Curr Biol. Author manuscript; available in PMC 2015 May 19.
Published in final edited form as:
Curr Biol. 2014 May 19; 24(10): R453–R462.
doi: 10.1016/j.cub.2014.03.034
PMCID: PMC4055301
NIHMSID: NIHMS586698
PMID: 24845678
ROS Function in Redox Signaling and Oxidative Stress
Michael Schieber and Navdeep S. Chandel*
Author information Copyright and License information PMC Disclaimer
The publisher's final edited version of this article is available free at Curr Biol
Abstract
Oxidative stress refers to elevated intracellular levels of reactive oxygen species (ROS) that cause damage to lipids, proteins and DNA. Oxidative stress has been linked to a myriad of pathologies. However, elevated ROS are also signaling molecules i.e. redox biology that maintain physiological functions. In this review we discuss the two faces of ROS, redox signaling and oxidative stress, and their contribution to both physiological and pathological conditions. Redox biology refers to low levels of ROS that activate signaling pathways to initiate biological processes while oxidative stress denotes high levels of ROS that incur damage to DNA, protein or lipids. Thus, the response to ROS displays hormesis. The In this review, we argue that redox biology, rather than oxidative stress, underlies physiological and pathological conditions.
산화 스트레스는
지질, 단백질, DNA에 손상을 일으키는
세포 내 활성산소종(ROS) 수치가 높아지는 것을 말합니다.
산화 스트레스는
무수히 많은 병리와 관련이 있습니다.
그러나
활성산소는 생리적 기능을 유지하는
신호 전달 분자, 즉 산화 환원 생물학이기도 합니다.
이 리뷰에서는
산화 환원 신호와 산화 스트레스라는
ROS의 두 가지 측면과 생리적 및
병리학적 상태에 대한 기여에 대해 설명합니다.
Redox biology 산화 환원 생물학은
생물학적 과정을 시작하기 위해 신호 경로를 활성화하는
낮은 수준의 ROS를 의미하며,
산화 스트레스는
DNA, 단백질 또는 지질을 손상시키는
높은 수준의 ROS를 의미합니다.
따라서 ROS에 대한 반응은 호르메시스를 나타냅니다. 이 리뷰에서는 산화 스트레스가 아닌 산화 환원 생물학이 생리적 및 병리학적 상태의 기초가 된다고 주장합니다.
Introduction
Reactive oxygen species (ROS) are byproducts of aerobic metabolism. ROS include the superoxide anion (O2−), hydrogen peroxide (H2O2), and hydroxyl radicals (OH·), all of which have inherent chemical properties that confer reactivity to different biological targets. ROS is often associated with the principle of oxidative stress which suggests ROS induce pathology by damaging lipids, proteins, and DNA [1]. However, in the past two decades it has become apparent that ROS also serve as signaling molecules to regulate biological and physiological processes [2]. It appears early in evolution, nature selected for ROS as a signal transduction mechanism to allow for adaptation to changes in environmental nutrients and the oxidative environment [3]. Indeed in prokaryotes, there are well-described mechanisms whereby ROS directly activate transcription factors for adaption to stress [4].
An understood mechanism of redox signaling involves H2O2-mediated oxidation of cysteine residues within proteins [5]. Cysteine residues exist as a thiolate anion (Cys-S-) at physiological pH and are more susceptible to oxidation compared to the protonated cysteine thiol (Cys-SH) [6]. During redox signaling, H2O2 oxidizes the thiolate anion to sulfenic form (Cys-SOH) causing allosteric changes within the protein that alter its function. The sulfenic form can be reduced to thiolate anions by the disulfide reductases, thioredoxin (Trx) and glutaredoxin(Grx), to return the protein function to its original state [7]. Hence, first degree oxidation of cysteine residues within proteins serves as a reversible signal transduction mechanism. It is estimated that thiolate oxidation in living cells occurs in nM range of H2O2 while higher levels of peroxide further oxidize thiolate anions to sulfinic (SO2H) or sulfonic (SO3H) species. Unlike sulfenic modifications, sulfinic and sulfonic can be irreversible alterations and results in permanent protein damage (i.e. oxidative stress). Thus, cells have professional enzymes dedicated to prevent buildup of intracellular H2O2, primarily peroxiredoxins and glutathione peroxidases. H2O2 is generated from superoxide produced by mitochondria and NADPH oxidases [8, 9].
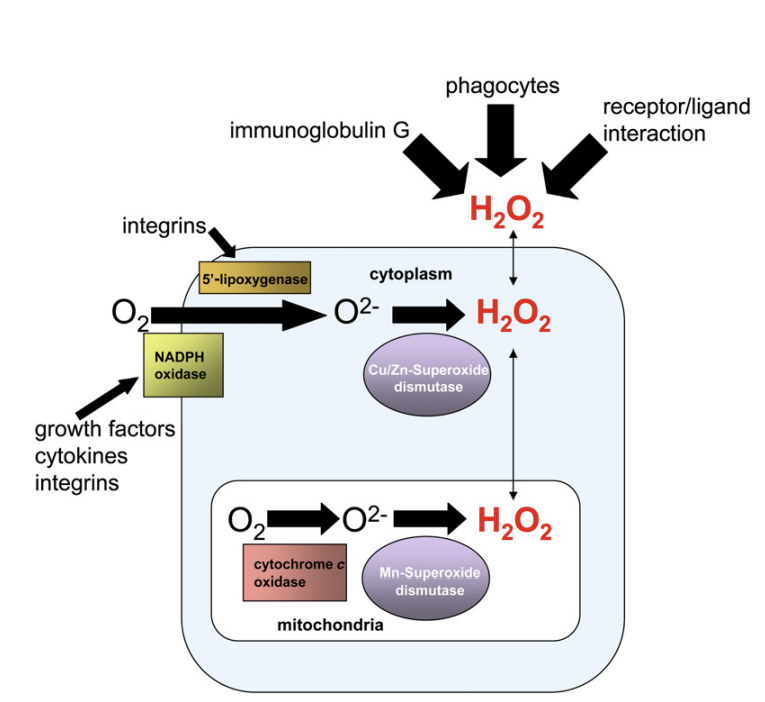
Superoxide forms from the one-electron reduction of molecular oxygen (O2) and, within the cell, is rapidly converted by superoxide dismutases 1 and 2 (SOD 1 and 2) into H2O2. SOD1 is primarily located in the cytosol and mitochondrial intermembrane space while SOD2 is located in the mitochondrial matrix. SODs prevent accumulation of superoxide that can damage and inactivate proteins containing iron-sulfur clusters [10]. Thus, accumulation of superoxide is more associated with oxidative stress than redox signaling. However, it is important to note that superoxide does not indiscriminately damage proteins. There are a specific set of proteins sensitive to inactivation by superoxide which activate signaling pathways promoting adaptation to elevated superoxide or, alternatively, initiating cell death [11]. This supports our current view of oxidative stress as a combination of cellular damage and stress responsive signaling. A third type of ROS is the extremely reactive hydroxyl radical which indiscriminately oxidizes lipids, proteins, and DNA, resulting in damage or genomic instability [12]. Typically, hydroxyl radicals are generated from H2O2 in the presence of ferrous ions (i.e. the Fenton reaction). Therefore, cells have multiple mechanisms to maintain iron homeostasis to prevent the formation of toxic hydroxyl radicals. It is important to note, that the changes in H2O2 required for signaling do not cause significant changes in intracellular ratio of oxidized glutathione (GSSG)/reduced glutathione (GSH) or NADPH/NADP+ [13]. In fact, large changes in these parameters are usually a sign of oxidative stress causing toxicity rather than signaling associated with redox biology [14].
Aside from the specificity and selectively of ROS on their targets, the compartmentalization of ROS production within cells is an important determinant of whether damage or redox signaling occurs. In order for effective redox signaling, the H2O2dependent oxidation of a given protein is likely to be close to the source of H2O2 production. For example, the protein targets of H2O2 generated from plasma membrane NADPH oxidases are also located on plasma membrane. Mitochondria are known to dynamically move towards their targets thus allowing mitochondrial generated H2O2 to activate signaling pathways [15]. Similarly, superoxide accumulation in mitochondrial matrix has different outcomes than superoxide accumulation in the cytosol. This is in part due to a high content of iron sulfur cluster protein located in the mitochondrial matrix. Indeed, SOD2 knockout mice have a dramatically severe pathological phenotype compared to SOD1 knockout mice. Accordingly, both the type of ROS and it's local concentration collectively determine whether redox signaling or oxidative stress induced damage occurs.
In this review we will discuss the two faces of ROS biology: redox signaling and oxidative stress. We will focus on both physiological and pathological conditions using the examples of (1) normal and cancer cell proliferation, (2) beneficial and pathological inflammation, and (3) the normal and accelerated aging process.
활성 산소종(ROS)은
유산소 대사의 부산물입니다.
ROS에는
슈퍼옥사이드 음이온(O2-),
과산화수소(H2O2),
하이드 록실 라디칼(OH-)이 포함되며,
이들은 모두 다양한 생물학적 표적에 반응성을 부여하는
고유한 화학적 특성을 가지고 있습니다.
superoxide anion (O2−), hydrogen peroxide (H2O2), and hydroxyl radicals (OH·)
https://www.cell.com/molecular-cell/pdf/S1097-2765(07)00186-4.pdf

ROS는
지질, 단백질, DNA를 손상시켜 병리를 유발하는
산화 스트레스의 원리와 관련이 있는 경우가 많습니다[1].
