of TLRs expressed by B cells may aid in autoreactive B cell activation and possibly can activate these cells in a T cell-independent fashion, for example in SLE.
T 세포에 의존하는 B 세포의 활성화를 위해서는
BCR에 결합할 수 있는 항원이 있어야 하고,
이후 세포 표면의 MHC-II 분자에 의해 내재화, 처리 및 제시되어
항원 특이적인 TH 세포를 활성화해야 합니다.
또한 TLR 및 CR과 같은 선천성 면역 수용체의 자극은
B 세포의 활성화를 촉진하는 데 도움이 될 수 있습니다.
대부분의 연구에서
자가 반응성 B 세포 자체의 활성화를 구체적으로 조사하지 않았기 때문에
자가 반응성 B 세포의 활성화와
이것이 정상적인 B 세포 활성화에서 벗어나는지에 대한 데이터는 부족합니다.
따라서 이러한 메커니즘을 간접적으로 조사한 연구를 중심으로 활성화 메커니즘을 요약합니다.
B 세포는
이차 림프 기관을 통해 순환하는 세포 외 항원을 만나거나
항원을 원래 상태로 제시하는
특수 APC에 의해 제시됩니다 [51].
자가 항원은
질병 유형과 표적 자가 항원에 따라
다양한 세포 내 및 세포 외 위치에 존재할 수 있습니다(표 1).
RA는 시트룰린화 피브리노겐과 II형 콜라겐을 포함한 대부분의 시트룰린화 펩타이드가 세포 외부에 위치하기 때문에 자가항원이 지속적으로 이용 가능한 예입니다[52,53]. 그러나 많은 자가면역질환에서 자가항원은 일반적으로 세포외에 위치하지 않으므로 자가반응 B 세포에서 사용할 수 없습니다. 감염, 부상, 염증은 이러한 자가 항원의 가용성을 높이는 중요한 요인일 가능성이 높습니다. 이러한 요인은 세포 손상과 자가 항원의 방출을 유발합니다. 중요한 것은 세포 파편 제거, 괴사, 호중구 세포 외 트랩의 분해 장애가 자가 항원의 세포 외 존재 증가로 인한 자가 면역 질환에 대한 감수성 증가와 관련이 있다는 점입니다 [54]. 특히 SLE에서는 식세포 작용 감소로 인해 아포토시스 세포 제거가 부족하여 아포토시스 세포의 이차 괴사를 초래하는 것으로 보고되었습니다 [55]. 그 후 세포 내 항원이 방출됩니다. 또한, 호중구 세포 외 트랩의 분해가 SLE에서 손상되어 세포 내 항원이 세포 외로 더 오래 존재하게 됩니다 [55]. 또한 세포 내 자가 항원은 염증 과정 중에 세포가 정상 기능의 일부로 방출할 수 있습니다. 예를 들어, GPA의 자가 항원인 프로테아제 3(PR3)은 세포 외 기질 단백질의 분해에 관여하는 프로테아제로, 염증 중 활성화 시 호중구에서 방출되어 PR3 특이적 B 세포를 자가 항원에 노출시킵니다 [56,57,58].
B 세포의 T 세포 의존적 활성화는 완전한 활성화를 위해 T 세포의 도움이 필요하기 때문에 자가 반응성 B 세포와 동일한 항원 특이성을 가진 T 세포가 자가 면역 질환에 존재하여 자가 반응성 B 세포 활성화를 지원해야 합니다. 자가 항원 특이적 T 세포는 RA, SLE, GPA 및 천포창을 포함한 여러 자가 면역 질환에서 발견되었습니다 [59,60,61,62]. 예를 들어 RA 환자의 경우 말초 혈액과 림프 조직에서 다양한 RA 관련 시트룰린화 펩티드에 반응하는 T 세포가 검출되었습니다[59]. 검출을 위해 시트룰린화 펩타이드의 결합 및 제시와 관련된 HLA 클래스 II RA 위험 대립 유전자에 의해 코딩되는 HLA-DRB1*04:01에 결합된 시트룰린화 펩타이드로 구성된 테트라머를 사용했습니다. 자가항원-MHC-II와 T 세포의 결합은 MHC-II가 표시하는 자가항원을 인식하는 T 세포가 존재하며, 따라서 자가반응성 B 세포와 동일한 항원 특이성을 갖는다는 것을 시사합니다. 이러한 연구는 이러한 자가면역 질환에서 B 세포 내성에 문제가 있을 뿐만 아니라 T 세포 내성에도 문제가 있음을 시사합니다.
B 세포는 BCR의 공동 수용체 역할을 하는 여러 TLR을 포함한 다양한 패턴 인식 수용체를 발현합니다. B 세포에 의해 발현되는 TLR 중 TLR7 및 TLR9의 활성화는 자가면역과 관련이 있습니다 [63]. TLR7과 TLR9은 세포의 소포체 구획에 위치하며, 각각 ssRNA와 DNA가 메틸화되지 않은 CpG와 결합하면 활성화됩니다 [64]. 핵 항원과 함께 BCR 및 TLR7 또는 TLR9의 시너지 활성화는 자가 반응성 류마티스 인자(RF) 특이적 쥐 B 세포를 활성화하는 것으로 나타났습니다 [65,66,67]. 흥미롭게도 TLR7/TLR9 활성화 및 BAFF 과발현은 T 세포와 무관하게 SLE 유사 마우스 모델에서 자가 반응성 B 세포의 활성화와 관련이 있습니다 [68,69]. SLE의 자가 항원은 대부분 BCR과 TLR 모두에 대한 리간드이기 때문에 이러한 항원에 의한 자극만으로도 T 세포 신호와 무관하게 자가 반응성 B 세포를 활성화할 수 있다는 가설을 세울 수 있습니다 [53]. 이러한 데이터는 세포 외 핵 항원이 존재할 경우 TLR을 동시에 활성화하여 자가 반응성 B 세포 활성화에 기여할 수 있음을 시사합니다.
