Omics 시대가 오면서
에너지 대사는
치료연구의 핵으로 떠오르고 있다!!
- Review Article
- Open access
- Published: 18 February 2025
Energy metabolism in health and diseases
- Hui Liu,
- Shuo Wang,
- Jianhua Wang,
- Xin Guo,
- Yujing Song,
- Kun Fu,
- Zhenjie Gao,
- Danfeng Liu,
- Wei He &
- Lei-Lei Yang
Signal Transduction and Targeted Therapy volume 10, Article number: 69 (2025) Cite this article
Abstract
Energy metabolism is indispensable for sustaining physiological functions in living organisms and assumes a pivotal role across physiological and pathological conditions. This review provides an extensive overview of advancements in energy metabolism research, elucidating critical pathways such as glycolysis, oxidative phosphorylation, fatty acid metabolism, and amino acid metabolism, along with their intricate regulatory mechanisms. The homeostatic balance of these processes is crucial; however, in pathological states such as neurodegenerative diseases, autoimmune disorders, and cancer, extensive metabolic reprogramming occurs, resulting in impaired glucose metabolism and mitochondrial dysfunction, which accelerate disease progression. Recent investigations into key regulatory pathways, including mechanistic target of rapamycin, sirtuins, and adenosine monophosphate-activated protein kinase, have considerably deepened our understanding of metabolic dysregulation and opened new avenues for therapeutic innovation. Emerging technologies, such as fluorescent probes, nano-biomaterials, and metabolomic analyses, promise substantial improvements in diagnostic precision. This review critically examines recent advancements and ongoing challenges in metabolism research, emphasizing its potential for precision diagnostics and personalized therapeutic interventions. Future studies should prioritize unraveling the regulatory mechanisms of energy metabolism and the dynamics of intercellular energy interactions. Integrating cutting-edge gene-editing technologies and multi-omics approaches, the development of multi-target pharmaceuticals in synergy with existing therapies such as immunotherapy and dietary interventions could enhance therapeutic efficacy. Personalized metabolic analysis is indispensable for crafting tailored treatment protocols, ultimately providing more accurate medical solutions for patients. This review aims to deepen the understanding and improve the application of energy metabolism to drive innovative diagnostic and therapeutic strategies.
초록
에너지 대사는 생물체의 생리적 기능을 유지하는 데 필수적이며, 생리적 및 병리적 조건 전반에서 핵심적인 역할을 합니다.
이 리뷰는 에너지 대사 연구의 최신 진전을 포괄적으로 개괄하며,
글리코시스,
산화적 인산화,
지방산 대사,
아미노산 대사 등 주요 대사 경로와 그 복잡한 조절 메커니즘을 설명합니다.
glycolysis, oxidative phosphorylation, fatty acid metabolism, and amino acid metabolism, along with their intricate regulatory mechanisms
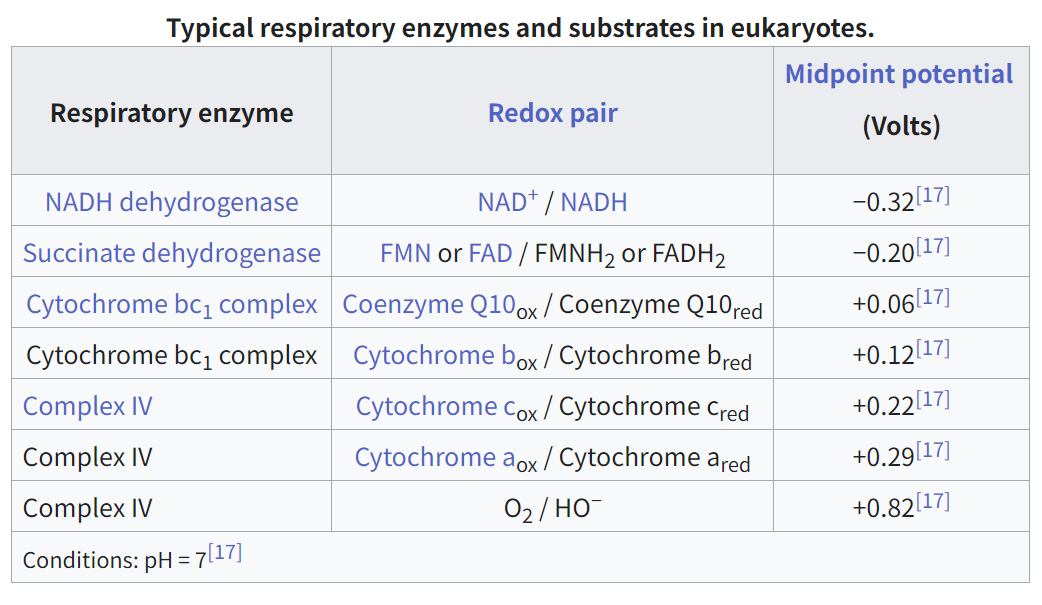
호흡 효소 전자 전달에 관여하는 효소 (예: 복합체 I–IV)
산화환원 쌍 전자(산화/환원 형태)를 주고받는 분자 쌍
중간 전위 (볼트) 산화환원 쌍의 절반이 산화되고 절반이 환원되는 전압. 전자 친화도를 나타냅니다.
📊 표의 주요 내용
효소 산화환원 쌍 중간 전위 (V) 역할
NADH 탈수소효소 NAD⁺ / NADH −0.32 V 복합체 I – 낮은 전위에서 전자 전달 체계를 시작
수크신산 탈수소효소 FAD / FADH₂ 또는 FMN / FMNH₂ −0.20 V 복합체 II – FADH₂를 통해 전자 전달 체계에 진입
사이토크롬 bc₁ 복합체 CoQ (Qₒₓ / Qᵣₑd), 사이토크롬 b +0.06 V, +0.12 V 복합체 III – 사이토크롬 c로 전자 전달
복합체 IV 사이토크롬 c / a / O₂ +0.22 V, +0.29 V, +0.82 V 산소로의 최종 전자 전달
🧪 생화학적 의미
- 전자들은 저전위(더 음의)에서 고전위(더 양의) 환원-산화 쌍으로 흐릅니다.
- 이 전자 흐름은 프로톤(H⁺)을 미토콘드리아 막을 통해 펌핑하는 데 사용됩니다 → 프로톤 그라디언트를 생성 → ATP 합성효소를 통해 ATP 합성을 촉진합니다.
- 최종 전자 수용체는 O₂로, 가장 높은 전위(+0.82 V)를 갖습니다 → 전자 흐름의 일방성을 보장합니다.
🔋 비유: 전기 회로
- NADH와 FADH₂는 저전압(음극)의 고에너지 배터리와 같습니다.
- 산소는 고전압의 전자를 필요로 하는 최종 전구와 같습니다.
- 전자들은 에너지적으로 '내리막길'을 따라 흐르며 에너지를 방출해 세포의 에너지 화폐인 ATP를 생성합니다.
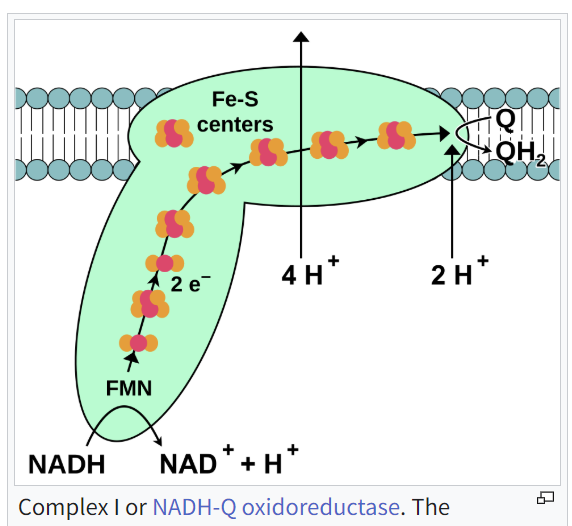
이 다이어그램은 미토콘드리아의 전자 전달 사슬 (ETC)에서 Complex I (NADH:ubiquinone oxidoreductase라고도 함)의 기능을 설명합니다.
다이어그램에서 발생하는 과정을 간략히 설명합니다:
🔬 Complex I의 단계별 기능
1. NADH가 전자를 기부합니다
NADH는 Complex I에 2개의 전자 (2e⁻)를 제공합니다.
NADH는 산화되어 NAD⁺ + H⁺로 변환됩니다.
2. FMN이 전자를 받아들입니다
전자는 먼저 FMN(플라빈 모노뉴클레오티드)로 이동합니다.
FMN은 환원되어 전자를 사슬 아래로 전달합니다.
3. Fe-S 중심이 전자를 전달합니다
전자는 Fe-S(철-황) 클러스터를 통해 이동하며, 이는 전자를 복합체 깊숙이 전달하는 '계단' 역할을 합니다.
4. 전자들이 코엔자임 Q(유비퀴논)를 환원합니다
전자들은 Q(CoQ 또는 유비퀴논)에 도달하여 이를 QH₂(유비퀴놀)로 환원합니다.
이 QH₂는 다음으로 복합체 III로 이동합니다.
🧪 프로톤 펌핑(ATP 생산의 핵심)
전자들이 이동하면서 복합체 I은 프로톤(H⁺)을 미토콘드리아 매트릭스에서 인터멤브레인 공간으로 펌핑합니다:
4 H⁺가 막을 통해 펌핑됩니다(위쪽 화살표).
2 H⁺가 Q를 QH₂로 환원하는 데 사용됩니다.
이 과정에서 프로톤 그라디언트가 생성되며, 이는 ETC의 최종 단계에서 ATP 합성효소를 구동합니다.
🧾 요약
과정 효과
NADH → NAD⁺ 전자 전달
전자 → FMN → Fe-S 복합체 I을 따라 이동
CoQ → QH₂ 복합체 III로의 전자 운반체
4 H⁺ 펌핑으로 ATP 합성을 위한 프로톤 그라디언트 형성


아미노산은 단백질 합성의 기본 단위입니다. 이들은 세포의 구조적 요소이자 에너지 공급원으로, 정상적인 세포 성장, 분화 및 기능에 필수적입니다. 아미노산 대사 장애는 대사 질환, 심혈관 질환, 면역 질환, 암 등 다양한 병리적 상태와 연관되어 있습니다. 종양의 경우, 아미노산 대사 변화는 암 진행의 임상적 지표로 활용될 뿐만 아니라 치료 전략으로도 활용될 수 있습니다. 종양의 성장과 발달은 외부 아미노산의 섭취에 의존하기 때문에, 종양 관련 아미노산의 대사를 표적으로 삼아 종양 세포를 선택적으로 죽이는 연구가 점점 더 증가하고 있습니다. 또한 면역 관련 연구는 아미노산 대사가 효과 T 세포와 조절 T 세포의 기능을 조절하여 면역 세포의 기능에 영향을 미친다는 것을 확인했습니다. 따라서 질병과 관련된 아미노산 대사를 연구하고 아미노산 대사 경로 내의 표적을 식별하는 것은 질병 치료에 도움이 될 수 있습니다. 이 논문은 종양 관련 질환에서의 아미노산 대사 연구에 주로 초점을 맞추며, 아미노산 대사와 관련된 대사 질환, 심혈관 질환 및 면역 관련 질환의 연구 및 임상 연구 진전을 검토하여 아미노산 대사 표적 치료의 이론적 기반을 제공하기 위해 작성되었습니다.
이러한 과정의 항상성 균형은 매우 중요하지만,
신경퇴행성 질환, 자가면역 질환, 암과 같은 병리적 상태에서는
광범위한 대사 재프로그래밍이 발생하여
포도당 대사 장애와 미토콘드리아 기능 장애를 초래하며,
이는 질병 진행을 가속화합니다.
최근 메커니즘적 표적 라파마이신(mTOR),
시르투인(sirtuins),
아데노신 모노포스페이트 활성화 단백질 키나제(AMPK)와 같은
핵심 조절 경로에 대한 연구는 대사 장애의 이해를 크게 심화시켰으며,
치료 혁신을 위한 새로운 길을 열었습니다.
형광 프로브, 나노바이오재료, 대사체 분석과 같은 신기술은 진단 정확도의 상당한 개선을 약속합니다. 이 리뷰는 대사 연구의 최근 진전과 진행 중인 과제를 비판적으로 검토하며, 정밀 진단과 개인화 치료 개입의 잠재력을 강조합니다.
향후 연구는
에너지 대사 조절 메커니즘과 세포 간 에너지 상호작용의 역학을 규명하는 것을
우선순위로 삼아야 합니다.
최신 유전자 편집 기술과 다중 오믹스 접근법을 통합하여
기존 치료법(면역요법, 식이 개입 등)과 시너지 효과를 내는 다중 표적 약물의 개발은
치료 효과를 향상시킬 수 있습니다.
맞춤형 대사 분석은
환자 맞춤형 치료 프로토콜을 수립하는 데 필수적이며,
궁극적으로 환자에게 더 정확한 의료 솔루션을 제공할 수 있습니다.
이 리뷰는
에너지 대사 연구의 이해를 심화하고 적용을 개선하여
혁신적인 진단 및 치료 전략을 촉진하는 것을 목표로 합니다.
Similar content being viewed by others
Creatine metabolism: energy homeostasis, immunity and cancer biology
Article 03 June 2020

Mitochondrial dysfunction: mechanisms and advances in therapy
Article Open access15 May 2024
RNA-binding proteins as versatile metabolic regulators
Article Open access13 January 2025
Introduction
Energy metabolism, a cornerstone of physiological function, has been extensively scrutinized since von Helmholtz first articulated the concept in 1847.1,2 Over time, our comprehension of this vital process—which underpins life by supplying the essential energy required for diverse cellular activities—has expanded profoundly.3,4,5,6,7 Energy metabolism involves a series of sophisticated biochemical pathways that convert nutrients into adenosine triphosphate (ATP), the primary energy currency of cells. The meticulous regulation of these pathways is paramount for sustaining cellular homeostasis and ensuring the optimal functioning of organs and tissues. Dysregulation in energy metabolism is intricately associated with the pathogenesis of various disorders, encompassing neurological diseases, cardiovascular conditions, metabolic syndromes, autoimmune disorders, and cancer.8,9,10,11,12,13,14
Abnormal energy metabolism linked to the aforementioned diseases has been extensively studied.15,16 However, the mechanisms of intracellular energy conversion, dynamic changes in energy metabolic pathways, and the regulatory signals of different energy metabolic pathways remain unclear. Notably, the heterogeneous regulation of metabolism across different tissues and organ systems involving genetics, environment, and sex, among other factors, remains relatively under-studied. Recent studies are shifting focus towards intercellular energy transfer interactions, such as in pancreatic ductal adenocarcinoma, where lipid-rich cancer-associated fibroblasts transfer lipids to cancer cells, increasing oxidative phosphorylation (OXPHOS) to promote cancer cell growth.17 Nevertheless, owing to the complex interplay between immune and non-immune cells, mechanisms of energy interactions and regulatory strategies require further exploration. Mitochondria are central to energy metabolism; however, our understanding of mitochondrial dynamics, their role in regulating energy metabolism, and leveraging mitochondrial function to improve disease prognosis is still limited.
Therefore, a deeper understanding and research on energy metabolism can contribute to the diagnosis and treatment of various diseases. With technological advancements, detecting energy metabolic processes has become more feasible. However, in complex physiological and pathological microenvironments, the challenge lies in non-invasively and reliably monitoring the heterogeneous energy metabolism across different cell types using imaging, mass spectrometry, and biosensors. Although multi-omics technologies are evolving, the integration of metabolomics, spatial transcriptomics, imaging, and clustered regularly interspaced short palindromic repeats screening techniques for interdisciplinary diagnosis of diseases presents a promising yet under-explored research direction. Current treatments for energy metabolic diseases mainly target key pathways; however, owing to dynamic changes in disease energy metabolism, where early-stage tumors rely on glycolysis and advanced stages on fatty acid oxidation (FAO), treatment efficacy is often sub-optimal. This ongoing controversy underscores the importance of some studies supporting dietary modifications (such as ketogenic diets) to adjust energy metabolism, while others emphasize the significance of drug therapies. Future exploration lies in integrating targeted energy metabolism pathways with diet and/or standard care therapies like immunotherapy. Deep insights into the dynamic changes of energy metabolism in health and disease aid in discovering early diagnostic and therapeutic metabolic biomarkers. Studies on targeted therapeutic drugs for metabolic pathways are limited. Thus, future studies should focus on identifying specific and stable metabolic biomarkers and developing multi-targeted therapeutic drugs (Fig. 1).
서론
에너지 대사(energy metabolism)는 생리적 기능의 핵심 요소로, 1847년 폰 헬름홀츠가 이 개념을 처음 제시한 이래로 광범위하게 연구되어 왔습니다. 1,2 시간이 지나면서, 다양한 세포 활동에 필요한 필수 에너지를 공급함으로써 생명을 유지하는 이 중요한 과정에 대한 우리의 이해는 크게 확장되었습니다.3,4,5,6,7
에너지 대사 과정은
영양소를 세포의 주요 에너지 화폐인 아데노신 삼인산(ATP)으로 전환하는
복잡한 생화학적 경로들의 연속으로 이루어집니다.
이러한 경로의 세밀한 조절은
세포 내 환경의 균형 유지와 장기 및 조직의 최적 기능을 보장하는 데 필수적입니다.
에너지 대사 장애는
신경계 질환, 심혈관 질환, 대사 증후군, 자가면역 질환, 암 등
다양한 질환의 병리학적 과정과 밀접하게 연관되어 있습니다.8,9,10,11,12,13,14
위에서 언급된 질환과 관련된 이상적인 에너지 대사는 광범위하게 연구되어 왔습니다.15,16 그러나 세포 내 에너지 전환 메커니즘, 에너지 대사 경로의 동적 변화, 다양한 에너지 대사 경로의 조절 신호는 여전히 명확히 규명되지 않았습니다. 특히 유전적 요인, 환경, 성별 등 다양한 요인이 관여하는 조직 및 장기 시스템 간 대사 조절의 이질성은 상대적으로 덜 연구되었습니다. 최근 연구는 세포 간 에너지 전달 상호작용에 초점을 옮기고 있습니다.
예를 들어,
췌관 선암에서 지방이 풍부한 암 관련 섬유아세포가
암 세포에 지방을 전달하여 산화적 인산화(OXPHOS)를 증가시켜
암 세포 성장 촉진하는 현상이 관찰되었습니다.17
세포수준 연구에서 지방을 에너지로 사용하여 암세포가 성장하더라!!

임상의 대부분은 케톤대사는 암세포 굶기기 식단으로 효과가 좋다.
https://pmc.ncbi.nlm.nih.gov/articles/PMC8471358/


그러나
면역 세포와 비면역 세포 간의 복잡한 상호작용으로 인해
에너지 상호작용 메커니즘과 조절 전략은 추가 연구가 필요합니다.
미토콘드리아는
에너지 대사에서 중심적인 역할을 하지만,
미토콘드리아 동역학, 에너지 대사 조절에서의 역할, 미토콘드리아 기능을 활용해
질병 예후를 개선하는 방법에 대한 이해는 여전히 제한적입니다.
따라서
에너지 대사 연구는
다양한 질병의 진단과 치료에 기여할 수 있습니다.
기술적 진보로 에너지 대사 과정의 탐지가 더 가능해졌습니다. 그러나 복잡한 생리적 및 병리적 미세환경에서 다양한 세포 유형 간 이질적인 에너지 대사 과정을 비침습적이고 신뢰성 있게 모니터링하는 것이 과제입니다. 이미징, 질량 분석법, 생체 센서 등을 활용하는 접근법이 있지만, 다중 오믹스 기술의 발전에도 불구하고 대사체학, 공간 전사체학, 이미징, 클러스터드 정규 간격 단일 염기 반복(CRISPR) 스크리닝 기술의 통합을 통한 다학제적 질병 진단은 유망하지만 아직 충분히 탐구되지 않은 연구 방향입니다.
에너지 대사 질환의 현재 치료법은
주로 핵심 경로를 표적으로 삼지만,
질병의 에너지 대사 변화가 동적이며
초기 단계의 종양이 글리코시스(glycolysis)에 의존하고
진행 단계에서는 지방산 산화(FAO)에 의존한다는 점 때문에 치료 효과는 종종 미흡합니다.
이 지속적인 논쟁은 일부 연구가
식이 조절(예: 케톤 식이)을 통해 에너지 대사를 조정하는 것을 지지하는 반면,
다른 연구는 약물 치료의 중요성을 강조한다는 점을 강조합니다.
미래 연구 방향은
표적 에너지 대사 경로를 식이요법 및/또는 면역요법과 같은 표준 치료법과 통합하는 것입니다.
건강과 질병 상태에서 에너지 대사 동적 변화에 대한 깊은 이해는
조기 진단 및 치료를 위한 대사 바이오마커 발견에 기여합니다.
대사 경로를 표적으로 하는 치료제 연구는 제한적입니다.
따라서 향후 연구는 특정적이고 안정적인 대사 바이오마커를 식별하고
다중 표적 치료제 개발에 초점을 맞춰야 합니다(그림 1).
Fig. 1
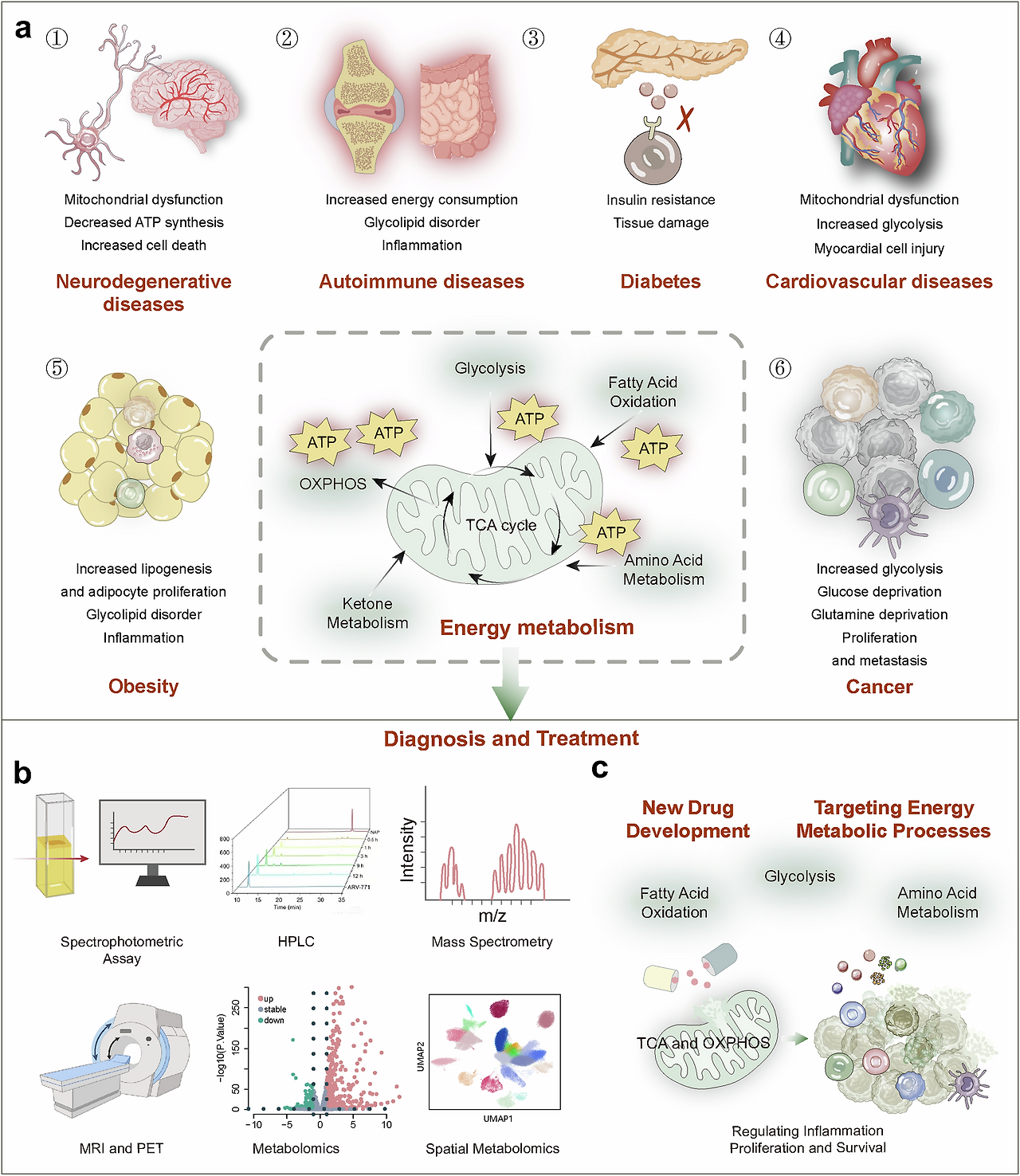
Diagram of energy metabolism alterations, detection, and therapeutics.
a Energy metabolic alterations accompany a variety of diseases, which include increased energy demands and shifts in energy production pathways, ultimately leading to mitochondrial dysfunction-based metabolic disorders that cause functional abnormalities or cell death in normal cells.
b Detection methods for altered energy metabolism encompass established spectroscopic assays, as well as advanced imaging techniques such as MRI and PET/CT, and the burgeoning field of metabolomics, including spatial omics technologies.
c Pharmacological interventions targeting changes in energy metabolism are directed at multiple stages of metabolic pathways, including glycolysis, fatty acid oxidation and mitochondrial oxidation, to ameliorate abnormal energy metabolic shifts
에너지 대사 변화, 진단, 및 치료법 다이어그램.
a 에너지 대사 변화는 다양한 질환과 동반되며, 이는 에너지 수요 증가 및 에너지 생산 경로의 변화로 이어져 결국 정상 세포의 기능 이상 또는 세포 사멸을 초래하는 미토콘드리아 기능 장애 기반 대사 장애를 유발합니다.
b 에너지 대사 변화의 검출 방법은 전통적인 분광 분석법뿐만 아니라 MRI 및 PET/CT와 같은 고급 영상 기술, 그리고 공간 오믹스 기술을 포함한 대사체학 분야가 급속히 발전하고 있습니다.
c 에너지 대사 변화에 대한 약리학적 개입은 글리코겐 분해, 지방산 산화 및 미토콘드리아 산화 등 대사 경로의 여러 단계에 초점을 맞춰 비정상적인 에너지 대사 변화를 완화하는 것을 목표로 합니다.
This comprehensive review explores the evolution and progress of energy metabolism research, providing an in-depth examination of its core pathways and regulatory mechanisms. We briefly summarize various energy metabolic pathways, including glycolysis, OXPHOS, FAO, and amino acid metabolism. A detailed analysis of the regulatory pathways is provided, encompassing hormone regulation, adenosine monophosphate-activated protein kinase and mechanistic target of rapamycin signaling, and the impact of the sirtuin (SIRT) protein family, emphasizing the roles of metabolic products or non-metabolic enzymatic functions in energy metabolism. Our analysis extends to the specific metabolic adaptations inherent in various pathophysiological states, including metabolic shifts in neurodegenerative diseases (ND), heart metabolic reprogramming in cardiovascular diseases, disturbances in metabolic pathways in obesity and diabetes syndromes, autoimmune diseases, and cancer, focusing on how disease treatment can be achieved through targeting energy metabolism. Throughout the discussion, we further analyzed the controversies and limitations of current research, identifying directions for future investigation. We introduced cutting-edge technologies such as fluorescent probes, chromatography, metabolomics, and nanobiomaterials, comparing the characteristics of these technologies, significantly enhancing the capacity to study and monitor metabolic processes. However, review articles may lack depth and detail in certain areas compared with research articles focused on a single theme, which can be a limitation. Ultimately, this review aims to provide a comprehensive understanding of the intricate patterns of energy metabolism, stimulate further exploration, and drive the development of innovative diagnostic and treatment strategies.
이 포괄적인 검토는
에너지 대사 연구의 진화와 발전을 탐구하며,
그 핵심 경로와 조절 메커니즘에 대한 심층적인 분석을 제공합니다.
우리는
글리코시스, OXPHOS, FAO, 아미노산 대사 등
다양한 에너지 대사 경로를 간략히 요약합니다.
규제 경로에 대한 상세한 분석을 제공하며,
호르몬 조절,
아데노신 모노포스페이트 활성화 단백질 키나제(AMP-activated protein kinase) 및
라파마이신 표적 단백질(mTOR) 신호전달 경로,
시르투인(SIRT) 단백질 가족의 영향을 포함합니다.
특히
에너지 대사에서 대사 산물이나
비대사적 효소 기능의 역할을 강조합니다.
분석은
신경퇴행성 질환(ND)에서의 대사 변화, '
심혈관 질환에서의 심장 대사 재프로그래밍,
비만 및 당뇨병 증후군,
자가면역 질환,
암에서의 대사 경로 장애 등 다양한 병리생리학적 상태에 내재된
특정 대사 적응에까지 확장됩니다.
특히 에너지 대사를 표적으로 삼아 질병 치료를 달성하는 방법에 초점을 맞췄습니다. 논의 전반에 걸쳐 현재 연구의 논란과 한계를 분석하고 미래 연구 방향을 제시했습니다. 우리는 형광 프로브, 크로마토그래피, 대사체학, 나노바이오재료 등 최첨단 기술을 소개했으며, 이러한 기술의 특성을 비교함으로써 대사 과정의 연구 및 모니터링 능력을 크게 향상시켰습니다. 그러나 리뷰 논문은 특정 분야에 대한 깊이와 세부 사항이 연구 논문보다 부족할 수 있으며, 이는 한계로 작용할 수 있습니다. 결론적으로, 이 리뷰는 에너지 대사 과정의 복잡한 패턴에 대한 포괄적인 이해를 제공하며, 추가 연구를 촉진하고 혁신적인 진단 및 치료 전략 개발을 이끌어내는 것을 목표로 합니다.
Chronicles of energy metabolism research
Energy metabolism encompasses a series of intricate and complex biochemical processes within organisms that involve the release, transfer, storage, and utilization of energy. Organisms must continually extract energy from food to sustain and promote growth. The metabolism of glucose, fats, and amino acids produces ATP, which is required for cellular energy processes. Notably, carbohydrate and lipid metabolism accounts for >90% of the energy requirements of the body. Among these metabolic pathways, aerobic oxidation plays a crucial role in ATP production, ensuring the efficient conversion and utilization of energy. This process is not only essential for maintaining fundamental biological functions but also plays a pivotal role in the onset, progression, and treatment of various diseases (Fig. 2).
에너지 대사 연구의 연대기
에너지 대사는
유기체 내에서 에너지의 방출, 전달, 저장, 및 이용과 관련된 복잡하고 정교한
생화학적 과정의 연속체입니다.
유기체는
성장과 유지에 필요한 에너지를 지속적으로
음식으로부터 추출해야 합니다.
포도당, 지방, 및 아미노산의 대사는
세포 에너지 과정에 필수적인 ATP를 생성합니다.
특히,
탄수화물과 지방 대사는
신체 에너지 요구량의 90% 이상을 차지합니다.
이 대사 경로 중 산소 호흡은
ATP 생산에 결정적인 역할을 하며,
에너지의 효율적인 전환과 이용을 보장합니다.
이 과정은
기본적인 생물학적 기능을 유지하는 데 필수적일 뿐만 아니라
다양한 질병의 발병, 진행, 치료에 중요한 역할을 합니다(그림 2).