그러나
지난 20년 동안
ROS가 생물학적 및 생리적 과정을 조절하는
신호 분자로도 작용한다는 사실이 밝혀졌습니다[2].
진화 초기에 자연은
환경 영양분과 산화 환경의 변화에 적응할 수 있도록
신호 전달 메커니즘으로
ROS를 선택한 것으로 보입니다 [3].
실제로
원핵생물에서는
ROS가 직접적으로 전사인자를 활성화하여
스트레스에 적응하는 메커니즘이 잘 설명되어 있습니다[4].
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6211135/

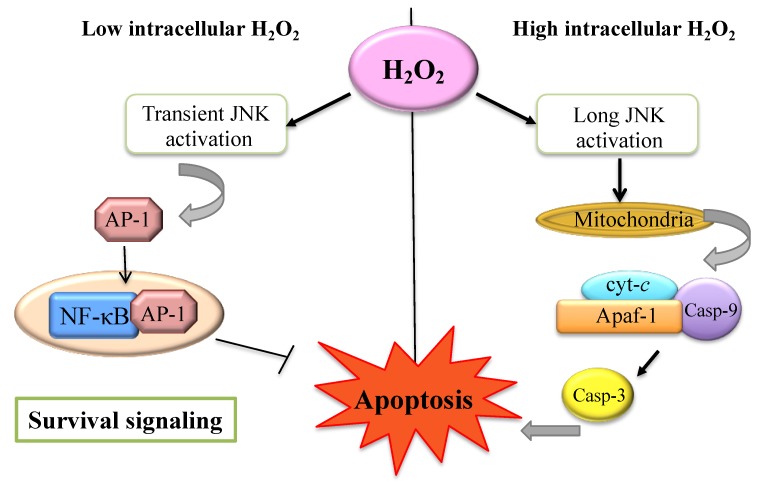
산화 환원 신호의 이해된 메커니즘은
단백질 내 시스테인 잔기의
H2O2 매개 산화를 포함합니다 [5].
시스테인 잔기는
생리적 pH에서 티올레이트 음이온(Cys-S-)으로 존재하며
양성자화된 시스테인 티올(Cys-SH)에 비해
산화에 더 취약합니다[6].
산화 환원 신호 중에
H2O2는 티올산염 음이온을 산화시켜
단백질 내에서 알로스테릭 변화를 일으켜
기능을 변화시킵니다(Cys-SOH).
설펜 형태는
이황화 환원 효소인 티오레독신(Trx)과
글루타레독신(Grx)에 의해
티올레이트 음이온으로 환원되어
단백질 기능을 원래 상태로 되돌릴 수 있습니다[7].
따라서
단백질 내 시스테인 잔기의 1도 산화는
가역적인 신호 전달 메커니즘으로 작용합니다.
살아있는 세포에서 티올레이트 산화는
nM 범위의 H2O2에서 발생하는 것으로 추정되며,
더 높은 수준의 과산화물은 티올레이트 음이온을 설핀(SO2H) 또는
설폰(SO3H) 종으로 추가 산화시킵니다.

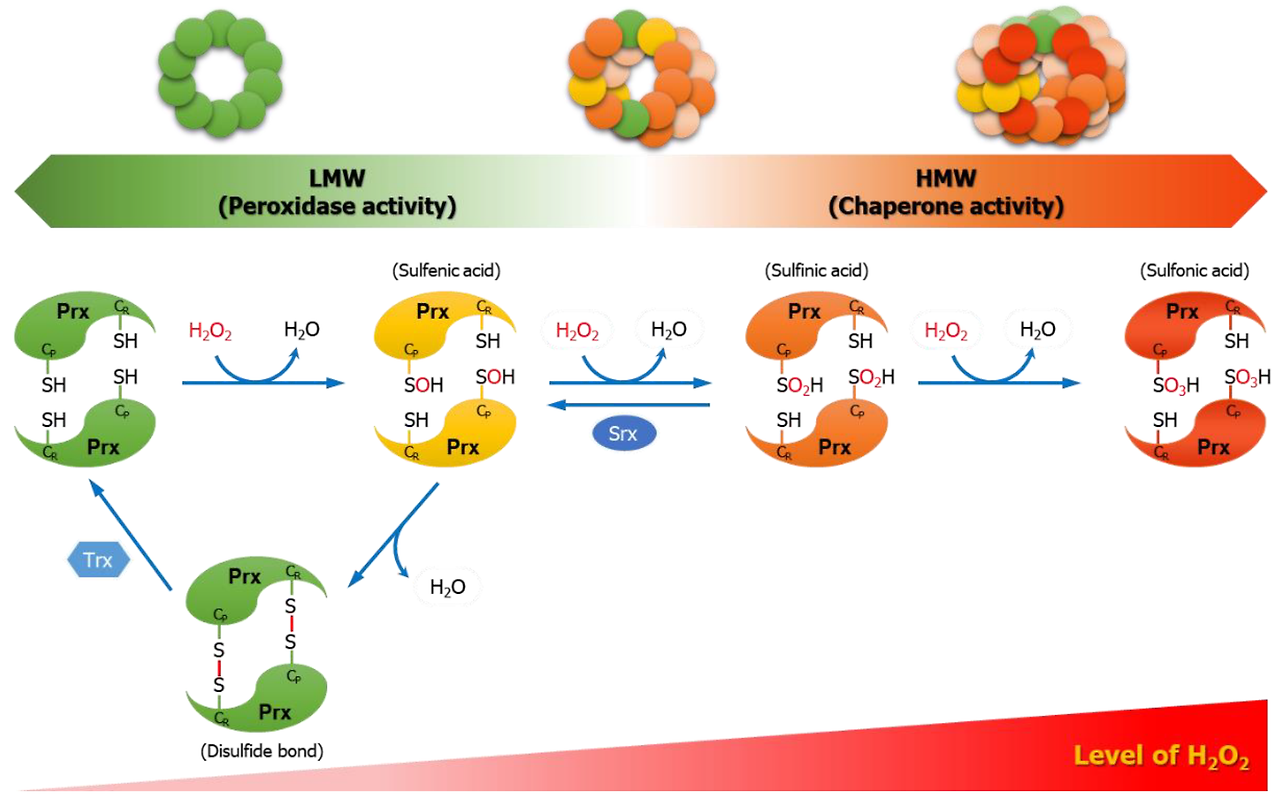
설펜 변형과 달리 설핀 및 설폰 변형은 비가역적인 변형으로
영구적인 단백질 손상(즉, 산화 스트레스)을 초래할 수 있습니다.
따라서
세포에는 세포 내 H2O2 축적을 방지하는 전문 효소,
주로 퍼옥시레독신과 글루타치온 퍼옥시다아제가 있습니다.
H2O2는
미토콘드리아와
NADPH 산화 효소에 의해 생성된 과산화물에서 생성됩니다[8, 9].
슈퍼옥사이드는
분자 산소(O2)의 1전자 환원으로부터 형성되며,
세포 내에서 슈퍼옥사이드 디스뮤타제 1 및 2(SOD 1 및 2)에 의해
H2O2로 빠르게 전환됩니다.
SOD1은
주로 세포질과 미토콘드리아 막간 공간에 위치하며,
SOD2는 미토콘드리아 매트릭스에 위치합니다.
SOD는
철-황 클러스터를 포함하는 단백질을 손상시키고
비활성화할 수 있는
슈퍼옥사이드의 축적을 방지합니다[10].
따라서
슈퍼옥사이드의 축적은
산화 환원 신호보다 산화 스트레스와 더 관련이 있습니다.
그러나
슈퍼옥사이드가
무차별적으로 단백질을 손상시키지는 않는다는 점에
유의하는 것이 중요합니다.
과산화물에 의해 비활성화되는 데 민감한
특정 단백질 세트가 있으며,
이 단백질은 신호 경로를 활성화하여
상승된 과산화물에 대한 적응을 촉진하거나 세포 사멸을 시작합니다[11].
이는 산화 스트레스를
세포 손상과 스트레스 반응 신호의 조합으로 보는
현재의 견해를 뒷받침합니다.
세 번째 유형의 ROS는
지질, 단백질, DNA를 무차별적으로 산화시켜
손상이나 게놈 불안정성을 초래하는
반응성이 매우 높은 하이드록실 라디칼입니다[12].
일반적으로
하이드록실 라디칼은
철 이온이 있을 때
H2O2에서 생성됩니다(즉, 펜톤 반응).
따라서
세포는
독성 수산기의 형성을 방지하기 위해
철 항상성을 유지하는 여러 가지 메커니즘을 가지고 있습니다.
신호 전달에 필요한 H2O2의 변화는
산화 글루타티온(GSSG)/환원 글루타티온(GSH) 또는
NADPH/NADP+의 세포 내 비율에
큰 변화를 일으키지 않는다는 점에 유의하는 것이 중요합니다 [13].
실제로 이러한 파라미터의 큰 변화는
일반적으로 산화 환원 생물학과 관련된 신호라기보다는
독성을 유발하는 산화 스트레스의 신호입니다 [14].
표적에 대한 ROS의 특이성 및 선택성 외에도
세포 내 ROS 생성의 구획화는
손상 또는 산화 환원 신호 발생 여부를 결정하는
중요한 요소입니다.
효과적인 산화 환원 신호 전달을 위해서는
특정 단백질의 H2O2 의존적 산화가
H2O2 생성원에 가까워야 합니다.
예를 들어,
원형질막에서 생성된 H2O2의 단백질 표적은
원형질막에 있는 NADPH 산화 효소입니다.