종합하면 자가면역 질환에 따라 세포 외에서 지속적으로 발현되는 자가 항원에 의해 자가 반응성 B 세포의 T 세포 의존적 활성화가 시작될 수 있습니다. 그러나 대부분의 질환에서 자가 항원은 세포 외적으로 존재하지 않으며 감염 및 세포 사멸 결함 등의 요인으로 인해 자가 항원이 방출되어 자가 반응성 B 세포에 접근할 수 있게 됩니다. 자가 항원 특이적 T 세포가 많은 자가 면역 질환에서 발견된다는 관찰은 이러한 질환에서 자가 반응성 B 세포와 자가 항원 특이적 T 세포의 상호 작용이 자가 반응성 B 세포의 활성화를 위해 일어날 수 있음을 시사합니다. 또한, B 세포에 의해 발현되는 TLR의 활성화는 자가 반응성 B 세포의 활성화를 도울 수 있으며, 예를 들어 SLE에서와 같이 T 세포와 독립적인 방식으로 이러한 세포를 활성화할 수 있습니다.
3.3. Escape of Tolerance
Self-tolerance is compromised in autoimmunity, but how autoreactive B cells escape self-tolerance checkpoints is largely unknown. Recent studies have shed some light on this issue.
Genetic predisposition and environmental triggers are known to play a central role in the breakdown of tolerance, leading to the escape of autoreactive B cells from tolerance mechanisms. Various genetic variants of HLA genes, B/T cell signaling genes, stimulatory/inhibitory signaling pathways, and cytokine/cytokine receptor genes have been identified as risk factors associated with loss of self-tolerance and development of autoimmunity [70]. To illustrate the role of genetic susceptibility, Joshua et al. analyzed the effect of carriage of the genetic variant of the PTPN22 gene (R620W) on the frequency of circulating citrullinated fibrinogen peptide-specific B cells in RA patients [71]. PTPN22 is a protein tyrosine phosphatase involved in BCR signaling. The R620W variant of the PTPN22 gene has been identified as a strong risk factor for several autoimmune diseases, possibly by affecting BCR signaling and selection of autoreactive B cells during development [72]. The authors reported a trend towards increased frequencies of circulating citrullinated fibrinogen peptide-specific B cells in RA patients carrying the R620W variant compared to non-R620W-carrying RA patients. Thereby, they suggested a link between genetic risk factors and breach of B cell self-tolerance.
Aberrant BCR signaling, in particular enhanced Bruton’s tyrosine kinase (BTK) activity, is possibly implicated in the breach of self-tolerance in autoimmunity. Components of the BCR signaling pathway control the activation threshold of B cells, therefore they play a crucial role in maintaining self-tolerance. BTK is a tyrosine kinase in the BCR signaling pathway that plays a central role in transducing BCR signals into downstream signals which result in B cell activation and survival [73]. It has been shown that autoimmunity is induced in transgenic mice overexpressing human BTK in B cells and that inhibition of BTK is effective in reducing or preventing autoimmunity in various murine autoimmune models [74]. These mouse studies suggest that BTK overexpression can breakdown B cell tolerance [74,75]. Interestingly, compared to healthy individuals, elevated BTK levels were observed in circulating B cell subsets, including naive B cells, of patients with ACPA-positive RA, Sjögren’s syndrome, and active GPA [76,77]. Moreover, it has been demonstrated in active GPA patients that the B cell subsets with elevated BTK levels (transitional and naive B cells) are hyperresponsive to BCR stimulation [77]. This could indicate that elevated BTK levels may increase BCR sensitivity, thus lowering the activation threshold of B cells. Furthermore, BTK is associated with contributing to disease pathogenesis since BTK levels of B cells were correlated with serum RF-antibody levels, and the degree of salivary gland T cell infiltration in Sjögren’s syndrome and ACPA positivity in RA patients [76]. Together, these studies demonstrate that elevated BTK levels are a characteristic of B cells across different autoimmune diseases. Elevated BTK levels could lower the activation threshold of B cells, suggesting that B cells with elevated BTK levels possess the capacity to breach tolerance.
A possible mechanism by which autoreactive B cells can escape from tolerance checkpoints in germinal centers is by the introduction of new N-linked glycans in the variable (Fab) region of BCRs, which potentially influence autoreactive B cell selection. N-linked glycosylation is a process in which N-linked glycans are attached to asparagine (N) sites of proteins [78]. SHM can introduce new N-linked glycosylation sites into the Fab region which are not present in germline-encoded antibodies, causing new glycosylation patterns [79,80,81]. It has been estimated that 15–25% of serum IgG in healthy individuals is Fab glycosylated [78,82,83]. Intriguingly, a hyperglycosylated Fab region pattern has been observed in several autoimmune diseases. In RA, it has been shown that more than 90% of ACPA-IgG in serum carry N-linked glycans in their Fab region, while this is only around 17% of ACPA-depleted IgG from the same patients [82]. In Sjögren’s syndrome, it was demonstrated that 24% of serum IgG carry N-linked glycans in the Fab region [84]. The exact mechanisms by which Fab glycosylation can alter selection during germinal center reactions, and thereby could aid in escaping tolerance checkpoints, are still elusive. However, it has been proposed that Fab glycosylation introduced by SHM in autoreactive B cells might change BCR affinity for antigens, which can give these cells a selective advantage when antigen binding affinity is increased [79,85]. Moreover, the interaction of Fab glycans with lectins derived from the microenvironment may cause crosslinking of BCRs. Crosslinking of BCRs can provide a survival signal, thus may give a survival advantage to autoreactive B cells [79,85].