Fig. 2
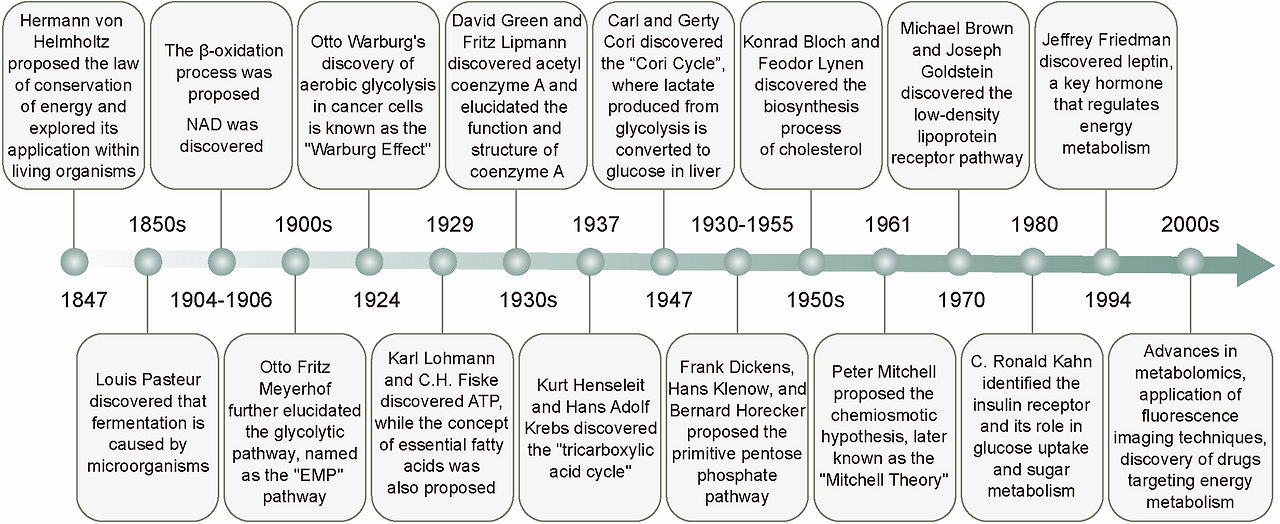
The milestones of energy metabolism development. The field of energy metabolism research originated in the mid-19th century and experienced rapid growth throughout the 20th century. Historical milestones, including the establishment of the law of energy conservation, the discovery of the Warburg effect, the identification of ATP, and the elucidation of the TCA cycle, have significantly advanced our understanding of energy metabolic processes and led to the revelation of multiple metabolic pathways. With the advent of the 21st century, researchers are extensively investigating the regulatory mechanisms of energy metabolism and actively exploring methods for its detection, as well as therapeutic strategies targeting energy metabolism for disease treatment
에너지 대사 발달의 주요 단계. 에너지 대사 연구 분야는 19세기 중반에 시작되어 20세기 내내 급속히 발전했습니다. 에너지 보존 법칙의 수립, 워버그 효과의 발견, ATP의 식별, TCA 순환의 규명 등 역사적인 주요 단계들은 에너지 대사 과정에 대한 이해를 크게 진전시켰으며, 다양한 대사 경로의 발견으로 이어졌습니다. 21세기 들어 연구자들은 에너지 대사 조절 메커니즘을 광범위하게 조사하고 있으며, 에너지 대사 검출 방법 및 질병 치료를 위한 에너지 대사 표적 치료 전략을 적극적으로 탐구하고 있습니다.
In 1847, von Helmholtz introduced the concept of energy metabolism and further developed this theory by exploring the application of the “law of conservation of energy” within biological systems, building on the work of Mayer and others.1 Further, the discovery of nicotinamide adenine dinucleotide (NAD) in 1906 provided critical insights into the roles of enzymes in energy metabolism.18,19 In 1929, Lohmann and Fiske identified ATP as a pivotal energy molecule.20,21 Moreover, since its discovery, it has been recognized as a central molecule in energy metabolism, catalyzing extensive scientific investigations.22
Glucose metabolism is the primary energy-producing mechanism in the body, contributing approximately 50–70% of the total energy supply. For over a century, studies on glucose metabolism, with an early focus on glycolysis, have broadened our understanding of the mechanisms underlying cellular energy production. In the 1850s, Louis Pasteur first unveiled the process of microbial fermentation, providing a foundation for understanding how cells convert glucose into energy.23 In 1897, Buchner discovered that cell-free extracts could also perform fermentation.24 Building on previous studies, including the isolation of adenosine monophosphate (AMP) and description of the glycolytic pathway by Embden, Parnas’s studies on phosphorylative processes, and Lohmann’s discovery of ATP, Meyerhof characterized the glycolytic pathway and identified the enzymes involved. This pathway was subsequently named the “Embden–Meyerhof–Parnas pathway” and became recognized as the first metabolic pathway ever discovered.25,26 During the 1920s and 1930s, Warburg further elucidated the glycolysis process in cancer cells, in which glucose is broken down into lactic acid.27,28,29,30 He observed that cancer cells prefer glycolysis for energy production even under aerobic conditions, a phenomenon later termed the “Warburg effect”.31,32,33,34 This discovery has provided new avenues for cancer research, contributing crucial insights into the metabolic adaptations in cancer cells.
In 1931, Warburg was awarded the Nobel Prize in Physiology or Medicine for his discovery of the nature of and mechanism associated with respiratory enzymes, establishing the groundwork to understand electron transfer mechanisms involved in cellular respiration. He later conceptualized the respiratory chain, determining that NAD+ serves as an electron carrier, and uncovered the existence of nicotinamide adenine dinucleotide phosphate (NADP+). This provided the basis for OXPHOS, which involves the transfer of electrons through a series of protein complexes to ultimately produce ATP.35,36 In 1937, Krebs and Henseleit discovered the tricarboxylic acid (TCA) cycle, which converts pyruvate into carbon dioxide and water under aerobic conditions while generating energy.37,38 This discovery is regarded as a milestone in metabolic research and provides crucial insights into the mechanisms underlying energy production in cells under aerobic conditions. The TCA cycle is a central pathway for the complete oxidation of carbohydrates, fats, and proteins (amino acids) and represents a pivotal link between their inter-conversion and energy release.39 In 1961, Mitchell proposed a chemiosmotic hypothesis to explain the formation and utilization of a proton gradient during OXPHOS. This theory, later known as the Mitchell hypothesis, revolutionized the understanding of bioenergetics.40 Further, in 1957, Boyer, Walker, and Skou elucidated the roles of ATP synthase and ATPases in energy production, via glucose metabolism.41 Their work provided key insights into the molecular mechanisms that drive ATP synthesis, further advancing knowledge on cellular energy metabolism.
Other pathways and regulatory molecules involved in glucose metabolism were also identified. In 1947, Carl and Gerty Cori identified the Cori cycle, a mechanism by which lactate, produced via anaerobic glycolysis in the muscles, is converted back to glucose in the liver.42 Between 1930 and 1955, substantial contributions by Dickens, Klenow, and Horecker led to the identification and subsequent refinement of the pentose phosphate pathway.43 In cells, this pathway generates NADPH, which supports their antioxidant responses.44 In the 1980s, Kahn elucidated the role of insulin receptor in glucose uptake and metabolism, advancing the understanding of insulin signaling and resistance.45,46,47,48 Moreover, in 1994, Friedman discovered leptin, a hormone that regulates energy balance and metabolism.49 Since the 2000s, studies have revealed how alterations in glucose metabolism promote cancer cell growth, uncovering reprogrammed metabolic pathways in cancer cells and elucidating the role of glucose metabolism in tumor development.50 These findings have suggested new strategies for cancer therapy, offering potential avenues for targeting metabolic alterations in cancer cells.
Concurrently, with ongoing advancements in glucose metabolism research, fatty acid metabolism processes have also been progressively elucidated. Fatty acid metabolism contributes to approximately 30–50% of the energy requirements of the body. In the 1920s, Bloor and Burr conducted preliminary studies on the role of lipids in cellular functions and nutrition. In 1929, George and Mildred Burr discovered the dietary necessity of certain fatty acids, highlighting that specific fats not only provide energy to the body but also play a critical role in sustaining life. These essential fatty acids, including the well-known ω-3 and ω-6 fatty acids, cannot be synthesized by the body and must be obtained through the diet.51,52,53,54 In the 1930s, Green and Lipmann discovered ATP-dependent acetylating enzymes and elucidated the role and structure of coenzyme A, revealing its critical role in fatty acid metabolism. This discovery facilitated an understanding of the activation and entry of fatty acids into the β-oxidation process.55,56,57 Specifically, acetyl-CoA generated from β-oxidation enters the TCA cycle for ATP production. In the 1950s, Bloch and Lynen uncovered the complex process of cholesterol biosynthesis.58,59 Moreover, in 1971, Corey and Skoulos successfully synthesized prostaglandins, and during the 1970s, Brown and Goldstein discovered the low-density lipoprotein receptor pathway, which not only provides cells with the cholesterol necessary for constructing cell membranes and other biomolecules but also helps to maintain cholesterol homeostasis in the plasma.60,61,62 In the 1980s, the role of oxidized low-density lipoprotein in atherosclerosis was identified, enhancing the understanding of the involvement of fatty acid metabolism in cardiovascular disease.63,64,65 Since the 2000s, the development of metabolomics has enabled the comprehensive analysis of lipids within biological systems, advancing research on the metabolic pathways linking lipid metabolism to cancer progression. Amino acid metabolism primarily supports the resynthesis of cellular components or the synthesis of bioactive substances, such as enzymes and hormones. Under normal conditions, the role of amino acid metabolism in energy provisions is limited.
As research on energy metabolism broadens, its links to various diseases have progressively been revealed, alongside continuous advancements in therapeutic approaches for metabolic disorders. In the 1950s, the discovery of the Cori cycle revealed the biochemical pathway by which lactate is converted back to glucose in the liver, enhancing our understanding of glucose metabolism in conditions such as diabetes.42,66 In the 1980s, Kahn provided crucial insights into how insulin regulates glucose uptake and metabolism, thereby enhancing our understanding of insulin resistance and type 2 diabetes (T2DM).45,48,67 Concurrently, in-depth studies of fatty acid metabolism have highlighted the critical role of FAO in energy metabolism, offering essential clues to understanding the role of fats in metabolic diseases.68,69
In 1994, the discovery of leptin, a hormone that regulates energy balance and body weight, provided new possibilities for treating obesity and cancer.70,71 In the 1990s, researchers began to focus on the central role of mitochondria in energy metabolism, identifying mitochondrial dysfunction as a contributing factor to various metabolic diseases,72,73 thus providing a theoretical foundation for developing mitochondria-targeted therapies. Since the 2000s, studies have increasingly emphasized metabolic pathways in cancer cells, revealing how altered glucose and lipid metabolism support cancer cell growth and survival.74,75 Additionally, studies on glutamine metabolism in cancer cells have identified new therapeutic targets for cancer treatment, further elucidating the relationship between metabolism and cancer progression.76 For example, researchers can influence tumor cell growth and survival by modulating energy metabolism pathways, and clinical studies integrating metabolic inhibitors into cancer treatment protocols are transforming metabolic insights into therapeutic strategies. With advancements in genomics and metabolomics, the application of metabolomic technologies has enabled more accurate diagnosis and monitoring of metabolic diseases, such as cancer, diabetes, and obesity.77,78 Recent studies have also demonstrated, for the first time, that alterations in cardiac energy metabolism can promote heart regeneration. By inhibiting FAO, cardiomyocyte energy metabolism shifts from FAO to glycolysis, thereby triggering the regenerative capacity of heart cells.79
1847년, 폰 헬름홀츠는 에너지 대사 개념을 도입하고, 마이어와 다른 연구자들의 작업을 바탕으로 생물학적 시스템 내에서 “에너지 보존의 법칙”을 적용하는 것을 탐구함으로써 이 이론을 더욱 발전시켰습니다.1
또한 1906년
니코틴아미드 아데닌 디뉴클레오티드(NAD)의 발견은
에너지 대사에서 효소의 역할을 이해하는 데 결정적인 통찰을 제공했습니다. 18,19
NAD합성 과정 : 니아신이나 트립토판같은 전구물질로부터 세포질, 미토콘드리아에서 합성.
NADH/NAD ratio : 세포 대사상태와 건강을 알려주는 지표. nad는 산화형태, nadh는 환원형.
이 비율이 낮아지면 세포기능이 떨어진다는 뜻

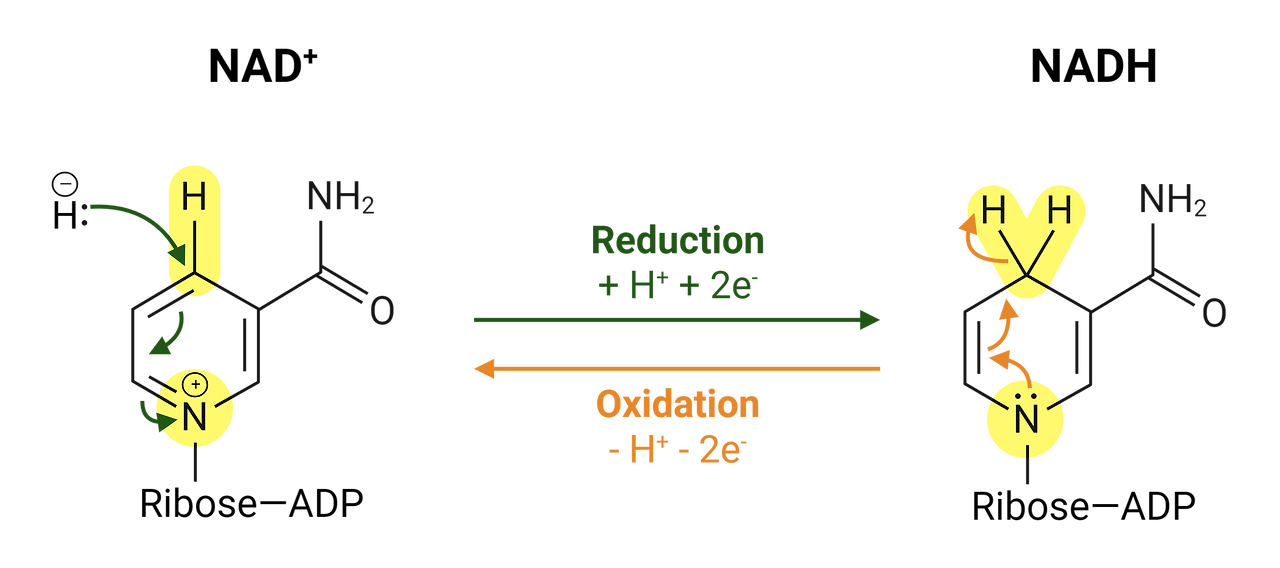
NAD⁺란 무엇인가요?
NAD⁺(니코틴아미드 아데닌 디뉴클레오티드의 약자)는 신체 내 모든 세포에 존재하는 작은 보조 분자입니다.
이것은 재충전 가능한 배터리나 에너지 전달자처럼 생각할 수 있으며, 신체에서 에너지를 생성하고 손상을 복구하는 데 도움을 줍니다.
NAD⁺는 어떤 역할을 하나요?
1. 에너지 생성
신체는 세포 호흡이라는 과정을 통해 음식을 에너지로 변환합니다.
NAD⁺는 음식에서 전자를 가져와 전달하여 에너지가 생성되는 곳으로 이동시킵니다.
이 과정은 세포가 ATP(세포의 에너지 화폐)를 생성하는 데 도움을 줍니다.
🧃 음식을 먹는 것을 휴대폰을 충전하는 것과 비교해 보세요—NAD⁺는 세포 내부의 전하를 이동시켜 세포가 정상적으로 작동할 수 있도록 돕습니다!
2. 세포 복구 및 보호
NAD⁺는 시르투인과 PARP라는 특수 단백질을 돕습니다.
이 단백질들은 DNA를 복구하고 세포의 수명을 연장하며 노화 효과를 줄입니다.
🛠️ NAD⁺는 세포 내부의 고장난 부분을 '수리'하는 도구처럼 작용합니다.
3. 운동이나 단식 시 증가합니다
운동과 건강한 스트레스(예: 간헐적 단식)는 몸이 더 많은 NAD⁺를 생성하도록 합니다.
이것은 더 나은 기분, 더 긴 수명, 그리고 건강 유지에 도움을 줄 수 있습니다.
1929년, 로흐만과 피스키는 ATP를 핵심 에너지 분자로 식별했습니다.20,21
또한, 그 발견 이후로 ATP는 에너지 대사에서 중심 분자로 인정받아 광범위한 과학적 연구를 촉진해 왔습니다.22
글루코스 대사는
신체에서 주요 에너지 생산 메커니즘으로,
전체 에너지 공급량의 약 50–70%를 차지합니다.
1세기 이상에 걸쳐 포도당 대사 연구는 초기에는 글리코lysis에 초점을 맞추어 세포 에너지 생산 메커니즘에 대한 이해를 넓혀왔습니다. 1850년대 루이 파스퇴르는 미생물 발효 과정을 최초로 밝혀내어 세포가 포도당을 에너지로 전환하는 과정을 이해하는 기반을 마련했습니다.23 1897년 부흐너는 세포 외 추출물도 발효를 수행할 수 있음을 발견했습니다. 24 이전 연구를 바탕으로, 아데노신 모노포스페이트(AMP)의 분리 및 글리코lysis 경로의 설명을 포함한 엠벤의 연구, 파르나스의 인산화 과정 연구, 로흐만의 ATP 발견을 바탕으로, 메이어호프는 글리코lysis 경로를 특성화하고 관련된 효소를 식별했습니다. 이 경로는 이후 “엠벤-메이어호프-파르나스 경로”로 명명되었으며, 최초로 발견된 대사 경로로 인정받았습니다.25,26
1920년대와 1930년대, 워버그는
암 세포에서 포도당이 젖산으로 분해되는 글리코lysis 과정을
그는 암 세포가 산소 존재 하에서도 에너지 생산에 글리코겐 분해를 선호한다는 현상을 관찰했으며,
이는 나중에 “워버그 효과”라고 명명되었습니다.31,32,33,34
이 발견은 암 연구에 새로운 방향을 제시했으며,
암 세포의 대사 적응 메커니즘에 대한 중요한 통찰을 제공했습니다.
1931년, 워버그는 호흡 효소의 본질과 메커니즘을 발견한 공로로 생리학 또는 의학 분야 노벨상을 수상했습니다. 이는 세포 호흡에 관여하는 전자 전달 메커니즘을 이해하는 기반을 마련했습니다. 그는 이후 호흡 사슬을 개념화했으며, NAD+가 전자 운반체로 작용함을 밝히고 니코틴아미드 아데닌 디뉴클레오티드 인산(NADP+)의 존재를 발견했습니다. 이는 OXPHOS의 기반을 제공했으며, 이는 단백질 복합체를 통해 전자 전달을 통해 최종적으로 ATP를 생성하는 과정입니다.35,36

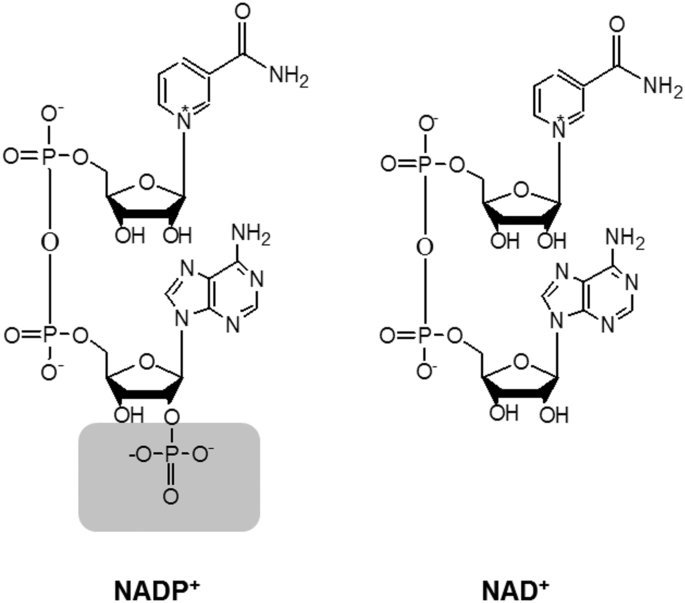
NADP+ .. NADPH
NAD 분자에 리보스 부분에 인산기 1개가 붙어 ..
NAD가 에너지 생성과정에서 전자전달체로 쓰이는 반면
NADP는 주로 생합성 경로나 항산화 작용, 해독반응에 사용
NADP전구체 NMN, NR 같은 보충제가 있음.

1937년, 크레브스와 헨셀라이트는
산소 조건 하에서 피루vate를 이산화탄소와 물로 전환하며 에너지를 생성하는
트리카르복실산(TCA) 사이클을 발견했습니다. 37,38
이 발견은
대사 연구의 이정표로 여겨지며,
산소 조건 하에서 세포 내 에너지 생산 메커니즘에 대한 중요한 통찰을 제공합니다.
TCA 순환은
탄수화물, 지방, 단백질(아미노산)의 완전한 산화 과정의 중심 경로이며,
이들의 상호 전환과 에너지 방출 사이의 핵심 연결 고리를 구성합니다.39

1961년, 미첼은 OXPHOS 과정에서 프로톤 그라디언트의 형성 및 활용을 설명하기 위해 화학삼투 가설을 제안했습니다.
이 이론은 나중에 미첼 가설로 알려졌으며, 생체 에너지학에 대한 이해를 혁명적으로 변화시켰습니다.40

또한 1957년 보이어, 워커, 스쿠는 포도당 대사 과정을 통해
ATP 합성효소와 ATPase의 에너지 생산 역할을 규명했습니다.41
그들의 연구는
ATP 합성을驱动하는 분자적 메커니즘에 대한 핵심적인 통찰을 제공했으며,
세포 에너지 대사 연구를 한 단계 발전시켰습니다.

포도당 대사 과정에 관여하는 다른 경로와 조절 분자들도 식별되었습니다.
1947년, 칼과 게르티 코리(Carl and Gerty Cori)는
근육에서 무산소 당분해(anaerobic glycolysis)를 통해 생성된 젖산(lactate)이
간에서 다시 포도당으로 전환되는 코리 순환(Cori cycle)을 발견했습니다.42
1930년부터 1955년까지
디킨스(Dickens), 클레노(Klenow), 호레커(Horecker)의 중요한 기여를 통해
펜토스 인산 경로(pentose phosphate pathway)가 발견되고 정교화되었습니다. 43
세포 내에서 이 경로는
NADPH를 생성하여
세포의 항산화 반응을 지원합니다.44
1980년대, 카ahn은
인슐린 수용체의 포도당 흡수 및 대사 역할을 규명하여
인슐린 신호전달과 저항성에 대한 이해를 진전시켰습니다.45,46,47,48
또한 1994년, 프리드먼은
에너지 균형과 대사를 조절하는 호르몬인 레プチ인을 발견했습니다. 49
2000년대 이후 연구는
포도당 대사 변화가 암 세포 성장 촉진 메커니즘을 밝히며,
암 세포 내 재프로그래밍된 대사 경로를 규명하고
포도당 대사가 종양 발달에 미치는 역할을 설명했습니다.50
이러한 발견은
암 치료를 위한 새로운 전략을 제시하며,
암 세포의 대사 변화 표적화 가능성을 열어주었습니다.
동시에 포도당 대사 연구의 지속적인 진전과 함께
지방산 대사 과정도 점차적으로 규명되었습니다.
지방산 대사는
신체 에너지 요구량의 약 30–50%를 차지합니다.
1920년대 Bloor와 Burr는 지질이 세포 기능과 영양에 미치는 역할을 연구하는 초기 연구를 수행했습니다. 1929년 George와 Mildred Burr는 특정 지방산의 식이 필수성을 발견했으며, 특정 지방이 단순히 신체에 에너지를 공급하는 것뿐 아니라 생명을 유지하는 데 필수적인 역할을 한다는 점을 강조했습니다.
이 필수 지방산에는 잘 알려진
ω-3 및 ω-6 지방산이 포함되며,
신체에서 합성되지 않아 식이를 통해 섭취해야 합니다.51,52,53,54
1930년대 그린과 립만은 ATP 의존성 아세틸화 효소를 발견하고 코엔자임 A의 역할과 구조를 규명하여 지방산 대사에서의 핵심적 역할을 밝혔습니다. 이 발견은 지방산의 활성화와 β-산화 과정으로의 진입 메커니즘을 이해하는 데 기여했습니다.55,56,57
구체적으로,
β-산화 과정에서 생성된 아세틸-코엔자임 A는
ATP 생성을 위해 트리카르복실산 회로(TCA 회로)로 진입합니다.
1950년대, Bloch와 Lynen은 콜레스테롤 생합성의 복잡한 과정을 밝혀냈습니다.58,59 또한 1971년 Corey와 Skoulos는 프로스타글란딘을 성공적으로 합성했으며, 1970년대 Brown과 Goldstein은 저밀도 지단백질 수용체 경로를 발견했습니다. 이 경로는 세포가 세포막 및 기타 생체 분자 구축에 필요한 콜레스테롤을 공급할 뿐만 아니라 혈장 내 콜레스테롤 균형을 유지하는 데도 도움을 줍니다. 60,61,62 1980년대에는 동맥경화증에서 산화 저밀도 지단백질의 역할이 밝혀지며, 지방산 대사 과정이 심혈관 질환에 미치는 영향에 대한 이해가 깊어졌습니다.63,64,65 2000년대 이후 대사체학의 발전은 생물학적 시스템 내 지질의 종합적 분석을 가능하게 하여, 지질 대사 과정과 암 진행을 연결하는 대사 경로 연구를 진전시켰습니다. 아미노산 대사는 주로 세포 구성 요소의 재합성이나 효소 및 호르몬과 같은 생물학적 활성 물질의 합성을 지원합니다. 정상 조건에서 아미노산 대사의 에너지 공급 역할은 제한적입니다.
에너지 대사 연구가 확대됨에 따라, 대사 장애의 치료 접근법이 지속적으로 발전하는 동시에 다양한 질환과의 연관성이 점차 밝혀지고 있습니다. 1950년대 코리 순환의 발견은 간에서 젖산이 포도당으로 전환되는 생화학적 경로를 밝혀내며, 당뇨병과 같은 질환에서의 포도당 대사 이해를 심화시켰습니다. 42,66
1980년대, Kahn은
인슐린이 포도당 흡수 및 대사를 조절하는 메커니즘을 밝혀내어
인슐린 저항성과 제2형 당뇨병(T2DM)에 대한 이해를 심화시켰습니다.45,48,67
동시에 지방산 대사 연구는
FAO가 에너지 대사에서 차지하는 핵심적 역할을 강조하며,
대사 질환에서 지방의 역할을 이해하는 데 필수적인 단서를 제공했습니다.68,69
1994년,
에너지 균형과 체중을 조절하는 호르몬인 렙틴의 발견은
비만과 암 치료에 새로운 가능성을 열었습니다.70,71
1990년대에는 연구자들이 에너지 대사에서
미토콘드리아의 중심적 역할을 주목하기 시작했으며,
미토콘드리아 기능 장애가 다양한 대사 질환의 원인 중 하나임을 밝혀내어
미토콘드리아 표적 치료법 개발의 이론적 기반을 마련했습니다.72,73
2000년대 이후 연구는 암 세포의 대사 경로에 점점 더 초점을 맞추며, 변형된 포도당 및 지방 대사 과정이 암 세포의 성장과 생존을 지원하는 메커니즘을 밝혀냈습니다.74,75 또한 암 세포의 글루타민 대사 연구는 암 치료를 위한 새로운 치료 표적을 식별했으며, 대사 과정과 암 진행 간의 관계를 더욱 명확히 했습니다. 76 예를 들어, 연구자들은 에너지 대사 경로를 조절함으로써 종양 세포의 성장과 생존을 영향을 미칠 수 있으며, 대사 억제제를 암 치료 프로토콜에 통합한 임상 연구는 대사학적 통찰을 치료 전략으로 전환시키고 있습니다.
유전체학 및 대사체학의 발전으로 대사체학 기술의 적용은 암, 당뇨병, 비만 등 대사 질환의 진단과 모니터링을 더욱 정확하게 가능하게 했습니다.77,78 최근 연구에서는 심장 에너지 대사 변화가 심장 재생 촉진에 기여한다는 것이 처음으로 입증되었습니다.
지방산 산화(FAO)를 억제하면
심근 세포의 에너지 대사가 FAO에서 글리코lysis로 전환되어
심장 세포의 재생 능력을 활성화합니다.79
Energy metabolism in physiology
Overview of energy metabolism
Living organisms rely on energy to support growth and reproduction, maintain structural integrity, and respond to environmental changes. Energy metabolism encompasses the intricate biochemical processes responsible for extracting energy from food sources and utilizing it for various physiological activities. Associated pathways consist of interconnected processes, such as glycolysis, the citric acid cycle (Krebs cycle), and OXPHOS, which are tightly regulated by enzymes and metabolic intermediates. The primary objective of energy metabolism is ATP generation, which is considered the cellular currency for energy transactions. ATP plays a critical role in essential functions, including muscle contraction, neuronal signaling, and metabolic synthesis. The subsequent sections provide a comprehensive overview of the different aspects of energy metabolism, including the fundamental steps of various energy metabolism types and the hormonal and signaling systems governing its regulation (Fig. 3).
생리학에서의 에너지 대사
에너지 대사의 개요
생물체는 성장과 번식을 지원하고 구조적 안정성을 유지하며 환경 변화에 대응하기 위해 에너지를 의존합니다. 에너지 대사는 음식원에서 에너지를 추출하고 다양한 생리적 활동에 활용하는 복잡한 생화학적 과정을 포괄합니다.
관련된 경로는
글리코시스, 시트르산 회로(크렙스 회로), OXPHOS와 같은 상호 연결된 과정으로 구성되며,
이는 효소와 대사 중간체에 의해 엄격히 조절됩니다.
에너지 대사의 주요 목적은 ATP 생성이며,
이는 세포 내 에너지 거래의 기본 단위로 간주됩니다.
ATP는
근육 수축, 신경 신호 전달, 대사 합성 등 필수 기능에 결정적인 역할을 합니다.
다음 섹션에서는 에너지 대사의 다양한 측면을 포괄적으로 설명하며,
다양한 에너지 대사 유형의 기본 단계와 그 조절을 지배하는 호르몬 및 신호 전달 시스템을 포함합니다(그림 3).