미토콘드리아는 표적을 향해 동적으로 이동하여
미토콘드리아에서 생성된 H2O2가
신호 전달 경로를 활성화하는 것으로 알려져 있습니다 [15].
마찬가지로
미토콘드리아 매트릭스에 과산화물이 축적되는 것은
세포질에 과산화물이 축적되는 것과는 다른 결과를 가져옵니다.
이는 부분적으로
미토콘드리아 매트릭스에 위치한
철 황 클러스터 단백질의 높은 함량 때문입니다.
실제로
SOD2 녹아웃 마우스는
SOD1 녹아웃 마우스에 비해
병리학적 표현형이 극적으로 심각합니다.
따라서
ROS의 유형과 국소 농도는
산화 환원 신호 또는
산화 스트레스에 의한 손상 발생 여부를 종합적으로 결정합니다.
이 리뷰에서는
산화 환원 신호와 산화 스트레스라는
ROS 생물학의 두 가지 측면에 대해 논의할 것입니다.
(1) 정상 및 암세포 증식,
(2) 유익한 염증과 병적인 염증,
(3) 정상 및 가속화된 노화 과정을
예로 들어 생리적 상태와 병리학적 상태에 대해 집중적으로 살펴볼 것입니다.
Redox signaling and oxidative stress: Regulation of normal and cancer cell proliferation
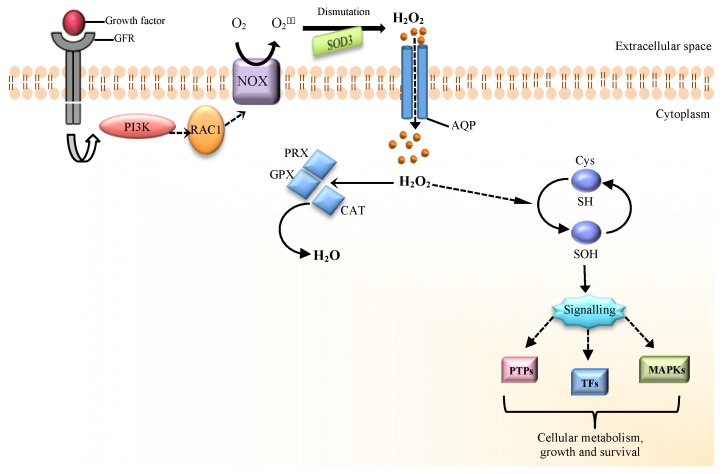
Metazoans use growth factors to coordinate mitogenic, survival, and nutrient uptake signals for cell growth and proliferation [16]. Growth factors such as epidermal growth factor (EGF) and platelet-derived growth factor (PDGF) activate the intrinsic tyrosine kinase activity of their receptors (RTK) that leads to the phosphorylation of specific tyrosine residues on the cytoplasmic tails of the receptor [17]. This recruits multiple proteins to the receptor resulting in activation of several key signal transduction pathways, notably PI3K-AKT and RAS-MEK-ERK to promote cell proliferation, nutrient uptake, and cell survival [18]. However, RTKs and PI3K are negatively regulated by protein-tyrosine phosphatases (PTPs) and PTEN, respectively, resulting in dampening of mitogenic signaling [19, 20]. Thus, to sustain signal transduction pathways it is necessary to inactivate these phosphatases.
Initial experiments demonstrated that PDGF and EGF can rapidly and transiently increase ROS generation through NADPH oxidases and that these ROS were required for growth factor induced receptor tyrosine phosphorylation [21]. Subsequently, it was demonstrated that H2O2 produced in response to EGF oxidized the catalytic cysteine of protein-tyrosine phosphatase 1B (PTP1B) to a sulfenic moiety causing inactivation of this phosphatase [22]. PTP1B dephosphorylates tyrosine residues of EGFR [23]. Thus, its inactivation by H2O2 results in increased tyrosine phosphorylation of EGFR and transmission of downstream growth signaling. The oxidized PTP1B is reactivated by thioredoxin, illustrating the reversibility of the redox signal [24]. Intracellular MAP-kinase signaling following PDGF stimulation is also reinforced through oxidation and inactivation of the PDGF-receptor associated phosphatase, SHP-2 [25]. In fact, H2O2 can reversibly oxidize a number of purified PTP family members in vitro resulting in their inactivation [26]. Purified human PTEN phosphatase, a tumor suppressor and negative regulator of phosphoinositide 3-kinase (PI3-K) signaling, is inactivated by H2O2 through oxidation and disulfide bond formation between Cys121 and Cys71, a modification reversed by thioredoxin activity [27]. Increased levels of oxidized PTEN can be measured in cells shortly after stimulation with various growth factors involved in PI3-K activation [28]. Normally, the abundance of peroxiredoxins quickly decreases H2O2 upon growth factor stimulation [29, 30]. However, recent data indicates that local pool of peroxiredoxin I (PRXI) associated with cell membranes is phosphorylated and inactivated upon growth factor stimulation to allow accumulation of local H2O2 and inhibition of phosphatase activity [31]. Since this PRX1 inactivation is localized to membranes it allows cellular PRX1 pools to remain active thus not allowing peroxide build up in the cells. These data support a model where growth factor activation must be accompanied by a localized burst in ROS production at the plasma membrane. ROS then inactivates the action of phosphatases, reinforcing proliferative signaling pathways. Recent studies indicate that in addition to NADPH oxidases, mitochondrial ROS can also cause inactivation of phosphates through oxidation [32].
Cancer cells “hijack” normal cell machinery by constitutively activating growth factor pathways to sustain cellular growth and proliferation [33]. This allows cancer cells to uptake abundant nutrients, survive stress, and continuously proliferate. Consequently, the hyper-metabolism of cancer cells causes abundant generation of ROS from mitochondria, endoplasmic reticulum, and NADPH oxidases [34]. Initial observations over two decades ago demonstrated that cancer cells generate higher levels of ROS than their non-transformed counterparts [35]. It was assumed that these elevated ROS levels caused genomic instability to promote tumorigenesis [36]. However, chromosomal instability is likely attributable to loss of p53 and other mechanisms that promote aneuploidy. Cancer cells driven by the MYC oncogene demonstrate no detectable increase in chromosomal instability and drive tumorigenesis through ROS dependent increase in signaling pathways [37]. Furthermore, treatment with the antioxidant N-acetyl-cysteine (NAC) or inhibitors of NADPH oxidase prevent mitogenic signaling pathways in oncogenic Kras driven mouse fibroblasts [38]. Human cancer cells driven by oncogenic Kras require mitochondrial ROS for proliferation [39].
Mitochondrial mutations resulting in TCA cycle or electron transport chain dysfunction generate ROS to activate tumorigenic signaling pathways including PI3K and MAPK pathways [40-42]. Another important target of ROS is the transcription factor NF-κ B known to control cell survival of tumor cells [43]. NF-κB was of one of the early transcription factors discovered to be responsive to ROS [44].
The high rate of ROS production is counterbalanced by an equally high rate of antioxidant activity in cancer cells to maintain redox balance [45]. If cancer cells do not control their ROS levels then they are susceptible to oxidative stress-induced cell death [46, 47]. Steady state ROS levels in cancer cells are determined both by rate of ROS production and also rate of ROS scavenging. Thus, at steady state, cancer cells can display either overall increase or decrease in ROS compared to normal cells. Additionally, the signaling pathways responsive to hydrogen peroxide are localized to the sources of ROS generation, allowing activation of these pathways despite the high overall antioxidant activity in cancer cells that protects against oxidative stress-induced cell death.
The major mechanism by which cancer cells increase their antioxidant proteins by is through activating the transcription factor nuclear factor erythriod 2-related factor 2 (NRF2) [48]. Normally NRF2 interacts with Kelch-like ECH-associated protein 1 (KEAP1) thus targeting NRF2 for proteasomal degradation. Elevated ROS oxidizes redox sensitive cysteine residues on KEAP1 resulting in dissociation of KEAP1 from NRF2. Subsequently, NRF2 translocates to the nucleus, heterodimerizes with small protein MAF and binds to antioxidant-responsive elements (AREs) within regulatory region of multiple antioxidant genes. Aside from elevated ROS, signaling pathways such as ERK MAPK and PI3K can activate NRF2. Furthermore, certain tumor cells display mutations of KEAP1 resulting in constitutively activation of NRF2 [49]. The loss of NRF2 in cancer cells increases oxidative stress resulting in diminished tumorigenesis [50]. It is important to note that loss NRF2 diminishes multiple antioxidant defense systems thus making multiple types of ROS (i.e. superoxide, peroxide and hydroxyl radicals) increase at a threshold that invokes damage to cancer cells. However, loss of a specific antioxidant defense system might result in elevation in ROS levels to levels below the threshold that causes damage. In this scenario, the elevated ROS levels hyper-activate signaling pathways to promote tumorigenesis as observed during the loss of peroxiredoxin I, which increases tumorigenesis [51, 52].