It cannot be excluded that autoreactive B cells display distinct gene expression profiles which make them more prone to escape self-tolerance. Indeed, studies have demonstrated that in several autoimmune diseases the autoreactive B cells show a distinct gene expression pattern compared to non-autoreactive B cells. In one study, the expression profile of Bcl-6/Bcl-xL-immortalized CCP2-specific memory B cells, generated from peripheral blood or synovial fluid of RA patients, was assessed by RNA-sequencing and/or flow cytometry [86]. Bcl-6/Bcl-xL-immortalized CCP2-specific memory B cells showed higher expression of the costimulatory receptors CD40 and C5aR1 compared to Bcl-6/Bcl-xL-immortalized non-CCP2-specific memory B cells generated from the same patients. In another study, higher expression of IL-15Rα and amphiregulin (AREG; an epidermal growth factor receptor ligand) was observed in circulating CCP1-specific B cells of RA patients compared to the circulating non-CCP1-specific B cells from the same patients [87]. Since costimulatory signals, cytokine receptors, and growth factors are crucial factors in B cell development and self-tolerance, it is tempting to speculate that their increased expression by autoreactive B cells contributes to the escape of tolerance induction. Theoretically, increased expression of costimulatory molecules could enhance B-TH/TFH cell interactions, providing a survival advantage and causing increased activation, proliferation, and differentiation of autoreactive B cells, in comparison with non-autoreactive B cells. Moreover, the upregulation of cytokine receptors and growth factors could have similar effects. Collectively, these factors may promote the breach of self-tolerance by autoreactive B cells. While this an interesting concept, research has yet to be conducted to causally link distinct genetic traits of autoreactive B cells and breach of self-tolerance.
Overall, various mechanisms have been proposed by which autoreactive B cells may breach self-tolerance checkpoints. These mechanisms include a break of self-tolerance influenced by genetic and environmental factors, enhanced BTK activity, altered glycosylation patterns of autoreactive BCRs, and possibly aberrant expression of stimulatory receptors by autoreactive B cells.
자가 면역에서 자기 관용은 손상되지만 자가 반응성 B 세포가 자기 관용 검사 지점을 어떻게 탈출하는지는 거의 알려지지 않았습니다. 최근 연구에서 이 문제를 어느 정도 밝혀냈습니다.
유전적 소인과 환경적 유발 요인이 내성 파괴에 중심적인 역할을 하여 자가 반응성 B 세포가 내성 메커니즘에서 탈출하는 것으로 알려져 있습니다. HLA 유전자, B/T 세포 신호 유전자, 자극/억제 신호 경로, 사이토카인/사이토카인 수용체 유전자의 다양한 유전적 변이가 자가 내성 상실 및 자가 면역 발생과 관련된 위험 요인으로 확인되었습니다 [70]. 유전적 감수성의 역할을 설명하기 위해 조슈아 등은 PTPN22 유전자의 유전적 변이(R620W)의 운반이 RA 환자에서 순환하는 시트룰린화 피브리노겐 펩타이드 특이적 B 세포의 빈도에 미치는 영향을 분석했습니다[71]. PTPN22는 BCR 신호 전달에 관여하는 단백질 티로신 포스파타제입니다. PTPN22 유전자의 R620W 변이는 발달 중 BCR 신호와 자가 반응성 B 세포의 선택에 영향을 미쳐 여러 자가 면역 질환의 강력한 위험 요인으로 확인되었습니다 [72]. 저자들은 R620W 변이를 보유한 RA 환자에서 순환하는 시트룰린화 피브리노겐 펩타이드 특이적 B 세포의 빈도가 비R620W 보유 RA 환자에 비해 증가하는 경향을 보고했습니다. 이를 통해 연구팀은 유전적 위험 요인과 B 세포 자가 내성 위반 사이의 연관성을 제시했습니다.
특히 브루톤 티로신 키나아제(BTK) 활성 증가와 같은 비정상적인 BCR 신호가 자가면역의 자기관용성 위반과 관련이 있을 수 있습니다. BCR 신호 경로의 구성 요소는 B 세포의 활성화 역치를 제어하므로 자가 내성을 유지하는 데 중요한 역할을 합니다. BTK는 BCR 신호 경로의 티로신 키나아제로서 BCR 신호를 다운스트림 신호로 변환하여 B세포 활성화와 생존을 유도하는 데 중심적인 역할을 합니다 [73]. B세포에서 인간 BTK를 과발현하는 형질전환 마우스에서 자가면역이 유도되며, 다양한 쥐 자가면역 모델에서 BTK를 억제하면 자가면역을 줄이거나 예방하는 데 효과적이라는 것이 밝혀졌습니다 [74]. 이러한 마우스 연구는 BTK 과발현이 B세포 내성을 무너뜨릴 수 있음을 시사합니다 [74,75]. 흥미롭게도 건강한 개인에 비해 ACPA 양성 RA, 쇼그렌 증후군 및 활성 GPA 환자의 순환 B 세포 서브셋(순진한 B 세포 포함)에서 BTK 수치가 상승하는 것이 관찰되었습니다 [76,77]. 또한 활성 GPA 환자에서 BTK 수치가 상승한 B 세포 하위 집합(과도기 및 순진한 B 세포)이 BCR 자극에 과민하게 반응한다는 것이 입증되었습니다 [77]. 이는 BTK 수치가 높아지면 BCR 민감도가 증가하여 B 세포의 활성화 역치가 낮아질 수 있음을 나타낼 수 있습니다. 또한, B세포의 BTK 수치가 혈청 RF 항체 수치, 쇼그렌 증후군에서 침샘 T세포 침윤 정도 및 RA 환자에서 ACPA 양성과 상관관계가 있는 것으로 보아 BTK는 질병 발병에 기여하는 것과 관련이 있습니다 [76]. 이러한 연구 결과를 종합하면 BTK 수치 상승은 다양한 자가면역 질환에 걸쳐 B 세포의 특징임을 알 수 있습니다. BTK 수치가 상승하면 B 세포의 활성화 역치가 낮아질 수 있으며, 이는 BTK 수치가 상승한 B 세포가 내성을 위반할 수 있는 능력을 가지고 있음을 시사합니다.