Fig. 3
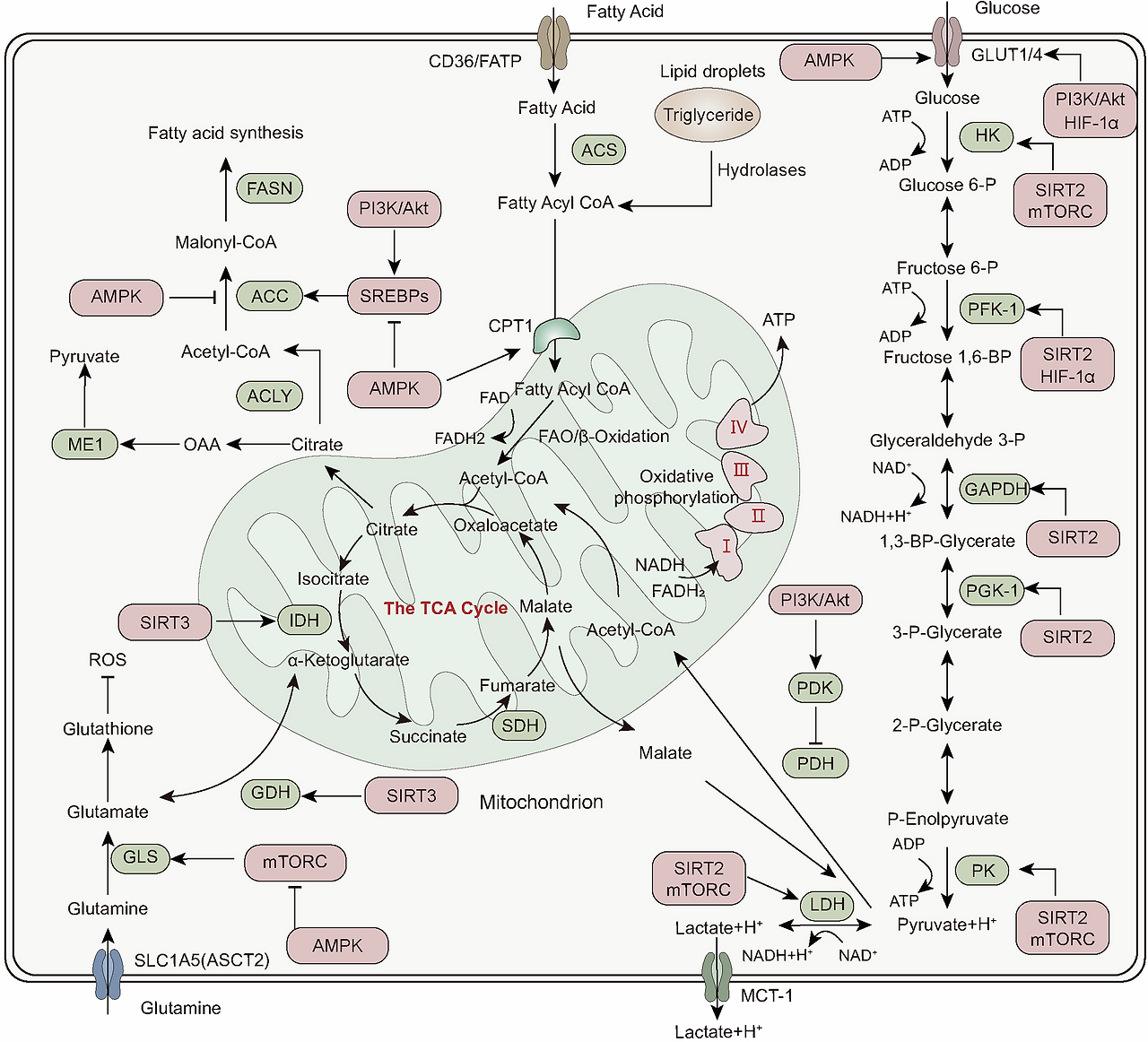
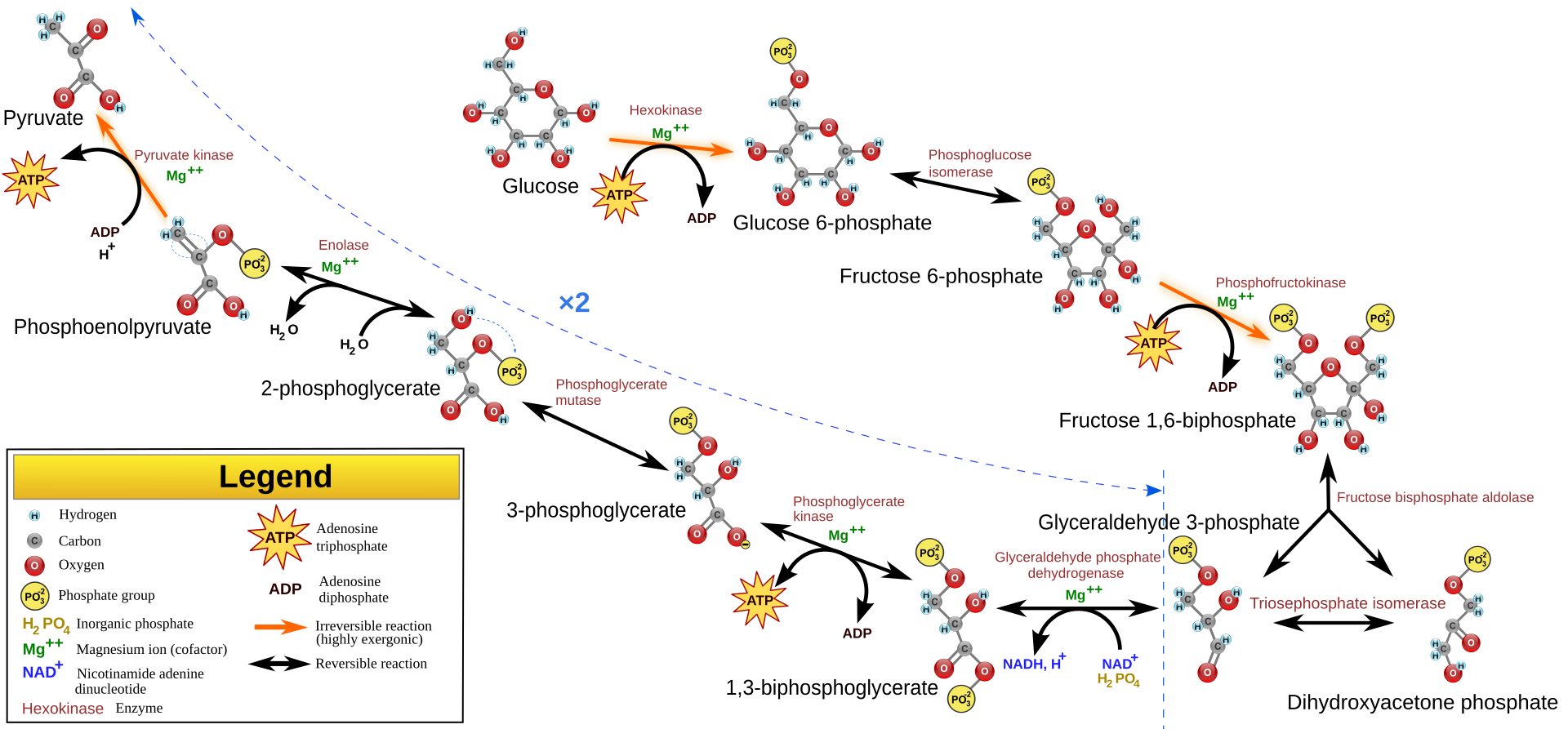
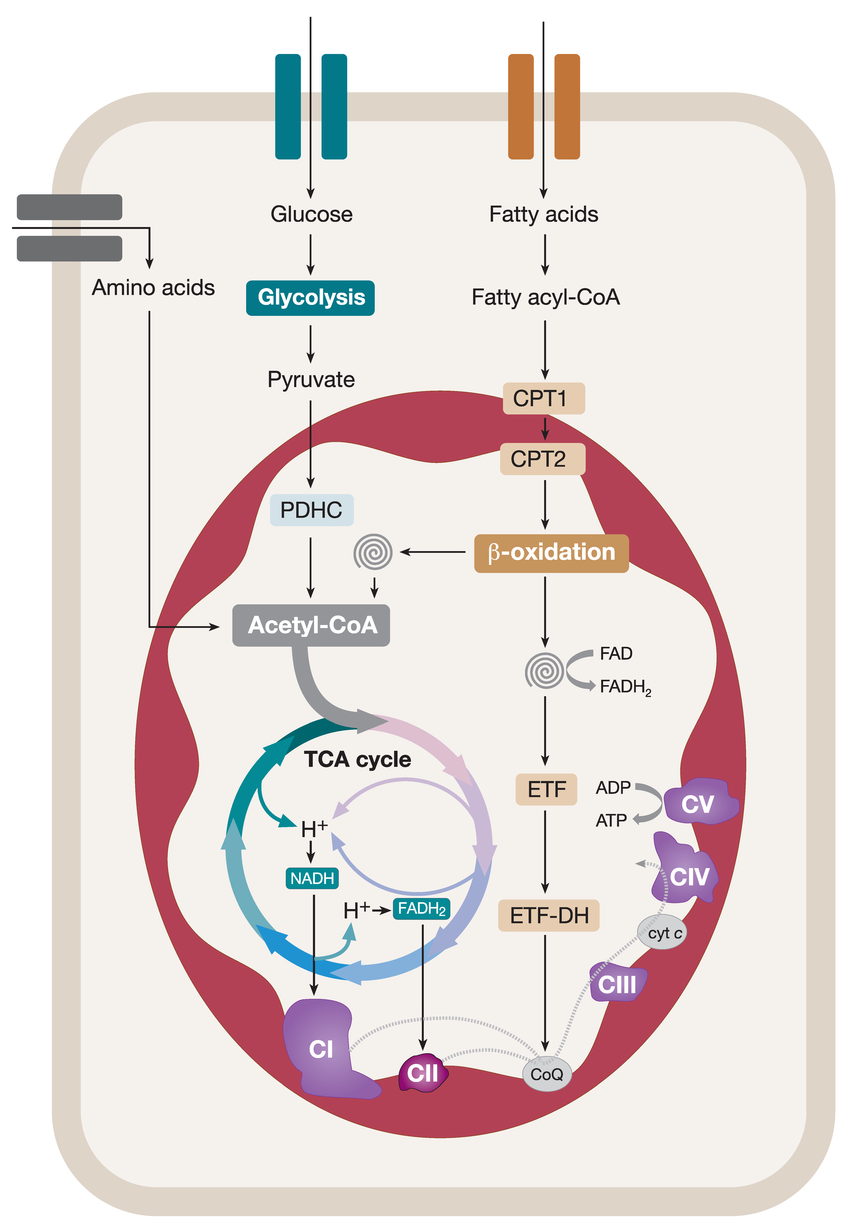
Main pathways for cellular energy production and molecular signal regulation. Glycolysis begins with the phosphorylation of glucose by hexokinase (HK), producing 6-phosphogluconate. Subsequently, phosphofructokinase-1 (PFK-1) converts 6-phosphofructose into 1,3-bisphosphoglycerate. In the subsequent cleavage reaction, aldolase (ALDO) cleaves 1,3-bisphosphoglycerate into two molecules of 3-phosphoglyceraldehyde. 3-phosphoglyceraldehyde is oxidized to 1,3-bisphosphoglycerate under the catalysis of glyceraldehyde-3-phosphate dehydrogenase (GAPDH), and reduced coenzyme II (NADH) is produced in the process. Thereafter, 1,3-bisphosphoglycerate is converted into 3-phosphoglycerate by phosphoglycerate kinase (PGK1), generating one molecule of ATP. 3-phosphoglycerate is then transformed into 2-phosphoglycerate by phosphoglycerate mutase (PGAM1), and then catalyzed by enolase (ENO1) to form 2-phosphoenolpyruvate. 2-phosphoenolpyruvate is ultimately converted into pyruvate under the action of pyruvate kinase (PK), releasing another molecule of ATP. Under anaerobic conditions, pyruvate is reduced to lactate by lactate dehydrogenase (LDH). Fatty acids first need to be activated into acyl-CoA (acyl-coenzyme A). After activation, the fatty acids are transferred from the cytoplasm to the mitochondrial matrix through Carnitine palmitoyltransferase I (CPT1). The fatty acids undergo a series of β-oxidation cycles, resulting in the production of acetyl-CoA and NADH. Glutamine enters the cell through ASCT2/SLC1A5 and is converted into glutamate by the deamination reaction catalyzed by glutaminase (GLS). Glutamate can be further converted into alpha-ketoglutarate (α-KG). Nutrient-derived acetyl-CoA enters the TCA cycle, which is catalyzed by enzymes such as succinate dehydrogenase (SDH), fumarate hydratase (FH), and isocitrate dehydrogenase (IDH), ultimately producing energy molecules ATP and reducing agents NADH and FADH2. NADH and FADH2 enter OXPHOS to further generate ATP. In the aforementioned process, various signaling molecules, such as AMP-activated protein kinase (AMPK), phosphatidylinositol-3-kinase/protein kinase B (PI3K/AKT), mechanistic target of rapamycin complex (mTORC), and sirtuins (SIRT), play crucial regulatory roles in controlling energy production under physiological conditions.
세포 에너지 생산 및 분자 신호 조절의 주요 경로.
글리코lysis는 글루코스의 헥소키나아제(HK)에 의한 인산화 반응으로 시작되어 6-포스포글루코네이트를 생성합니다. 이후 포스포프루토키나아제-1(PFK-1)은 6-포스포프루토스를 1,3-비스포스포글리세레이트로 전환합니다. 후속 분해 반응에서 알도라제(ALDO)는 1,3-비스포스포글리세레이트를 두 분자의 3-포스포글리세랄데히드로 분해합니다. 3-포스포글리세랄데히드는 글리세랄데히드-3-포스페이트 탈수소효소(GAPDH)의 촉매 작용으로 1,3-비스포스포글리세레이트로 산화되며, 이 과정에서 환원된 코엔자임 II(NADH)가 생성됩니다. 이후 1,3-비스포스포글리세레이트는 포스포글리세레이트 키나아제(PGK1)에 의해 3-포스포글리세레이트로 전환되며, 이 과정에서 ATP 한 분자가 생성됩니다. 3-포스포글리세레이트는 포스포글리세레이트 뮤타아제(PGAM1)에 의해 2-포스포글리세레이트로 변환된 후, 에놀아제(ENO1)의 촉매 작용으로 2-포스포엔올피루베이트를 형성합니다. 2-포스포엔올피루베이트는 피루베이트 키나아제(PK)의 작용으로 피루베이트로 전환되며, 이 과정에서 또 다른 분자의 ATP가 방출됩니다. 무산소 조건 하에서는 피루베이트가 젖산 탈수소효소(LDH)에 의해 젖산으로 환원됩니다. 지방산은 먼저 아실-코엔자임 A(아실-코엔자임 A)로 활성화되어야 합니다. 활성화 후 지방산은 카르니틴 팔미토일트랜스퍼레이즈 I(CPT1)을 통해 세포질에서 미토콘드리아 매트릭스로 이동합니다. 지방산은 β-산화 사이클을 거쳐 아세틸-코엔자임 A와 NADH를 생성합니다. 글루타민은 ASCT2/SLC1A5를 통해 세포 내로 들어와 글루타미나제(GLS)에 의해 탈아미노 반응을 통해 글루타메이트로 전환됩니다. 글루타메이트는 알파-케토글루타레이트(α-KG)로 추가 변환될 수 있습니다. 영양소 유래 아세틸-CoA는 TCA 회로에 진입하여 수크신산 탈수소효소(SDH), 푸마르산 수화효소(FH), 이소시트르산 탈수소효소(IDH) 등의 효소에 의해 촉매되어 에너지 분자 ATP와 환원제 NADH 및 FADH2를 생성합니다. NADH와 FADH2는 OXPHOS에 들어가 추가로 ATP를 생성합니다. 위 과정에서 AMP-활성화 단백질 키나제(AMPK), 포스파티딜인오실-3-키나제/단백질 키나제 B(PI3K/AKT), 라파마이신 복합체(mTORC), 시르투인(SIRT)과 같은 다양한 신호 전달 분자들은 생리적 조건 하에서 에너지 생산을 조절하는 데 중요한 역할을 합니다.
PDH, pyruvate dehydrogenase; PDK, pyruvate dehydrogenase kinase; GDH, glutamate dehydrogenase; ACS, acyl-CoA synthetase; ACC, acetyl-CoA carboxylase; FASN, fatty acid synthase; ACLY, ATP citrate lyase
Glycolysis
Glycolysis was the first metabolic route discovered, and the term “glycolysis” derives from the Greek word “glykys,” meaning sweetness, and “lysis,” meaning division or splitting.80 This refers to the breakdown of a single glucose molecule into two pyruvate molecules that act as glycolytic end products. Under aerobic conditions, pyruvate usually enters the mitochondria for oxidation to generate acetyl-CoA. Conversely, under anaerobic conditions, pyruvate is reduced to lactate. There are three essential components associated with glycolysis. First, it serves as the primary pathway for ATP generation when oxygen is limited or in cells without mitochondria, such as red blood cells. Second, under conditions of abundant oxygen, glycolysis produces pyruvate, which subsequently enters the mitochondrial TCA cycle to generate ATP. Third, glycolysis and TCA cycle yield various metabolites that can participate in anabolic pathways for NADPH synthesis and the generation of vital constituents.80
글리코lysis
글리코lysis는 최초로 발견된 대사 경로이며, “글리코lysis”라는 용어는 그리스어 “glykys”(달콤함)와 “lysis”(분해 또는 분열)에서 유래했습니다.80 이는 단일 글루코스 분자가 글리코lysis 최종 산물로 작용하는 두 개의 피루vate 분자로 분해되는 과정을 의미합니다. 산소 조건 하에서는 피루vate가 보통 미토콘드리아로 들어가 산화되어 아세틸-CoA를 생성합니다. 반면 산소 부족 조건에서는 피루vate가 젖산으로 환원됩니다. 글리코lysis와 관련된 세 가지 필수 구성 요소가 있습니다. 첫째, 산소가 부족하거나 미토콘드리아가 없는 세포(예: 적혈구)에서 ATP 생성의 주요 경로로 기능합니다. 둘째, 산소가 풍부한 조건에서 글리코lysis는 피루베이트를 생성하며, 이는 이후 미토콘드리아의 TCA 회로로 들어가 ATP를 생성합니다. 셋째, 글리코lysis와 TCA 회로는 NADPH 합성 및 필수 구성 요소 생성에 참여할 수 있는 다양한 대사물을 생성합니다.80
TCA cycle
In 1937, an important study titled “The Significance of Citric Acid in Animal Tissue Intermediate Metabolism” initially presented the notion of the TCA cycle, which was alternatively recognized as the Krebs cycle.81 The TCA cycle plays a vital role in eukaryotic cell metabolism by facilitating the entry of various molecules, such as fatty acids, amino acids, and pyruvate, into the cycle. Unlike linear pathways, the TCA cycle operates in a cyclic manner, with oxaloacetate serving as both the initial material for citrate synthesis, catalyzed by citrate synthase and the final product, produced by malate dehydrogenase, ensuring continuous renewal of the cycle. Notably, this pathway is considered amphibolic because it provides intermediates for macromolecule synthesis (such as lipids) and generates NADH and reduced flavin adenine dinucleotide (FADH2) molecules, which are necessary for ATP production via OXPHOS. Owing to its ability to accommodate multiple substrates, the TCA cycle plays a central role in cellular metabolism.
TCA 회로
1937년, “동물 조직 중간 대사에서 시트르산의 중요성”이라는 제목의 중요한 연구에서 TCA 회로의 개념이 처음 제시되었으며, 이는 이후 크렙스 회로로도 알려져 있습니다.81 TCA 회로는 지방산, 아미노산, 피루vate와 같은 다양한 분자가 회로에 진입하는 것을 촉진함으로써 진핵세포 대사에서 중요한 역할을 합니다. 선형 경로와 달리 TCA 순환은 순환적 방식으로 작동하며, 옥살아세트산은 시트르산 합성효소에 의해 시트르산 합성의 초기 물질로 작용하며, 말산 탈수소효소에 의해 생성되는 최종 제품으로 기능하여 순환의 지속적인 재생을 보장합니다. 특히 이 경로는 지질과 같은 거대분자 합성에 필요한 중간체를 제공하고, ATP 생산에 필수적인 NADH와 환원된 플라빈 아데닌 이중핵산(FADH2) 분자를 생성하기 때문에 암피볼릭 경로로 분류됩니다. 이 능력 덕분에 TCA 순환은 세포 대사에서 중심적인 역할을 합니다.
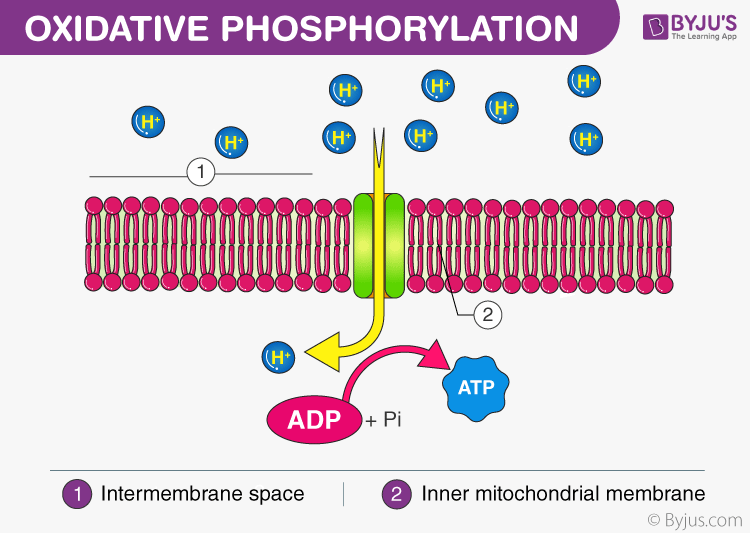
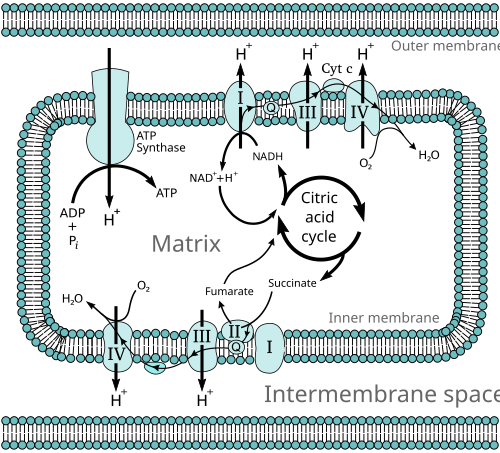
OXPHOS
OXPHOS is an essential process for ATP production within cells, particularly under aerobic conditions as part of cellular respiration. The key to OXPHOS lies in the electron transport chain (ETC), composed of a series of protein complexes that accept electrons from NADH and FADH2 and pass them to oxygen, the final electron acceptor, which combines with protons to form water. The energy released during this electron transport is converted into chemical energy, which leads to the combination of ADP and inorganic phosphate (Pi) to form ATP. This process is central to cellular energy metabolism and is crucial for maintaining the life activities of cells and organisms.
OXPHOS
OXPHOS는 세포 내 ATP 생산에 필수적인 과정으로, 특히 산소 존재 하에서 세포 호흡의 일부로 이루어집니다. OXPHOS의 핵심은 NADH와 FADH2로부터 전자를 받아 산소(최종 전자 수용체)로 전달하는 일련의 단백질 복합체로 구성된 전자 전달 사슬(ETC)에 있습니다. 산소는 프로톤과 결합하여 물을 형성합니다. 이 전자 전달 과정에서 방출되는 에너지는 화학 에너지로 전환되어 ADP와 무기 인산염(Pi)이 결합하여 ATP를 형성합니다. 이 과정은 세포 에너지 대사에서 중심적인 역할을 하며, 세포와 유기체의 생명 활동을 유지하는 데 필수적입니다.
Glutamine metabolism
Glutamine plays a crucial role as the primary source of energy for rapidly dividing cells, including hematopoietic stem cells and cancer cells. Glutamine is taken up by cells through specific transporters, such as SLC1A5, SLC38A1, and SLC38A2. Once inside the cell, it is used for various biosynthetic processes in the cytoplasm, including hexosamine production, nucleotide synthesis, and asparagine formation.82 Glutaminase (GLS) converts glutamine to glutamate by catalyzing its hydrolysis and releasing ammonium ions. The resulting mitochondrial glutamate can exit the mitochondria into the cytosol, where this exported glutamate plays a role in the synthesis of important molecules, such as glutathione and non-essential amino acids (NEAAs). Glutamate within mitochondria is further converted into α-ketoglutarate (α-KG). α-KG participates in fatty acid biosynthesis and NADH generation.83 It also serves as a substrate for both OXPHOS pathways, supporting the TCA cycle.84 Within the OXPHOS pathway, metabolites derived from glutamine contribute to the generation of electron donors, such as NADH or FADH2, which are utilized for ATP synthesis through the ETC.
글루타민 대사
글루타민은 혈액 줄기세포와 암세포를 포함한 빠르게 분열하는 세포의 주요 에너지 공급원으로 중요한 역할을 합니다. 글루타민은 SLC1A5, SLC38A1, SLC38A2와 같은 특정 운반체를 통해 세포에 흡수됩니다. 세포 내로 들어간 글루타민은 세포질에서 헥소사민 생산, 뉴클레오티드 합성, 아스파라긴 형성 등 다양한 생합성 과정에 사용됩니다.82 글루타민아제(GLS)는 글루타민을 글루타메이트로 전환하는 가수분해 반응을 촉매하며 암모늄 이온을 방출합니다. 미토콘드리아에서 생성된 글루타메이트는 미토콘드리아를 벗어나 세포질로 이동하며, 이 수출된 글루타메이트는 글루타티온과 비필수 아미노산(NEAAs)과 같은 중요한 분자의 합성에 역할을 합니다. 미토콘드리아 내의 글루타메이트는 α-케토글루타레이트(α-KG)로 추가 전환됩니다. α-KG는 지방산 생합성과 NADH 생성에 참여합니다.83 또한 OXPHOS 경로의 두 가지 경로 모두의 기질로 작용하여 TCA 사이클을 지원합니다.84 OXPHOS 경로 내에서 글루타민에서 유래한 대사산물은 NADH 또는 FADH2와 같은 전자 공급체를 생성하는 데 기여하며, 이는 ETC를 통해 ATP 합성에 활용됩니다.
FAO
FAO is one of the important pathways for cells to obtain energy, especially during prolonged fasting, starvation, or intense exercise when glycogen stores are depleted. The FAO process begins with acyl-CoA dehydrogenase. This enzyme forms a trans double bond between the alpha and beta carbons on acyl-CoA, yielding FADH2, subsequently contributing 1.5 ATP molecules via the ETC.85 The next step involves enoyl-CoA hydratase, which hydrates the double bond through the addition of a hydroxyl group to the beta carbon and a proton to the alpha carbon. In the third step, β-hydroxyacyl CoA dehydrogenase oxidizes the beta carbon, producing NADH, which generates 2.5 ATP molecules through the ETC.86 In the final step, catalyzed by β-keto thiolase, the α-β carbon bond is broken, resulting in the formation of acetyl-CoA and a shortened fatty acyl-CoA, allowing the cycle to repeat until all even-chain fatty acids are converted into acetyl-CoA.
FAO
FAO는 세포가 에너지를 얻는 중요한 경로 중 하나로, 특히 장시간 금식, 영양 결핍, 또는 강도 높은 운동으로 글리코겐 저장량이 고갈된 상황에서 중요한 역할을 합니다. FAO 과정은 아실-코엔자임 A 탈수소효소(acyl-CoA dehydrogenase)로 시작됩니다. 이 효소는 아실-코엔자임 A의 알파와 베타 탄소 사이에 트랜스 이중 결합을 형성하여 FADH2를 생성하며, 이는 전자 전달 체계(ETC)를 통해 1.5개의 ATP 분자를 공급합니다.85 다음 단계에서는 에노일-코엔자임 A 수화효소(enoyl-CoA hydratase)가 베타 탄소에 수산기 그룹을, 알파 탄소에 프로톤을 추가하여 이중 결합을 수화시킵니다. 세 번째 단계에서 β-하이드록시아실 코엔자임 A 탈수소효소는 베타 탄소를 산화시켜 NADH를 생성하며, 이는 ETC를 통해 2.5개의 ATP 분자를 생성합니다. 86 최종 단계에서 β-케토 티올라아제(β-keto thiolase)의 촉매 작용으로 알파-베타 탄소 결합이 끊어져 아세틸-CoA와 단축된 지방산-CoA가 형성되며, 이 과정은 모든 짝수 사슬 지방산이 아세틸-CoA로 전환될 때까지 반복됩니다.
Ketone metabolism
Ketones, including β-hydroxybutyrate (BHB), acetoacetate (AcAc), and acetone, are synthesized primarily from fatty acids by the liver.87 Catalyzed by β-ketoacyl-CoA synthase (HMGCS2), two acetyl-CoA molecules combine to form HMG-CoA, which is then broken down into AcAc and acetyl-CoA by HMG-CoA lyase (HMGCL). Most AcAc is then reduced back to BHB by β-hydroxybutyrate dehydrogenase (BDH1). Ketones are transported to target organs, metabolized in the mitochondria, and converted back to acetyl-CoA for energy generation. Ketone production increases mainly when the supply of glucose/acetoacetate decreases to maintain energy production. However, ketone production also leads to an increase in mitochondrial oxidative stress, a mechanism significantly incongruent with the anti-inflammatory effects of ketone supplementation.88 Ketone supplementation is generally believed to initially promote inflammation but gradually shifts toward anti-inflammatory and antioxidative mechanisms as cells adapt. This involves the regulation of NRF2, SIRT, and AMPK.89
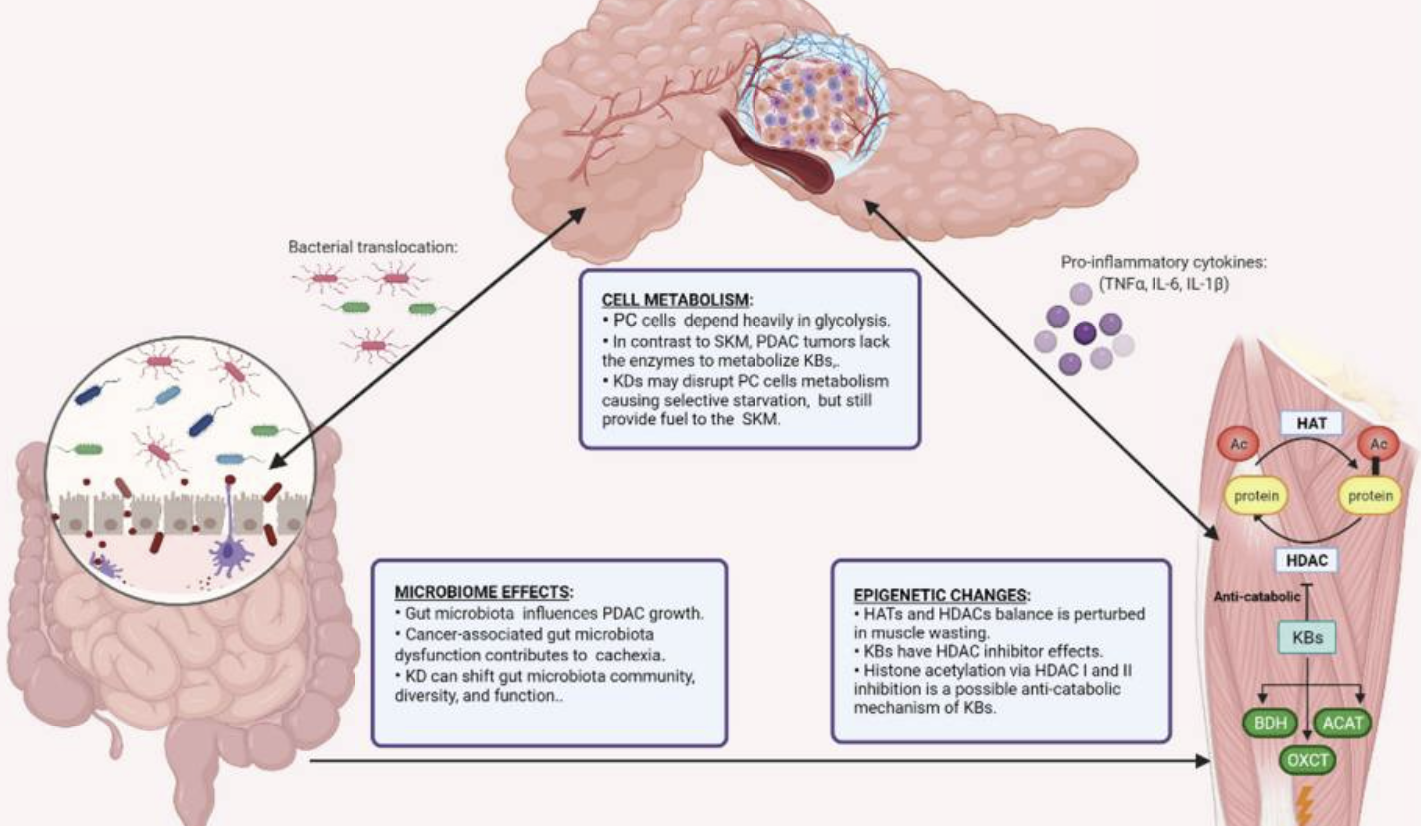
The relationship between ketone metabolism and pathophysiology is closely intertwined. In the nervous system, ketones can enter brain tissue through monocarboxylic acid transporters (MCTS) in endothelial and astrocytic cells. Neurons, astrocytes, and oligodendrocytes metabolize ketones at a higher rate than glucose dose, providing energy. Ketones protect neurons by improving mitochondrial respiration and reducing inflammation.90 In the cardiovascular system, ketones serve as an energy source for cardiac muscle and endothelial cells. While they do not enhance cardiac efficiency,91 they can improve cardiac inflammatory conditions by inhibiting the NLRP3 signal.92 In cardiovascular diseases, ketones provide a potential alternative fuel source for a failing heart.93 However, the role of ketone oxidation in myocardial infarction remains unclear. In cancer, ketones exhibit diverse effects, either promoting or inhibiting cancer cell proliferation. Studies suggest that ketones are essential for CD8+ T cells as an energy source, enhancing tumor killing effects and strengthening effector functions.94 However, other research indicates that ketones may support tumor growth and metastasis as energy sources. In pancreatic ductal adenocarcinoma, ketone metabolism, particularly BHB as an energy source, promotes tumor growth and progression.95 Further research is needed to understand how ketones regulate cancer progression.
케톤 대사
케톤은 주로 간에서 지방산으로부터 β-하이드록시부티레이트 (BHB), 아세토아세테이트 (AcAc), 및 아세톤으로 합성됩니다. 87 β-케토아실-CoA 합성효소(HMGCS2)의 촉매 작용으로 두 분자의 아세틸-CoA가 결합하여 HMG-CoA를 형성하며, 이는 HMG-CoA 리아제(HMGCL)에 의해 아세토아세테이트와 아세틸-CoA로 분해됩니다. 대부분의 AcAc는 β-하이드록시부티레이트 디히드로게나제(BDH1)에 의해 BHB로 환원됩니다. 케톤은 목표 기관으로 운반되어 미토콘드리아에서 대사되어 에너지 생성을 위해 아세틸-CoA로 전환됩니다. 케톤 생산은 주로 글루코스/아세토아세테이트 공급이 감소하여 에너지 생성을 유지하기 위해 증가합니다.
그러나 케톤 생산은
미토콘드리아 산화 스트레스 증가를 유발하며,
이는 케톤 보충의 항염증 효과와 크게 상반되는 메커니즘입니다.88
케톤 보충은
일반적으로 초기에는 염증을 촉진하지만
세포가 적응함에 따라 점차 항염증 및 항산화 메커니즘으로 전환된다고 알려져 있습니다.
이는 NRF2, SIRT, AMPK의 조절을 포함합니다.89
케톤 대사 및 병리생리학 간의 관계는 밀접하게 연관되어 있습니다. 신경계에서 케톤은 내피 세포와 아스트로사이트 세포의 단일 카르복시산 운반체(MCTS)를 통해 뇌 조직으로 들어갈 수 있습니다. 신경세포, 아스트로사이트, 올리고덴드로사이트는 포도당 투여량보다 높은 속도로 케톤을 대사하여 에너지를 공급합니다. 케톤은 미토콘드리아 호흡을 개선하고 염증을 감소시켜 신경세포를 보호합니다.90 심혈관 시스템에서 케톤은 심근 세포와 내피 세포의 에너지원으로 작용합니다. 심장 효율을 향상시키지는 않지만,91 NLRP3 신호를 억제함으로써 심장 염증 상태를 개선할 수 있습니다.92 심혈관 질환에서 케톤은 기능이 저하된 심장에 대한 잠재적 대체 연료원으로 작용합니다.93 그러나 심근경색에서 케톤 산화의 역할은 여전히 명확하지 않습니다.
암에서는 케톤이 암 세포 증식을 촉진하거나 억제하는 다양한 효과를 나타냅니다.