If increased ROS levels are essential to promote and reinforce proliferative signals, one could predict tumor suppressors (TSs) could serve as antioxidants, reducing cellular ROS to levels which do not support proliferation. The highly mutated tumor suppressor p53 controls the expression of a variety of antioxidant genes [53]. Tumor formation in p53-/- mouse models can be suppressed through supplementation of NAC in the diet, suggesting a primary tumor suppressive function of p53 in certain cancers is to decrease ROS [54]. Furthermore, a recent study suggested that the major tumor suppressive function of p53 might be through regulating antioxidant and metabolism genes rather than apoptosis and cell cycle arrest [55]. The induction of TIGAR gene is one mechanism that p53 regulates metabolism to control antioxidant function [56]. TIGAR functions as a fructose-2,6-bisphosphatase which lowers fructose-2,6-bisphosphate levels, a positive regulator of phosphofructokinase-1 [57]. This results in a decrease glycolytic flux and shunting of glucose carbons into the pentose phosphate pathway to produce NADPH, which is required to maintain many antioxidant systems. Other tumor suppressor genes such as FOXOs also repress tumorigenesis by inducing antioxidants [58].
While ROS play a key role in maintaining mitogenic signals to drive cancer cell proliferation, they also are integral in adapting to the metabolic stress that occurs when highly proliferative tumors outstrip their blood supply [59]. The resulting tissue hypoxia stabilizes the family of transcription factors termed hypoxia-inducible factors (HIFs) [60]. HIFs are a heterodimer consisting of a constitutively stable subunit, HIFβ, and an oxygen sensitive subunit, HIFα. HIFα is hydroxylated at proline residues by prolyl hydroxylases (PHDs). Hydroxylated proline residues of HIFα protein are recognized by the E3 ubiquitin ligase von Hippel-Landau protein (pVHL), which targets HIFα to the proteasome [61]. Under hypoxia, HIFα is not hydroxylated by PHDs thereby preventing pVHL targeting HIFα to the proteasome. Subsequently, HIFα translocates to the nucleus and dimerize with HIFβ regulating metabolic adaption to hypoxia and pro-angiogenic genes such as VEGF [62]. Hypoxia increases ROS production leading to HIFα stabilization through the inhibition of prolyl hydroxylases (PHDs) [63, 64]. ROS induction of HIFs can promote tumorigenesis of certain cancer cells [37, 65, 66]. Furthermore, the sirtuin protein SIRT3 is a tumor suppressor by upregulating antioxidant defenses to prevent HIF activation [67, 68].
In summary, we support a model in which tumorigenic cells generate high levels of ROS to activate proximal signaling pathways that promote proliferation, survival and metabolic adaptation (i.e. redox biology). At the same time, cancer cells maintain a high level of antioxidant activity to prevent buildup of ROS to levels that could induce cell death (i.e. oxidative stress) (Figure 2). This presents a conundrum in how to approach ROS therapy in cancer: should treatments focus on lowering ROS levels to prevent signaling or increasing ROS to selectively kill cancer cells? A systematic review of randomized control studies with the antioxidants β-carotene, vitamin A, and vitamin C concluded no significant benefit, and possibly a detrimental effect, of these agents in cancer prevention [69]. However, it is possible that more targeted antioxidants that specifically enrich in cancer cells or prevent localized ROS production from mitochondria and NADPH oxidases may provide a clinical benefit. While randomized control human trials with pro-oxidant cancer therapy have not yet been completed, there is accumulating evidence that raising ROS levels through small molecules can selectively induce cancer cell death by disabling antioxidants [70-72]. There are two caveats to this approach, First, if the ROS levels are not sufficiently raised within the cancer cell then the therapy would simply further activate NF-κB, PI3K, HIFs and MAPKs to promote tumorigenesis. Additionally, agents should disable only the antioxidants utilized by cancer cells compared to normal cells. Since the role of ROS and sensitivity to both oxidants and antioxidants likely differs between cancer types, continuing to test both oxidants and antioxidants in vivo will hopefully yield new agents to add to existing chemotherapy regimens.
산화 환원 신호와 산화 스트레스: 정상 세포 및 암세포 증식 조절
메타조류는 성장 인자를 사용하여
세포 성장과 증식을 위한 유사 분열,
생존 및 영양소 흡수 신호를 조정합니다[16].
표피 성장 인자(EGF) 및 혈소판 유래 성장 인자(PDGF)와 같은 성장 인자는
수용체(RTK)의 고유한 티로신 키나제 활성을 활성화하여
수용체의 세포질 꼬리에서 특
정 티로신 잔기의 인산화를 유도합니다[17].
이는
여러 단백질을 수용체에 모집하여
여러 주요 신호 전달 경로, 특히
PI3K-AKT 및 RAS-MEK-ERK를 활성화하여
세포 증식, 영양소 흡수 및 세포 생존을 촉진합니다 [18].
그러나
RTK와 PI3K는 각각
단백질 티로신 포스파타제(PTP)와 PTEN에 의해
음성적으로 조절되어 유사 분열 신호가 약화됩니다 [19, 20].
따라서
신호 전달 경로를 유지하려면
이러한 포스파타제를 비활성화해야 합니다.
초기 실험에서는
PDGF와 EGF가 NADPH 산화 효소를 통해
ROS 생성을 빠르고 일시적으로 증가시킬 수 있으며
이러한 ROS가 성장 인자 유도 수용체 티로신 인산화에 필요하다는 것이 입증되었습니다 [21].
그 후, EGF에 반응하여 생성된 H2O2가 단백질-티로신 포스파타제 1B(PTP1B)의 촉매 시스테인을 황화 모이티브로 산화시켜 이 포스파타제를 비활성화한다는 사실이 입증되었습니다[22]. PTP1B는 EGFR의 티로신 잔기를 탈인산화합니다 [23]. 따라서 H2O2에 의한 비활성화는 EGFR의 티로신 인산화를 증가시키고 다운스트림 성장 신호 전달을 증가시킵니다. 산화된 PTP1B는 티오레독신에 의해 다시 활성화되어 산화 환원 신호의 가역성을 보여줍니다 [24]. PDGF 자극에 따른 세포 내 MAP-키나제 신호는 또한 PDGF 수용체 관련 포스파타제인 SHP-2의 산화 및 비활성화를 통해 강화됩니다 [25].
실제로
H2O2는 시험관 내에서 다수의 정제된 PTP 계열을
가역적으로 산화시켜 비활성화할 수 있습니다 [26].
종양 억제제이자 포스포이노시타이드 3-키나아제(PI3-K) 신호의 음성 조절자인 정제된 인간 PTEN 포스파타제는 티오레독신 활성으로 역전되는 변형인 Cys121과 Cys71 사이의 산화 및 이황화 결합 형성을 통해 H2O2에 의해 비활성화됩니다 [27]. PI3-K 활성화에 관여하는 다양한 성장 인자로 자극한 직후 세포에서 산화된 PTEN의 증가된 수준을 측정할 수 있습니다 [28]. 일반적으로 퍼옥시레독신의 풍부함은 성장 인자 자극 시 H2O2를 빠르게 감소시킵니다 [29, 30]. 그러나 최근 데이터에 따르면 세포막과 관련된 퍼옥시레독신 I(PRXI)의 국소 풀은 성장인자 자극 시 인산화되고 비활성화되어 국소 H2O2의 축적과 포스파타제 활성을 억제합니다[31]. 이러한 PRX1 비활성화는 세포막에 국한되어 있기 때문에 세포의 PRX1 풀이 활성 상태를 유지하여 세포에 과산화물이 쌓이지 않도록 합니다. 이러한 데이터는 성장 인자 활성화가 혈장막에서 국소적인 ROS 생산의 폭발을 동반해야 한다는 모델을 뒷받침합니다. 그런 다음 ROS는 포스파타제의 작용을 비활성화하여 증식성 신호 경로를 강화합니다. 최근 연구에 따르면 미토콘드리아 ROS는 NADPH 산화 효소 외에도 산화를 통해 인산염의 비활성화를 일으킬 수 있습니다 [32].
암세포는
세포 성장과 증식을 유지하기 위해
성장 인자 경로를 지속적으로 활성화하여
정상 세포 메커니즘을 "탈취"합니다 [33].
이를 통해 암세포는
풍부한 영양분을 흡수하고 스트레스를 견디며
지속적으로 증식할 수 있습니다.
결과적으로
암세포의 과대 대사는
미토콘드리아, 소포체 및 NADPH 산화효소로부터
ROS를 풍부하게 생성합니다 [34].
20여 년 전의 초기 관찰에 따르면
암세포는 변형되지 않은 세포보다
더 높은 수준의 ROS를 생성하는 것으로 나타났습니다 [35].
이러한 높은 ROS 수치가
게놈 불안정성을 유발하여
종양 형성을 촉진한다고 가정했습니다[36].
그러나 염색체 불안정성은 p53의 손실 및 이수성을 촉진하는 다른 메커니즘에 기인할 가능성이 높습니다. MYC 발암 유전자에 의해 구동되는 암세포는 염색체 불안정성이 감지할 수 없을 정도로 증가하지 않으며 신호 전달 경로의 ROS 의존적 증가를 통해 종양 형성을 촉진합니다 [37]. 또한 항산화제인 N-아세틸 시스테인(NAC) 또는 NADPH 산화효소 억제제로 처리하면 발암성 Kras 유도 마우스 섬유아세포에서 발암성 신호 전달 경로를 방지할 수 있습니다 [38]. 발암성 Kras에 의해 구동되는 인간 암세포는 증식을 위해 미토콘드리아 ROS가 필요합니다 [39].
TCA 주기 또는 전자 수송 사슬 기능 장애를 초래하는 미토콘드리아 돌연변이는
PI3K 및 MAPK 경로를 포함한
종양 유발 신호 경로를 활성화하기 위해
ROS를 생성합니다 [40-42].
ROS의 또 다른 중요한 표적은 종양 세포의 세포 생존을 조절하는 것으로 알려진 전사인자 NF-κ B입니다 [43]. NF-κB는 ROS에 반응하는 것으로 밝혀진 초기 전사인자 중 하나였습니다 [44].