자가 반응성 B 세포가 배아 중심부의 내성 체크포인트에서 벗어날 수 있는 가능한 메커니즘은 BCR의 가변(Fab) 영역에 새로운 N-연결 글리칸이 도입되어 잠재적으로 자가 반응성 B 세포 선택에 영향을 미칠 수 있다는 것입니다. N-결합 글리코실화는 단백질의 아스파라긴(N) 부위에 N-결합 글리칸이 부착되는 과정입니다[78]. SHM은 생식세포 인코딩 항체에는 존재하지 않는 새로운 N-연결 당화 부위를 Fab 영역에 도입하여 새로운 당화 패턴을 유발할 수 있습니다 [79,80,81]. 건강한 사람의 혈청 IgG의 15-25%는 Fab 당화되어 있는 것으로 추정됩니다[78,82,83]. 흥미롭게도 여러 자가면역 질환에서 고당화 Fab 영역 패턴이 관찰되었습니다. RA의 경우, 혈청 내 ACPA-IgG의 90% 이상이 Fab 영역에 N-결합 글리칸을 가지고 있는 반면, 같은 환자의 ACPA가 고갈된 IgG는 약 17%에 불과한 것으로 나타났습니다 [82]. 쇼그렌 증후군에서는 혈청 IgG의 24%가 Fab 영역에 N-결합 글리칸을 가지고 있는 것으로 나타났습니다 [84]. Fab 글리코실화가 배아 중심 반응 중에 선택을 변경하여 내성 체크포인트를 탈출하는 데 도움이 될 수 있는 정확한 메커니즘은 아직 밝혀지지 않았습니다. 그러나 자가 반응성 B 세포에서 SHM에 의해 도입된 Fab 글리코실화가 항원에 대한 BCR 친화성을 변화시켜 항원 결합 친화성이 증가하면 이러한 세포에 선택적 이점을 제공할 수 있다고 제안되었습니다 [79,85]. 또한 Fab 글리칸과 미세 환경에서 유래한 렉틴의 상호 작용은 BCR의 가교를 유발할 수 있습니다. BCR의 가교는 생존 신호를 제공할 수 있으므로 자가 반응성 B 세포에 생존 이점을 제공할 수 있습니다 [79,85].
자가 반응성 B 세포는 뚜렷한 유전자 발현 프로파일을 보여 자가 내성에서 벗어나기 쉽다는 점도 배제할 수 없습니다. 실제로 여러 자가 면역 질환에서 자가 반응성 B 세포는 비자가 반응성 B 세포와 비교하여 뚜렷한 유전자 발현 패턴을 보인다는 사실이 여러 연구를 통해 입증되었습니다. 한 연구에서는 RA 환자의 말초 혈액 또는 활액에서 생성된 Bcl-6/Bcl-xL 불멸화 CCP2-특이적 기억 B 세포의 발현 프로파일을 RNA 시퀀싱 및/또는 유세포 분석법으로 평가했습니다 [86]. Bcl-6/Bcl-xL 불멸화 CCP2 특이적 기억 B 세포는 같은 환자에서 생성된 Bcl-6/Bcl-xL 불멸화 비-CCP2 특이적 기억 B 세포에 비해 비용 자극 수용체 CD40 및 C5aR1의 발현이 더 높은 것으로 나타났습니다. 또 다른 연구에서는 동일한 환자의 순환하는 비-CCP1 특이적 B 세포에 비해 RA 환자의 순환하는 CCP1 특이적 B 세포에서 IL-15Rα와 암피레귤린(AREG; 표피 성장 인자 수용체 리간드)의 높은 발현이 관찰되었습니다 [87]. 비용 자극 신호, 사이토카인 수용체 및 성장 인자는 B 세포 발달과 자가 내성에 중요한 요소이므로 자가 반응성 B 세포에 의한 이들의 발현 증가가 내성 유도의 탈출에 기여한다고 추측할 수 있습니다. 이론적으로 비용 자극 분자의 발현 증가는 B-TH/TFH 세포 상호작용을 강화하여 비자가 반응성 B 세포에 비해 생존 이점을 제공하고 자가 반응성 B 세포의 활성화, 증식 및 분화를 증가시킬 수 있습니다. 또한 사이토카인 수용체와 성장 인자의 상향 조절도 비슷한 효과를 가져올 수 있습니다. 종합적으로 이러한 요인들은 자가 반응성 B 세포의 자기 내성 위반을 촉진할 수 있습니다. 이는 흥미로운 개념이지만, 자가 반응성 B 세포의 뚜렷한 유전적 특성과 자가 내성 위반을 인과적으로 연결하는 연구는 아직 수행되지 않았습니다.
전반적으로 자가 반응성 B 세포가 자기 내성 체크포인트를 위반할 수 있는 다양한 메커니즘이 제안되었습니다.
이러한 메커니즘에는
유전적 및 환경적 요인에 의해 영향을 받는
자가 내성 파괴,
BTK 활성 강화,
자가 반응성 BCR의 당화 패턴 변화,
자가 반응성 B 세포에 의한 자극 수용체의 비정상적인 발현 등이 있습니다.
3.4. Effector Functions of Autoreactive B Cells
Autoreactive B cells that have escaped self-tolerance mechanisms may drive the inflammatory manifestations through the exertion of various effector functions. Rituximab targets CD20, which is expressed by B cells from the pre-B cell stage through the pre-plasma stage, thus excluding the cells that produce antibodies, i.e., plasmablasts and plasma cells [88]. Clinical outcomes following rituximab provide insights into the functional characteristics of autoreactive B cells.