연구 결과, 케톤은 CD8+ T 세포의 에너지 원으로 필수적이며, 종양 살상 효과를 강화하고 효과기 기능을 강화합니다.94
그러나
다른 연구에서는 케톤이 에너지 원으로 종양 성장과 전이를 지원할 수 있음을 나타냅니다.
췌관 선암에서 케톤 대사, 특히 BHB를 에너지 원으로 사용 시 종양 성장과 진행을 촉진합니다.95
케톤이 암 진행을 조절하는 메커니즘을 이해하기 위해 추가 연구가 필요합니다.
Non-metabolic functions of energy metabolism enzymes and metabolites
During energy metabolism, several metabolic enzymes and metabolites play crucial non-metabolic roles, referred to as “moonlighting” functions, to regulate gene transcription, translation, and epigenetic modifications. These functions exert considerable impacts under various physiological and pathological conditions.
Regulation of gene expression
Metabolic enzymes influence gene expression through diverse mechanisms. For instance, metabolic enzymes localized in the cytoplasm or mitochondria can translocate to the nucleus and modify chromatin structure by interacting with histones and DNA, thereby directly regulating gene expression. HK2 interacts with nuclear proteins such as Max, Sirt1, IWS1, CTR9, and Spin1, increasing chromatin accessibility to regulate gene expression.96 Upon activation, phosphofructokinase 1 (PFK1) can bind to the transcription factor TEAD, stabilizing its interaction with YAP/TAZ in the nucleus, promoting YAP/TAZ transcriptional output, and impacting breast cancer progression.97 In glioblastoma cells with aberrant epidermal growth factor receptor (EGFR) signaling, PFK1 undergoes acetylation at the K395 site and translocates to the plasma membrane, recruiting and binding with p85α, leading to sequential activation of PI3K/AKT, PFK2, and PFK1.98 Moreover, pyruvate kinase M2 (PKM2) can enter mitochondria to maintain its function and translocate to the nucleus to regulate gene expression.99 Once in the nucleus, it can activate various transcription factors such as hypoxia-inducible factor 1 alpha (HIF-1α), histone H3, NRF2, and STAT3, influencing the activation of downstream target genes. Fructose-1,6-bisphosphatase inhibits the activity of HIF-1α in the nucleus, decreases the expression of HIF target genes, and promotes its degradation by binding to Notch1, regulating tumor formation.100 Nuclear translocation of fructose-1,6-bisphosphatase 2 inhibits c-Myc-mediated gene expression, thereby suppressing mitochondrial biogenesis and respiration.101 Phosphorylation of fumarate hydratase (FH) by p38 in response to TGF-β signaling leads to its binding to the transcription factor CSL/p53 complex on the p21 promoter, which inhibits histone H3K36 demethylation, enhances p21 transcription, and induces cell growth arrest.102 Phosphoenolpyruvate carboxykinase 1 utilizes GTP as a phosphate donor to phosphorylate INSIG1 and INSIG2 in the endoplasmic reticulum (ER), leading to the transcription of downstream lipid synthesis genes mediated by SREBP.103 NADPH directly inhibits the activation of HDAC3 through interaction, disrupting the binding of HDAC3 with its coactivating factors NCOR1/2, thereby regulating cellular epigenetics.104 Recent studies revealed that in macrophages, the mediator binds to 2-ketoacid dehydrogenases, generating acetyl-CoA, which increases histone acetylation levels in specific chromatin regions, thereby regulating gene transcription. However, the exact molecular mechanism of this binding requires further investigation.105
DNA repair
HK2 exhibits moonlighting functions in the nucleus, with its overexpression increasing chromatin accessibility at DNA repair sites, thereby reducing DNA double-strand breaks.96 Tumor-inducing EGFR signaling induces the phosphorylation of phosphoglycerate kinase 1 (PGK1) at S256 by casein kinase 2α (CK2α), leading to the binding of phosphorylated PGK1 to CDC7, converting local adenosine diphosphate produced by CDC7 into ATP, facilitating the recruitment of DNA helicase to the replication origin, and promoting DNA replication.106 Nuclear ATP-citrate lyase can be phosphorylated and activated in response to DNA damage, providing acetyl-CoA to promote the recruitment of BRCA1 and DNA repair.107 Under DNA damage stress, activation of tyrosine kinase SRC leads to phosphorylation of glyceraldehyde-3-phosphate dehydrogenase (GAPDH), crucial for its nuclear translocation. Nuclear GAPDH is recruited to DNA damage sites and interacts with DNA polymerase (PAR), participating in DNA repair mechanisms.108 Fumarate is involved in DNA damage response; it translocates to the nucleus after DNA damage, produces fumarate esters that inhibit histone lysine demethylase KDM2B, and promotes protein binding during DNA repair.109 Additionally, metabolites such as 2-hydroxyglutarate, succinate, and fumarate derived from mutations in IDH1/2, FH, and SDH inhibit DNA repair pathways by repressing lysine demethylase KDM4B, resulting in abnormal excessive H3K9 methylation at DNA break sites, impairing recruitment of TIP60 and ATM, reducing end resection, and decreasing recruitment of downstream repair factors.110
Post-translational protein modifications
Post-translational modifications of proteins, including acetylation, phosphorylation, and lactylation, are extensively influenced by metabolic processes. Acetyl-CoA serves as a primary driver for histone acetylation, a process facilitated by lactate dehydrogenase A (LDHA). In T cells lacking LDHA, histone acetylation is reduced at H3K9AC.111 Under oxidative stress, LDHA undergoes a transition from a tetramer to a dimer, facilitating its translocation to the nucleus. The atypical enzymatic activity of nuclear LDHA catalyzes the conversion of α-KB to α- hydroxybutyrate (α-HB), leading to α-HB accumulation. This accumulation, through enhanced histone methylation modifications, regulates gene expression, enhances cell resistance to reactive oxygen species (ROS), and promotes cell survival.112 Lactate accumulation influences protein transcription or physicochemical characteristics through lactylation. Lactylation of lysine 62 (K62) on PKM2 plays a role in regulating the feedback signaling of glycolysis.113 Lactylation also modulates immune cell functions; for instance, lactylation of MOESIN at Lys72 enhances its interaction with TGF-β receptor I, activating the TGF-β-SMAD3 signaling pathway and regulating Treg cell functions.114 Lactate-fueled histone lactylation facilitates the activation of repair genes in macrophages; however, reduced histone lactylation weakens gene expression in repair macrophages.115
In addition to affecting lactylation, lactate also impacts acetylation. Lactate inhibits the deacetylase SIRT1 through Hippo/YAP-mediated lactylation and G protein-coupled receptor 81-dependent β-arrestin2-mediated recruitment of p300/CBP in the nucleus, driving HMGB1 acetylation.116 Lactate also inhibits histone deacetylase activity, leading to excessive H3K27 acetylation at Tcf7 super-enhancer sites, increasing Tcf7 expression. This results in an increased proportion of CD8 + T cells expressing stem cell-like Tcf-1 in tumors, enhancing the efficacy of immunotherapy.117 Succinate indirectly influences the expression of histone deacetylases by stabilizing HIF-1α.118 Accumulation of fumarate inhibits the KDM5 family of histone demethylases, increasing the levels of the active gene transcription marker H3K4me3.119 Furthermore, fumarate accumulation can activate the PI3K/AKT signaling pathway through PTEN succinylation, exerting oncogenic effects.120 Acetylation of PKM2 at the K433 site by acetyltransferase p300 leads to the accumulation of the dimeric form of PKM2 in the nucleus, functioning as a protein kinase, phosphorylating STAT3 and activating downstream signaling pathways.121 However, the debate over whether PKM2 exhibits protein kinase activity continues. PGK1 can undergo auto-phosphorylation at the Y324 site to achieve maximal activation. Conversely, PTEN dephosphorylates self-phosphorylated PGK1, inhibiting its activity and thereby leading to a reduction in glycolysis in brain tumors.122
Extensive research on post-translational protein modifications has led to the discovery of new modification modes, such as histone tyrosine sulfation. SULT1B1 catalyzes the sulfation modification of histone H3 at the Y99 site directly using PAPS as a substrate, thereby regulating H4R3me2a and gene transcription.123 However, the specific sites and types of protein residue modifications remain partially defined, and the exact roles of protein residue modifications remain unclear. While protein modifications are reportedly involved in the development of many diseases, their precise roles and molecular mechanisms in different biological processes need further elucidation. A deeper understanding of the “writers,” “erasers,” and “readers” of these modifications on proteins, as well as their mechanisms of action in various biological processes, remains a focal point and challenge in current research.
Signaling pathways that regulate energy metabolism
To maintain balanced energy intake and expenditure, which are crucial for the overall well-being and health of an organism,124,125 energy metabolism must be regulated. The absorption and utilization of nutrients, as well as the release of stored energy from fuel sources, are significantly influenced by signaling molecules and pathways.
Hormonal regulation of energy metabolism
Hormones are critical in energy metabolism, acting as messengers produced by various organs to regulate physiological processes. Key hormones like insulin, glucagon, leptin, and ghrelin influence metabolism by binding to specific receptors on target cells. Insulin, crucial during feeding for energy balance, modulates glucose metabolism and overall homeostasis. Its effects include (i) suppressing liver glucose production; (ii) enhancing glucose uptake in muscles, liver, and fat cells; (iii) inhibiting lipolysis, decreasing plasma fatty acids, and reducing liver glucose output; and (iv) promoting vasodilation in muscles to increase glucose disposal.126
Insulin’s role is mediated through signaling pathways associated with its receptor.126 The PI3K/Akt pathway is pivotal, where insulin activation phosphorylates phosphatidylinositol 4,5-bisphosphate via PI3K to produce phosphatidylinositol 3,4,5-triphosphate, triggering Akt.127 Akt also hinders ATP-citrate lyase activity, impedes fatty acid synthesis, and disrupts mammalian target of rapamycin complex 1 (mTORC1) function, thereby enhancing protein synthesis. Additionally, it activates sterol regulatory element binding proteins (SREBPs), which mediate the transcription of genes related to cholesterol and fatty acid synthesis. The PI3K/Akt pathway also regulates the translocation of glucose transporter 4 (GLUT4) from intracellular vesicles to muscle and fat cell membranes, facilitating glucose uptake upon insulin stimulation.128 Any interference in the sequential steps involved in GLUT4 transport could cause diabetes mellitus and insulin resistance.
In addition to insulin, pancreatic islets release glucagon, somatostatin, and other pancreatic polypeptides hormones, which collaborate to maintain ideal glucose levels in the bloodstream and control energy metabolism.129 Insulin secretion is stimulated by glucagon, whereas somatostatin acts as an inhibitor.129 Consequently, β-cells integrate multiple regulatory inputs to ensure adequate insulin secretion and maintain glucose homeostasis.129 Additionally, the brain is affected by the hormone leptin, which is released by fat cells and regulates overall energy balance by reducing food intake and increasing energy utilization.130 Leptin signaling involves a receptor located on the cell membrane that initiates downstream signaling pathways, such as the Janus kinase (JAK)/signal transducer and activator of transcription (STAT) pathway, to stimulate energy expenditure while suppressing appetite.131
AMPK signaling and energy metabolism
AMPK, which comprises an enzyme complex, is critical for controlling cellular metabolism and maintaining energy equilibrium.132 Moreover, it functions in the detection of the cellular energy level and adjusts to variations in baseline conditions, stressors, and pathological conditions.132 In mammals, the energy status is sensed by AMPK through its ability to monitor cellular AMP, ADP, and ATP levels.133 AMPK is activated when ATP levels decrease and AMP levels increase,133 and its activation aids in restoring energy equilibrium by inhibiting ATP-consuming processes and promoting ATP-generating processes.133 Furthermore, by regulating cellular metabolic functions, AMPK can prime organs and tissues to defend against ischemic damage and promote the prompt resolution of inflammatory responses.
AMPK regulation is pivotal in responding to energy stress by sensing changes in intracellular AMP, ADP, and ATP levels.134 AMPK activation occurs through a three-step mechanism: first, AMP or ADP binds to the γ subunit, leading to Thr172 phosphorylation in the α subunit’s kinase domain via upstream kinases.134 Liver-kinase-B1 (LKB1), a serine/threonine kinase, is the main kinase for Thr172 phosphorylation during energy stress,135,136,137 a critical step that increases AMPK activity 100-fold in vitro.138,139,140 Second, AMP or ADP binding causes a structural change that protects Thr172 from dephosphorylation, although the specific phosphatases involved under physiological conditions are largely unidentified, with some suggested by recent studies.141,142 Lastly, AMP (but not ADP) significantly boosts allosteric activity, increasing it potentially by 10-fold.138 Notably, ATP inhibits all three mechanisms. In addition to variations in adenine nucleotide levels, there are alternative and noteworthy mechanisms that control AMPK. One extensively investigated strategy for regulating AMPK, which does not rely on nucleotides, involves Thr172 phosphorylation mediated by calcium/calmodulin-dependent protein kinase kinase 2 (CAMKK2).143,144,145 CAMKK2 is triggered by elevations in intracellular Ca2+ level. Therefore, although CAMKK2 does not directly sense the cellular energy status, it plays a crucial role in regulating various aspects of overall metabolism through AMPK.
Beyond the traditional AMP/ADP-dependent mechanisms, AMPK can be activated through alternative pathways.146,147 The glycolytic process involves the conversion of glucose into fructose-1,6-bisphosphate (FBP), a molecule that is subsequently metabolized by FBP aldolases. Research indicates that glucose deprivation leads to a decrease in FBP-bound aldolase, which in turn initiates the phosphorylation and subsequent activation of LKB1, a key upstream kinase of AMPK. Consequently, this study reveals the pivotal role of FBP as a metabolic signal for glucose levels and identifies FBP aldolases as metabolic sensors that communicate the status of glucose availability to AMPK.146,147
In recent years, our knowledge of how AMPK regulates metabolism has advanced significantly, owing to the discovery of numerous substrates that are targeted by this kinase. This progress was greatly facilitated by the identification of a specific motif recognized by AMPK.148,149 Additionally, innovative proteomic techniques have expanded the list and range of substances that are potentially regulated by AMPK.150,151,152 As previously mentioned, AMPK plays a crucial role in replenishing ATP levels under metabolic stress by temporarily suppressing ATP utilization in biosynthetic processes, while simultaneously activating pathways that facilitate ATP production. AMPK phosphorylates various transcription factors (or cofactors) that serve as key regulators of biosynthetic pathways and metabolism.153 In this manner, AMPK can promptly reinstate energy equilibrium while transcriptionally reprogramming cellular metabolism in response to extended periods of reduced energy levels.
Glucose and lipid metabolism
Glucose and lipids play crucial roles in providing and storing energy within cells. Through separate mechanisms, AMPK stimulates glucose uptake by phosphorylating TBC domain family member 1 (TBC1D1) and thioredoxin-interacting protein (TXNIP), which regulate the movement and surface expression of GLUT4 and GLUT1.154,155 AMPK also plays a role in the acute regulation of glycolysis in certain tissues through the phosphorylation of PFKFB3. Additionally, it inhibits glucose storage in specific tissues by targeting and inhibiting multiple GYS isoforms involved in glycogen synthesis.154,156 It also regulates cellular lipid metabolism by directly phosphorylating acetyl-CoA carboxylase (ACC)1 and ACC2, thereby inhibiting fatty acid synthesis and promoting FAO. This is achieved by alleviating the inhibition of carnitine palmitoyltransferase 1 (CPT1), which is mediated by the local production of malonyl-CoA at the outer membrane of the mitochondria, via ACC2. Notably, the amino terminus of ACC2 contains a sequence that targets it to the mitochondria. Additionally, 3-hydroxy-3-methyl-glutaryl-CoA (HMG-CoA) reductase (HMGCR) is phosphorylated and inhibited by AMPK. These combined effects on ACC1, ACC2, and HMGCR result in the preprogramming of lipid and sterol synthesis within the cell. AMPK was initially discovered for its ability to suppress fatty acid and cholesterol synthesis. This is achieved through direct phosphorylation, leading to the inhibition of ACC and HMG-CoA reductase. Furthermore, AMPK enhances food intake and reduces liver steatosis, fibrosis, insulin resistance, platelet dysfunction, renal fibrosis, and hepatocellular carcinoma. Moreover, the promotion of FAO in response to pharmacological activators also relies on the phosphorylation and inhibition of ACC, whereas alternative mechanisms, such as AKAP1 phosphorylation, might play a significant role during exercise. AMPK activity hinders the activation of gluconeogenesis-related genes by phosphorylating cyclic-AMP-regulated transcriptional co-activator 2 (CRTC2) and histone deacetylases (HDACs), which are essential cofactors for gene transcription and are involved in de novo glucose synthesis.157,158 In addition, AMPK phosphorylates and inhibits transcription factors that stimulate glycolytic and lipogenic transcriptional processes, particularly SREBP1, a key regulator of lipid synthesis,159 as well as hepatocyte nuclear factor-4α (HNF4α) and carbohydrate-responsive element binding protein (ChREBP).160,161 Hence, the immediate stimulation of AMPK facilitates the absorption of glucose to facilitate ATP replenishment, whereas its prolonged activation alters cellular functions to restrict glucose and lipid synthesis while promoting the utilization of fatty acids for energy production.
Protein metabolism
The suppression of protein synthesis, mediated by AMPK, is primarily achieved through direct inhibition of mTORC1. Mammalian TOR serves as a central regulator that integrates signals from nutrients and growth factors, triggering various biosynthetic pathways, particularly the protein translation process, which promotes cellular expansion. Moreover, AMPK and mTORC1 play opposing roles in controlling cellular metabolism, and the activity of mTORC1 is suppressed by AMPK through a dual mechanism involving the phosphorylation-induced activation of tuberous sclerosis complex 2 (TSC2)162 and the phosphorylation-mediated inhibition of regulatory-associated protein of mTOR (Raptor), a component of the mTORC1 complex.148 In addition to inhibiting mTOR, AMPK limits protein synthesis by hindering ribosomal RNA production. This is accomplished through the phosphorylation and inhibition of TIF-IA, which is responsible for initiating RNA polymerase I activity.163 AMPK impedes protein elongation by activating eEF2K, an enzyme that suppresses the elongation process.164 Crucially, mTORC1 also acts as a primary regulator of eEF2K,165 exemplified by the numerous downstream targets of AMPK that are directly phosphorylated by mTORC1 or S6K1, to counteractively modulate their functions in relation to AMPK phosphorylation. Thus, AMPK and mTOR govern anabolism and catabolism by traversing the cellular environment and activating or deactivating a limited set of pivotal metabolic switches.
Autophagy
Autophagy is a cellular process involving the breakdown of proteins, organelles, and other large molecules via lysosomal transport. When energy availability is low, cells utilize this mechanism for regular turnover and to generate nutrients. AMPK plays a crucial role in promoting autophagy through various mechanisms. One such mechanism involves the phosphorylation and activation of unc-51-like autophagy-activating kinase 1 (ULK1) by AMPK, which initiates a cascade that triggers autophagy.166,167,168 Moreover, mTOR effectively inhibits autophagy by directly phosphorylating and suppressing ULK1.167 Consequently, AMPK promotes autophagy not only through direct ULK1 activation but also by inhibiting mTORC1 and preventing its suppressive effect on ULK1. Furthermore, ULK1 comprises another critical juncture where AMPK and mTOR regulate specific metabolic processes in contrasting manners. AMPK also differentially regulates VPS34-containing complexes that contribute to the initiation of autophagy.169 These complexes are essential for autophagosome formation and initiation. In addition, AMPK directly inhibits VPS34 by phosphorylating non-autophagic complexes lacking autophagic adaptor proteins. Simultaneously, it enhances VPS34 activity through the direct phosphorylation of beclin-1 in pro-autophagic complexes.169 Thus, AMPK is thought to hinder the unnecessary movement of vesicles and instead direct membrane trafficking towards the autophagic pathway in the absence of nutrients. However, many unanswered questions remain regarding the precise regulation and coordination of autophagy initiation in response to different stressors, as both AMPK and ULK1 can directly phosphorylate distinct regions within beclin-1 and Vps34. Additionally, both AMPK and ULK1 can phosphorylate Atg9, a transmembrane protein involved in the early formation of autophagosomes, thereby exerting control over its localization.168,170,171
mTOR signaling and energy metabolism
Mammalian TOR is widely recognized as a critical regulator of homeostasis, particularly in maintaining energy balance and mTORC1 plays a pivotal role in balancing growth-promoting factors with nutrient availability.172 At the cellular level, mTORC1 functions as a nutrient sensor, coordinating the equilibrium between anabolism and catabolism in response to external conditions.173 In mammals, changes in energy levels are closely tied to dietary intake. Under restricted feeding conditions, mTORC1 is activated to promote tissue growth and energy storage in organs, such as the liver and muscles. Conversely, during fasting, mTORC1 activity is suppressed to conserve resources.173 Activation of mTORC1 enhances metabolic pathways, including glycolysis, the oxidative branch of the pentose phosphate pathway, and de novo lipid biosynthesis.174
While mTORC2 signaling is less understood than mTORC1 signaling is, recent research has indicated that mTORC2 is involved in cellular metabolism and the organization of the actin cytoskeleton. It also enhances cell viability by activating the survival kinase Akt.175,176 Mammalian TORC1 is implicated in regulating the size, morphology, and synaptic plasticity of neurons and maintaining energy balance within the central nervous system.177 Additionally, mTORC1 is sensitive to intrinsic and extrinsic factors that inhibit cell growth, such as reduced ATP levels, oxygen deprivation, and genetic damage.173 A decrease in cellular energy, such as inadequate glucose supply, triggers the activation of AMPK, a metabolic regulator involved in the stress response, leading to the inhibition of mTORC1.178
Molecular structure of mTOR
The serine-threonine kinase mTOR, a member of the PI3K-related kinase (PIKK) family, is evolutionarily conserved. It forms two distinct protein complexes, mTORC1 and mTORC2, each with unique roles, regulatory mechanisms, and rapamycin sensitivity.174 Thus, mTORC1 consists of mTOR, Raptor, GβL/mLST8, DEPTOR, and PRAS40,174 whereas mTORC2 includes mTOR, GβL/mLST8, Rictor, Protor/PRR5, DEPTOR, and mSIN1. The primary role of mTORC1 is to integrate signals from growth factors and nutrients to drive cellular growth, under conditions of energy abundance or nutrient-scarcity-triggered catabolism.174 While mTORC1 is well-recognized for its role in cellular growth and metabolism, mTORC2 is more associated with the regulation of cell proliferation and survival.173
mTOR as an energy sensor
The mTORC1 pathway detects and regulates cellular growth and survival in response to environmental, extracellular, and intracellular stresses, such as hypoxia, reduced ATP levels, and DNA damage.172 Under glucose-deprived conditions, cellular energy levels decrease significantly, leading to the activation of AMPK, a metabolic regulator that senses energy stress. AMPK activation inhibits mTORC1 activity by directly phosphorylating Raptor or indirectly through TSC2 phosphorylation.148 Moreover, low glucose levels suppress mTORC1 signaling by inhibiting RAG GTPase activity, particularly in AMPK-deficient cells. A study by Dai et al. demonstrated that mTORC1 activation in response to glucose can be modulated via the AMPK-mediated phosphorylation of WDR24.179
Amino acids not only are essential for protein synthesis but also serve as vital reservoirs of carbon and energy for various metabolic signaling pathways.180 The modulation of amino acid levels in response to dietary changes is closely linked to mTORC1 activation. The discovery of RAG GTPases as key components in mTORC1 signaling, particularly in amino acid detection, has significantly advanced our understanding of mTOR signaling.181 These findings suggest that mTORC1 can effectively perceive glucose- and energy-related challenges through multiple molecular pathways.182,183
Energy regulation of mTOR
Mammalian TORC1 regulates cellular energy requirements through AMPK, which functions as an intracellular energy sensor. Enhanced glucose metabolism triggers mitochondrial activity by increasing AMP levels, disrupting the ATP:AMP ratio and subsequently activating AMPK. This activation leads to the phosphorylation of TSC2, which increases its GAP activity toward Rheb and suppresses mTORC1 activity.184 Additionally, AMPK directly phosphorylates Raptor, further reducing mTORC1 activity under low energy conditions.148
In addition, mTORC1 promotes cellular proliferation by shifting glucose metabolism from OXPHOS to glycolysis, a phenomenon known as the Warburg effect.185 mTORC1 signaling promotes the Warburg effect by upregulating and activating key glycolytic enzymes, including PKM2, HK2, and lactate dehydrogenase A (LDHA). This upregulation enhances glycolytic flux, supplying energy and essential building blocks for cell growth and division.185,186
Under hypoxic stress or oxygen deprivation, LDH catalyzes the conversion of pyruvate to lactate via NADH, potentially increasing lactic acid levels and leading to lactic acidosis. Lactic acidosis can promote oncogenesis by altering the tumor microenvironment (TME). HIF-1α is a well-known regulator that enhances the expression of glucose transporters and glycolytic enzymes and,186,187 facilitates glucose entry into cells and catabolism, respectively.185,186,187
Furthermore, mTORC1 can enhance the translation of HIF-1α. The activation of SREBP by mTORC1 also increases flux through the pentose phosphate pathway, leading to the production of NADPH and other intermediate metabolites necessary for cellular proliferation and growth.188
SIRT protein family and energy metabolism
Sirtuins are enzymes that remove acetyl groups from histones and depend on NAD for their function. They play a vital role in regulating key signaling pathways in both prokaryotes and eukaryotes and are involved in various biological processes.189 By modulating fat and glucose metabolism in response to energy fluctuations, sirtuins serve as essential regulators of the complex network responsible for maintaining energy balance.190 In mammals, the sirtuin family comprises seven proteins (SIRT1–SIRT7), each with distinct tissue specificity, subcellular localization, enzymatic activity, and target genes.190 Here, we discuss the specific roles of sirtuin family members in regulating cellular energy metabolism.
Cellular NAD availability, which regulates SIRT1, is influenced by various environmental signals.191 For example, SIRT1 activity is stimulated under conditions that elevate cellular NAD levels due to decreased energy status, such as fasting, caloric restriction, and physical exercise.192,193,194,195 Conversely, SIRT1 activity is reduced in high-energy states, which lower cellular NAD levels, such as during high-fat diet consumption or acute inflammatory responses.196,197,198 Additionally, modifications in NAD synthesis and breakdown can affect cellular NAD levels and, consequently, SIRT1 activity. SIRT1 is crucial for regulating liver metabolism, controlling fat release, promoting the conversion of white adipose tissue into brown fat, regulating insulin secretion, sensing nutrient availability by the hypothalamus, influencing obesity-related inflammatory responses in macrophages, and modulating circadian clock function in metabolic tissues.199 An intricate regulatory network that is responsive to nutritional, hormonal, and environmental signals operates at multiple levels to govern SIRT1 expression and activity and modulate cellular NAD levels.200,201 This network is vital for maintaining appropriate SIRT1 levels in response to diverse environmental stimuli.
SIRT2, the primary sirtuin localized in the cytoplasm, shows decreased expression during the transition from mitochondrial OXPHOS to glycolysis when cells transform into cancer cells. This reduction leads to increased acetylation and activation of key glycolytic enzymes.202 Hamaidi et al. demonstrated that SIRT2 interacts with eight glycolytic enzymes: HK1, phosphofructokinase (PFK), ALDOA, GAPDH, PGK1, ENO1, PKM, and LDH.203 Cha et al. found that SIRT2, rather than SIRT1, regulates the acetylation levels and activities of glycolytic enzymes (ALDOA, PGK1, ENO1, and GAPDH), highlighting the crucial role of SIRT2 in the glycolytic pathway. Moreover, GCK facilitates glucose consumption through glycolysis by converting glucose to G6P.204 SIRT2 activates GCK by deacetylating GKRP, which regulates GCK expression. This deacetylation causes GKRP to dissociate from GCK, thereby promoting hepatic glucose uptake.204
Numerous studies have shown that SIRT3 is expressed predominantly in mitochondria-rich tissues such as the liver, muscle, heart, brain, and kidney.205 SIRT3 functions as a deacetylase, regulating mitochondrial acetylation levels, which are closely linked to metabolic processes such as OXPHOS, FAO, and the TCA cycle across various organs. The enzymatic core of SIRT3 includes an NAD binding domain, a zinc-binding motif, and substrate-binding sites. SIRT3 activity is directly regulated by its metabolic cofactor (NAD+) and the resulting byproduct, nicotinamide. NAD+ facilitates the deacetylation process of SIRT3,206 whereas nicotinamide inhibits it by promoting the reverse reaction through binding to the reaction product.207 SIRT3 expression is significantly enhanced by caloric restriction, a critical factor in its regulation. The essential role of SIRT3 during fasting was confirmed by the identification of multiple deacetylation sites in 3-hydroxy-3-methylglutaryl CoA synthase 2, an enzyme regulating ketone body production, crucial energy sources for the brain when blood glucose levels are low.208 Additionally, SIRT3 regulates metabolism by facilitating the deacetylation and activation of isocitrate dehydrogenase 2 (IDH2), a key enzyme in the TCA cycle. SIRT3 also enhances the deacetylation of components within the OXPHOS complexes I53, II54, and III55, which are involved in the final stage of mitochondrial aerobic respiration.206 SIRT3 further plays a role in the deacetylation and activation of GDH within the TCA cycle, although the physiological significance of this function remains unclear.209
SIRT4 modulates the efficiency of OXPHOS by regulating ANT2 and its overexpression is associated with elevated ATP levels, while its deficiency is linked to mitochondrial uncoupling, resulting in increased oxygen consumption.210 This effect may be mediated by retrograde signaling from the mitochondria to the nucleus, which regulates ATP levels via the ANT2/AMPK/PGC-1α (peroxisome proliferator-activated receptor-γ coactivator-1α) signaling pathway in cases of SIRT4 deficiency.210 The potential involvement of other SIRT proteins in energy metabolism has not been extensively explored and warrants further investigation. Given the significant impact of sirtuins on energy metabolism, the development of sirtuin modulators is considered a promising avenue for drug discovery to address unmet needs in both common and rare diseases.211
Electrolytes and energy metabolism
Electrolytes such as potassium (K+), calcium (Ca2+), and magnesium (Mg2+) play crucial roles in modulating various metabolic pathways and enzyme activities, thereby regulating cellular energy metabolism. Among these ions, Mg2+ serves as an essential cofactor for numerous enzymes, including those pivotal in glycolysis and the TCA cycle, such as HK2, PFK, and PK.212 Disruption of Mg2+ homeostasis within mitochondria not only impacts the morphology and dynamics of mitochondria but also disturbs ATP synthesis and energy metabolism. K+ participates in diverse cellular metabolic processes, such as protein synthesis, carbohydrate metabolism, and the maintenance of enzyme activity.213 Adequate K+ concentration is indispensable for amino acid transport into cells during protein synthesis. In carbohydrate metabolism, both glycogen synthesis and glucose oxidation necessitate the involvement of K+. Furthermore, K+ activates ATPase in the TCA cycle and provides energy for the Na+-K+ pump to maintain normal cellular membrane function. The accumulation of Ca2+ within mitochondria serves not only as a buffering system but also as a signaling pathway regulating energy metabolism.214 Under physiological conditions, increased Ca2+ levels within mitochondria activate OXPHOS, stimulating ATP synthesis, inducing depolarization of the mitochondrial membrane, enhancing cellular respiration, activating Ca2+-dependent mitochondrial enzymes, and subsequently facilitating energy accumulation within mitochondria.
Energy metabolism and cell death
Energy metabolism is closely related to cell death in a way in which energy and energy mutually influence each other. In the execution process of apoptosis, the activation of caspases triggers the cleavage of nuclear DNA and cytoplasmic shrinkage, requiring significant energy support. Apoptotic cells typically upregulate the glycolytic pathway to provide additional energy, and mitochondria also participate in regulating apoptosis. Additionally, the functionality and activity of glucose transporter (GLUT) proteins can influence the cell’s apoptotic process.215 There is a close connection between lipid metabolism and ferroptosis. Intracellular iron overload can lead to increased oxidative stress reactions, resulting in the accumulation of lipid peroxides, which in turn impair mitochondrial function and cause abnormal cell energy metabolism. As the primary source of ROS, mitochondria can stimulate cells to undergo ferroptosis. Intermediate metabolites in the TCA cycle, such as alpha-ketoglutarate, oxaloacetate, succinate, and malate, can effectively replace glutamine and induce ferroptosis.216 On the other hand, mitochondrial metabolic pathways such as fatty acid beta-oxidation can reduce lipid peroxidation, thereby inhibiting ferroptosis.217 The glycolytic pathway affects ferroptosis by regulating the redox reactions of cells. Aberrant expression of G-6-P dehydrogenase may induce ferroptosis by disrupting NADPH and GSH metabolism.218 L Lactic acid, a product of glycolysis, can upregulate the expression of the ferroptosis-related proteins GPX4 and FSP1 and increase the levels of NADH, NADPH, and GSH, thereby inhibiting ferroptosis.219,220 Many regulatory molecules of energy metabolism also affect cell death. For instance, the activation of AMPK can inhibit ferroptosis,221 whereas the interaction between AKT and p53 can influence the execution of apoptosis.222 These interconnected molecules and pathways collectively form a complex regulatory network between cell death and energy metabolism, revealing the intricacy and diversity of cellular activities.