높은 ROS 생성 속도는 산화 환원 균형을 유지하기 위해 암세포에서 똑같이 높은 항산화 활성으로 상쇄됩니다 [45]. 암세포가 ROS 수치를 조절하지 못하면 산화 스트레스로 인한 세포 사멸에 취약해집니다 [46, 47]. 암세포의 정상 상태 ROS 수준은 ROS 생성 속도와 ROS 제거 속도에 의해 결정됩니다. 따라서 정상 상태에서 암세포는 정상 세포에 비해 전반적으로 ROS가 증가하거나 감소할 수 있습니다. 또한 과산화수소에 반응하는 신호 전달 경로는 ROS 생성원에 국한되어 있어 암세포의 전반적인 항산화 활성이 높더라도 이러한 경로가 활성화되어 산화 스트레스로 인한 세포 사멸을 방지할 수 있습니다.
암세포가 항산화 단백질을 증가시키는 주요 메커니즘은 전사인자 핵인자 에리스리드 2 관련 인자 2(NRF2)를 활성화하는 것입니다[48]. 일반적으로 NRF2는 켈치 유사 ECH 관련 단백질 1(KEAP1)과 상호 작용하여 프로테아좀 분해를 위해 NRF2를 표적으로 삼습니다. 상승된 ROS는 KEAP1의 산화 환원 민감성 시스테인 잔기를 산화시켜 NRF2에서 KEAP1의 해리를 초래합니다. 그 후 NRF2는 핵으로 이동하여 작은 단백질 MAF와 이합체화되고 여러 항산화 유전자의 조절 영역 내에서 항산화 반응 요소(ARE)에 결합합니다. 상승된 ROS 외에도 ERK MAPK 및 PI3K와 같은 신호 전달 경로는 NRF2를 활성화할 수 있습니다. 또한, 특정 종양 세포는 KEAP1의 돌연변이를 일으켜 NRF2를 지속적으로 활성화합니다 [49]. 암세포에서 NRF2가 손실되면 산화 스트레스가 증가하여 종양 형성이 감소합니다 [50]. NRF2의 손실은 여러 항산화 방어 시스템을 감소시켜 암세포의 손상을 유발하는 역치에서 여러 유형의 ROS(예: 슈퍼옥사이드, 과산화물 및 하이드록실 라디칼)를 증가시킨다는 점에 유의하는 것이 중요합니다. 그러나 특정 항산화 방어 시스템의 손실로 인해 ROS 수치가 손상을 유발하는 임계치 이하로 상승할 수 있습니다. 이 시나리오에서 상승된 ROS 수치는 종양 형성을 촉진하는 신호 경로를 과도하게 활성화하여 퍼옥시레독신 I이 손실되는 동안 관찰된 것처럼 종양 형성을 증가시킵니다 [51, 52].
증식 신호를 촉진하고 강화하기 위해 증가된 ROS 수치가 필수적이라면 종양 억제제(TS)가 항산화제 역할을 하여 세포 ROS를 증식을 지원하지 않는 수준으로 감소시킬 수 있을 것으로 예측할 수 있습니다. 고도로 돌연변이된 종양 억제 유전자 p53은 다양한 항산화 유전자의 발현을 조절합니다[53]. p53-/- 마우스 모델에서 종양 형성은 식이에서 NAC를 보충함으로써 억제될 수 있으며, 이는 특정 암에서 p53의 주요 종양 억제 기능이 ROS를 감소시키는 것임을 시사합니다 [54]. 또한, 최근 연구에서는 p53의 주요 종양 억제 기능이 세포 사멸과 세포 주기 정지가 아닌 항산화 및 대사 유전자 조절을 통한 것일 수 있다고 제안했습니다 [55]. TIGAR 유전자의 유도는 p53이 대사를 조절하여 항산화 기능을 조절하는 한 가지 메커니즘입니다 [56]. TIGAR은 포스포프락토키나제-1의 양성 조절자인 과당-2,6-비스포스페이트 수치를 낮추는 과당-2,6-비스포스파타제로서 기능합니다 [57]. 그 결과 해당 작용이 감소하고 포도당 탄소가 펜토오스 인산염 경로로 전환되어 많은 항산화 시스템을 유지하는 데 필요한 NADPH를 생성하게 됩니다. FOXO와 같은 다른 종양 억제 유전자도 항산화 물질을 유도하여 종양 형성을 억제합니다[58].
ROS는 암세포 증식을 유도하는 유사 분화 신호를 유지하는 데 중요한 역할을 하지만, 증식성이 높은 종양이 혈액 공급을 초과할 때 발생하는 대사 스트레스에 적응하는 데도 필수적인 역할을 합니다 [59]. 그 결과 조직 저산소증은 저산소증 유도 인자(HIF)라고 하는 전사 인자군을 안정화시킵니다[60]. HIF는 구성적으로 안정적인 하위 단위인 HIFβ와 산소에 민감한 하위 단위인 HIFα로 구성된 헤테로다이머입니다. HIFα는 프롤린 잔기에서 프롤릴 하이드 록실화 효소(PHD)에 의해 하이드 록실화됩니다. HIFα 단백질의 수산화된 프롤린 잔기는 E3 유비퀴틴 리가제 폰 히펠-란다우 단백질(pVHL)에 의해 인식되어 프로테아좀에 HIFα를 표적으로 삼습니다[61]. 저산소증에서는 HIFα가 PHD에 의해 수산화되지 않으므로 pVHL이 HIFα를 프로테아좀으로 표적화하는 것을 방지할 수 있습니다. 그 후, HIFα는 핵으로 이동하여 저산소증에 대한 대사 적응과 VEGF와 같은 혈관 신생성 유전자를 조절하는 HIFβ와 이량체화됩니다 [62]. 저산소증은 ROS 생성을 증가시켜 프롤릴 하이드 록실 라제(PHD)의 억제를 통해 HIFα를 안정화시킵니다 [63, 64]. HIF의 ROS 유도는 특정 암세포의 종양 형성을 촉진할 수 있습니다 [37, 65, 66]. 또한 시르투인 단백질 SIRT3는 항산화 방어를 상향 조절하여 HIF 활성화를 방지하는 종양 억제제입니다 [67, 68].
요약하면, 우리는 종양 유발 세포가 높은 수준의 ROS를 생성하여 증식, 생존 및 대사 적응을 촉진하는 근위 신호 경로(즉, 산화 환원 생물학)를 활성화하는 모델을 지지합니다. 동시에 암세포는 높은 수준의 항산화 활성을 유지하여 세포 사멸(즉, 산화 스트레스)을 유발할 수 있는 수준까지 ROS가 축적되는 것을 방지합니다(그림 2). 이는 암에서 ROS 치료에 접근하는 방법에 대한 수수께끼를 제시합니다. 신호 전달을 막기 위해 ROS 수치를 낮추는 데 중점을 두어야 할까요, 아니면 암세포를 선택적으로 죽이기 위해 ROS를 증가시키는 데 중점을 두어야 할까요? 항산화제인 β-카로틴, 비타민 A, 비타민 C를 사용한 무작위 대조 연구에 대한 체계적인 검토에 따르면 이러한 약제는 암 예방에 큰 이점이 없으며 오히려 해로운 영향을 미칠 수 있다고 결론지었습니다[69]. 그러나 암세포에 특이적으로 풍부하거나 미토콘드리아 및 NADPH 산화효소로부터 국소화된 ROS 생성을 막는 표적 항산화제가 임상적 이점을 제공할 수 있습니다. 항산화제 암 치료에 대한 무작위 대조군 임상시험은 아직 완료되지 않았지만, 저분자를 통해 ROS 수준을 높이면 항산화제를 무력화하여 암세포 사멸을 선택적으로 유도할 수 있다는 증거가 축적되고 있습니다[70-72]. 이 접근법에는 두 가지 주의 사항이 있는데, 첫째, 암세포 내에서 ROS 수치가 충분히 높아지지 않으면 치료제가 NF-κB, PI3K, HIF 및 MAPK를 더욱 활성화하여 종양 형성을 촉진할 수 있다는 것입니다. 또한, 치료제는 정상 세포에 비해 암세포가 사용하는 항산화 물질만 비활성화해야 합니다. ROS의 역할과 산화제와 항산화제에 대한 민감도는 암 유형에 따라 다를 수 있으므로 생체 내에서 산화제와 항산화제를 계속 테스트하면 기존 화학 요법에 추가할 수 있는 새로운 약제를 찾을 수 있을 것입니다.
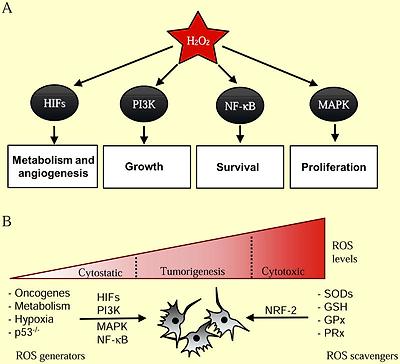
ROS regulation of normal and cancer cell proliferation
(A) Hydrogen peroxide (H2O2) is required for activation of a number of cellular pathways involved in cellular proliferation. (B) Cancer cells generate higher levels of ROS that are essential for tumorigenesis. Genetic alterations leading to activation of oncogenes (PI3K, MAPK, HIFs, NF-κB) and loss of tumor suppressors (p53) coordinate an elevated redox state. ROS is also generated from increased oxidative metabolism and hypoxia in rapidly expanding tumors. Cancer cells also express elevated levels of cellular antioxidants (SODs, GSH, GPx, PRx) in part through NRF-2 to protect against oxidative stress-induced cell death.