Autoantibodies have an important effector function in some diseases. For example, in pemphigus, anti-Dsg1 and anti-Dsg3 antibodies bind to keratinocyte adhesion proteins desmoglein-1 and -3, respectively [11]. Consequently, adhesion is lost between keratinocytes, leading to blister formation in the skin or oral mucosa [11]. In SLE, ANAs bind to nuclear antigens and subsequently form antigen-antibody complexes [9]. These immune complexes deposit in various tissues, which cause inflammation and tissue damage [9]. Given that rituximab targets CD20, the contribution of short-lived plasma cells and long-lived plasma cells to autoantibody production can be identified. It has been demonstrated that rituximab is effective in diseases such as RA, GPA, and pemphigus and can lead to a reduction of autoantibody levels [89,90,91,92,93,94,95]. These observations indicate that short-lived plasma cells contribute to the production of pathogenic autoantibodies in these diseases. In pemphigus in particular, the high efficacy of rituximab, the dramatic reduction of anti-Dsg1/anti-Dsg3 serum levels following rituximab, and the correlation of these autoantibodies with clinical response indicate that short-lived plasma cells are the main source of autoantibodies in this disease [93,94,95]. In RA and GPA however, autoantibodies levels are not always drastically reduced following rituximab, implying that long-lived plasma cells also contribute to autoantibody production [89,92]. In contrast, long-lived plasma cells may be the major source of autoantibodies in diseases in which the efficacy of rituximab is low. Rituximab has low efficacy in Sjögren’s syndrome, an autoimmune disease characterized by the presence of anti-SSA(Ro) and anti-SSB(La) autoantibodies [96,97]. Various studies have demonstrated that rituximab does not impact anti-SSA(Ro)/anti-SSB(La) levels in patients supporting that long-lived plasma cells are likely the predominant autoantibody producers [98,99,100].
B cells may promote inflammatory processes through antibody-independent functions [5,6]. As an example, cytokine expression by autoreactive B cells has been linked to disease pathogenesis in pemphigus vulgaris. In pemphigus vulgaris patients, the frequency of IL-1β-expressing circulating Dsg1/Dsg3-specific B cells was significantly higher at baseline compared to healthy individuals and decreased to healthy control levels when in remission following rituximab treatment. Conversely, no change in the frequencies of IL-1β-expressing circulating non-Dsg1/Dsg3-specific B cells was observed in the same patients, suggesting that autoreactive B cells possibly mediate autoimmune processes by the secretion of specific cytokines [101]. Furthermore, as another example, crosstalk between autoreactive B cells and CD8+ T cells may promote pathogenic processes as well. While not specifically focused on the autoreactive B cells themselves, it has been demonstrated in GPA patients that rituximab lowered the frequency of circulating CD8+ TEMRA cells, whereas the frequencies of different CD4+ T cell subsets and Treg cells were unaffected. Importantly, rituximab reduced cytokine and chemokine production of CD8+ T cells and coculturing of B cells from untreated active GPA patients with CD8+ T cells from the same patients enhanced CD8+ T cell proinflammatory cytokine production, suggesting a pathogenic crosstalk between these cells [102].
Collectively, autoreactive B cells can drive autoimmune disease by pathogenic autoantibodies of which the contribution of antibodies produced by short-lived versus long-lived plasma cells vary between diseases. Additionally, autoreactive B cells may influence disease processes by antibody-independent mechanisms, such as by the production of specific cytokines and crosstalk with T cells.
자가 내성 메커니즘에서 벗어난 자가 반응성 B 세포는
다양한 이펙터 기능을 발휘하여
염증 증상을 유발할 수 있습니다.
리툭시맙은
B 세포 전 단계부터 혈장 전 단계까지
B 세포에 의해 발현되는 CD20을 표적으로 하여
항체를 생성하는 세포,
즉 형질 모세포와 혈장 세포를 배제합니다 [88].
리툭시맙 투여 후 임상 결과는 자가 반응성 B 세포의 기능적 특성에 대한 통찰력을 제공합니다.
자가항체는 일부 질병에서 중요한 이펙터 기능을 합니다. 예를 들어, 천포창에서 항-Dsg1 및 항-Dsg3 항체는 각각 각질 세포 부착 단백질 데스모글린-1 및 -3에 결합합니다 [11]. 결과적으로 각질 세포 사이의 접착력이 상실되어 피부 또는 구강 점막에 물집이 형성됩니다 [11]. SLE에서 ANA는 핵 항원에 결합한 후 항원-항체 복합체를 형성합니다 [9]. 이러한 면역 복합체는 다양한 조직에 침착되어 염증과 조직 손상을 일으킵니다 [9]. 리툭시맙이 CD20을 표적으로 한다는 점을 감안할 때, 단수명 형질세포와 장수명 형질세포가 자가항체 생성에 기여하는 바를 확인할 수 있습니다. 리툭시맙은 RA, GPA, 천포창과 같은 질환에 효과적이며 자가항체 수치를 감소시킬 수 있음이 입증되었습니다[89,90,91,92,93,94,95]. 이러한 관찰은 단수명 형질세포가 이러한 질환에서 병원성 자가항체 생성에 기여한다는 것을 나타냅니다. 특히 천포창에서 리툭시맙의 높은 효능, 리툭시맙 투여 후 항-Dsg1/항-Dsg3 혈청 수준의 급격한 감소, 이러한 자가항체와 임상 반응의 상관관계는 단수명 형질세포가 이 질환에서 자가항체의 주요 공급원임을 나타냅니다[93,94,95]. 그러나 RA와 GPA에서는 리툭시맙 투여 후 자가항체 수치가 항상 급격히 감소하는 것은 아니며, 이는 수명이 긴 혈장 세포도 자가항체 생성에 기여한다는 것을 의미합니다 [89,92]. 반대로, 리툭시맙의 효능이 낮은 질환에서는 장수 혈장 세포가 자가 항체의 주요 공급원이 될 수 있습니다. 리툭시맙은 항-SSA(Ro) 및 항-SSB(La) 자가항체의 존재를 특징으로 하는 자가면역질환인 쇼그렌 증후군에서 효능이 낮습니다[96,97]. 다양한 연구에 따르면 리툭시맙은 환자의 항SSA(Ro)/항SSB(La) 수치에 영향을 미치지 않는 것으로 나타나 장수 혈장 세포가 주요 자가항체 생산자일 가능성이 높다는 것을 뒷받침합니다[98,99,100].