Role of the gut‒liver‒brain axis in energy metabolism
The intricate multi-directional connections formed between the intestines, liver, and brain constitute the gut‒liver‒brain axis. This axis regulates the body’s complex energy metabolism network through the exchange of hormones, cytokines, and nutritional metabolites. Initially proposed for regulating hepatic glycogen metabolism and maintaining energy balance, the gut‒liver‒brain axis plays critical roles in metabolic regulation. When blood glucose levels rise, the hypothalamus activates the parasympathetic nervous system, stimulating hepatic glycogen synthesis, and promoting insulin release from the pancreas to lower blood glucose levels. Conversely, when blood glucose levels decrease, the hypothalamus triggers the sympathetic nervous system, facilitating hepatic glycogen breakdown to maintain stable blood glucose levels. Moreover, the hypothalamus influences feeding and glucose metabolism regulation through appetite suppression and promotion signals.223 In the central nervous system, leptin enhances triglyceride outflow via the vagus nerve, reducing hepatic fat synthesis and thereby inhibiting hepatic fat deposition.224
Moreover, the liver and intestines can secrete metabolites that mutually influence each other and modulate brain signaling. For instance, the intestinal secretion of glucagon-like peptide 1 (GLP-1) influences the hypothalamus via the gut-brain axis, inducing insulin secretion, regulating appetite, and energy metabolism.225 Additionally, the gut microbiota plays crucial roles in the physiological functions of the gut‒liver axis and the gut‒brain axis. The gut microbiota produces short-chain fatty acids that enter the liver through the bloodstream, serving as an energy source for liver cells.226 These short-chain fatty acids can activate vagal afferent neurons, inhibiting food intake.227 Bile acids, synthesized in the liver through the cholesterol metabolism pathway, act as ligands for the transmembrane G protein-coupled receptor 5 (TGR5) and increase intestinal GLP-1 secretion. Bile acids not only regulate central metabolism and immune balance in the central nervous system but also modulate food intake through direct or indirect actions on the brain via TG5.228 Bile acids secreted by the liver promote FGF15 production by activating the FXR hormone receptor. FGF15 controls bile acid synthesis and hepatic lipid metabolism, impacting gluconeogenesis by inhibiting the CREB-PGC-1α pathway.229 During fasting, the liver perceives glycogen deficiency and transmits signals for fat breakdown through the hepatic vagus nerve, converting the energy source from carbohydrates to triglycerides to maintain energy balance.230 The complex interactions of the gut‒liver‒brain axis pose challenges for research but also offer potential targets for treating metabolic disorders.
Synergy of signaling pathways regulating energy metabolism across physiological states
The interconnected effects of signaling pathways can sense the energy status and regulate the storage and consumption of energy to ensure a balance between energy supply and demand, which is highly important for nearly all physiological states, including cell growth and differentiation, maintenance of biological rhythms, adaptation to environmental changes, and responses to multiple stresses.231 Under basal metabolic conditions, such as during rest, energy metabolism primarily supports essential functions through glucose oxidation in the brain and red blood cells, alongside FAO in muscle tissues.232,233 In exercise states, energy metabolism shifts dynamically: during high-intensity or short-duration exercise, glucose metabolism dominates as glycogen stores are rapidly utilized,233,234 while prolonged or lower-intensity activity increasingly relies on FAO to conserve glycogen.235 As exercise commences, the cellular AMP-to-ATP ratio increases, leading to the allosteric activation of AMPK.236 Once activated, AMPK stimulates FAO and glycolysis, which are essential for energy generation.236 AMPK also activates SIRT1, which then deacetylates and activates PGC-1α, increasing mitochondrial biosynthesis levels and enhancing energy production.195,237 Concurrently, AMPK activation curbs the activity of mTORC1, thereby inhibiting energy-consuming processes like gluconeogenesis, lipid synthesis, and protein synthesis.238 This strategic suppression aids in the reallocation of resources, ensuring that the energy balance is maintained during periods of scarcity. This coordinated regulation by the AMPK, SIRT, and mTOR pathways ensures energy balance and maintains cellular and systemic energy equilibrium during physical exertion.
In cold environments, energy metabolism undergoes distinct changes to maintain body temperature.239 The body increases thermogenesis, primarily through non-shivering thermogenesis in brown adipose tissue, which involves increased FAO and uncoupling of OXPHOS to generate heat rather than ATP.240 Within non-shivering thermogenesis, the β3-adrenergic signaling pathway plays a pivotal role.241 When exposed to cold, the sympathetic nervous system is activated, leading to the release of norepinephrine, which binds to β3-adrenergic receptors (β3-ARs) on the surface of brown adipocytes.241 This binding activates adenylate cyclase, increasing the production of cyclic AMP (cAMP), leading to the upregulation of thermogenic genes such as uncoupling protein 1 (UCP1).242 UCP1 facilitates proton leakage into the mitochondrial matrix, bypassing ATP synthesis and releasing energy as heat, which is essential for thermoregulation under cold conditions.243 Furthermore, energy-boosting pathways, including the AMPK and SIRT pathways, enhance FAO and glycolysis, increasing heat generation beyond ATP production under the influence of UCP1. Shivering thermogenesis, driven by muscle contractions, also amplifies glucose and lipid metabolism to meet the heightened energy demands of muscle activity.244
Under conditions of starvation or fasting, the body adeptly shifts its energy reliance towards stored fat, with increased FAO and ketogenesis providing energy to the brain and other tissues, while gluconeogenesis from amino acids sustains blood glucose levels.245 A coordinated series of signaling pathways are activated to regulate energy metabolism and preserve energy balance for vital physiological functions.245 Initially, during fasting, signaling pathways mediated by mTOR, insulin, and leptin are suppressed.245,246 This suppression reduces anabolic effects on glycogen, fat, and protein synthesis, while simultaneously promoting gluconeogenesis and lipolysis to furnish the body with energy. The diminished leptin levels invigorate the hypothalamus, subsequently increasing appetite and stimulating food intake behaviors.247 Furthermore, the hypothalamus releases corticotropin-releasing hormone, which prompts the pituitary gland to secrete adrenocorticotropic hormone. This sequence culminates in increased cortisol production by the adrenal cortex. Cortisol, in turn, stimulates gluconeogenesis and lipolysis, bolstering the body’s energy supply.248 Similar to the state during exercise, fasting increases a rise in the cellular AMP-to-ATP ratio, thereby activating AMPK signaling.249 This activation promotes catabolic processes that generate energy and, concurrently, inhibits mTORC1, halting energy-consuming processes such as gluconeogenesis, lipid synthesis, and protein synthesis. Additionally, in response to nutrient deprivation, AMPK phosphorylates the Unc-51-like kinase 1 (ULK1), initiating autophagy—a process that degrades and recycles cellular components to sustain energy balance.167
In summary, the interactive effects of signaling pathways are pivotal in orchestrating the complex dance of energy metabolism within cells, which allows the body to adapt swiftly to environmental changes, such as fluctuations in nutrient availability, shifts in oxygen levels, and temperature variations. Moreover, the signaling pathways also contribute to the intricate timing of biological rhythms, such as circadian rhythms, which regulate sleep patterns, hormone release, and other physiological processes.250,251 Additionally, the intricate balance maintained by these signaling pathways is vital for the prevention and treatment of a myriad of diseases. Disorders of energy metabolism, such as obesity and diabetes, are often linked to the dysregulation of these pathways. Understanding how these pathways interact can lead to the development of targeted therapies that can restore metabolic balance and improve patient outcomes.
Energy metabolism disorders in the development of diseases
Energy metabolism in neurodegenerative diseases
Neurodegenerative diseases such as Alzheimer’s disease (AD) and Parkinson’s disease (PD) are frequently linked to abnormal changes in energy metabolism and oxidative damage during their progression. These alterations, including impaired glucose uptake, may drive the onset and progression of these diseases.252,253,254,255 Disruptions in energy metabolism can result in insufficient cellular energy, oxidative stress, and cellular injury, leading to neuronal malnutrition, structural changes, and functional loss. Before the diagnosis of AD, there is generally a decrease in glucose uptake, but at this stage, brain oxygen, lactate, and ketone metabolism remain normal. As the disease progresses, reduced cerebral glucose metabolism is caused by a variety of factors, including decreased neuronal glucose uptake, impaired aerobic glycolysis and the tricarboxylic acid cycle, disrupted axonal transport function, and glial cell failure to supply energy to neurons. Additionally, neuroinflammation can promote glial cells to compete for glucose, further exacerbating neuronal glucose hypometabolism. This ultimately leads to synaptic loss and neuronal death.
Energy metabolism in AD
The high energy demand of the brain makes it particularly vulnerable to fluctuations in energy supply and mitochondrial function. In AD, a progressive neurodegenerative disorder, disruptions in energy metabolism contribute to the gradual degeneration and apoptosis of neuronal cells in the brain, ultimately causing severe cognitive impairment. Glucose oxidation serves as the primary energy foundation for the brain, with ketones, glycogen, and amino acids acting as supplementary sources. The metabolic processes of these substances are interlinked through the mitochondrial TCA cycle. The pathological progression of AD primarily results from mitochondrial dysfunction, characterized by reduced glucose uptake by neurons, alterations in glucose receptors, and changes in the metabolic phenotype of astrocytes. These dysfunctions are collectively regulated by metabolites, transport proteins, receptors, and enzymes. Such disruptions in energy metabolism facilitate the accumulation of brain amyloid-beta (Aβ) and aggregation of tau, leading to oxidative stress, inflammation, impaired autophagy, and various disease-causing cascades.113
Glycolysis
The brain accounts for approximately 25% of total body glucose consumption, primarily generating energy through OXPHOS. Research shows a significant reduction in brain glucose metabolism in patients with AD, shifting energy production from efficient aerobic oxidation to a less efficient glycolytic pathway. This shift decreases ATP generation efficiency, likely leading to an inadequate energy supply for neurons and accelerating pathological progression.256,257 Neurons may depend on lactate from neighboring astrocytes via the astrocyte–neuron lactate shuttle to meet their energy needs. Recent research indicates a significant inhibition of lactate production in astrocytes in AD, leading to impaired neuronal glucose metabolism. Inhibiting IDO1 activity can promote glycolysis and lactate production in astrocytes, thereby improving the energy metabolism state of neurons.258 Reduced glucose metabolism often precedes clinical AD symptoms, initially affecting the parietal-temporal lobes and posterior cingulate cortex before spreading to other regions, such as the frontal lobes.259,260 In the frontal lobe, glycolytic activity significantly increases in the inner, outer, and prefrontal areas but remains relatively low in the cerebellum and medial temporal lobe.261 Typically, the locus coeruleus activates astrocytes by releasing norepinephrine, which induces calcium influx, increases cAMP levels, and enhances aerobic glycolysis in astrocytes.262,263 However, as AD progresses, degeneration of the locus coeruleus shifts the energy source of the brain from glucose metabolism to a greater dependence on ketone bodies.264 Ketone bodies help compensate for ATP deficiencies and stabilize synaptic function by promoting mitochondrial biogenesis.265,266 Consequently, ketogenic diets may mitigate AD symptoms267,268 and have been shown to be effective in treating epilepsy.269
Declines in glucose metabolism in neuronal cells are often linked to changes in the expression of transport proteins and enzymes. Neurons rely on GLUTs to transport glucose from the bloodstream to critical brain regions, such as the hippocampus, as they cannot produce or store glucose. In AD patients, GLUT1 expression is often reduced in the hippocampus and frontal cortex,270,271,272 which may limit glucose entry and affect cellular energy supply. Research has also shown decreased GLUT3 expression in the hippocampus and parietal cortex, potentially further impairing glucose metabolism in AD.272,273 Conversely, GLUT4 expression increases in the hippocampus of AD patients, possibly as a compensatory mechanism to address impaired glucose uptake.274,275 Notably, β-amyloid peptides interact with these GLUTs, promoting their internalization and reducing glucose transport efficiency,276,277 which is considered a key factor in metabolic dysregulation. Additionally, key glycolytic enzymes such as aldolase, triosephosphate isomerase, GAPDH, phosphoglycerate mutase 1, and enolase become functionally impaired due to oxidative stress in AD, reducing glucose metabolism efficiency.272
A hallmark of AD is the abnormal accumulation of Aβ peptides and the associated inflammatory responses in brain tissue. Under physiological conditions, microglia are essential for maintaining neuronal energy, with the expression of Trem2 playing a crucial role in microglia-mediated synaptic refinement.278 However, in disease progression, microglia play a critical role in this process by exhibiting metabolic changes distinct from those in neuronal cells. Stimulation by LPS and Aβ induces microglial polarization towards the M1 phenotype, which involves significant reprogramming of glucose metabolism from OXPHOS to glycolysis, which is dependent on the mTOR-HIF-1α pathway.279 Activated M1 microglia demonstrate show enhanced glucose uptake and increased activities of HK, G6P dehydrogenase (G6PDH), PFK1, and LDH, leading to elevated lactate release.280 This increased glucose uptake is due to the upregulation of GLUTs, with GLUT1, GLUT4, and GLUT6 being critical for M1 microglia activation.281,282 Furthermore, the glycolytic pathway leads to G6P accumulation, which can enhance ROS production via the pentose phosphate pathway.283 This metabolic shift mirrors the “Warburg effect” observed in tumor cells.284 Under glucose scarcity, microglia utilize lactate as an alternative energy source to meet their metabolic needs.285 In contrast, M2 microglia maintain a functional TCA cycle and higher OXPHOS levels, supporting their phagocytic activity and potentially mitigating AD. In the future, conducting more metabolism-related polarization studies on microglia to delineate their roles and mechanisms may be a crucial research direction.286 Furthermore, due to the complexity of microglia in immune metabolism regulation, further research on potential therapeutic drugs and treatment methods involving metabolic remodeling regulation is warranted. Given the close relationship between microglial cells and neurons, targeting different cell types simultaneously for treatment will also be a future research focus.
In AD, multiple signaling molecules regulate glycolysis, with inflammation and oxidative stress playing significant roles. The inflammatory environment in AD triggers the release of cytokines such as interleukin-1β (IL-1β) and tumor necrosis factor-α (TNF-α), which can reduce GLUT expression in brain cells.287,288 Oxidative stress damages cell membranes and disrupts proteins, including GLUTs, crucial for energy metabolism.289 Throughout AD progression, a shift from OXPHOS to glycolysis occurs, driven by the mTOR-HIF-1α signaling pathway. Activation of this pathway induces metabolic changes that may impair normal microglial function.290,291 Furthermore, genetic risk factors for AD, such as the loss of TREM2, reduce the expression of GLUTs, glycolytic enzymes, and HIF-1α. However, interferon-γ (IFN-γ) can reverse glycolytic defects and inflammatory dysfunctions in microglia, potentially alleviating AD-related symptoms.292 The activation of STAT3 and STAT6 is also crucial for energy metabolism and microglial polarization, which are essential for maintaining cellular homeostasis and function.293,294
AMPK regulates GLUT3 translocation to the membrane; however, Aβ inhibits AMPK activity, affecting glucose transport.295,296 Additionally, studies have shown a correlation between PGC-1α expression levels and amyloid protein generation.297 In vitro experiments have found that oligomeric Aβ increases PGC-1α and SIRT1 levels, disrupting their interaction, which is crucial for maintaining metabolic balance. Insulin is vital in the hippocampus for promoting GLUT4 translocation to the plasma membrane, enhancing glycolysis, and improving spatial memory in rats.298 Insulin stimulates glucose uptake and glycogen accumulation in astrocytes by activating GLUT4.299 The pathogenesis of AD is often linked to insulin resistance, with PI3K playing a central role in insulin signaling, including GLUT translocation, glycogen synthesis, lipid and protein synthesis, anti-lipolytic effects, and hepatic gluconeogenesis.300 PI3K exerts effects through the Akt/PKB and protein kinase C cascades,300 with Akt activation inhibiting GSK3 via mTOR and downstream elements, promoting glycogen synthesis.301 ApoE2 protects against AD, whereas ApoE4 is associated with impaired glucose metabolism.302 This difference may be due to insufficient expression of insulin signaling-related genes, such as insulin receptor substrate, peroxisome proliferator-activated receptor γ (PPAR-γ), and insulin-degrading enzyme.303 Under normal conditions, SIRT3 levels are high in the brain but decrease in the frontal cortex of AD patients. SIRT3, located in the inner mitochondrial membrane, plays a crucial role in maintaining mitochondrial function.304 As a deacetylase, SIRT3 regulates p53-induced mitochondrial damage, preventing ROS accumulation and protecting neurons. Dysregulation of SIRT3 may serve as a biomarker for mitochondrial damage in AD, suggesting that modulating SIRT3 activity could be a novel therapeutic strategy.305 However, the efficacy of the aforementioned experimental animal model studies in transitioning to clinical trials still needs improvement, emphasizing the complexity of molecular regulatory signals. Research on the regulatory mechanisms of AD remains an enduring focal point.
Aerobic oxidation
In AD, cerebral features extend beyond impaired glucose metabolism to include microglia-driven inflammatory responses, mitochondrial dysfunction and heightened oxidative stress.306,307,308 Compared with controls, AD patients show elevated lactate and pyruvate levels and decreased succinate, fumarate, and glutamine levels, suggesting disrupted mitochondrial glucose metabolism.309 Pyruvate, a glycolysis product, typically enters mitochondria to participate in the TCA cycle. As a critical substrate for the pyruvate dehydrogenase (PDH) complex, pyruvate links glycolysis to the TCA cycle; however, PDH flux is reduced in AD.306 The activity of mitochondrial respiratory chain complexes I-IV is significantly diminished in AD, likely due to Aβ-mediated inhibition. Mitochondrial energy metabolism disorders, such as decreased TCA cycle enzyme activity, impaired respiratory chain function, reduced ATP production, and increased free radical and ROS levels, support the “mitochondrial cascade hypothesis” in AD pathogenesis.310
Pro-inflammatory activation further disrupts the TCA cycle, leading to substrate accumulation (e.g., citrate, succinate, and aconitate). This exacerbates intracellular inflammation, inhibits OXPHOS, reduces ATP production efficiency, and increases saturated fatty acid synthesis. The accumulation of fatty acids can affect membrane phospholipids, increasing cell susceptibility to lipid peroxidation and inflammatory signaling, thus enhancing neuroinflammation.311,312 NADPH is crucial for energy supply and redox balance. Mitochondrial NADPH production depends on nicotinamide nucleotide transhydrogenase (NNT), IDH2, and malic enzymes, with NNT contributing up to 50% of total NADPH output.313 In AD mouse models, NNT expression is reduced,314 and the activities of key metabolic enzymes, such as aconitase, α-KG dehydrogenase, and malic dehydrogenase (MDH), are inhibited,315,316 further impairing energy metabolism and antioxidant capacity. Under pathological conditions, NNT no longer catalyzes NADPH production but promotes NADH generation in reverse mode, leading to a reduced clearance of hydrogen ions by the redox system.317 However, further in-depth research is needed to understand the specific reverse mode, particularly its role in the brain.
Oxidative stress is crucial in neurodegenerative diseases, especially in the early stages of AD, where it is closely linked to mitochondrial dysfunction.318 Dysfunctional mitochondria produce less ATP and excessive ROS, contributing to the oxidative imbalance observed in AD. Enzymes essential for glycolysis, the TCA cycle, and the ETC are oxidized in the AD brain, leading to decreased activity. This inhibition disrupts glucose metabolism, reduces ATP synthesis, impairs neuronal function, causes synaptic loss, and promotes neurodegeneration.319 Variations in TREM2 are associated with several neurodegenerative diseases, including AD. Cells lacking TREM2 show impaired energy metabolism reduced mitochondrial quality, and abnormal organelle structure.278 Tau protein-activated microglia can cause metabolic abnormalities, such as abnormal succinate accumulation and TCA cycle disruption, which exacerbate neuroinflammation.320 The SIRT family of proteins regulates processes related to AD pathogenesis, including tau protein aggregation, mitochondrial dysfunction, oxidative stress, and neuroinflammation.321 Additionally, Aβ peptides and tau proteins can activate the PI3K/Akt/mTOR signaling pathway in microglia, which is crucial for regulating energy metabolism and ATP production.322,323 In light of these findings, current research is also exploring the therapeutic potential of antioxidants in neurodegenerative diseases. However, the efficacy of individual antioxidants is quite limited, suggesting that antioxidants are more suitable as adjunctive treatments, further prompting us to consider whether oxidative stress imbalance is a cause or a consequence of neurodegenerative diseases.
FAO
In AD, lipid metabolism dysregulation is closely linked to disease onset and progression. Long-chain fatty acids in the brain are synthesized de novo from acetyl-CoA.324 Fatty acid synthase (FASN) levels are elevated in AD patients, particularly around amyloid plaques.325 ACC activity is increased in the brains of AD mice.326 Elevated free fatty acids in the cerebrospinal fluid of AD patients can be neurotoxic, causing mitochondrial uncoupling and bioenergetic dysfunction.327 β-Oxidation of fatty acids is essential for their entry into the TCA cycle for energy production. This pathway helps meet the increased energy demands of the AD brain.328 Therefore, the timely degradation of free fatty acids, especially peroxidized fatty acids, is crucial for maintaining brain health because of their potential negative impact on cellular function.327 In patients with AD and elderly individuals, β-oxidation is increased to address increased energy demands.329 Increased FAO reduces triglyceride accumulation and lipid-induced inflammatory responses.330,331 There is controversy regarding the role of fatty acids as fuel substrates in meeting the brain’s energy demands.328 This discrepancy may stem from mitochondrial FAO and its association with oxidative stress and ROS generation compared to the oxidative breakdown of glucose. Neurons are particularly susceptible to the oxidative-redox environment due to their limited antioxidant capacity. In this scenario, transferring fatty acids from neurons to astrocytes, thereby utilizing the capacity of the cells for fatty acid degradation, may be an effective therapeutic approach.332 In this process, APOE4 exerts inhibitory regulatory effects, reducing transport efficiency, whereas APOE3 acts as a facilitator.333 Apart from ApoE, other AD risk factor genes like ABCA1, ABCA7, and PICALM are involved in fatty acid clearance.334 Further research on the regulatory molecules involved in fatty acid clearance in AD, specifically inhibitory factors, may mitigate the pathological progression of AD by alleviating oxidative stress in neurons.
FAO is also linked to insulin levels. In AD patients, cerebrospinal fluid insulin levels are significantly lower than plasma insulin levels. This insulin deficiency may cause substantial lipolysis in adipose tissue, releasing large amounts of fatty acids that, if not efficiently utilized, could impair cellular functions. In mouse models, neuron-specific insulin receptor knockout resulted in increased body weight, insulin resistance, and impaired glucose tolerance.335 Furthermore, pro-inflammatory factors such as LPS or IFN-γ may suppress genes involved in FAO.336 The PI3K/Akt signaling pathway regulates carbohydrate and lipid metabolism, affecting glucose uptake and utilization. PPAR-γ agonists have been shown to improve lipid and glucose metabolism and may reduce the pathological burden of Aβ plaques. These findings highlight the importance of maintaining proper insulin levels and regulating fatty acid metabolism in the prevention and treatment of AD.
AD is a complex pathological and physiological cascade disorder primarily caused by disrupted glucose metabolism. While research on neurodegenerative diseases has advanced significantly, with increasing insights from cell lines and murine animal models highlighting the bidirectional link between energy metabolism and brain inflammatory responses, the specificity of human specimens is limited to postmortem brain tissues. This limitation contributes to discrepancies often observed between clinical trial outcomes and animal models. The specific metabolic processes, such as the role of glucose and lipid metabolism disorders in driving neurodegenerative disease progression, and whether neuroinflammation is the cause of energy metabolism disorders, or merely a consequence, remain subjects of debate. Currently, the focus tends towards oxidative stress disturbances, inflammation, and other mechanisms as common pathological factors. Research on the multifactorial nature of the disease has made some progress over the past few decades, but the true etiology remains intricate, and involves genetic and lifestyle factors, necessitating further exploration.
Energy metabolism in PD
PD is a chronic, progressive neurodegenerative disorder primarily characterized by the loss of dopaminergic (DA) neurons in the substantia nigra (SN), resulting in reduced DA activity in the nigrostriatal pathway. Aging is a major risk factor for the development of PD and leads to significant alterations in energy metabolism,337,338 particularly manifested as mitochondrial dysfunction and impairments in the TCA cycle and ETC.339
Glycolysis
Disruption of glucose metabolism is closely linked to the progression of PD, potentially because of the negative impact of impaired glucose metabolism on the DA system.340 Although aerobic glycolysis is less efficient at producing ATP than OXPHOS is, rapid ATP production is critical for neuronal synapse formation.341 However, in the pathological state of PD, neurons exhibit significantly reduced glucose utilization efficiency.342 Studies suggest that activating glycolysis to supplement energy can provide neurons with the necessary ATP, thereby alleviating the symptoms of PD.343
Similar to observations in AD, the reprogramming of microglial energy metabolism is implicated in the pathogenesis of PD.344 The neuroinflammation induced by M1-type microglia has been closely linked to the progression of PD.345 Inhibiting specific metabolic pathways in M1-type microglia or promoting their shift to the M2 phenotype can significantly reduce neuroinflammation, thereby protecting DA neurons from damage. In M1-type microglia, increased pentose phosphate pathway activity and aerobic glycolysis are associated with excessive production of ROS. The accumulation of ROS can activate the nuclear factor κB (NF-κB) signaling pathway, a critical inflammatory signaling pathway that promotes neuroinflammation and may facilitate the onset and progression of PD.346,347,348 Nr1h4 inhibits the activation of NF-κB, thereby suppressing neuroinflammation in PD.349 In PD animal models and brain tissues of PD patients, the activation of microglia along with the infiltration of inflammatory factors such as TNFα, IL-6, Nos2, and COX2 can be observed, attracting T cells to cross the blood-brain barrier.350,351 This phenomenon may be associated with the activation of the JAK/STAT pathway,352 providing in vivo evidence for the involvement of inflammation and immune responses in the progression of PD.
In PD, the expression of GLUTs in neurons significantly changes. Although there are differing viewpoints within the scientific community, the prevailing research suggests that GLUT expression levels are downregulated.353 Thus, increasing GLUT expression levels may help mitigate the progression of PD.354 Critical glycolytic enzymes, such as PGK1, HK2, and PFK, exhibit insufficient activity in the pathogenesis of PD.355 Notably, the overexpression of HK2 has been shown to alleviate PD symptoms by enhancing glycolysis.356 Furthermore, pharmacological agents such as meclizine, which activates the glycolytic enzyme PFK, can increase glycolytic rates, suggesting a potential therapeutic strategy against PD.357
PARK7, a protein closely associated with PD, harbors mutations in familial PD cases that may reduce protection against damage induced by 1,3-BPG. 1,3-BPG can form reactive intermediates and react with amino groups, but PARK7 disrupts these intermediates, preventing improper modifications of proteins and metabolites.358 The Wnt/β-catenin signaling pathway is a critical factor in the development of many neurodegenerative diseases.359 In PD, downregulation of this pathway is a hallmark of the disease.359 Impaired function of the Wnt/β-catenin pathway can lead to restricted expression of its target genes, such as pyruvate dehydrogenase kinase 1 (PDK1), MCT1, c-Myc, cyclin D1, and LDHA, which play important roles in enhancing glucose metabolism.360 Additionally, studies have shown that the expression of PGC-1α-responsive genes is downregulated in the brains of PD patients, possibly due to hypermethylation of PGC-1α transcriptional elements, leading to bioenergetic dysregulation.361 Disruption of glycolytic metabolism is a significant energy metabolic alteration in PD. However, current research on its regulation remains limited. Future studies should integrate metabolomics, spatial transcriptomics, CRISPR screening, and other technologies to identify more potential regulatory factors, thereby offering novel diagnostic and therapeutic targets.
Aerobic oxidation
Current research widely acknowledges that impaired mitochondrial energy production plays a crucial role in promoting the progression of PD. Multiple factors can lead to mitochondrial dysfunction in PD, ultimately affecting ATP synthesis. Genes associated with PD, such as PTEN-induced kinase 1 (PINK1), Parkin, DJ-1, LRRK2, and ATP13A2, are directly or indirectly involved in maintaining mitochondrial integrity and function. Mutations in these genes are linked to familial PD and are closely associated with mitochondrial dysfunction.362,363,364,365 PD-related gene mutations may inactivate PINK1/Parkin, inhibit mitophagy, and result in the accumulation of dysfunctional mitochondria, thereby triggering apoptosis.366 Given the high energy demands of DA neurons and the reliance on OXPHOS for energy, impairments in mitochondrial OXPHOS are particularly critical in the progression of PD.367,368 Sustained mitochondrial OXPHOS increases the risk of oxidative damage, and impaired mitochondria in PD release more ROS. Excessive ROS accumulation damages cellular components, including proteins, lipids, and DNA, exacerbating mitochondrial dysfunction and potentially initiating a cascade of neurodegenerative processes.369,370,371 The abnormal aggregation of α-synuclein (α-Syn) into Lewy bodies is closely associated with DA neuron dysfunction. Mechanistically, α-Syn oligomers interact with mitochondrial respiratory chain complex I, impairing its function, leading to mitochondrial membrane potential depolarization, and promoting the release of proapoptotic factors into the cytoplasm. Once released, these factors can interact with intracellular antiapoptotic and survival-promoting proteins, inducing mitochondria-mediated apoptosis.372,373 Recent clinical studies suggest that the initiation of deformation in DA neurons is mediated by the TAU, rather than the aggregation of α-Syn.374 The pathological similarities observed in these neurodegenerative diseases imply the existence of potential common pathogenic mechanisms that may impact the progression of each disease. In the pathogenesis of α-Syn, HSPB6 inhibits the aggregation of α-Syn through a lipid-dependent mechanism.375 Additionally, environmental toxins can contribute to PD by disrupting the mitochondrial ETC, thereby affecting OXPHOS.376,377 The opening of the mitochondrial permeability transition pore (MPT) can lead to the collapse of the mitochondrial membrane potential, impaired OXPHOS, and eventual cell death.378
In DA neurons, reduced expression of PGC-1α has been observed, suggesting that PGC-1α dysfunction may play a role in the clinical pathogenesis of PD.379 In PGC-1α knockout mouse models, DA neurons exhibit increased sensitivity to the neurotoxin MPTP, potentially due to the lack of PGC-1α-mediated induction of antioxidant responses.380 The sirtuin protein family plays crucial regulatory roles in neurodegenerative diseases. Specifically, SIRT2 is involved primarily in the development of the nervous system, whereas SIRT1 is closely linked to cellular energy metabolism and survival. Moreover, SIRT3 and SIRT4 are central to the regulation of mitochondrial metabolism.381,382 SIRT1 has been shown to activate antioxidant pathways through the deacetylation of NRF2, thereby exerting neuroprotective effects in various PD models.383 In MPP+-induced PD cell models and primary DA neurons, SIRT1 enhances mitochondrial biogenesis through the deacetylation of PGC-1α, providing neuroprotection.384 Activation of AMPK can increase SIRT activity or directly phosphorylate PGC-1α. This is attributed to the elevation in NAD+ levels. Mitochondrial SIRT is also capable of activating the AMPK signaling pathway, increasing autophagic function, and accelerating the clearance of the α-Syn protein.385 SIRT3 can reduce cell death, inhibit the generation of inflammatory cytokines, and eliminate mitochondrial ROS through mitochondrial pathways, thereby exerting neuroprotective effects. Additionally, reduced levels of dynamin-related protein 1 (DRP1) have been observed in the astrocytes of PD patients. A decrease in DRP1 levels alters mitochondrial morphology, indicating that mitochondrial dynamics may be impaired in the astrocytes of PD patients.386 Mutations in the mitochondrial inner membrane protein CHCHD2 result in severe mitochondrial damage and a subsequent metabolic shift towards glycolysis, leading to the destruction of DA neurons.387 Recent studies have further emphasized the role of mitochondrial dysfunction in promoting the pathology of PD, with various regulatory mechanisms being uncovered. However, in-depth exploration of these mechanisms is still crucial for unraveling the pathogenesis of PD.