Redox signaling and oxidative stress: Regulation of inflammation
The innate and adaptive immune systems are critical for pathogen-specific defense and immunological memory. Furthermore, the immune system is crucial for tissue repair. However, if immune system either fails to be properly activated or is persistently activated it can contribute to multiple diseases including autoimmunity, cardiovascular disease, and accelerate the normal aging process. In the past two decades, there is substantial evidence that ROS are essential second messengers in innate and adaptive immune cells [73, 74]. Yet, increased levels of ROS within immune cells can result in hyperactivation of inflammatory responses resulting in tissue damage and pathology [75].
The innate immune system responds to microorganism-derived pathogen-associated molecular patterns (PAMPs) and endogenous cell-derived damage-associated molecular patterns (DAMPs) to tissue injury [76]. PAMPs and DMAPs bind to specific receptors including Toll-like receptors (TLR), RIG-I-like receptors (RLRs), and NOD-like receptors (NLRs) to generate cytokines that are essential for fighting against pathogen or repair tissue damage [77-79]. Initial studies implicating ROS in innate immune system came from the observations that LPS activates inflammatory cytokines through the generation of NADPH oxidase and mitochondrial generated ROS [80, 81]. More recent studies have shown that mitochondrial ROS are essential for other TLR-initiated pathways including TLR1, TLR2, and TLR4 and for optimal bactericidal activity of macrophages [82]. The RIG-I-like receptors (RLRs) also signal through mitochondrial ROS [83]. This might not be surprising since the outer mitochondrial membrane serves as a platform for formation of the RLR molecular complex [84]. The NLRP3, a NLR that is a component of the inflammasome, also requires NADPH and mitochondrial ROS for activation [85-89]. Interestingly, patients with tumor necrosis factor receptor-associated periodic syndrome (TRAPS) have heightened responsiveness to LPS due to increased mitochondrial ROS production which promotes inflammation, again suggesting redox biology, and not oxidative stress, is regulating inflammatory diseases [90].
Adaptive immunity involves the expansion of T cells and B cells specific for pathogens via rapid proliferative responses. Initial evidence for redox signaling in the process stems from the observations that treatment of primary T cells with pharmacologic antioxidants inhibits proliferation and production of the cytokine IL-2 following T-cell receptor stimulation in vitro [91]. Antioxidants also diminished the expansion of T cells in vivo [92]. The major initial source of ROS required for T cells activation is mitochondria [93, 94]. Pharmacological or genetic manipulation dampening mitochondrial ROS generation can diminish T cell activation in vitro and in vivo. However, NADPH oxidase can be invoked in response to mitochondrial ROS to further sustain ROS levels to maintain T cell activation [95]. NADPH oxidase and mitochondrial generated ROS have been implicated for B cell activation and proliferation upon stimulation of the B-cell receptor (BCR) [96, 97]. Thus, both TCR and BCR signaling requires ROS generation to mount the proper inflammatory response.
What happens when ROS levels are elevated during immune responses? The simple answer is that this depends on the degree to which ROS levels are elevated beyond what is expected during a normal immune response. A slight elevation could be beneficial under certain conditions or detrimental [98]. For example, uncoupling Protein 2 (UCP2) knockout mice feature higher levels of mitochondrial ROS and possess increased immunity to bacterial pathogens [99]. This suggests a low elevated level of ROS in the immune system might enhance normal immune function. Indeed, mice heterozygous for Mclk1 (a mitochondrial hydroxylase necessary for ubiquinone synthesis) display increased mitochondrial ROS with elevated normal innate and adaptive inflammatory responses to fight pathogens without incurring tissue damage [100]. By contrast, high levels of ROS generation due to loss of NRF2 lead to elevated levels of pro-inflammatory cytokines [101]. NRF2 deficient mice display exacerbated innate immune derived inflammation responses to pathogens resulting in worsened pneumonia and sepsis [102]. Antioxidants improve survival of NRF2 deficient mice in these models of sepsis. Antigen-specific adaptive immunity induced by sensitization to ovalbumin model of asthma is also intensified by Nrf2 deficiency [103]. Thus, slightly elevated ROS levels may enhance immune system function while high levels of ROS could promote a pathological inflammatory response.
The finding that ROS functions both in the normal innate and adaptive immunity presents a challenge as to when antioxidants could be utilized as an immunomodulatory therapy. Clearly antioxidants should not be administered in healthy individuals that have robust antoxidant defense and a healthy immune system, since ROS are intimately tied to optimal pathogen clearance. However, when the immune system becomes dysregulated, as observed in autoimmune disease, antioxidants could be helpful in ameliorating the heightened inflammation response. As with ROS therapy in cancer, there are obstacles that need to be considered when using antioxidants for immunomodulation. For example, the dosing of antioxidants should not be so high as to interfere with normal immune responses. Furthermore, the timing of antioxidants is crucial during the progression of an inflammatory disease. This is certainly the case in critically ill patients in the intensive care unit (ICU). These patients often display signs of elevated ROS and heightened inflammatory responses that result in multi-organ failure and mortality. Even in the cases of an acute infection, it is possible that a pro-inflammatory cytokine storm is primarily responsible for admission to the ICU. Yet, multiple clinical trials have consistently showed no efficacy or an increase in mortality in patients with critical illness in the ICU treated with antioxidants [104]. Reasons for this failure are not fully understood but we speculate that the antioxidants might interfere with the normal responses to pathogens in certain immunosuppressed populations. It is possible that these immunosuppressed patients might even benefit from pro-oxidant therapy to boost their immune system.
Going forward, it will be important to characterize how different inflammatory cells respond to changes in ROS levels. It is now more appreciated that different T cell and macrophage subsets can be pro- or anti-inflammatory. But, it is not fully understood whether these different subsets have differential responses to ROS. Along these lines, it might be beneficial to increase a particular subset of T cells or macrophages by either increasing or decreasing ROS levels to ameliorate immune system pathologies.
Redox signaling and oxidative stress: Regulation of aging
The ability to regenerate tissues as well as to prevent damage to existing tissues are two key determinants of aging. One of the original theories of aging formulated over 50 years ago is Denham Harman's Free Radical Theory of Aging, which proposes ROS contribute to aging through their reactivity towards cellular macromolecules, particularly in the mitochondria [105]. Damaged mitochondria, through inefficient oxidative phosphorylation, produce escalating amounts of ROS inevitably impairing cellular function [106]. However, interventions in reducing ROS levels have had mixed results and it not clear whether ROS induced damage is the underlying cause of aging [107]. On the contrary, recent evidence suggests ROS-signaling is required for maintenance of tissues and increasing ROS can activate cellular stress pathways to dampen tissue degeneration to promote healthy aging [108].
Longevity studies in multiple model organisms have not consistently demonstrated that antioxidants prevent aging. Early studies in Drosophila suggested that increasing SOD and catalase activity in the cytosol extend longevity [109]. However, other investigators could not duplicate these experiments [110]. Furthermore, careful measurements of ROS in Drosophila do not find any correlation between ROS levels and longevity. In mice, overexpression of cytosolic SOD with catalase or mitochondrial SOD also does not increase longevity [111]. By contrast, the overexpression of mitochondrial matrix catalase (CATmm) but not cytosolic or nuclear catalase in mice does extend longevity [112]. The conventional interpretation is that CATmm detoxified mitochondrial matrix hydrogen peroxide to water preventing peroxide induced oxidative damage to mitochondria. An alternative explanation is detoxification of matrix generated hydrogen peroxide prevented leakage of hydrogen peroxide into the cytosol thereby interfering with normal ROS signaling pathways that prevent pathologies such as cancer, a major cause of death in laboratory mice. Since mitochondria are a major source of ROS in the cell, the mitochondrial genome is often thought to be the particularly susceptible to oxidative damage. Yet, deletion of mitochondrial matrix SOD in mice increases mitochondrial DNA damage and increases cancer incidence, but does not accelerate aging [113, 114]. Interestingly, loss of superoxide dismutase enzymes in C. elegans can even extend lifespan [115].
An observation that further questions the Free Radical Theory of Aging is that elevation of ROS through signaling mechanisms can increase longevity from yeast to mice [116]. In yeast, inhibition of TOR (target of rapamycin) or caloric restriction extends chronological lifespan by increasing mitochondrial ROS [117, 118]. In C. elegans, glucose restriction, mitochondrial electron transport mutations, and diminished insulin-growth factor (IGF) signaling extend lifespan by increasing mitochondrial ROS [119-121]. Paraquat, a direct generator of mitochondrial ROS, is sufficient to increase lifespan in C. elegans [122]. Sirtuin dependent extension of lifespan in C. elegans has also been shown to be dependent on an increase in ROS production. This result was unexpected as sirtuin contribution to lifespan was previously thought primarily to be mediated by deactylation of proteins including histones [123]. There is also increasing evidence for a conserved mitochondrial longevity pathway in mammals. Mice heterozygous for MCLK1, a protein required for proper electron transport, have increased mitochondrial ROS [124]. However, these mice feature less oxidative damage to cytosolic proteins and are long-lived, supporting a model where elevated ROS levels are paradoxically protective through induction of stress pathways [125]. A common model of human aging in cell culture is examining replicative senescence. Initial studies to support the free radical theory comes from the observation that hypoxia increase human diploid fibroblasts replicative life span [126]. The original interpretation was that hypoxia decreased ROS resulting in less accumulation of oxidative damage to increase replicative lifespan. However, later studies have demonstrated a paradoxical increase in mitochondrial ROS during hypoxia resulting in activation of HIFs to increase replicative lifespan of human fibroblasts [127]. The long-lived mitochondrial mutants in C. elegans also depend on ROS-dependent activation of HIF for increased lifespan [119]. Beyond HIF, there are likely to be multiple signaling pathways that ROS activates to increase lifespan.