B 세포는 항체와 독립적인 기능을 통해 염증 과정을 촉진할 수 있습니다[5,6]. 예를 들어, 자가 반응성 B 세포에 의한 사이토카인 발현은 천포창의 질병 발병과 관련이 있습니다. 천포창 환자에서 IL-1β를 발현하는 순환 Dsg1/Dsg3 특이 B 세포의 빈도는 건강한 사람에 비해 기저시점에서 유의하게 높았으며, 리툭시맙 치료 후 관해 상태가 되면 건강한 대조군 수준으로 감소했습니다. 반대로, 동일한 환자에서 IL-1β를 발현하는 순환하는 비-Dsg1/Dsg3 특이 B 세포의 빈도에는 변화가 관찰되지 않았는데, 이는 자가 반응성 B 세포가 특정 사이토카인의 분비를 통해 자가 면역 과정을 매개할 가능성이 있음을 시사합니다 [101]. 또한, 또 다른 예로 자가 반응성 B 세포와 CD8+ T 세포 사이의 누화가 병원성 과정을 촉진할 수도 있습니다. 자가 반응성 B 세포 자체에 특별히 초점을 맞춘 것은 아니지만, 리툭시맙이 순환하는 CD8+ TEMRA 세포의 빈도를 낮추는 반면, 다른 CD4+ T 세포 하위 집합과 Treg 세포의 빈도는 영향을 받지 않는 것으로 GPA 환자에서 입증되었습니다. 중요한 것은 리툭시맙이 CD8+ T 세포의 사이토카인과 케모카인 생산을 감소시켰으며, 치료받지 않은 활성 GPA 환자의 B 세포와 같은 환자의 CD8+ T 세포를 공배양하면 CD8+ T 세포의 염증성 사이토카인 생산이 증가하여 이들 세포 간의 병원성 교차가 있음을 시사한다는 점입니다[102].
종합적으로
자가 반응성 B 세포는
병원성 자가 항체에 의해 자가 면역 질환을 유발할 수 있으며,
이 중 단수명과 장수명 형질 세포에 의해 생성되는 항체의 기여도는
질환에 따라 다릅니다.
또한 자가 반응성 B 세포는
특정 사이토카인의 생성 및
T 세포와의 누화와 같은
항체 독립적 메커니즘을 통해 질병 과정에 영향을 미칠 수 있습니다.
4. Implications for Therapy and Perspectives
Currently, clinical remission is often successfully induced in most patients through broad suppression of the immune system by corticosteroids, immunosuppressants, and biologics. Despite remission induction, many patients experience relapses upon cessation of therapy. Moreover, chronic treatment of patients with broad immune-suppressing agents may lead to adverse effects such as increased susceptibility to infections and diminished effectiveness of vaccines. Therefore, there is a need for novel therapeutic strategies that more specifically target autoreactive B cells and prevent the reconstitution of these cells.
Intervening with the mechanisms involved in the breach of self-tolerance is crucial for preventing autoreactive B cell development. The fact that tolerance checkpoints in germinal centers are breached among various autoimmune diseases suggests that restoring these checkpoints could hold promise for preventing autoreactive B cell development. Since tolerance of B cells is regulated by BCR signaling thresholds, targeting modulators of B cell signaling may restore tolerance. Some important inhibitory checkpoints of B cells that could be targeted are for example programmed cell death 1 (PD1), low-affinity immunoglobulin-γ Fc region receptor IIb (FcγRIIb), and CD22 [103]. Examples of important stimulatory checkpoints are CD40, TLRs, and B cell-activating factor receptor (BAFFR) [103]. Furthermore, the finding that BTK levels of B cells are elevated in various autoimmune diseases, and BTK overexpressing is associated with breaching tolerance and development of autoimmunity, makes BTK a potential therapeutic target. Inhibition of BTK may enhance the activation threshold of autoreactive B cells, thereby preventing the breach of tolerance. Clinical trials are currently underway to investigate the effect of BTK inhibition in autoimmune diseases including RA, SLE, pemphigus, Sjögren’s syndrome, and multiple sclerosis (www.clinicaltrials.gov, accessed on 12 May 2021). These trials will soon provide information regarding the efficacy of BTK inhibition. Moreover, Fab glycosylation may also be an interesting therapeutic target, although more research is needed to decipher the exact role of Fab glycosylation in autoimmunity.