FAO
Dysregulated lipid metabolism is closely associated with PD.388,389 Intracellular lipid accumulation can lead to mitochondrial dysfunction, which not only reduces the number of mitochondria but also exacerbates lipid accumulation, creating a vicious cycle.390 The abnormal aggregation of α-syn is a hallmark of PD, and the accumulation of lipids, including polyunsaturated fatty acids (PUFAs) and cholesterol, plays a crucial role in this process.391 In PD, hyperactive neurons may produce toxic fatty acids, which are transported to astrocyte lipid droplets via APOE-positive lipid particles for detoxification through mitochondrial β-oxidation.392 However, impaired mitochondrial function in astrocytes reduces their capacity to metabolize fatty acids, leading to neurotoxicity from fatty acids, which could further exacerbate the pathological process of PD. In the brains of PD patients, decreased activity of SIRT6 has been observed, which may be associated with mitochondrial dysfunction.393 Studies indicate that a lack of SIRT6 leads to reduced mitochondrial gene expression, increased production of ROS, and accelerated mitochondrial decay, collectively triggering mitochondrial dysfunction.394 ACSL4 promotes lipid peroxidation, whereas the inhibition of ACSL4 can prevent the death of DA neurons.395 Recent studies have underscored the potential of the Lrrk2 gene as a diagnostic marker.396 However, to date, there are no biomarkers that can predict which individuals are more likely to develop PD. Over the past few years, research has increasingly recognized the critical role of fatty acid metabolism in energy disturbances in PD, with fatty acid-binding proteins such as FAP3 being targeted for therapy and serving as alternative treatment strategies for PD.397
Electrolytes
In the nervous system, ions such as Mg2+ and K+ play pivotal roles in establishing and conducting neuronal membrane potentials. These electrolyte ions maintain ion balance in neurons through ion channels and are pumped on the neuronal cell membrane, supporting normal neuronal function and neural signal transmission. Abnormal potassium ion channels may accelerate pathological α-Syn accumulation, increasing the risk of PD.398 Magnesium ions can inhibit neuroinflammation mediated by neuroglial cells by downregulating pro-inflammatory cytokines and oxidative stress, a phenomenon closely associated with age-related chronic disease development.399
Due to the high clinical, pathological, and genetic heterogeneity of PD, the disease can last for several decades, significantly increasing the difficulty in studying its etiology and treatment. Despite some progress, the exact causes and pathogenic mechanisms of PD remain incompletely understood. Clinical treatments only offer symptomatic relief and not a halt to disease progression. Therefore, there is a growing emphasis on personalized diagnosis and treatment where the prodromal phase of the disease can provide a therapeutic window for early intervention. Subsequent crucial research involves the development of improved non-invasive biomarkers to detect and monitor pathological progression, including pathways related to genetics (such as mitochondrial or synaptic dysfunction). Studying how factors like exercise, diet, and sleep affect energy metabolism in PD patients can lead to effective interventions through lifestyle modifications. Additionally, combining drug screening technologies to explore new applications for existing drugs may provide feasible short-term improvements in PD treatment. The emergence of new technologies like spatial transcriptomics and spatial metabolomics offers hope for identifying more potential metabolites in animal models, although effectively applying these methods to human samples remains a significant challenge.
Energy metabolism in cardiovascular diseases
The heart, which is essential for life as a powerful pump, constantly requires ATP to maintain continuous beating.400,401 Fatty acids are the primary energy source, contributing 60–90% of the heart’s energy, and are transported into cardiomyocytes by CD36 and FATP, where they undergo β-oxidation to produce acetyl-CoA for the TCA cycle and ATP generation.9,402 In cardiovascular diseases, an increase in fatty acids is associated with abnormal lipid metabolism changes, leading to mitochondrial oxidative stress and the accumulation of toxic lipids. This lipid oxidation-reduction imbalance can promote ferroptosis. Glucose also plays a key role, as it enters cells through GLUT1 and GLUT4, with insulin enhancing glucose uptake by promoting GLUT4 translocation to the cell membrane.403 Additionally, ketone bodies, which constitute only 15–20% of the energy supply, are crucial for heart protection because they improve heart failure and hypertrophy and are easily converted to acetyl-CoA for energy.378 Ketone supplementation can mitigate negative heart remodeling and enhance cardiovascular function.404,405 The high mitochondrial content of the heart is pivotal for energy production, but it also generates significant amounts of ROS during OXPHOS.406 Mitochondrial dysfunction contributes to cardiac damage, leading to impaired mitophagy, altered enzyme activity in the ETC, and reduced ATP production.407 In the early and middle stages of cardiovascular diseases such as heart failure, myocardial energy metabolism remains relatively normal, with little or slight increase in fatty acid utilization; however, in the end-stage, myocardial cells transition from predominantly relying on fatty acid metabolism to predominantly relying on glucose metabolism for energy, yet glucose utilization rates are low. Moreover, decreased fatty acid utilization leads to the accumulation of lipid peroxides, resulting in myocardial cell apoptosis. As heart failure progresses to the end stage, the levels of metabolic products related to ketone oxidation and metabolic enzymes in the body usually increase.
In the cardiovascular system, Mg2+ promotes the release of NF-κB through its antioxidant effect by eliminating ROS, impacting downstream inflammatory signaling pathways, and regulating inflammation.408 Inflammation triggered by hypomagnesemia may affect lipid profiles by altering lipid peroxidation, leading to increased triglycerides in lipoproteins, accumulation of apolipoprotein B, and decreased high-density lipoprotein cholesterol, resulting in disrupted lipid metabolism. Recent studies have indicated that potassium regulation can counteract detrimental ion accumulation induced by hypoxia and ischemia (such as Ca2+ and Na+), maintain stable myocardial membrane potentials, and prevent mitochondrial dysfunction, which is crucial.409
Brown adipose tissue (BAT), a crucial thermogenic organ, plays a key role in promoting glucose and lipid metabolism, enhancing energy expenditure, and maintaining cardiac metabolic health. The presence of BAT can reduce the incidence of dyslipidemia and cardiovascular diseases.410 BAT with multilocular lipid droplets is rich in mitochondria and significantly expresses uncoupling protein 1 (UCP1), PGC-1α, and PRDM16.411 Active BAT promotes cardiac metabolism health by burning triglycerides and free fatty acids derived from glucose, thereby preventing lipid metabolism abnormalities, obesity, and insulin resistance.412 BAT increases glucose uptake and utilization and lowers blood glucose levels, thus exerting a positive effect on T2DM and cardiovascular diseases. Through the SIRT1-PGC1α-PPAR-γ pathway, BAT inhibits NF-κB to suppress inflammation.413 Factors secreted by BAT, such as NRG4, contribute to improving atherosclerosis by inhibiting endothelial inflammation and macrophage accumulation in plaques, protecting endothelial vessels from damage.414 Furthermore, BAT and its secreted protein BMP3b can alleviate cardiac ischemia-reperfusion injury.415 However, besides promoting energy expenditure and thermogenesis, BAT also inhibits adipose tissue inflammation and promotes mitochondrial amino acid catabolism, providing protective nutrients for crucial glucose metabolism.416 Beige adipose tissue, which is primarily distributed in epicardial fat tissue, is characterized by both brown and white adipose tissue. Like brown adipocytes, beige adipocytes possess multiple small lipid droplets, high mitochondrial content, and the expression of thermogenic genes such as UCP1 and Pgc1a.417 Under specific environmental, genetic, or pharmacological stimuli, beige adipocytes can develop in white adipose tissue (WAT). Upon stimulus withdrawal, the gene expression profile of beige adipocytes is altered, reverting to WAT characteristics.418 Brown and beige adipose tissues play significant roles in regulating energy balance and providing protection against cardiovascular diseases. Research into their potential roles and mechanisms holds crucial importance for disease treatment and prevention.
Multiple signaling molecules regulate cardiovascular energy metabolism, playing key roles in the transcription of metabolism-related genes, including AMPK, PGC-1α, SIRT, and PPARs. When the energy demand increases, AMPK is activated, increasing CD36 translocation to the membrane and promoting FAO.400 AMPK also induces mitochondrial biogenesis via PGC-1α phosphorylation and SIRT1 activation.419 In mouse models, PGC-1α regulates lipid metabolism by upregulating genes involved in the TCA cycle and mitochondrial FAO.420,421 PPARs facilitate cardiac fatty acid uptake and oxidation by binding FAs in the cytoplasm, transferring them to the nucleus, and interacting with the retinoid X receptor (RXR) to regulate FA metabolism genes.422 In addition to upregulating FA uptake proteins, PPAR also activates genes involved in OXPHOS, thus modulating mitochondrial functions.423 Angiopoietin-like proteins (ANGPTL) influence the clearance of FAs by inhibiting lipoprotein lipase activity, a process regulated by transcription factors such as PPAR and HIF-1α.424 Suppressing the expression of ANGPTL4 can improve abnormal glucose metabolism by enhancing insulin sensitivity; however, the specific regulatory pathways involved in cardiac energy metabolism still require exploration. In the heart, SIRT3 enhances glucose transport by activating GLUT1 and GLUT4 while regulating PFK activity, reducing TP53-induced glycolysis and apoptosis regulator (TIGAR) expression to increase glucose metabolism.425,426 SIRT3 deficiency impairs the mitochondrial membrane potential, induces ROS accumulation, and triggers NLRP3 inflammasome activation, exacerbating cardiac damage.427 Cardiac energy metabolism is further regulated by hormones. Insulin increases glucose uptake in cardiomyocytes by promoting GLUT4 translocation, whereas thyroid hormones increase energy production via mitochondrial OXPHOS.400 Adrenaline responds to increased energy demands under physiological or pathological conditions by activating the oxidation of various energy substrates in the heart.400
Owing to advancements in metabolomics, research on metabolic pathways in cardiovascular diseases is currently extensive, leading to the discovery of more biomarkers. Research on the regeneration of cardiac tissue based on energy metabolism has become feasible. However, translating findings from basic research into clinical applications still poses challenges.
Energy metabolism in ischemic heart disease
Ischemic heart disease, caused by reduced coronary blood flow, profoundly affects cardiac energy metabolism (Fig. 4a). These effects include altered substrate utilization, OXPHOS dysfunction, impaired ATP synthesis, and mitochondrial dysfunction. These metabolic disturbances not only contribute to the onset of the disease but also accelerate its progression, leading to significant impairment of cardiomyocyte function. When blood flow to the heart is restricted, cardiomyocytes shift toward glycolysis to maintain their energy supply. This shift is driven by increased glucose uptake, GLUT translocation, reduced levels of glycolytic intermediates to stimulate glycogen breakdown, and elevated AMP levels, which activate AMPK. These changes increase GLUT4 translocation and PFK activation, increasing fructose 2,6-bisphosphate production.428,429 AMPK activation further promotes glycolysis and increases fatty acid oxidation, although fatty acid oxidation levels remain below normal under low blood flow. Despite this, FAs continue to be a primary energy source. Under conditions of low or absent cardiac blood flow, CD36 expression on the sarcolemma is downregulated,430 reducing or halting fatty acid β-oxidation, and glucose becomes the predominant substrate for ATP production.431 The diminished capacity of the heart to oxidize fatty acids results in lower citrate levels, which in turn enhances glucose uptake and glycolysis.432 Citrate, an allosteric regulator, typically inhibits PFK1 and PFK2, thereby reducing glycolysis.433
Fig. 4
Energy metabolism in ischemic heart disease and ischemia-reperfusion injury. Under physiological conditions, the heart primarily relies on the oxidation of fatty acids entering the TCA cycle for energy supply. In various disease states (a ischemic heart disease and b ischemia-reperfusion injury), changes in cardiac energy metabolism led to increased production of ROS, ultimately affecting mitochondrial function and resulting in decreased ATP production, which impacts the heart’s ability to perform its functions. FAO fatty acid oxidation, PFK phosphofructokinase, BCAA branched-chain amino acids, HK hexokinase, PK pyruvate kinase, ACS acyl-CoA synthetase
Under normal oxygen conditions, glycolysis accounts for approximately 5% of the energy requirements of the heart.434 In contrast, during hypoxia, glycolysis and lactate metabolism predominate.435 Reduced coronary blood flow exacerbates ischemia, where increased glycolysis temporarily supports ATP production and cardiac ion balance.433 However, severe ischemia leads to the accumulation of harmful waste, such as intracellular protons, negating the benefits of glycolysis. The accumulation of long-chain fatty acids within mitochondria impairs PDH activity, reducing glucose oxidation and increasing lactate and proton levels, which worsens myocardial injury.436 This also induces mitochondrial membrane hyperpolarization and increases ROS generation, further damaging cardiac tissue.437 In ischemic heart disease, mitochondrial function is hindered by depleted TCA cycle intermediates and free CoA.438 This dysfunction elevates succinate, a hallmark of ischemia, which is a primary source of ROS during reperfusion.439
Multiple signaling pathways regulate energy metabolism during heart ischemia. During heart ischemia, the AMPK/SIRT1/PGC-1α pathway exhibits abnormalities. Therefore, this pathway may serve as a crucial target for treating cardiovascular diseases. The ALDH2/SIRT3/HIF-1α axis plays a myocardial protective role, reducing 4-HNE levels post myocardial ischemia/reperfusion (I/R), resulting in decreased apoptosis, a reduced myocardial infarct size, and decreased ROS levels. Depletion of ALDH2 eliminates these beneficial effects.440
Energy metabolism during ischemia‒reperfusion injury
Ischemia‒reperfusion injury occurs when blood flow restoration to the heart exacerbates dysfunction and structural damage instead of recovery (Fig. 4b). During ischemia, oxygen deprivation suppresses critical metabolic pathways such as those involving fatty acids, ketone bodies, branched-chain amino acids (BCAAs), and glucose oxidation, drastically reducing ATP production. To compensate, cardiomyocytes shift to glycolysis for basic energy needs, although this source is insufficient for normal cardiac activity.9 In response to energy scarcity, 5’AMP accumulation activates the AMPK pathway, increasing phosphoinositide-dependent kinase-2 (PDK2) activity and GLUT1/4 expression to increase glycolysis. However, AMPK also suppresses the fatty acid transporter CD36, limiting fatty acid uptake and oxidation and thus aggravating metabolic imbalance.400 Furthermore, impaired BCAA metabolism triggers intermediate metabolite buildup, inhibiting PDH and disrupting glucose oxidation, potentially leading to increased reliance on glycolysis.441
During myocardial reperfusion, although reoxygenation and nutrient supply should theoretically restore OXPHOS, the glycolytic pathway initiated during ischemia provides only short-term energy and is unsustainable. This shift results in NAD+ depletion, lactate buildup, and a decrease in the intracellular pH, negatively impacting cellular function.442 The rapid reintroduction of oxygen and normalization of pH during reperfusion can lead to increased ROS production, causing mitochondrial damage and impeding ATP synthesis. The generation of ROS after ischemia‒reperfusion activates multiple pathways, such as the ATF4-CHOP, TLR4/TRIF, and USP7/p53/TfR1 pathways, triggering ferroptosis and promoting immune cell recruitment, further exacerbating inflammation.443,444,445 Moreover, calcium overload, mitochondrial dysfunction, and disrupted signaling pathways during reperfusion exacerbate cardiac damage. The activation of Nrf2 plays a role in protecting the heart from ischemia/reperfusion injury. Notably, OXPHOS impairment during ischemia limits oxygen utilization, decreasing glucose oxidation. Research highlights the importance of enhancing glucose oxidation to reduce ischemia‒reperfusion injury.446 Despite sustained high glycolytic rates, impaired glucose oxidation decreases ATP production, reducing cardiac efficiency.447
In ischemia-reperfusion injury, high-density lipoprotein (HDL) inhibits BID-mediated mitochondrial apoptosis activation by regulating the expression and phosphorylation of the anti-apoptotic factor BCL-XL. This is primarily mediated by the anti-apoptotic properties of sphingosine-1-phosphate (S1P) in HDL, which prevents the opening of mitochondrial permeability transition pores by activating the SAFE (Survivor Activating Factor Enhancement) and RISK (Reperfusion Injury Salvage Kinase) pathways.448,449 HDL may serve as a potential therapeutic strategy for ischemia-reperfusion injury.
Energy metabolism in heart failure
Heart failure is a critical clinical syndrome characterized by a reduced ability of the heart to pump blood and failure to meet the body’s metabolic demands (Fig. 5a). This condition involves multiple disruptions in cardiac energy metabolism, including substrate utilization, mitochondrial function, and ATP synthesis. Chronic illnesses such as hypertension, diabetes, and obesity can trigger adverse metabolic shifts, resulting in Na+, H+, and Ca2+ overload, leading to cellular acidosis and damage, and increasing energy consumption in cardiac myocytes, leading to inadequate energy supply and potentially progressing to heart failure.450 A hallmark of heart failure is the reprogramming of myocardial energy metabolism, which includes decreased oxidation of fatty acids, glucose, and BCAAs, with a compensatory increase in glycolysis. Despite increased glycolysis attempting to offset reduced FAO, this approach yields approximately 30% less ATP than a healthy heart does, which is insufficient to meet energy needs.451,452 Moreover, impaired OXPHOS in mitochondria, often uncoupled from glucose oxidation, further compromises ATP production and cardiac efficiency in heart failure.450 This mitochondrial dysfunction, characterized by increased ROS and abnormal dynamics, is closely linked to the progression of heart failure.453 Elevated free fatty acid levels in the heart correlate with heart failure severity.400 The inhibition of PGC-1α transcription and deactivation of the PPAR/PGC-1α pathway results in increased activity of the crucial regulatory factor CPT-1 for fatty acid uptake, while the synthesis of the FAO regulator β-oxidase is inhibited.454,455 This leads to reduced FAO and increased lipid accumulation in cardiac myocytes. Aberrant lipid peroxides, through TLR4/NOX4 involvement, contribute to ferroptosis, mediating myocardial cell death and exacerbating heart failure. Inhibitors of ferroptosis can significantly improve the survival rate in a heart failure mouse model.456 Inadequate oxygen supply in failing hearts shunts some free fatty acids into nonoxidative pathways, producing toxic lipids such as ceramides and diacylglycerol, which further damage mitochondrial function and exacerbate heart failure progression.457 Disturbances in BCAA metabolism also play a critical role, with elevated BCAA levels detected in failing hearts, likely due to impaired BCAA oxidation during heart failure.458,459 Impaired BCAA catabolism/oxidation is associated with contractile dysfunction and heart failure development,433 and these metabolic disturbances are linked not only to cardiac dysfunction but also to cardiac remodeling and insulin resistance in heart failure patients.433 In heart failure, metabolic defects in fatty acid and glucose metabolism shift cardiac myocytes towards ketone body metabolism. Ketone bodies could serve as a potential alternative fuel for failing hearts, bypassing disrupted metabolic pathways such as β-oxidation and converting acetoacetate to acetyl-CoA. Recent studies have indicated that elevated levels of ketone bodies can increase the myocardial energy supply and alleviate cytokine-induced mitochondrial dysfunction and fibrosis.460 Enhancing ketone metabolism in heart failure patients is a feasible strategy for regulating cardiac function and improving prognosis, but further exploration is needed on the regulatory mechanisms governing the interconversion of ketone bodies and glucose‒lipid metabolism.
Fig. 5
Energy metabolism in heart failure and diabetic heart disease. Under physiological conditions, the heart primarily relies on the oxidation of fatty acids entering the TCA cycle for energy supply. In various disease states (a heart failure and b diabetic heart disease), changes in cardiac energy metabolism led to increased production of ROS, ultimately affecting mitochondrial function and resulting in decreased ATP production, which impacts the heart’s ability to perform its functions. FAO fatty acid oxidation, PFK phosphofructokinase, BCAA branched-chain amino acids, HK hexokinase, PK pyruvate kinase, ACS acyl-CoA synthetase
During heart failure, cardiac energy metabolism undergoes reprogramming to meet shifting metabolic demands. The expression of GLUT1 increases, paralleling enhanced glycolysis, indicating that GLUT1 may drive this increased glycolytic activity.400 Conversely, GLUT4 expression decreases, potentially reducing myocardial glucose utilization efficiency. In a normal adult heart, PKM1 levels are high, whereas PKM2 levels are low. PKM1 is crucial for maintaining a stable hemodynamic response, regulating mitochondrial energy production through pyruvate generation.461 However, during heart failure, decreased PKM1 and increased PKM2 levels are observed. The lack of PKM1 diminishes pyruvate production, inhibiting PDH activity. Since pyruvate is pivotal for entering the TCA cycle, its reduction decreases TCA flux, impairing mitochondrial energy output. This metabolic disruption exacerbates heart dysfunction and fibrosis caused by pressure overload. In contrast, overexpression of PKM1 offers cardioprotection against contractile dysfunction, which is essential for maintaining glucose uptake and glycolysis, thereby supporting ATP production and biosynthesis for cardiac function.461 In sharp contrast, PKM2 elevation is associated with pathological cardiac remodeling, acting as a detrimental factor in heart failure progression. Increased PKM2 activity may drive harmful metabolic reprogramming, leading to adverse structural and functional cardiac changes.461
In heart failure, energy metabolism changes involve complex regulatory pathways. The uncoupling of glycolysis from glucose oxidation is mediated by mitochondrial uncoupling protein-2 (UCP-2), which disrupts the entry of pyruvate into the mitochondria for oxidation. On the other hand, FTO maintains cellular glucose uptake and stabilizes glycolysis. Metabolic disturbances cause BCAA accumulation, activating mTOR signaling and promoting cardiac hypertrophy.462 Targeting BCAA metabolism or using mTOR inhibitors such as rapamycin may improve heart function.463 AMPK is pivotal in heart failure and is activated under stress to increase cardioprotection by enhancing GLUT4 translocation and glucose uptake.464,465 It also increases fatty acid uptake by moving CD36 to the membrane, inhibits ACC, reduces malonyl-CoA, and increases CPT1 activity.466 PGC-1α is key for mitochondrial biogenesis,467 with AMPK enhancing activity through direct phosphorylation or SIRT1-mediated deacetylation.468 SIRT3, which is crucial in cardiovascular health, is downregulated in heart failure, increasing PDH and ATP synthase acetylation and reducing mitochondrial activity.469,470 SIRT3 overexpression enhances SOD2 activity, lowering ROS,471 and suppresses fibrosis pathways such as the TGF-β/Smad pathway.472,473 PPAR-α, which is upstream of SIRT3, influences fatty acid metabolism. Its suppressed signaling in heart failure affects FAO, leading to functional impairment. Activating PPAR-α increases metabolic balance, reduces ischemia/reperfusion damage, and improves heart function and antioxidant capacity.474,475 Nrf2 inhibits oxidative stress sensitivity and cardiac damage in heart failure, thereby attenuating the progression of heart failure. The activation of Nrf2 is regulated by the Akt/GSK-3β/Fyn signaling pathway.476
Improving myocardial energy metabolism provides a new perspective for the prevention and treatment of heart failure. Metabolic therapies that lower fatty acid intake and oxidation, enhance glucose oxidation, and increase ketone and branched-chain amino acid oxidation offer hope for improving the prognosis of heart failure patients. However, the regulation of metabolic pathways in heart failure is diverse, while increased glycolysis may increase cardiac workload, it also serves as an energy source in the state of heart failure. The alterations in substrate metabolism during heart failure, whether they result from the disease or represent residual compensatory responses within the body, remain controversial. The debate continues regarding whether the reduction in FAO in heart failure is protective or maladaptive. There is still much work to be done in fully elucidating the changes in energy metabolism signals and their role in heart failure, advancing the development of relevant metabolic therapeutic drugs.
Energy in diabetic cardiomyopathy
Diabetic cardiomyopathy is a severe diabetes complication linked to profound cardiac metabolic alterations (Fig. 5b). In diabetic cardiomyopathy, an increase in lipid metabolism and a decrease in glucose metabolism disrupt the fatty acid and glucose balance. This shift elevates the oxygen demand and leads to mitochondrial dysfunction, resulting in cardiomyocyte death and ventricular dysfunction.477 Insulin resistance or impaired signaling in diabetes activates FOXO-1 and PDK4, reducing mitochondrial glucose oxidation. FOXO-1 also increases CD36 expression, enhancing lipid uptake and shifting energy reliance from glucose to lipids. This shift increases metabolic stress and dependence on fatty acids, resulting in excessive oxygen use and metabolic imbalance, with the accumulation of lipid intermediates and ROS. ROS are critical factors in the progression of diabetic cardiomyopathy.478 ROS accumulation causes lipotoxicity and mitochondrial dysfunction, impairing signaling pathways and leading to myocardial damage and contractile dysfunction. TGR5, a bile acid receptor, may limit fatty acid influx into cardiomyocytes, maintaining energy balance and mitigating diabetic cardiomyopathy risk.479
Under normal physiological conditions, insulin signaling through Akt and PKA phosphorylates PFK2, increasing glycolysis and balancing fatty acid and glucose metabolism. In diabetes, impaired insulin signaling decreases glucose uptake. Type 1 diabetes, which is characterized by insulin deficiency, results in reduced cardiac glucose uptake and oxidation, as the heart’s glucose regulation is primarily insulin dependent. In T2DM, hyperglycemia and hyperinsulinemia, coupled with decreased GLUT4 expression,480 weaken insulin signaling and diminish glucose metabolism. Without insulin signaling, PFK2 is degraded via lysosomes,481 further disrupting cardiac energy metabolism. In circulation, Klotho inhibits insulin and insulin-like growth factor-1 (IGF-1) intracellular signaling by blocking the tyrosine phosphorylation of the insulin and IGF-1 receptors.482 Mice lacking Klotho exhibit improved glucose tolerance and insulin sensitivity, along with decreased energy expenditure and storage. PPAR-γ plays a pivotal role in glucose and lipid metabolism and overall homeostasis; Klotho, being a target gene of PPAR-γ, suggests its involvement in the partial metabolic regulation mediated by PPAR-γ. Recombinant Klotho can enhance cholesterol efflux by inhibiting the Wnt/β-catenin signaling pathway, thereby reducing lipid accumulation in foam cells.483
Despite the increasing depth of research on cardiovascular diseases in recent years, there are still numerous controversies and challenges. The debate on whether energy metabolism overload or deprivation plays a more crucial role in the occurrence and progression of cardiovascular diseases continues. The aberrations in energy metabolism in cardiovascular diseases are influenced by various factors such as genetics, lifestyle, and environmental factors, integrating these elements into metabolic studies poses a challenge. Alterations in energy metabolism in cardiovascular diseases often involve systemic pathophysiological changes across multiple systems, while the key energy metabolic targets (AMPK, SIRT) exhibit functional variances in different tissues, suggesting the need for in-depth mechanistic exploration using tissue-specific animal models and expanding clinical research in the future. Despite advancements in metabolomics, which reveal changes in metabolites, a deeper understanding of the biological mechanisms behind these changes and metabolic regulatory processes is still lacking. Metabolomic studies need to consider the dynamic changes in metabolism during the development of cardiovascular diseases; addressing how comprehensive monitoring and analysis of the metabolome can be achieved at different time points is crucial. Despite the numerous challenges associated with the evolution of multi-omics technologies, the maturation of 3D culture models, and advancements in biosensor technologies, exploring metabolic alterations and regulatory signals under pathological cardiac conditions in vitro holds promise for further elucidating the molecular mechanisms of diseases.
Energy metabolism and metabolic diseasesEnergy metabolism in obesity
Obesity, a chronic condition, is fundamentally tied to energy imbalance, where intake surpasses expenditure, leading to excess fat storage. In obesity, disrupted free fatty acid metabolism is vital, with increased synthesis and storage expanding adipose tissue. Although β-oxidation converts fatty acids to energy, this process is often underactive in obese individuals, resulting in insufficient energy release.
Hormones play a pivotal role in energy metabolism regulation in obesity. Obesity is often accompanied by insulin resistance, a state in which insulin signaling is impaired, hindering glucose uptake and utilization. This not only disrupts energy balance but also accelerates fatty acid metabolism, intensifying metabolic disorders in individuals with obesity. Mitochondrial function, which is essential for energy metabolism, is compromised in the skeletal muscle and adipose tissue of obese individuals, contributing to insulin resistance, T2DM, and obesity.484,485 In individuals with obesity, elevated glucose and free fatty acid levels directly induce mitochondrial dysfunction.486 This dysfunction is driven by ROS, oxidative lipids, endoplasmic reticulum (ER) stress, and genetic predispositions, all of which are worsened by caloric excess.485 Oxidative stress damages insulin signaling and incites inflammation, leading to adipocyte dysfunction, whereas inflammation further impairs mitochondrial function in white adipose tissue.487,488,489 Consequently, mitochondrial dysfunction hampers lipid processing and ATP production in adipocytes, contributing to adipocyte hypertrophy.490
Leptin, which is secreted by adipose tissue, regulates energy homeostasis, with increased fat reserves typically leading to increased circulating leptin levels.491 It influences lipid metabolism not only by regulating food intake but also by reducing triglyceride storage in white adipocytes and the liver, thereby increasing FAO.492 Leptin further stimulates FAO in skeletal muscle by activating AMPK, preventing the accumulation of lipotoxic metabolites,493 which suppresses appetite and supports efficient energy use. However, in individuals with obesity, this regulatory mechanism is often impaired.494 Studies have shown that obese individuals have increased fatty acid uptake in skeletal muscle, yet the ability of leptin to promote FAO is limited, potentially leading to intramuscular triglyceride accumulation.495 This metabolic shift suggests potential leptin signaling resistance in obesity, likely contributing to energy metabolism imbalances in muscle tissue.
In obesity, energy metabolism regulation is complex and involves many signaling pathways. The cAMP-dependent protein kinase (PKA) pathway is critical for controlling fatty acid synthesis and breakdown via metabolic enzymes such as ACC. Disruption of PKA signaling is linked to altered fat and glucose metabolism in obesity.496,497 PGC-1α is essential for mitochondrial biogenesis and function, driving DNA replication and protein expression through TFAM and NRF1. In obesity, decreased PGC-1α activity reduces mitochondrial function, impairing FAO and energy output.484 SIRT1 and AMPK are vital for lipid metabolism. SIRT1 promotes mitochondrial function by deacetylating targets such as PGC-1α, whereas AMPK adjusts fatty acid processes according to energy needs. Obesity-induced inflammation decreases SIRT1 and AMPK activity,498,499 diminishing the role of PGC-1α and affecting mitochondrial efficiency. SIRT1 enhances insulin secretion through NAD+-dependent deacetylation and counteracts inflammatory signals to prevent pancreatic β-cell apoptosis.500 Inflammation driven by excess lipids is crucial in metabolic disorders associated with obesity. It triggers the release of cytokines such as TNF-α and IL-6 from macrophages.501 These cytokines inhibit insulin signaling, causing insulin resistance,486,502 which disrupts fatty acid handling and accelerates obesity.
Epigenetic modifications influence energy metabolism processes in obese patients. Histone deacetylases (HDACs) regulate two main transcription factors, PRDM16 and PGC-1α, through acetylation and deacetylation, playing crucial roles in the pathophysiology of obesity. In mice, the absence of HDAC6 contributes to the onset of obesity.503 HDAC3 enhances the expression of PPAR-γ target genes such as adiponectin and AP2, thereby ameliorating obesity.504 Inhibiting HDAC5 and HDAC11 can alter adipocyte phenotypes, reducing obesity.505,506 Additionally, HDAC5 regulates energy metabolism by affecting BAT activity and UCP1 levels through the inhibition of the hypothalamic STAT5b-TH axis.507 HDAC1 influences energy expenditure, obesity, and glucose tolerance by suppressing Pgc1α/Ucp1 transcription.508 Inhibiting HDAC8 enhances mitochondrial biosynthesis and function through increased PGC1α, preventing obesity.509 HDAC inhibitors improve the intestinal epithelial integrity of obese patients by increasing SCFA levels and activating the Notch signaling pathway.510 High methylation levels of FAO -related genes (such as Acaa2, Acsl1, and Cox7a1) are closely associated with abnormal brown fat metabolism.511 Enhanced DNA demethylation in the Prdm16 gene promoter may be a significant factor in obesity, and supplementing with α-ketoglutarate can combat obesity in mice.512 Folic acid supplementation lowers overall methylation levels by influencing the distribution of differentially methylated regions in adipocytes of obese mice, thereby improving insulin resistance and obesity-related metabolic disruptions.504 Recently, substantial progress has been made in understanding the role and mechanisms of epigenetics in regulating thermogenesis in adipose tissue. However, challenges remain: most studies are conducted in vitro or in animals, lacking sufficient clinical trial support for the regulation of adipose tissue thermogenesis, transitioning from laboratory research to clinical applications presents ongoing challenges. Furthermore, different HDAC types have varying effects on obesity regulation, and even within the same class, HDACs exhibit diverse impacts on obesity, necessitating further exploration into the underlying molecular mechanisms.