Aging is accelerated when the tissues that are damaged are not repaired [128]. The maintenance of adult tissue and organ systems requires removal of damaged cells and replenishment from undifferentiated stem cell populations. Stem cells have to both self-renew to maintain the stem cell pool and also differentiate to generate specialized tissue. The best-studied example is the hematopoietic stem cell (HSC), which differentiate to provide myeloid and lymphoid progenitors throughout lifespan. An emerging model in HSCs is that generation of low levels of ROS through NADPH oxidases or from mitochondria is required to activate proliferative pathways, serving as a “go” signal to support stem cell proliferation. By contrast, high levels of ROS impair stem cell function by activating signaling pathways that limit self-renewal, but do not necessarily cause cellular damage. For example, HSCs from mice that lack the ataxia telangiectasia mutated (Atm) gene have higher ROS levels that activate p38 MAPK and p16INK4a to reduce HSC repopulating capacity and exhaustion of the stem cell population [129]. Interestingly, the rise in p16INK4a expression increases with age and has been directly shown to limit stem cell renewal and function [130]. The antioxidant NAC rescues defects due to loss of ATM [131]. Other defects caused by loss of ATM include development of thymic lymphoma and innate and adaptive immune dysregulation [132]. These defects are alleviated by the expression of mitochondrial catalase in ATM null mice suggesting that the normal function of ATM might to be to control ROS levels [132]. Indeed, ROS oxidize a specific cysteine residue on ATM to generate disulfide-linked activated ATM dimers that promote anti-oxidant responses by regulating NADPH production from the pentose phosphate pathway (PPP) [134, 134]. ATM also maintains a low level of ROS to maintain stem cell function in part through the Bcl2 protein BID [135].
Consistent with the ATM observation on HSCs is that the loss of FOXOs, Bmi1, or Tsc1 triggered increase in ROS levels in HSCs, which limited HSC repopulating capacity [136-138]. Aside from HSCs, neural stem cells are also sensitive to increase in ROS. The loss of PRDM16, a transcription factor that regulates brown fat, triggers defects in HSCs and neural stem cell function [139]. PRDM16 is highly expressed in HSCs and neural stem cells. However, NAC only rescued neural stem cell defects but not HSCs suggesting that redox biology is context dependent. Thus, ROS levels have to be maintained in a range that allows for stem cells to function properly, and that concentration may differ between tissues.
ROS are also essential for stem cell differentiation. Mouse HSCs deficient in both AKT1 and AKT2 have reduced levels of ROS that impaired differentiation [140]. In Drosophila hematopoietic progenitors, increasing ROS triggers differentiation while decreasing ROS impairs differentiation [141]. Furthermore, human bone marrow mesenchymal stem cells also require ROS for differentiation in to adipocytes and mitochondrial ROS generated from complex I can trigger muscle differentiation [142, 143]. Within the skin epidermis, undifferentiated cells along the basement membrane undergo a regulated transformation into mature apical epidermal cells. Lowering mitochondrial ROS impairs this differentiation process which can surprisingly be restored by supplementing exogenous H2O2 [144]. Similarly, lowering ROS levels decreases the regenerative capacity of neural stem cells and spermatogonial stem cells [145, 146]. However, aberrant elevation of ROS levels impairs cardiac myocyte differentiation [147]. This raises two questions: (1) What levels of ROS are required for stem cell renewal compared to stem cell differentiation? (2) What levels of ROS inhibit stem cell renewal compared to impaired stem cell differentiation? We speculate that quiescent stem cells reside at low levels of ROS and a slight increase in ROS provides the signal for self-renewal and cellular differentiation. ROS levels above those required for self-renewal or differentiation impairs these critical two stem cell properties.
The idea that stem cell and tissue impairment are not a consequence of oxidative stress-induced damage is further supported from experiments in mice harboring mitochondrial mutations. Mice engineered to accumulate mitochondrial DNA mutations due to defective DNA polymerase proofreading prematurely age and display impairment of NSC and HSC function beginning in utero [148]. Remarkably, administration of NAC rescued both NSC and HPC dysfunction, suggesting the genetic mutations caused by the error-prone polymerase were dispensable for self-renewal. Other mutations of mitochondrial DNA, such as A1555G, result in maternally inherited phenotypes by increases in ROS [149]. However, these ROS cause their pathologies through activation of signaling pathways and apoptosis, rather than oxidative damage. Consequently, the stem cell, tissue degeneration, and aging communities have undergone an evolution in the past two decades from viewing ROS as simply toxins that cause cellular damage to molecules that regulate cellular signaling pathways to invoke beneficial or detrimental effects.
Conclusion
In the past two decades, ROS have undergone a shift from being molecules that invoke damage (i.e. oxidative stress) to regulating signaling pathways that impinge on normal physiological and biological responses (i.e. redox biology). The levels and compartmentalization of hydrogen peroxide dictate redox biology while high levels of superoxide or hydroxyl radicals invoke oxidative stress. Redox signaling is required for numerous cellular processes, as indicated by the role of ROS in proper cellular differentiation, tissue regeneration, and prevention of aging. On the other hand, we propose that redox signaling, and not oxidative stress, is also crucial in regulating signaling pathways that control various disease states, including tumorigenesis, automimmunity, and loss of tissue regeneration with age. This conceptual shift makes it difficult to interfere with redox biology by administering antioxidants, which would affect redox biology of both normal and abnormal responses. To date, physical exercise is one strategy that increases ROS resulting in activation of beneficial pathways that diminish cancer, diabetes, and ageing [150]. But, since most of us find it difficult to spend time at the gym, it will be important to identify the molecular effectors of redox biology that keep normal biological and physiological responses functioning from those that are promote human pathologies. This would allow for selective therapies that would alleviate disease, but not interfere with healthy tissue.
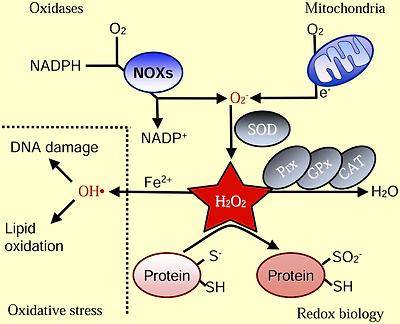
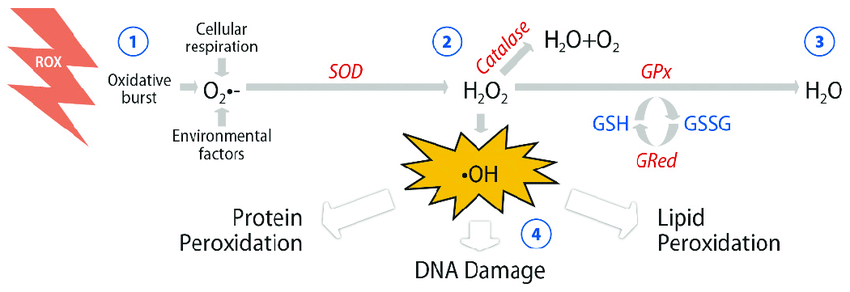
Basics of ROS
Intracellular superoxide (O2-) is primarily produced from the oxidation of NADPH by oxidase enzymes (NOX) or from electron leak from aerobic respiration in the mitochondria. Superoxide is rapidly converted into hydrogen peroxide (H2O2) by compartment-specific superoxide dismutases (SODs). H2O2 is capable of oxidizing cysteine residues on proteins to initiate redox signaling. Alternatively, H2O2 may be converted to H2O by cellular antioxidant proteins, such as peroxiredoxins (PRx), glutathione peroxidase (GPx), and catalase (CAT). When H2O2 levels increase uncontrollably, hydroxyl radicals (OH·) form through reactions with metal cations (Fe2+) and irreversibly damage cellular macromolecules
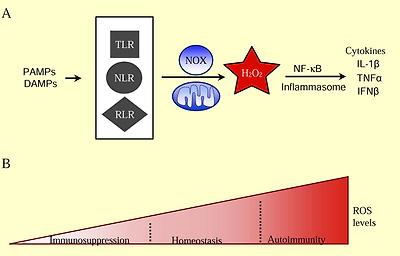
ROS Regulation of Inflammation
(A) Activation of the innate immune system requires ROS signaling. Common features of pathogens and cell damage (PAMPs, DAMPs) activate surveillance receptors (TLR, NLR, RLR) which increase ROS through NAPDH oxidase enzymes and the mitochondria. ROS is required for release of pro-inflammatory cytokines (IL-1β, TNFα, IFNβ) for a proper immune response. (B) Low levels of ROS maintain a healthy immune system. Decreasing ROS levels inhibits activation of proper immune responses, leading to immunosuppression. Elevated ROS levels contribute to autoimmunity through increased release of pro-inflammatory cytokines and proliferation of specific subsets of adaptive immune cells.