The observation that autoreactive B cells have a distinct gene expression profile compared to autologous non-autoreactive B cells is interesting and may provide clues to new targets for therapy, although it is unlikely that such targets will be specific for autoreactive B cells. Recently, novel cell-based therapies have been developed for the treatment of autoimmune diseases that aim to specifically eliminate autoreactive B cells [104,105]. Chimeric autoantibody receptor T (CAAR-T) cells are T cells that are genetically engineered to express a chimeric receptor that consists of an extracellular antigen domain fused to T cell cytoplasmic signaling domains. CAAR-T cells are activated upon binding of B cells to the extracellular antigen domain, resulting in cytotoxic T cell-mediated elimination of B cells. Ellebrecht and colleagues have shown that Dsg3-CAAR-T cells could be used to specifically target Dsg3-specific B cells for the treatment of pemphigus vulgaris. Dsg3-CAAR-T cells were constructed of which the extracellular domain of the chimeric receptor consisted of Dsg3 and the intracellular signaling domains of CD137-CD3ξ. In vitro, they showed that Dsg3-CAAR-T cells had specific cytotoxic activity against Dsg3-specific hybridomas. In vivo, Dsg3-CAAR-T cells also had specific cytotoxic activity against Dsg3-specific hybridomas in NOD-scid-gamma mice, which were injected with a mix of Dsg3-specific hybridomas. In addition, Dsg3-CAAR-T cell treatment of mice lowered Dsg3-specific IgG autoantibody levels and prevented the formation of mucosal blisters [106]. In a subsequent pre-clinical study, they demonstrated that Dsg3-CAAR-T cells could also specifically eliminate circulating Dsg3-specific B cells from patients ex vivo [107]. Zhang and colleagues used a different approach to specifically deplete autoreactive B cells. In this approach, a combination of chimeric antigen receptor T (CAR-T) cells and fluorescently labeled autoantigens was used. Anti-fluorescein isothiocyanate (FITC) CAR-T cells, combined with various FITC-labeled RA-related citrullinated protein epitopes, were found to specifically eliminate hybridoma cells in vitro and autoreactive B cells from RA patients ex vivo [108].
Together, possible therapeutic targets for depleting autoreactive B cells and/or preventing the reconstitution of these cells include stimulatory and inhibitory checkpoints of B cells, BTK, Fab glycosylation, and targets based on the unique gene expression profile of autoreactive B cells. Ultimately, CAAR-T cell- and CAR-T cell-based strategies may be the most attractive approach for eliminating autoreactive B cells without targeting non-pathogenic cells (Figure 3).
현재 대부분의 환자에서 코르티코스테로이드, 면역억제제 및 생물학적 제제를 통해 면역 체계를 광범위하게 억제하여 임상적 관해가 성공적으로 유도되는 경우가 많습니다. 관해 유도에도 불구하고 많은 환자가 치료를 중단하면 재발을 경험합니다. 또한 광범위한 면역 억제제를 만성적으로 투여하면 감염에 대한 감수성이 증가하고 백신의 효과가 감소하는 등의 부작용이 발생할 수 있습니다. 따라서 자가 반응성 B 세포를 보다 구체적으로 표적으로 삼고 이러한 세포의 재구성을 방지하는 새로운 치료 전략이 필요합니다.
자가 내성 위반과 관련된 메커니즘에 개입하는 것은 자가 반응성 B 세포 발생을 예방하는 데 매우 중요합니다. 다양한 자가면역 질환에서 배아 중추의 내성 체크포인트가 위반된다는 사실은 이러한 체크포인트를 복원하면 자가반응성 B 세포 발달을 예방할 수 있다는 가능성을 시사합니다. B세포의 내성은 BCR 신호 역치에 의해 조절되기 때문에 B세포 신호의 조절자를 표적으로 삼으면 내성을 회복할 수 있습니다. 표적으로 삼을 수 있는 B 세포의 중요한 억제 체크포인트에는 프로그램된 세포 사멸 1(PD1), 저친화성 면역글로불린-γ Fc 영역 수용체 IIb(FcγRIIb), CD22 등이 있습니다[103]. 중요한 자극 체크포인트의 예로는 CD40, TLR, B세포 활성화 인자 수용체(BAFFR)가 있습니다[103]. 또한, 다양한 자가면역질환에서 B세포의 BTK 수치가 상승하고 BTK 과발현이 내성 위반 및 자가면역 발달과 관련이 있다는 사실이 밝혀지면서 BTK는 잠재적인 치료 표적이 되고 있습니다. BTK를 억제하면 자가 반응성 B 세포의 활성화 역치가 향상되어 내성 위반을 방지할 수 있습니다. 현재 RA, SLE, 천포창, 쇼그렌 증후군, 다발성 경화증 등 자가면역 질환에서 BTK 억제의 효과를 조사하기 위한 임상시험이 진행 중입니다(www.clinicaltrials.gov, 2021년 5월 12일에 액세스). 이러한 임상시험은 곧 BTK 억제의 효능에 관한 정보를 제공할 것입니다. 또한, 자가 면역에서 Fab 당화의 정확한 역할을 규명하기 위해서는 더 많은 연구가 필요하지만 Fab 당화도 흥미로운 치료 표적이 될 수 있습니다.
자가 반응성 B 세포가 자가 비반응성 B 세포와 비교하여 뚜렷한 유전자 발현 프로필을 가지고 있다는 관찰은 흥미롭고 새로운 치료 표적에 대한 단서를 제공할 수 있지만, 이러한 표적이 자가 반응성 B 세포에 특이적일 가능성은 낮습니다. 최근 자가면역질환 치료를 위해 자가반응성 B세포를 특이적으로 제거하는 것을 목표로 하는 새로운 세포 기반 치료법이 개발되고 있습니다[104,105].