Adjusting diet and lifestyle is crucial for managing obesity, as excessive energy intake can lead to weight gain. Western high-fat diets (HFDs) can irreversibly disrupt the diversity of the microbiota, disturbing the host’s circadian rhythm and metabolism, thereby promoting obesity.513 Compared with the Mediterranean diet and Jiangnan dietary patterns, increasing protein intake in the diet can enhance the abundance of folate-producing bacteria, upregulating folate-mediated one-carbon metabolism and FAO pathways.514 Although the ketogenic diet aids in obesity control, it can lead to dyslipidemia in obese mice.515 Studies, both in vivo and clinically, have demonstrated that supplementing with prebiotics along with physical exercise can lead to reduced BMI and plasma cholesterol levels in obese subjects and high-fat diet mice, ultimately improving glucose tolerance. Capsaicin regulates fatty acid and glucose metabolism, and its intake promotes thermogenic fat oxidation and aids in weight management.516 Currently, intermittent fasting helps improve compliance and represents a promising treatment method for obese patients.517
Energy metabolism in T2DM patients
In T2DM, energy metabolism is disrupted, which is characterized by chronic hyperglycemia and impaired carbohydrate, fat, and protein metabolism.518 A fundamental issue is insulin resistance, which reduces the efficacy of insulin in lowering blood glucose. This resistance is prominent in skeletal muscle, which handles more than 80% of insulin-stimulated glucose uptake. When insulin resistance occurs, glucose uptake and glycogen synthesis in muscle are compromised.519,520,521 This metabolic dysfunction stems from factors such as decreased GLUTs,522 diminished insulin-induced ATP production,523 and reduced expression of mitochondrial genes.524,525 In the liver, insulin suppresses the gluconeogenic enzymes PEPCK and G6 Pase via the Akt and FOXO pathways,526,527 but resistance impairs this process, increasing glucose output. T2DM progression further weakens β-cell insulin secretion, worsening insulin scarcity. Persistent hyperglycemia catalyzes ROS production through glucose autoxidation and pathways such as the polyol route, as well as AGE formation.528 High ROS levels induce oxidative stress and potentially trigger β-cell apoptosis,529 undermining insulin secretion and blood sugar control. The hallmark of T2DM is reduced mitochondrial OXPHOS capacity and decreased mitochondrial content in skeletal muscle cells and liver cells. Activation of PGC-1α has been shown to increase mitochondrial OXPHOS capacity, restore mitochondrial ATP production, promote insulin secretion in pancreatic β-cells, enhance insulin sensitivity in skeletal muscle and liver.530 Metrnl, an adipokine, has been identified as a key modulator in the insulin signaling pathway, improving impaired insulin responses in myotubes and skeletal muscle through AMPK/PPAR-δ-mediated signaling.531 In type 2 diabetic mice, Metrnl has been found to enhance β-cell function by inhibiting β-cell apoptosis and activating β-cell proliferation through the Wnt/β-catenin pathway.532 Treatment of C2C12 myotubes with Metrnl increases glucose uptake through the calcium-dependent p38 MAPK and AMPKα2 pathways and regulates the binding of HDAC5 to the GLUT4 promoter in an AMPKα2-dependent manner, promoting GLUT4 transcriptional activation.533 These findings underscore the role of Metrnl in regulating energy homeostasis in diabetes.
Epigenetic modifications influence the energy metabolism processes of T2DM patients. T2DM is closely associated with HDACs. HDAC inhibitors promote the differentiation of pancreatic β-cells, increase the levels of glycolytic and gluconeogenic enzymes, and improve insulin resistance.534 HDAC-3 promotes FOXO1 deacetylation leading to insulin resistance in T2DM.535 HDAC-3 negatively regulates PPAR-γ signaling, causing signaling disruption, altering glucose homeostasis, and increasing hepatic glucose and lipid metabolism.536 SREBP1 recruits HDAC-8 to activate the Wnt signaling pathway resulting in insulin resistance and hyperglycemia.537 HDAC-1 recruited by ATF6 inhibits SREBP2-mediated gene transcription, contributing to glucose homeostasis.538 Activation of CaMK signaling leads to the release of HDAC-5, which in turn activates GLUT4 in skeletal muscle to stimulate glucose uptake; any dysregulation can lead to insulin resistance.539 The intestinal microbiota composition differs in T2DM patients, and these microbial changes may lead to decreased methylation of CpG sites in the TLR4 exon and TLR2 promoter, affecting genetic epigenetic regulation.540 Currently, HDAC inhibitors are primarily used in clinical cancer treatment. Although laboratory studies suggest a potential role in alleviating diabetes, the development and translation of HDAC inhibitors for diabetes therapy are still in the early stages, highlighting the importance of future research in unraveling their mechanisms, optimizing drug delivery processes, and evaluating long-term safety through clinical trials to assess their efficacy in combating T2DM effectively.
Pharmacological strategies for treating T2DM aim to increase insulin effectiveness and secretion. These include oral hypoglycemic agents and injectables such as insulin and GLP-1 receptor agonists, which are designed to improve insulin sensitivity or supplement deficient insulin secretion. Among oral options, metformin is popular because of its antihyperglycemic effects. Metformin primarily reduces hepatic gluconeogenesis, thereby lowering glucose output.541 It also modestly enhances the skeletal muscle insulin response, increasing glucose uptake.542 Metformin acts by activating insulin receptors and IRS-2, increasing the activity of GLUTs, such as GLUT1, which facilitates glucose uptake.543 This action intensifies the inhibitory effect of insulin on gluconeogenesis.544 Additionally, metformin increases glucose uptake in muscle by activating insulin receptor tyrosine kinase, partly by promoting GLUT4 translocation to the cell membrane.545
The efficacy of metformin largely arises from the activation of AMPK, which is pivotal for maintaining the energy equilibrium of the cell. The activation of AMPK increases insulin receptor and IRS-2 activity, facilitating GLUT movement to the cell surface and thereby increasing glucose uptake.543 Furthermore, AMPK is vital for regulating lipid metabolism by curbing fatty acid synthesis and enhancing β-oxidation, which impacts metabolic balance.546,547 This action reduces the amount of intracellular free fatty acids that hinder insulin signaling and glucose transport548 and can impair insulin secretion in β-cells.549 Additionally, β-oxidation byproducts such as acetyl-CoA and citrate can inhibit glycolytic enzymes.550 Thus, metformin helps lower glucose and free fatty acids in the blood, preventing lipid buildup in insulin-sensitive tissues and enhancing insulin secretion and sensitivity.
Restoring mitochondrial functionality is becoming a strategic focus in T2DM treatment. By targeting ETC activity, certain compounds can increase mitochondrial energy output, improving metabolic states in T2DM patients. Imeglimin, a novel antidiabetic agent, positively impacts organs such as skeletal muscle, the liver, and the pancreas by restoring mitochondrial function.518 Mitochondrial biogenesis, which involves the renewal and replication of mitochondria, is vital for cellular energy. SIRT1, an NAD+-dependent deacetylase, activates PGC-1α to promote this process.551 Increased SIRT1 activity increases mitochondrial function and insulin sensitivity, which are crucial for better metabolic control in T2DM patients. Mitochondria are primary sources of ROS, which can lead to damage if they accumulate excessively. Antioxidants and radical scavengers help reduce oxidative damage, safeguarding mitochondrial integrity and ensuring proper function. In addition to pharmacological control, recent studies have revealed that, owing to the pathogenic role of mitochondrial abnormalities in diabetes, transferring healthy mitochondria to pancreatic β-cells to increase their insulin secretion function has emerged as a therapeutic strategy552 and has garnered significant attention. However, several key challenges persist, with mitochondrial storage remaining a hurdle.553 Isolated mitochondria can maintain activity on ice for 1–2 h, but the efficiency of mitochondrial transplantation is often suboptimal. Although various methods have been developed to increase transplantation efficiency, mitochondrial function is still affected. Mitochondrial DNA can be inherited through cytoplasmic genetics, raising safety concerns. The use of cross-species mitochondrial transplantation has raised concerns regarding the immune response. Future research needs to address these issues further to facilitate the clinical application of these therapies.
Physical activity combined with dietary interventions may represent a promising strategy for improving metabolic disorders. High-sugar diets can lead to insulin resistance and disrupted glucose metabolism. Among individuals with metabolic syndrome, interventions combining exercise training with dietary supplementation of polyunsaturated fatty acids have shown significant improvements in insulin sensitivity, serum C-reactive protein levels, and high-density lipoprotein concentrations.554 Dietary fiber can promote the growth of acetate- and butyrate-producing bacteria; increased production of short-chain fatty acids (SCFAs) can stimulate glucagon-like peptide 1 (GLP-1) secretion, thereby enhancing insulin secretion, improving glucose homeostasis, reducing inflammation, and alleviating T2DM.555 Due to its high content of unsaturated fatty acids, fiber, and antioxidants, the Mediterranean diet has a significant positive impact on the treatment of T2DM, improving the release of inflammatory factors and insulin resistance.556 However, further research is needed to elucidate the potential molecular mechanisms underlying how dietary components impact insulin sensitivity to better understand their effects. Intermittent fasting helps T2DM patients break down and utilize endogenous fats, regulating blood glucose levels, and thus enabling the body to regain energy balance.557
In metabolic disorders, magnesium deficiency is prevalent in individuals with obesity, diabetes, or metabolic syndrome, with magnesium supplementation shown to improve insulin sensitivity and blood glucose control in T2DM patients.558,559 Magnesium acts as a cofactor for enzymes like creatine kinase, adenylate cyclase, and Na+-K+-ATPase, directly participating in blood glucose regulation. Additionally, magnesium enhances insulin sensitivity, ameliorates insulin resistance and glucose metabolism, mediates beta cell insulin release, and modulates intracellular signal transduction downstream of the insulin receptor. However, hyperinsulinemia and insulin resistance in diabetic patients can impair insulin-stimulated cellular uptake of magnesium, leading to decreased magnesium levels in the body. Elevated blood glucose levels increase insulin secretion, affecting the transport of Na+ and K+, and influencing cellular energy metabolism status.
In metabolic disorders, the intricate interplay within the gut–brain–adipose axis, which includes neural and endocrine regulation, has garnered substantial attention. The influence of the gut microbiota on energy metabolism is highly important and warrants further exploration in future research directions. Given the complexity of pathogenic mechanisms involving multiple organs and systems, current studies face challenges in pinpointing singular therapeutic targets, posing a significant hurdle to drug development. Future endeavors should concentrate on merging advancements in multi-omics technologies to monitor interindividual metabolic variabilities, emphasizing early diagnosis and personalized therapeutic strategies.
Energy metabolism in autoimmune diseases
Autoimmune diseases arise when the immune system mistakenly attacks the body’s own tissues, producing pathological reactions to self-antigens. Altered energy metabolism plays a crucial role in these conditions, particularly affecting the activation, differentiation, and function of macrophages, T cells, and B cells. For example, metabolic reprogramming in T and B cells during autoimmune diseases results in heightened activation and distinct energy profiles compared with those of normal cells.560 Rheumatoid arthritis (RA) is a chronic autoimmune disorder characterized by inflammation of the synovial tissue and the presence of autoantibodies.561 Typically, the synovium is infiltrated by T cells, B cells, and macrophages, triggering a damaging response in fibroblast-like synoviocytes (FLSs), leading to excessive proliferation and destruction of cartilage and bone.562 Inflammatory bowel disease (IBD), which includes Crohn’s disease and ulcerative colitis, is characterized by chronic relapsing inflammation and epithelial injury in the gastrointestinal tract.563 Systemic lupus erythematosus (SLE) is an autoimmune disorder involving multiple organ systems and is characterized by clinical and serological heterogeneity and dysregulated interferon responses.564 Although the specific etiology of these diseases remains unclear, altered energy metabolism and the resulting immune dysregulation are believed to significantly contribute to their development and persistence. In the early stages, the body is in an inflammatory state, leading to an increase in metabolic rate to support the energy required for inflammation and immune responses. During this time, activation of the glucose metabolism pathways is typically observed. As the disease progresses, the body may experience impaired mitochondrial function, increased production of thiol proteins and free radicals, leading to disruptions in energy metabolism. With the continued development of the disease, exacerbation of inflammation further affects energy metabolism. Some autoimmune diseases such as RA may lead to joint destruction and tissue damage, consuming more energy to meet the demands of repair and recovery. Understanding these dynamic changes in energy metabolism is crucial for initiating early treatment or preventive measures during the window period of autoimmune diseases (Fig. 6).
Fig. 6
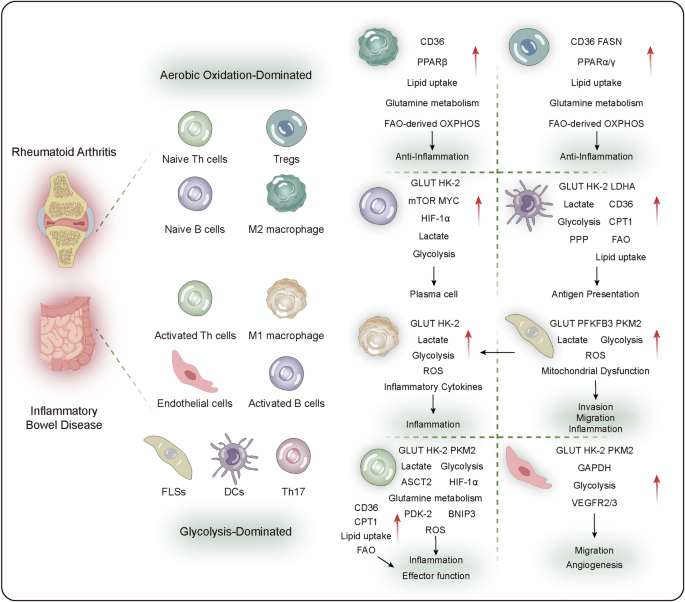
Energy metabolism in autoimmune diseases. In autoimmune diseases, metabolic abnormalities in immune cells are a key factor. In some situations, CD4+ T cells enhance glycolysis and mitochondrial OXPHOS, while in rheumatoid arthritis (RA), T cells suffer from impaired mitochondrial OXPHOS, turning to the pentose phosphate pathway. Naive Th cells and B cells tend to undergo aerobic oxidation, while activated Th cells and B cells tend to undergo glycolysis. Treg cells can utilize glycolysis and lactate to maintain their functions in the TME. M1 macrophages tend to have aerobic glycolysis, while M2 macrophages rely more on OXPHOS. FLSs undergo metabolic reprogramming, shifting towards enhanced glycolysis. As antigen-presenting cells, DCs significantly alter their metabolic pathways, such as OXPHOS and glycolysis, during activation. These changes in energy metabolism promote the abnormal proliferation of fibers and blood vessels and exacerbate the inflammatory process. DC dendritic cells, FLS fibroblast-like synoviocytes, Treg regulatory T cells, MDSC myeloid-derived suppressor cells
Autoimmune disease-related autoinflammatory responses require significant amounts of energy, leading to increased metabolism of fats, glucose, and glutamine and a shift from OXPHOS to glycolysis. Inflammation linked to autoimmunity involves diverse immune cell subsets within disease-specific environments, each with unique metabolic needs.565 Before encountering antigens, naive Th cells rely primarily on FAO and OXPHOS for energy. In contrast, activated Th cells depend more on glycolysis, increasing glucose uptake via transporters to support proliferation. Enhanced glucose uptake and glycolysis are critical in Th17 cell differentiation.566,567 Effector T cells rely on glycolytic metabolism for growth and function, whereas regulatory T cells (Tregs) use lipids through mitochondrial β-oxidation and generate ATP via OXPHOS.568 In various autoimmune diseases, proinflammatory T cells have higher lipid contents than healthy individuals do.569 T cells utilize lipids for membrane biosynthesis, cell division, and migration rather than energy production through β-oxidation.570,571 Fatty acid and cholesterol biosynthesis mediate Th17 cell formation, whereas FAO supports Treg differentiation.572 Amino acid metabolism, particularly glutaminolysis, is pivotal in Th17 cell development.573 B cells also exhibit altered energy metabolism under autoimmune conditions. Naïve B cells maintain a low metabolic state, whereas their activation relies on OXPHOS-driven metabolic programming.574 Autoimmune diseases feature diverse macrophage subsets with various functions.575 Generally, inflammatory macrophages depend on glucose as an energy source, whereas anti-inflammatory macrophages require less glucose and rely on mitochondrial OXPHOS for their bioenergetic and biosynthetic demands.576 Specifically, M1 macrophages utilize glycolysis, whereas anti-inflammatory M2 macrophages typically rely on β-oxidation.577
Glycolysis
In autoimmune diseases, the upregulation of GLUT1, which facilitates glucose transport, has been reported.578 Key glycolytic enzymes, such as HK, PKM2, GAPDH, and LDHA, exhibit increased activity in inflamed joints.579,580 HK catalyzes the initial step in glucose metabolism, enhancing the migration and invasion capabilities of FLSs.579 Elevated HK2 levels in Th17 cells, dendritic cells, and FLSs promote glycolysis. This enhanced glycolysis results in high lactate and low glucose concentrations in inflamed joints,581 disrupting immune cell balance. Specific inhibition of HK2 with 3-bromopyruvate reduces joint swelling and histological damage in SKG mice, a RA model, by suppressing glycolysis-dependent dendritic cell activation and Th17/Treg imbalance.582 Overactive HK2 promotes synovial cell proliferation and secretion by mediating AMPK activation of NF-κB signaling.583 Inhibitors of HK2 effectively suppress the production of inflammatory factors.584
Studies have shown that increased glucose uptake and glycolysis in the lymph nodes and thymus of patients with autoimmune diseases such as RA are correlated with disease severity and therapeutic response.585 Inadequate vascularization often creates hypoxic conditions in local tissues, which play crucial regulatory roles in cellular processes.586 Hypoxia can extensively alter mitochondrial structure, dynamics, and mtDNA stability, leading to impaired respiration, excessive ROS production, increased oxidative damage, and reduced ATP production.587 Driven by the hypoxic joint environment, Tregs can quickly transform into pathogenic Th17 cells,588 exacerbating inflammation.
Under normal oxygen conditions, HIF-1α is degraded, but under hypoxia, it accumulates and promotes the expression of glycolytic enzymes. HIF-1α acts as a key regulator of the anaerobic metabolic switch, enhancing processes that increase the production of glycolytic energy, including GLUTs and glycolytic enzymes.589 It also induces synovial fibroblasts to produce proinflammatory cytokines such as IL-1β, IL-6, IL-8, and TNF-α and cell adhesion molecules such as VCAM-1, TSP1, and CXCL12, thereby promoting inflammation.590 HIF-1α increases glycolytic flux and glucose consumption to produce energy and synthesize biosynthetic precursors.591 By deriving more energy from anaerobic glycolysis via HIF-1α, in addition to generating biomass, cells become inflammatory effectors.586,592 Mitochondrial dysfunction in T cells causes increased glucose flow into the pentose phosphate pathway, a process that is exacerbated under hypoxic synovial conditions.
The activation of the PI3K/Akt/mTOR signaling pathway is crucial during immune cell activation. CD28 costimulation triggers the PI3K/Akt axis in T cells, inducing GLUT1 expression, which enhances glucose uptake and utilization.593 This metabolic shift provides the energy and biosynthetic precursors necessary for T-cell proliferation and activation. mTORC1 and mTORC2 play distinct but complementary roles in regulating T and B-cell metabolism. Activated by the PI3K/Akt pathway, mTORC1 is closely linked to the expansion of proinflammatory T cells, supporting glycolysis and differentiation into Th1 and Th17 cells while limiting their differentiation into Treg cells.593 In contrast, mTORC2 is associated with cell survival and metabolic homeostasis.560 In SLE patients, B cells exhibit increased mTORC1 activity, which is correlated with increased lactate production, indicating a metabolic shift toward glycolysis.593
Owing to inflammation, macrophages, T cells, B cells, and stromal cells within tissues remain activated and under high metabolic stress, creating a microenvironment low in oxygen and glucose but rich in metabolic intermediates such as lactate. Lactate regulates T-cell differentiation, a process critical for sustaining chronic inflammation.594,595 It reprograms T cells, promoting proinflammatory Th17 phenotypes and aggressive CD8+ T-cell transformation.596,597 Th17 cells, a subset of helper T cells, secrete IL-17, which plays a central role in driving inflammation and autoimmune disease progression. Research indicates that lactate enhances IL-17 production through lactate-dependent metabolic pathways in CD4+ T cells, involving the activation of PKM2 and STAT3, which together facilitate Th17 differentiation and functionality.598 Tissue-resident T cells exhibit metabolic profiles similar to those of proinflammatory macrophages, which utilize nuclear PKM2/STAT3 signaling to maintain persistent IL-1 and IL-6 production.582 These findings suggest that lactate not only affects T-cell differentiation but also may modulate T-cell effector functions, particularly in tissue microenvironments. Furthermore, tissue lactate levels impact the migration of CD4+ and CD8+ effector T cells. The accumulation of extracellular lactate and its metabolites can inhibit T-cell motility, potentially limiting their infiltration into sites of inflammation and modulating immune responses.582 By sensing tissue lactate, synovial T cells become immobilized and trapped within the tissue microenvironment.599 These findings highlight the multifaceted role of lactate in immune cell behavior.
OXPHOS
OXPHOS, as a critical energy supply mechanism, involves metabolic reprogramming in autoimmune diseases. In patients with SLE and experimental animal models, CD4+ T cells exhibit increased mTOR signaling activation alongside increased glycolysis and mitochondrial OXPHOS. Pathogenic Th17 cells exhibit increased aerobic glycolysis and TCA activity, as suggested by single-cell RNA sequencing data, which can be suppressed through inhibition of CaMK4.578 Blocking the ETC and inhibiting OXPHOS can disrupt the effector functions of Th17 cells at sites of colitis inflammation, indicating that the secretion of cytokines by Th17 cells relies on ETC-mediated OXPHOS to induce inflammation in IBD and psoriasis models.600 Untreated RA patients show significant changes in OXPHOS transcriptional regulation in CD8+ TEM cells.601 Furthermore, Treg cells have been demonstrated to rely on OXPHOS for survival and functionality, serving as crucial regulatory cells in autoimmune diseases.
FA metabolism
Aberrant fatty acid metabolism exerts significant influence on the activation of immune cells, including Treg cells, naïve Th cells, and anti-inflammatory macrophages. Unlike effector T cells, Treg cells predominantly rely on FAO for energy production. Foxp3 suppresses Myc and glycolysis while enhancing OXPHOS, enabling Treg cells to survive in low-glucose environments.602 Alterations in fatty acid levels or composition can impact Treg function, potentially leading to immune dysregulation in autoimmune diseases.603 In RA, cells exhibit mitochondrial dysfunction and enhanced fatty acid metabolism, contributing to tissue invasiveness and pro-inflammatory characteristics. In an IBD mouse model, the upregulation of serum butyric acid, which enhances CPT1A activity, is considered a crucial pathway for reactivating FAO and subsequently inducing Treg differentiation.604 The activation of FAO and Treg cell differentiation through AhR activation alleviateS colitis and arthritic symptoms in mice.605 There is a potential association between abnormal fatty acid metabolism and the pathogenic activation of Th cells in autoimmune diseases. During Th17 cell differentiation, fatty acid synthesis is significantly upregulated. TRM cells play a central role in arthritis recurrence, resulting in increased fatty acid consumption and CCL5 production, aiding in recruiting immune effector cells during arthritis flare-ups.606 In RA patients, CD8+ TEM and CD8+ T cells show increased fatty acid uptake, characterized by the upregulation of fatty acid transport and sensing-related genes (FABP and CD36) as well as FAO-related genes (ACADVL).607 Studies suggest that inhibiting lipid metabolism can suppress the effector functions of CD8+ T cells in RA.606 However, the metabolic reprogramming of TRM cells in autoimmune diseases remains unclear.
Amino acid metabolism
The glutamine supply is essential for immune cell proliferation and is a key driver of angiogenesis.608 Amino acid transporters such as SLC1A5 enable efficient glutamine uptake in T cells, which is crucial for responding to T-cell receptor and CD28 costimulation signals. The activity of SLC1A5 links these signals to the mTORC1 signaling cascade, promoting T-cell expansion and differentiation into effector Th1 and Th17 cells, which are central to RA pathogenesis.609 When glutamine is limited, T-cell proliferation and cytokine secretion are inhibited, potentially shifting differentiation toward a Treg cell phenotype.610,611 Deficiency in glutamine inhibits the activation of mTORC1, subsequently downregulating NKT cell proliferation through mTORC1-c-Myc signaling.612 Elevated levels of glutamine metabolism have been observed in splenic mononuclear cells of MRL/lpr mice prone to lupus as well as in peripheral blood mononuclear cells of individuals with SLE, promoting differentiation of Th17 cells.613 The promotion of Th1, Th17 functions, and cytokine secretion play crucial roles in the pathogenesis of Crohn’s disease.614 Conversely, arginine deficiency promotes ATF4-mediated SLC7A11 transcription and Treg cell differentiation.615 Recent studies suggest that glutamine metabolism may control Th cell differentiation through epigenetic regulation. Aberrant activation of GLS-mediated glutamine breakdown triggers H3K9Ac and H3K27Ac epigenetic modifications within the IL17A gene promoter region of Th17 cells, enhancing chromatin accessibility of RORC, thereby exacerbating IL-17A expression.616 Blocking GLS1 can promote Th2 cell differentiation, suppress Th17 cell differentiation via mTORC1 pathway inactivation, while maintaining Th1 cell differentiation unaffected.617 The metabolite α-KG from glutamine breakdown has been identified as a crucial player involved in epigenetic reshaping mediated by glutamine breakdown, acting as a co-factor for peroxidases and regulating histone and DNA methylation levels in glutamine breakdown-mediated epigenetic reshaping.618 Currently, alterations in amino acid metabolism are considered potential biomarkers and therapeutic targets for autoimmune diseases. However, the complexity of regulatory mechanisms due to multiple metabolic changes poses challenges in interpreting and validating these metabolic alterations.
Interplay between epigenetic modifications and energy metabolism
Immune cells undergo energy metabolism reprogramming upon activation, and metabolic‒epigenetic changes play crucial roles in autoimmune diseases. The conversion of glutamine to α-KG, a substrate for the TCA cycle intermediate and histone demethylases, can influence T-cell differentiation by regulating chromatin accessibility.619 Overall, DNA methylation levels decrease in SLE patients, and disease activity and progression in lupus patients are negatively correlated with methylation patterns,620,621 primarily due to the reduced expression/activity of DNMTs.622 Global hypomethylation in RA may lead to immune dysregulation.623 Treg cells in RA patients exhibit an aberrant DNA methylation pattern, particularly in the promoter region of the CTLA-4 gene. Elevated methylation in this region reduces CTLA-4 expression, causing dysfunctional Tregs to be unable to activate immune regulatory pathways.624 The MEK-ERK kinase pathway primarily regulates methylation, and the inhibition of any protein in the ERK pathway could ultimately lead to the downregulation of DNMTs. T cells from active SLE patients show reduced phosphorylation of all three signaling molecules: ERK, MEK, and RAF.625 Intermediate products of energy metabolism, such as acetyl-CoA, are mediated by LDHA and promote histone acetylation, enhancing translation of Ifng during Th1 differentiation.111 GLUT3 is highly expressed in Th17 cells and converts citrate in the cytoplasm to acetyl-CoA, promoting histone acetylation around the Il-17 gene.626 SCFA butyrate inhibits histone deacetylases, promoting high histone acetylation in the promoter region of follicular regulatory T cell (Tfr) signature genes, leading to Tfr expansion;627 in RA patients, decreased butyrate levels may lead to Tfr suppression and synovial inflammation.628 Aberrant histone acetylation has been observed in MRL/lpr mice prone to lupus, where increased HDAC9 activity is noted; thus, defects in HDAC9 in MRL/lpr mice can mitigate autoimmune responses.629 Disruption of HAT/HDAC in the synovial tissue of RA patients ultimately leads to an imbalance in histone acetylation and deacetylation.630 HDAC inhibitors can serve as a therapeutic strategy to control pathological conditions characterized by functional expansion of Th1 and Th17 cells.622 The future challenge lies in discovering new, safe immunomodulatory drugs that act specifically on epigenetics, as epigenetic modifications are highly subject to variations influenced by micronutrients, diet, and/or physical activity.
Reprogramming of energy metabolism in autoimmune diseases serves as both a pathogenic factor and a potential therapeutic target. The conceptual framework of disease classification, intervention, and biomarker discovery suggests that reprogramming metabolism through epigenetics could be a novel strategy for developing protection against inflammation in autoimmune diseases. Furthermore, dietary management, recognized as a promising source for the accumulation or elimination of metabolic substrates, has been identified as a promising origin for new drug targets. Current research has focused primarily on key metabolic pathways and molecules such as mTOR, AMPK, and HIF-1α, with many targets for metabolic therapy under early stages of investigation in preclinical studies or clinical trials. However, the translation from research to clinical application has a long journey. Future studies should delve deeper into the potential mechanisms of immune cell metabolic dysregulation and identify potential therapeutic targets, with particular attention to the limited research on tissue-resident memory T (TRM) cells and their regulation in diseases such as RA, SLE, and IBD.
Energy metabolism in cancer
During cancer initiation and progression, tumor cells develop a distinct and aberrant metabolic phenotype known as metabolic reprogramming. This specialized energy metabolism encompasses several intracellular pathways, prominently featuring increased glycolysis, increased glutamine metabolism, alterations in the TCA cycle and OXPHOS, and aberrant FAO. Collectively, these metabolic adaptations form the foundation of metabolic reprogramming in tumor cells, which not only drives their rapid proliferation and metastatic potential but also significantly impacts their stemness, plasticity, and other critical biological properties.27,631,632,633,634,635 During the early stages of tumor growth, metabolic activity of tumor cells increases to support their rapid growth and proliferation demands. At this stage, tumor cells tend to generate energy through the glycolytic pathway, known as “aerobic” metabolism. As the tumor progresses, tumor cells may enhance glucose uptake and utilization while also relying on other metabolic pathways for energy production, such as fatty acid oxidation and protein metabolism. This mixed mode of “aerobic” and “anaerobic” metabolism may help tumor cells adapt to various microenvironmental conditions, such as low oxygen levels and nutrient deficiencies. With tumor growth and metastasis, internal tumor tissues might experience uneven blood supply and oxygen distribution, leading to localized hypoxia. This hypoxic environment can induce tumor cells to adjust their metabolic pathways, favoring a more anaerobic-dependent energy production method, facilitating tumor growth and invasion.