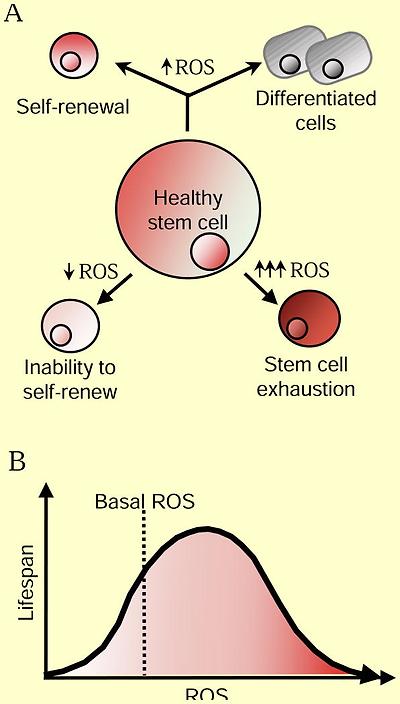
ROS Regulation of Aging
(A) Moderate ROS levels are required for proper stem cell differentiation and renewal through activation of signaling pathways. On one hand, decreased ROS levels impair stem cell properties, but ROS levels that are too high lead to stem cell exhaustion and premature aging through activation of signaling pathways. (B) Increased ROS are not detrimental to lifespan. Activation of cellular responses due to slight increases in ROS can increase signaling pathways that counter the normal aging process. However, high ROS levels can hyper-activate signaling pathways that promote inflammation, cancer and cell death leading to an accelerated aging phenotype.
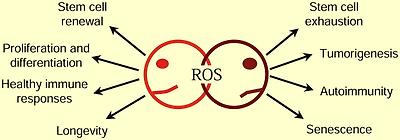
Janus of ROS: A Therapeutic Conundrum
Redox biology encompasses both the physiological and pathological roles of ROS. Determining whether to use prooxidant therapy to promote physiological ROS responses or antioxidant therapy to prevent ROS pathologies remains the central question in redox biology.
Acknowledgments
This work was supported by NIH grants T32-HL76139 (M.S.), 5P01HL071643 (N.S.C. and RO1CA123067 (N.S.C.).
Footnotes
The authors declare no competing financial interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
1. Cross CE, Halliwell B, Borish ET, Pryor WA, Ames BN, Saul RL, McCord JM, Harman D. Oxygen radicals and human disease. Annals of internal medicine. 1987;107:526–545. [PubMed] [Google Scholar]
2. Finkel T. Signal transduction by reactive oxygen species. J Cell Biol. 2011;194:7–15. [PMC free article] [PubMed] [Google Scholar]
3. Wood ZA, Poole LB, Karplus PA. Peroxiredoxin evolution and the regulation of hydrogen peroxide signaling. Science. 2003;300:650–653. [PubMed] [Google Scholar]
4. Kiley PJ, Storz G. Exploiting thiol modifications. PLoS Biol. 2004;2:e400. [PMC free article] [PubMed] [Google Scholar]
5. Rhee S. Cell signaling. H2O2, a necessary evil for cell signaling. Science. 2006;312:1882–1883. [PubMed] [Google Scholar]
6. Finkel T. From sulfenylation to sulfhydration: what a thiolate needs to tolerate. Sci Signal. 2012;5:pe10. [PubMed] [Google Scholar]
7. Winterbourn CC, Hampton MB. Thiol chemistry and specificity in redox signaling. Free Radic Biol Med. 2008;45:549–561. [PubMed] [Google Scholar]
8. Lambeth JD. NOX enzymes and the biology of reactive oxygen. Nat Rev Immunol. 2004;4:181–189. [PubMed] [Google Scholar]
9. Brand M. The sites and topology of mitochondrial superoxide production. Exp Gerontol. 2010;45:466–472. [PMC free article] [PubMed] [Google Scholar]
10. Fridovich I. Superoxide anion radical (O2-.), superoxide dismutases, and related matters. J Biol Chem. 1997;272:18515–18517. [PubMed] [Google Scholar]
11. Chen Y, Azad MB, Gibson SB. Superoxide is the major reactive oxygen species regulating autophagy. Cell Death Differ. 2009;16:1040–1052. [PubMed] [Google Scholar]
12. Dizdaroglu M, Jaruga P. Mechanisms of free radical-induced damage to DNA. Free Radic Res. 2012;46:382–419. [PubMed] [Google Scholar]
13. Morgan B, Sobotta MC, Dick TP. Measuring E (GSH) and H(2)O(2) with roGFP2-based redox probes. Free Radic Biol Med 2011 [PubMed] [Google Scholar]
14. Murphy MP. Mitochondrial thiols in antioxidant protection and redox signaling: distinct roles for glutathionylation and other thiol modifications. Antioxid Redox Signal. 2012;16:476–495. [PubMed] [Google Scholar]
15. Al-Mehdi AB, Pastukh VM, Swiger BM, Reed DJ, Patel MR, Bardwell GC, Pastukh VV, Alexeyev MF, Gillespie MN. Perinuclear mitochondrial clustering creates an oxidant-rich nuclear domain required for hypoxia-induced transcription. Sci Signal. 2012;5:ra47. [PMC free article] [PubMed] [Google Scholar]
16. Thompson CB. Rethinking the regulation of cellular metabolism. Cold Spring Harb Symp Quant Biol. 2011;76:23–29. [PubMed] [Google Scholar]
17. Lemmon MA, Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2010;141:1117–1134. [PMC free article] [PubMed] [Google Scholar]
18. Cantley LC. The phosphoinositide 3-kinase pathway. Science. 2002;296:1655–1657. [PubMed] [Google Scholar]
19. Tonks NK. Protein tyrosine phosphatases: from genes, to function, to disease. Nat Rev Mol Cell Bio. 2006;7:833–846. [PubMed] [Google Scholar]
20. Wishart MJ, Dixon JE. PTEN and myotubularin phosphatases: from 3-phosphoinositide dephosphorylation to disease. Trends in Cell Biology. 2002;12:579–585. [PubMed] [Google Scholar]
21. Sundaresan M, Yu Z, Ferrans V, Irani K, Finkel T. Requirement for generation of H2O2 for platelet-derived growth factor signal transduction. Science. 1995;270:296–299. [PubMed] [Google Scholar]
22. Bae YS, Kang SW, Seo MS, Baines IC, Tekle E, Chock PB, Rhee SG. Epidermal growth factor (EGF)-induced generation of hydrogen peroxide. Role in EGF receptor-mediated tyrosine phosphorylation. J Biol Chem. 1997;272:217–221. [PubMed] [Google Scholar]
23. Adachi M, Fischer EH, Ihle J, Imai K, Jirik F, Neel B, Pawson T, Shen S, Thomas M, Ullrich A, et al. Mammalian SH2-containing protein tyrosine phosphatases. Cell. 1996;85:15. [PubMed] [Google Scholar]
24. Lee SR, Kwon KS, Kim SR, Rhee SG. Reversible inactivation of protein-tyrosine phosphatase 1B in A431 cells stimulated with epidermal growth factor. J Biol Chem. 1998;273:15366–15372. [PubMed] [Google Scholar]
25. Meng T, Fukada T, Tonks N. Reversible oxidation and inactivation of protein tyrosine phosphatases in vivo. Mol Cell. 2002;9:387–399. [PubMed] [Google Scholar]
26. Denu JM, Tanner KG. Specific and reversible inactivation of protein tyrosine phosphatases by hydrogen peroxide: evidence for a sulfenic acid intermediate and implications for redox regulation. Biochemistry. 1998;37:5633–5642. [PubMed] [Google Scholar]
27. Lee SR, Yang KS, Kwon J, Lee C, Jeong W, Rhee SG. Reversible inactivation of the tumor suppressor PTEN by H2O2. J Biol Chem. 2002;277:20336–20342. [PubMed] [Google Scholar]
28. Leslie NR, Bennett D, Lindsay YE, Stewart H, Gray A, Downes CP. Redox regulation of PI 3-kinase signalling via inactivation of PTEN. EMBO J. 2003;22:5501–5510. [PMC free article] [PubMed] [Google Scholar]
29. Choi MH, Lee IK, Kim GW, Kim BU, Han YH, Yu DY, Park HS, Kim KY, Lee JS, Choi C, et al. Regulation of PDGF signalling and vascular remodelling by peroxiredoxin II. Nature. 2005;435:347–353. [PubMed] [Google Scholar]
30. Kwon J, Lee SR, Yang KS, Ahn Y, Kim YJ, Stadtman ER, Rhee SG. Reversible oxidation and inactivation of the tumor suppressor PTEN in cells stimulated with peptide growth factors. Proc Natl Acad Sci U S A. 2004;101:16419–16424. [PMC free article] [PubMed] [Google Scholar]
31. Woo HA, Yim SH, Shin DH, Kang D, Yu DY, Rhee SG. Inactivation of peroxiredoxin I by phosphorylation allows localized H(2)O(2) accumulation for cell signaling. Cell. 2010;140:517–528. [PubMed] [Google Scholar]
32. Connor KM, Subbaram S, Regan KJ, Nelson KK, Mazurkiewicz JE, Bartholomew PJ, Aplin AE, Tai YT, Aguirre-Ghiso J, Flores SC, et al. Mitochondrial H2O2 regulates the angiogenic phenotype via PTEN oxidation. J Biol Chem. 2005;280:16916–16924. [PubMed] [Google Scholar]
33. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. [PubMed] [Google Scholar]
34. Cairns RA, Harris IS, Mak TW. Regulation of cancer cell metabolism. Nat Rev Cancer. 2011;11:85–95. [PubMed] [Google Scholar]