키메라 자가항체 수용체 T(CAAR-T) 세포는 T세포 세포질 신호 전달 도메인에 융합된 세포 외 항원 도메인으로 구성된 키메라 수용체를 발현하도록 유전공학적으로 조작된 T세포입니다. CAAR-T 세포는 B 세포가 세포 외 항원 도메인에 결합하면 활성화되어 세포 독성 T 세포를 매개로 B 세포를 제거합니다. 엘브레히트와 동료들은 Dsg3-CAAR-T 세포가 천포창 치료를 위해 Dsg3 특이적 B 세포를 특이적으로 표적으로 삼는 데 사용될 수 있음을 보여주었습니다. 키메라 수용체의 세포 외 도메인이 Dsg3와 CD137-CD3ξ의 세포 내 신호 도메인으로 구성된 Dsg3-CAAR-T 세포를 제작했습니다. 시험관 내에서 연구진은 Dsg3-CAAR-T 세포가 Dsg3-특이적 하이브리드종에 대해 특정 세포 독성 활성을 갖는다는 것을 보여주었습니다. 생체 내에서도 Dsg3-CAAR-T 세포는 Dsg3-특이적 하이브리드종을 혼합하여 주입한 NOD-scid-gamma 마우스에서 Dsg3-특이적 하이브리드종에 대해 특정 세포 독성 활성을 나타냈습니다. 또한, 생쥐에 Dsg3-CAAR-T 세포를 처리하면 Dsg3 특이 IgG 자가항체 수치가 낮아지고 점막 물집 형성이 방지되었습니다[106]. 후속 전임상 연구에서 연구진은 Dsg3-CAAR-T 세포가 생체 외 환자에서 순환하는 Dsg3 특이적 B 세포도 특이적으로 제거할 수 있음을 입증했습니다[107]. 장과 동료들은 자가 반응성 B 세포를 특이적으로 고갈시키기 위해 다른 접근법을 사용했습니다. 이 접근법에서는 키메라 항원 수용체 T(CAR-T) 세포와 형광 표지된 자가 항원을 조합하여 사용했습니다. 항-플루오레세인 이소티오시아네이트(FITC) CAR-T 세포는 다양한 FITC 표지 RA 관련 시트룰린화 단백질 에피토프와 결합하여 시험관 내 하이브리드종 세포와 생체 외 RA 환자의 자가 반응성 B 세포를 특이적으로 제거하는 것으로 밝혀졌습니다 [108].
이와 함께 자가 반응성 B 세포를 고갈시키거나 이러한 세포의 재구성을 방지하기 위한 가능한 치료 표적에는 B 세포의 자극 및 억제 체크포인트, BTK, Fab 당화, 자가 반응성 B 세포의 고유 유전자 발현 프로파일에 기반한 표적 등이 있습니다. 궁극적으로 CAAR-T 세포 및 CAR-T 세포 기반 전략은 비병원성 세포를 표적으로 하지 않고 자가 반응성 B 세포를 제거하는 데 가장 매력적인 접근 방식일 수 있습니다(그림 3).
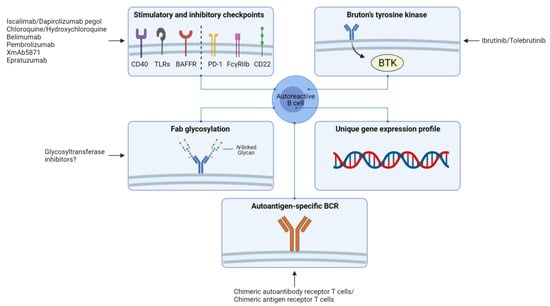
Figure 3. Overview of potential therapeutic approaches for targeting autoreactive B cells. Stimulatory and inhibitory checkpoints of B cells such as CD40, Toll-like receptors (TLRs), B cell-activating factor receptor (BAFFR), programmed cell death 1 (PD1), low-affinity immunoglobulin-γ Fc region receptor IIb (FcγRIIb), and CD22; Bruton’s tyrosine kinase (BTK); glycosylation patterns of the variable (Fab) region of the B cell receptor (BCR); targets based on the unique gene expression profiles of autoreactive B cells compared to autologous non-autoreactive B cells; and the autoantigen-specific BCR of autoreactive B cells. Figure was created with BioRender.
자가 반응성 B 세포를 표적으로 하는 잠재적 치료 접근법에 대한 개요.
CD40, 톨유사수용체(TLR), B세포 활성화 인자 수용체(BAFFR), 프로그램된 세포 사멸 1(PD1), 저친화성 면역글로불린-γ Fc 영역 수용체 IIb(FcγRIIb), CD22, 브루톤 티로신 키나제(BTK) 등 B세포의 자극 및 억제 체크포인트; B세포 수용체(BCR)의 가변(Fab) 영역의 당화 패턴, 자가 비자가 반응성 B세포와 비교한 자가 반응성 B세포의 고유 유전자 발현 프로파일을 기반으로 한 표적, 자가 반응성 B세포의 자가 항원 특이적 BCR. 그림은 BioRender로 제작되었습니다.
5. Conclusions
Overall, multiple mechanisms are likely involved in the development and activation of autoreactive B cells in B cell-mediated autoimmune diseases. Some mechanisms overlap between different diseases, suggesting also overlap in potential targets for therapy. Eventually, deciphering these mechanisms may further lead to the identification of specific targets as a basis for novel therapeutic strategies for autoimmune diseases.
전반적으로
B 세포 매개 자가면역 질환에서
자가 반응성 B 세포의 발달과 활성화에는
여러 메커니즘이 관여할 가능성이 높습니다.
일부 메커니즘은 서로 다른 질환 간에 겹치는 부분이 있어 잠재적인 치료 표적도 겹칠 수 있습니다. 궁극적으로 이러한 메커니즘을 해독하면 자가면역 질환에 대한 새로운 치료 전략의 기초가 되는 특정 표적을 식별할 수 있습니다.
Author Contributions
Conceptualization, C.G.B., W.H.A., A.R., P.H. and N.A.B.; Writing—Original Draft Preparation, C.G.B.; Writing—Review and Editing, W.H.A., A.R., P.H. and N.A.B.; and Supervision, W.H.A., A.R., P.H. and N.A.B. All authors have read and agreed to the published version of the manuscript.
Funding
This collaboration project was realized within the Target-to-B consortium, financed by the PPP Allowance made available by Top Sector Life Sciences & Health to Samenwerkende Gezondheidsfondsen (SGF) under project number LSHM18055-SGF to stimulate public–private partnerships and co-financing by health foundations that are part of the SGF.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References