The impact of alterations in energy metabolism on tumor proliferation, stemness, and other facets of tumor cell biologyGlycolysis
In various cancers, dysregulated energy metabolism, particularly the activation of glycolysis, is a prominent characteristic. This phenomenon, known as the Warburg effect,636 indicates that cancer cells preferentially utilize glycolysis instead of OXPHOS to meet their energy demands, even under aerobic conditions (Fig. 7). This metabolic shift is crucial for the growth and maintenance of cancer cells, especially in tumor stem cells (CSCs) or cells with stem-like properties, which often exhibit increased glycolytic activity.637,638
Fig. 7
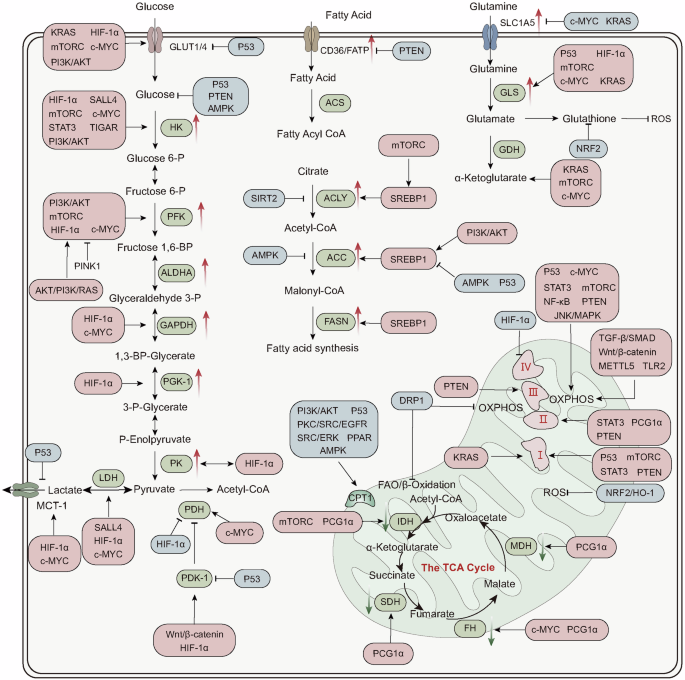
Energy metabolism in cancer. Cancer cells undergo metabolic reprogramming in their energy metabolism, characterized by enhanced glycolysis, glutamine metabolism, and FAO, but the TCA cycle and OXPHOS are suppressed. The activity of glycolysis-related transport proteins and enzymes such as GLUT, HK, PFK, PK, and LDH is increased. The activity of glutamine transporters and GLS is increased, catalyzing the production of glutamate for biosynthesis or energy synthesis. The expression of fatty acid transport proteins (CD36) and synthetic proteins (ACLY, ACC, FASN) is increased. However, the activity of key enzymes in the TCA cycle, such as IDH, SDH, FH, and MDH, is suppressed. A variety of signaling molecules undergo changes in expression during this process and regulate the energy metabolism of cancer cells. Overall, HIF1-α, KRAS, SALL4, c-MYC, PI3K/AKT, and mTOR, among others, play pro-oncogenic roles mainly by promoting glycolysis and glutaminolysis while inhibiting the TCA cycle and oxidative phosphorylation. P53, PTEN, AMPK, NRF2, PCG1α, and others play tumor-suppressive roles, inhibiting glycolysis and fatty acid transport and synthesis while promoting the mitochondrial TCA cycle and oxidative phosphorylation processes (red boxes: promoting signaling molecules; blue boxes: inhibitory signaling molecules). HK hexokinase, PFK phosphofructokinase, PK pyruvate kinase, ALDHO aldehyde dehydrogenase, GAPDH glyceraldehyde-3-phosphate dehydrogenase, PGK1 phosphoglycerate kinase 1, LDH lactate dehydrogenase, PDH pyruvate dehydrogenase, PDK1 pyruvate dehydrogenase kinase 1, ACLY ATP citrate lyase, ACC acetyl-CoA carboxylase, FASN fatty acid synthase, ACS acetyl-CoA synthase, GLS glutaminase, GDH glutamate dehydrogenase, IDH isocitrate dehydrogenase, SDH succinate dehydrogenase, FH fumarate hydratase, MDH malate dehydrogenase, CPT1 carnitine palmitoyltransferase 1
Compared with physiological aerobic oxidation, glycolytic alterations in tumor cells promote crucial biological processes such as proliferation, stemness, and metastasis. First, glycolysis provides a swift method for ATP production, potentially surpassing the traditional efficiency of the TCA cycle.639 This pathway also endows tumor cells with abundant precursors,640,641 which are essential for the biosynthesis of macromolecules such as lipids, nucleic acids, and proteins, thereby supporting rapid tumor growth and proliferation and inducing mesenchymal transformation.642 Additionally, lactate secretion during glycolysis lowers the pH of the TME,643 increasing invasive capacity.644 This local acidification increases the expression of vascular endothelial growth factor A, further promoting cancer cell proliferation645 and modifying other metabolic pathways.646,647 Furthermore, the preferential conversion of pyruvate to lactate not only reduces oxidative stress in mitochondria but also regenerates NAD+ from NADH, helping to alleviate the electron load and preventing disruption of the ETC caused by excess electrons.27,36
In tumor cells, key enzymes and transport proteins within the glycolytic pathway, including GLUTs, HK2, PFK1, and PKM2, are markedly overexpressed.648 This upregulation enhances glycolytic activity, providing the essential energy and biosynthetic precursors required for the rapid growth and proliferation of tumors.642 In contrast to that in normal cells, the increased expression of GLUTs in cancer cells leads to increased glucose uptake.649 HK2 is consistently upregulated across various cancers, driving the glycolytic process.650,651,652,653 Similarly, ALDOA is overexpressed in numerous tumors and is linked to cytoskeletal integrity and epithelial‒mesenchymal transition (EMT).654 PKM2 is particularly pivotal in metabolic reprogramming, modulating the ATP production necessary for tumor cell proliferation and activating the HIF-1 transcription factor, which further encourages glycolytic metabolism.655 In addition to its role in glycolysis and ATP production, PKM2 also regulates the redirection of glycolytic intermediates to the pentose phosphate pathway, thereby generating NADPH, which is critical for cellular redox homeostasis,656 thus shielding cancer cells from oxidative stress.657
The glycolytic process in tumor cells is intricately regulated by various signaling molecules. HIF-1α is pivotal in this regulation, promoting the glycolytic pathway by activating the transcription of genes encoding GLUTs and glycolytic enzymes.658,659 Under normal conditions, HIF-1α stability and activity are tightly controlled by oxygen levels660 and inhibited by PINK1,661 making it rarely detectable.662 However, in tumor cells, PINK1 is often deficient,661 and HIF-1α can be activated through nonhypoxic mechanisms even in oxygen-rich conditions.663 This is partly due to mutations in the von Hippel‒Lindau protein in some aggressive tumors, which impairs the degradation of HIF-1α. Additionally, oncogenes such as Akt, PI3K, and Ras can induce the nonhypoxic expression of HIF-1α.663 When metastatic cells enter the circulatory system, they encounter a more intense oxygen environment, potentially increasing oxidative stress. To counter this, circulating tumor cells (CTCs) exhibit unique adaptations: they tend to cluster together, forming cell aggregates.664 This clustering creates a hypoxic core where HIF-1α accumulates, thereby providing a survival advantage under high oxidative stress.665 HIF-1α enhances the expression of the GLUTs GLUT1, LDH, PKM2, and PDH kinase (PDK1).666,667 PDK1 inhibits the activity of the PDH complex through phosphorylation, reducing the oxidation of pyruvate to acetyl-CoA in the Krebs cycle and increasing lactate production.668,669 Furthermore, glycolytic enzymes themselves can influence HIF-1α; for example, PKM2 activates the HIF-1 transcription factor by modulating ATP levels required for tumor cell proliferation, thereby promoting glycolysis.655 pSTAT3 enhances the expression of HIF-1, GLUT1, and HK2, thereby increasing glucose consumption and lactate production, likely through the upregulation of HIF-1-mediated glycolytic genes. PGC-1α is overexpressed in many tumors, increasing GLUT4 and HK2 protein levels.670 Additionally, the overexpression of mTORC1 in numerous tumors increases GLUT1 and PFK1 mRNA levels and glucose consumption. The mTOR-induced activation of glycolysis is also associated with HIF-1 activation.188
The PI3K/Akt signaling pathway is crucial for regulating key cellular processes in cancer, including cell survival, proliferation, angiogenesis, and metabolic reprogramming.671,672 Upon activation, Akt enhances glucose uptake in cancer cells by promoting the surface localization of GLUT1 and inhibiting its internalization.666 Additionally, Akt stimulates mTORC1, which upregulates HK2 expression.673 Through increasing GLUT1 and PFK1 levels, mTORC1 facilitates increased glucose uptake and glycolysis, which is significantly tied to HIF-1α activation.188 Several other molecular players also modulate this metabolic shift. Krüppel-like factor 8 (KLF8) and insulin gene enhancer protein 1 (ISL1) increase GLUT4 expression.674,675 The stem cell factor SALL4 augments the expression of HK2 and LDH.676 The oncogene c-Myc directly upregulates a suite of glycolytic genes (HK2, PFK1, TPI, GAPDH, ENO, LDHA, and MCT1), intensifies glycolytic flux in cancer cells,663,677,678 and stabilizes HIF-1α under normoxic conditions by inhibiting its proteasomal degradation through interaction with the von Hippel–Lindau protein.679 KRAS, a prominent oncogene frequently mutated in cancers, characteristically leads to decreased OXPHOS, elevated glycolysis, and increased ROS production.680,681 This effect is mediated through the Raf/MAPK/ERK/c-Myc and PI3K/Akt pathways, which increase GLUT1 and HK expression.682 PTEN serves as a metabolic gatekeeper by inhibiting HK2 expression via inactivation of the Akt/mTORC1/4E-BP1 signaling pathways.683 Consequently, the loss of PTEN can reprogram glucose metabolism in cancer cells. Elevated PTEN levels, on the other hand, diminish glucose uptake while promoting mitochondrial biogenesis and multidrug resistance.684
The tumor suppressor p53 counteracts the Warburg effect and favors mitochondrial OXPHOS through multiple sophisticated mechanisms. A notable mechanism involves the direct interaction between p53 and HIF-1, where p53 sequesters HIF-1, thereby abrogating its metabolic functions. Under normoxic conditions, p53 directly limits glucose uptake by repressing the transcription of GLUT1 and GLUT4, and induces TIGAR expression, thereby reducing PFK1 activity.685 In hypoxic environments, p53 overexpression leads to an increase in glycolytic proteins (GLUT1 and GLUT3) while keeping HK2 levels unchanged, thereby markedly reducing glycolytic flux.686 When p53 is mutated, increased glycolysis is associated with increased protein levels of GLUT1, GLUT3, HK1, and HK2. The nutrient-deprived state of tumor cells can directly activate AMPK, a critical cellular energy sensor that helps restore energy homeostasis.687 AMPK, in turn, stabilizes and activates p53, creating a positive feedback loop that enhances its tumor-suppressive functions.666 Moreover, tumor suppressors such as PTEN inhibit metabolic adaptation by negatively regulating the PI3K/Akt and MAPK/ERK pathways.688
However, owing to the significant differences between intra- and extracellular energy metabolic processes, genetic, environmental, and cellular phenotypic dynamics influence metabolism during tumor development and metastasis.3 The metabolic plasticity in this context poses challenges for cancer therapies targeting the Warburg effect, as complete blockade of glucose uptake often proves to be unfeasible. In the quest for novel metabolic targets, approaches such as CRISPR–Cas9-mediated synthetic lethality screens targeting metabolic genes offer a pathway for identifying specific metabolic targets of interest, particularly in vivo. A genome-scale external library of sgRNAs revealed the critical role of the PBAF complex—involved in regulating HIF-1α metabolism markers—in resisting T-cell-mediated killing of B16F10 melanoma cells.689
Aerobic oxidation
The Warburg theory posits that for many tumor types, cancer cells preferentially generate energy through aerobic glycolysis rather than mitochondrial OXPHOS. However, this notion has been widely contested. Pioneering researchers such as Weinhouse, utilizing isotopic labeling experiments, have demonstrated that the OXPHOS rates of normal and tumor cells are comparable, indicating that mitochondrial functionality in cancer cells is largely preserved.660,690 In the presence of oxygen, tumor cells employ both aerobic glycolysis and OXPHOS to support their rapid proliferation.691 Glycolysis surpasses OXPHOS to become the dominant energy source only under hypoxic conditions, such as within the tumor core.660 This finding underscores that aerobic oxidation remains a vital ATP-generating process in cancer cells. The ability of cancer cells to toggle between glycolysis and oxidation on the basis of their microenvironmental conditions enables them to sustain high proliferation rates.
The process of aerobic oxidation in tumor cells is a complex metabolic pathway that involves the expression of various enzymes and is finely regulated by multiple signaling molecules. During aerobic oxidation, pyruvate produced from glycolysis is converted into acetyl-CoA by the PDH, which then enters the TCA cycle. However, in several cancers, the expression and activity of PDH are disrupted, impairing the normal aerobic oxidation process.692,693 Mutations in key TCA cycle enzymes, such as SDH, fumarate hydratase, and IDH, lead to TCA cycle dysfunction and mitochondrial metabolic defects across various human cancers.694,695 SDH plays a critical role in tumor suppression. Heterozygous mutations in SDH genes result in complete loss of protein function and are associated with hereditary paragangliomas and pheochromocytomas.696 Compared with normal tumors, tumors harboring SDH mutations typically exhibit increased aggressiveness and a significantly faster proliferation rate.697 Moreover, mutations in SDH subunits are also linked to other tumor types, including renal cell carcinoma (RCC), neuroblastoma, gastrointestinal stromal tumors, thyroid cancer, and seminomas.698 In many cancers, FH expression is downregulated or its function is lost, undermining its role as a tumor suppressor. Reduced FH expression leads to the accumulation of HIF-1α699 and high levels of fumarate, a cancer-associated oncometabolite, often resulting in cellular dysfunction in SDH- or FH-deficient cells.700 This can also trigger EMT.701 IDH is another critical enzyme in the TCA cycle, with mutations observed in multiple solid tumors.702 Mutations in IDH impair its ability to catalyze the conversion of isocitrate to α-KG and instead confer neomorphic activity, whereby it reduces α-KG to D-2-hydroxyglutarate (D-2HG) in an NADPH-dependent manner. The excessive accumulation of D-2HG, a tumor-specific metabolite, contributes to the formation of malignant gliomas.703
During the progression of malignant tumors, aberrant activation of multiple signaling pathways plays a crucial role in the process of aerobic oxidation. A prominent example is hyperactivation of the Wnt/β-catenin pathway, which promotes tumor angiogenesis by acting on PDK1.704 Within this pathway, lipoprotein receptor-related protein 5 (LRP5) serves as a significant coreceptor for signal transduction, facilitating Wnt signaling and further promoting cancer progression by increasing aerobic glycolysis.705 Under hypoxic conditions, the expression of HIF-1α is upregulated, promoting glycolysis and adapting to the low-oxygen environment by inhibiting mitochondrial function. Specifically, in Burkitt lymphoma, HIF-1α increases PDK mRNA levels, downregulates OXPHOS,706 and induces a reduction in COX4-1 protein levels, thereby decreasing mitochondrial stability. In contrast, the tumor suppressor p53 enhances mitochondrial metabolism and OXPHOS by promoting the assembly of cytochrome oxidase (COX) and glycolytic enzymes.707 p53 maintains mitochondrial integrity and positively regulates OXPHOS by upregulating the expression of cytochrome c oxidase 2 (SCO2) and apoptosis-inducing factor (AIF), both of which are essential for the assembly of ETC complexes.708
Although cancer cells can generate ATP through aerobic glycolysis, many cancer types, particularly in advanced stages, rely more heavily on mitochondrial OXPHOS to meet their energy demands.709 Mitochondria play a critical role in aerobic oxidation, converting various forms of cellular energy into ATP, which is essential for sustaining the metabolic needs of cancer cells.710 The overexpression of SALL4 promotes cancer cell metastasis by activating the TGF-β/SMAD signaling pathway and increasing mitochondrial OXPHOS levels.711 Cancer cells depend on these metabolic hubs to fulfill their heightened energy requirements and maintain ROS levels, which are crucial for their proliferation, migration, invasion, and metastasis. The overexpression of PTEN in cancer cells increases the protein levels of all respiratory chain complexes and OXPHOS flux while also increasing the levels of mitochondrial transcription factors such as PGC1 and p53.712 Activation of the NRF2/HO-1 signaling pathway suppresses ROS expression levels.713 Through the TCA cycle, cancer cells can secrete succinate into the extracellular environment, inducing the polarization of tumor-associated macrophages (TAMs), which facilitates EMT.714 Specific mitochondrial energy metabolism regulators, such as L-carnitine, BP, PA-2, and DOX, as well as active compounds such as PAB, have shown efficacy in inhibiting tumor cell growth and metastasis.661 These molecules function by inhibiting the PI3K/Akt signaling pathway and activating mitochondrial apoptotic pathways. Notably, melatonin increases p53 expression through the PI3K/Akt/mTOR signaling pathway, which plays a significant role in tumor suppression.715 Conversely, the oncogene MST1 promotes mitochondrial fission and apoptosis by inhibiting the AMPK-SIRT3 pathway, further impacting cancer cell survival.661 Within mitochondria, STAT3 directly interacts with and increases the activity of ND1 and SDH, enhancing OXPHOS flux.670 STAT3 protects ND1 and SDH activity from ischemic damage, acting as a ROS scavenger by reducing ROS production from respiratory chain complexes I and II. Moreover, c-Myc upregulates OXPHOS in various cancer cell lines, with high levels of endogenous c-Myc observed under normoxic conditions;716 however, its mitochondrial function is severely impaired under hypoxic conditions.
Fatty acid metabolism
Cancer cells frequently exhibit significant alterations in lipid metabolism, particularly with increased lipogenesis and fatty acid uptake. Fatty acid synthesis primarily depends on key rate-limiting enzymes: ATP-citrate lyase (ACLY), ACC, and FASN. Studies indicate that reduced expression of ACLY impairs the ability of cells to metabolize glucose into lipids, thereby inhibiting tumor growth.717 Elevated expression of FASN is observed in various cancers, including breast and prostate cancers.718 Fatty acid uptake is equally crucial in cancer cells and is facilitated by fatty acid transport proteins (FATP) and CD36, two key fatty acid transporters.719 In PTEN-deficient prostate cancer, CD36 enhances fatty acid uptake, accelerating cancer progression and suggesting a reliance on exogenous lipid intake.720 The increased expression of CD36 is correlated with fatty acid uptake, which supports the energy and structural demands of cancer cells. SREBP1, a primary transcriptional regulator of lipogenesis, is overexpressed in many cancer types. Its activation elevates the expression of key lipogenic genes, such as FASN, ACLY, and ACC.721,722,723 The activation of SREBPs is stimulated by the PI3K/Akt/mTOR signaling pathway, one of the most frequently activated oncogenic pathways in cancer.724 ACLY can be directly activated by binding to Akt, and its phosphorylation and acetylation increase its stability, promoting the production of acetyl-CoA, while SIRT2 deacetylates ACLY.725 FASN activity is increased through the enhancement of epidermal growth factor signaling via the MAPK and PI3K signaling cascades.726
When cellular energy demand increases, fatty acids are transported into the mitochondria for β-oxidation, where they are converted into acetyl-CoA and subsequently enter the TCA cycle to generate ATP. CPT1 facilitates this critical and rate-limiting step of FA transport into mitochondria. The role of augmented FAO oxidation in cancer cell proliferation is a subject of debate, with different cancers responding variably to alterations in FAO levels. In certain cancers, enhanced FAO might exert an inhibitory effect by depleting FA availability, thereby restraining tumor growth. Conversely, increased FAO may increase ATP production, supplying cancer cells with surplus energy and potentially supporting their proliferation. Consequently, targeting FAO represents a viable strategy to prevent cancer progression. Pharmacological agents such as etomoxir, a CPT1 inhibitor, and ranolazine, which indirectly inhibits FAO, have been shown to effectively induce apoptosis in cancer cells.690,727,728
In cancer cells, the cytosolic NADPH generated from FAO is crucial for mitigating oxidative stress. Under metabolic stress, FAO not only sustains ATP levels but also enhances NADPH production, which is vital for cancer cell survival and proliferation. Studies indicate that inhibiting FAO in glioma cells leads to a marked decrease in NADPH levels and increased accumulation of ROS, ultimately resulting in cell death.728 This process is regulated by AMPK, which promotes FAO by inhibiting the phosphorylation of ACC,729 thereby maintaining a balance between NADPH consumption in fatty acid synthesis and NADPH production via FAO. Additionally, PPAR activation can enhance the FAO pathway.730
Glutamine metabolism
In cancer, a prevalent metabolic alteration is the upregulation of glutamine metabolism. This amino acid is indispensable for cell proliferation, a fundamental discovery made by Eagle in 1955, demonstrating that proliferative capability is curtailed in its absence. Glutamine is integral to mitochondrial oxidative metabolism, facilitating ROS generation. Its conversion into glutathione (GSH) via the enzyme GCL provides antioxidative properties, which are crucial for maintaining cellular redox homeostasis. In mammalian cells, glutamine is a principal energy substrate. Through its catabolism, α-KG is generated, subsequently entering the TCA cycle, thus furnishing tumor cells with critical energy and biosynthetic precursors.731 Elevated glutamine levels are frequently observed in cancer patients. During glutaminolysis, glutamine is converted to glutamate by GLS.732 Wnt2 signaling pathway activation further augments glutamine metabolism, thereby underpinning the energetic demands of cancer cell proliferation and metastasis.733 Glutamate undergoes further degradation through the TCA cycle (via conversion to α-KG) or acts as a substrate for glutathione synthesis. Glutathione, a pivotal antioxidant,734 is instrumental in mitigating oxidative stress. Increased glutamine metabolism bolsters mitochondrial NADPH production through glutathione synthesis, efficiently quenching ROS.735,736
Research indicates that cancer cells exhibit elevated levels of glutamine transporters, particularly ASCT2 (SLC1A5). High ASCT2 expression is associated with increased disease aggressiveness and reduced patient survival.737 Under conditions of low glutamine availability, the transcription factor p53 plays a crucial role in prosurvival signaling by stimulating the expression of transporters to increase the uptake of other amino acids, thereby assisting cancer cells in overcoming nutrient scarcity. With p53 influence, aspartate and arginine uptake is increased, with the latter activating mTORC1 to promote tumor growth.738,739 Additionally, KRAS can enhance the expression of glutamine metabolism-related genes, including GLS, GLUD, SLC1A5, and transaminases, through the action of Myc.740 Myc further activates the expression of genes involved in glutamine uptake and catabolism. Specifically, Myc acts on the promoter regions of glutamine transporters at the transcriptional level, such as SLC1A5 and SLC38A5, facilitating increased glutamine uptake.741 Furthermore, PI3K/Akt pathway activation can stimulate NRF2, which in turn upregulates the expression of glutathione synthetase and glutamate-cysteine ligases, which are essential for glutathione production.742
Electrolytes
Electrolytes such as K+ and Mg2+ play crucial roles in the metabolic reprogramming of tumor cells, impacting cell proliferation and survival.743,744 Magnesium facilitates the killing of target cells by promoting CD8+ T cell activation-induced glycolysis through conformational changes in LFA-1. The activity of HK2 is influenced by intracellular K+ levels; severe K+ depletion disrupts HK2-dependent glycolysis, triggering an energy stress response. In tumors, elevated extracellular K+ is a characteristic of the TME, which may lead to a significant reduction in glycolysis intermediates and essential amino acids inside T cells, inhibiting T cell effector function while maintaining cell survival. However, studies suggest that when tumor-infiltrating lymphocytes exhibit higher intracellular K+ levels, they can suppress the AKT-mTOR signaling pathway driven by T-cell receptors (TCRs), enhancing their antitumor capabilities. Conversely, excessively high intracellular K+ in tumor cells may inhibit the antitumor abilities of tumor-associated macrophages, a process possibly achieved through the readjustment of OXPHOS and glycolysis.745
Telomere-telomerase system
The link between the telomeres-telomerase system and cancer development is close. Although research on how the telomeres-telomerase system regulates energy metabolism is still insufficient, evidence suggests that telomeres can directly or indirectly regulate the metabolic processes of cancer cells, which, in turn, are influenced by cancer metabolism. The tight connection between telomeres and mitochondrial function is evident in the activation of p53 when telomere function is impaired, leading to the suppression of PGC1α and PGC1β expression.746 Reduced expression of PGC1α/β results in mitochondrial biogenesis and functional decline, reducing the expression of genes involved in oxidative defense and thereby affecting the TCA and OXPHOS processes. Additionally, the expression of nuclear respiratory factor 1 and estrogen-related receptor α (NRF-1 and ERRα), which are downstream of PGC, is also inhibited, further exacerbating mitochondrial dysfunction and impeding gluconeogenesis.747 Enhancing the expression of TERT or PGC1α or gene knockout of p53 can increase the expression levels of PGC1α/β, G-6-P, and PEPCK, restoring gluconeogenesis.746 Elevated TERT expression levels significantly enhance the activity of genes in the glycolysis pathway.748 Therefore, telomerase activity may impact cancer cell uptake and utilization of glucose, consequently influencing the glycolysis process. The regulatory role of metabolic enzymes in telomerase function is also crucial. When the expression level of FBP1 increases, FBP1 can interact directly with TERT and induce dephosphorylation of the TERT S227 site. Dephosphorylated TERT cannot translocate to the nucleus, inhibiting telomerase activity in mice and shortening telomere length, thereby suppressing tumor cell proliferation and growth.749 These findings provide potential targets for the development of novel anticancer treatment strategies.
Interplay between energy metabolism and epigenetic modifications
The intermediate products of energy metabolism impact the process of epigenetic modifications; conversely, epigenetic alterations regulate energy metabolic processes. In cancer research, a recent focus lies on the interplay between energy metabolism and histone modifications, particularly those involving lactylation, palmitoylation, succinylation, and others.750 Another crucial aspect is how energy metabolism regulates nucleic acid modifications, including their influence on non-coding RNAs. Genome-wide analyses in cancer have unveiled widespread hypomethylation patterns in DNA, which when present, accompany the activation of transcription, repetitive sequences, transposable elements, and oncogenes, potentially leading to aneuploidy and genomic instability, hallmark features of cancer.751 Conversely, tumor suppressor genes undergo significant methylation alterations, which suppress their expression.752 Acetyl-CoA, a critical product of energy metabolism pathways, undergoes modulation in cancer cells, impacting gene expression by altering the acetylation status. For instance, KRAS mutations promote acetyl-CoA production, enhancing histone acetylation and boosting glucose uptake via an AKT-dependent mechanism.753 The role of lactate in chromatin modifications has been historically overlooked, but recent discoveries suggest that lactate promotes histone lactylation, potentially aiding cancer cell invasion.754 Despite ongoing research, significant unresolved questions persist. A key challenge is understanding how to coordinate these epigenetic processes to promote tumor development through metabolic regulation. Additionally, the efficacy of epigenetic drugs has been limited primarily to hematological malignancies, proving largely ineffective in solid tumors, possibly due to metabolic and epigenetic heterogeneity within tumors. Despite these challenges, high-throughput technologies exploring the bidirectional crosstalk between energy metabolism and epigenetic regulation, to identify specific epigenetic or metabolic vulnerabilities, remain promising endeavors.
Effects of changes in energy metabolism on the immune response within the TME
The TME is a complex ecosystem consisting of tumor cells, surrounding nontumor cells, and stromal components.755 Changes in the energy metabolism of tumor cells significantly impact various immune cells within the microenvironment (Fig. 8). In the TME, tumor cells, effector T cells, and M1 macrophages tend to upregulate glycolysis and glutaminolysis, whereas memory T cells, Tregs, and M2 macrophages primarily rely on FAO.756 This metabolic configuration gives Tregs a survival advantage within the TME.757 The similarity in energy metabolism demands between tumor and immune cells results in competition for essential energy substrates. However, immune cells fundamentally may not possess the same robust metabolic flexibility as tumor cells. Cytotoxic immune cells are impeded in the TME, leading to an overall skew towards immunosuppressive conditions within the TME, thereby promoting malignancy.
Fig. 8
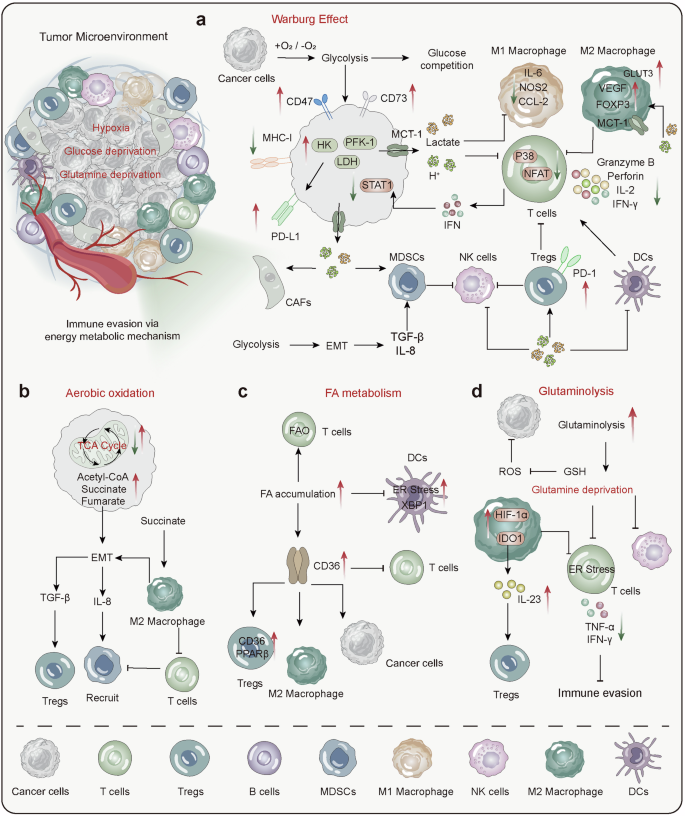
Energy metabolism-driven alterations in the TME. a The enhancement of the Warburg effect in cancer cells leads to glucose scarcity in the microenvironment, triggering competition for glucose between immune and cancer cells, which suppresses energy production in immune cells. Increased glycolysis results in the accumulation of lactate and a decrease in pH within the microenvironment. Lactic acid and low pH inhibit the function of M1 macrophages, activated T cells, and NK cells, reducing the secretion of inflammatory cytokines, perforin, and granzymes, thereby diminishing their cytotoxic capabilities. However, lactate promotes the growth of M2 macrophages, MDSCs, Tregs, and CAFs, potentially due to increased expression of glucose transporters and MCT-1, thus facilitating adaptation to the microenvironment. b Metabolic shifts in mitochondrial OXPHOS lead to the accumulation of intermediates such as acetyl-CoA, succinate, and fumarate. These intermediates promote epithelial-mesenchymal transition (EMT) in cancer cells and further recruit suppressive cells like MDSCs and Tregs through the release of TGF-β and IL-8. c Enhanced FAO promotes the expression of CD36, which facilitates energy production in Tregs, M2 macrophages, and cancer cells but exerts an inhibitory effect on activated T cells and DCs. d Increased glutaminolysis leads to glutamine depletion, thereby inhibiting the function of activated T cells and NK cells, reducing the release of pro-inflammatory cytokines such as TNF-α and IFN-γ, and promoting immune evasion. HK hexokinase, PFK phosphofructokinase, LDH lactate dehydrogenase, DC dendritic cells, Treg regulatory T cells, MDSC myeloid-derived suppressor cells, GSH glutathione
Glycolysis
Tumor cells increase glycolysis to meet their proliferative energy needs, facilitating EMT.758 This metabolic adaptation also affects stromal cancer-associated fibroblasts (CAFs), influencing their oxidative stress and aerobic glycolysis. Tumor cells then utilize lactate and pyruvate secreted by CAFs to sustain increased proliferation.759,760 Mutations, such as those in KRAS, enable cancer cells to withstand nutrient deprivation under hypoxic conditions, allowing them to evade immune surveillance through CD47 regulation.761 High glycolytic activity in tumor cells reduces glucose concentrations within the TME. Such glucose and oxygen deprivation can lead to decreased MHC class I antigen presentation in cancer cells, rendering them unresponsive to IFN-mediated cytotoxic effects due to STAT1 dysfunction.762,763 On the other hand, upon antigen stimulation, T lymphocytes shift from a quiescent state to a highly anabolic state, increasing their proliferation and differentiation into tumor-killing effector cells. However, glucose scarcity can impair the metabolic fitness and functionality of tumor-infiltrating lymphocytes (TILs) within the TME764 and diminish the effector function and viability of NK cells,765 thereby constraining the energy metabolism and antitumor capabilities of these immune cells.766,767,768 Effector T cells require substantial amounts of glutamine during activation and proliferation, and deprivation of glutamine in the TME may also impair T-cell functionality.757,768,769
In addition to energy competition, key glycolytic enzymes, such as HK, PFK, and LDH, play crucial roles in tumor immune evasion. The increased activity of HK and PFK not only enhances glycolysis in tumor cells but is also associated with the expression of immune checkpoint molecules, such as the upregulation of PD-L1.770,771 LDHA is highly expressed in glycolytic tumor cells, and its elevated expression is linked to increased tumor aggressiveness and poor prognosis. Inhibition of LDHA can enhance T-cell-mediated immune surveillance against tumors.772
Glycolytic byproducts directly suppress antitumor immunity by influencing inflammation, immune evasion, and tumor angiogenesis. High glycolysis in tumor cells causes lactate accumulation, which is known to suppress all antitumor immune cells. External lactate exposure reduces cytotoxic T-cell proliferation and decreases IL-2, IFN-γ, perforin, and granzyme B levels.773 Owing to concentration gradients, excess tumor-derived lactate in the TME inhibits lactate secretion by activated T cells,774 leading to endogenous lactate buildup that impairs effector T-cell function.775 An acidic pH in the TME can reduce the antigen-presenting capability of dendritic cells, impairing T-cell activation.756,776 The accumulation of lactate lowers the intracellular pH and inhibits effector T cells by reducing NFAT nuclear translocation and the activity of p38 and c-JNK/c-JUN.666 High lactate levels directly limit NK cell function and indirectly suppress it by increasing myeloid-derived suppressor cell numbers.777 Lactate reduces M1 macrophage activity by lowering IL-6, iNOS, and CCL2 expression.778 It also shifts M2 macrophages and increases PD-L1 expression to aid in immune evasion.661 Lactate induces a tumor-promoting phenotype in M2 macrophages.779 Lactate sustains Treg suppressive function by increasing FOXP3 and MCT1 expression.567 Increased FOXP3 expression reprograms Treg metabolism by inhibiting c-Myc and glycolysis, increasing OXPHOS, and increasing NAD+ oxidation, enabling Tregs to adapt to low-glucose, high-lactate TMEs,602 thus suppressing antitumor immunity.602,780,781 Another crucial focus lies in F-1,6-BP, which is converted into F-6-P by FBP1, impacting the balance between glycolysis and gluconeogenesis and thereby regulating the equilibrium of these processes. Furthermore, F-1,6-BP serves as a vital precursor in the pentose phosphate pathway and is crucial for maintaining cellular redox balance and synthesizing precursor molecules for nucleic acids. In the realm of tumor biology, F-1,6-BP plays a pivotal role. It can enhance immune responses and bolster the cytotoxic effects on tumor cells. F-1,6-BP activates a positive feedback loop of key metabolic enzymes like PFK1, PI3K/Akt, and PFK2/PFKFB3, enhancing aerobic glycolysis to sustain effector T cell activity while inhibiting oxidative metabolism.782 Conversely, studies suggest that F-1,6-BP inhibits AMPK-mediated SENP1-SIRT3 axis activation, leading to reduced T cell memory development.783 Moreover, intracellularly accumulated F-1,6-BP can bind with HMGB1, decreasing the affinity of HMGB1 for DNA and DNA adducts and sensitizing cancer cells to DNA replication stress and damage induced by chemotherapeutic agents, thereby promoting cancer cell apoptosis.784 F-1,6-BP also induces the expression of apoptotic proteins, facilitating apoptosis in liver cancer cells.785 These findings underscore the multifunctionality of F-1,6-BP in tumor metabolism and immune regulation, suggesting novel potential targets for cancer therapy.
Lipid metabolism
Lipid metabolism is pivotal in modulating immune responses within the TME. The accumulation of lipids enhances the expression of the scavenger receptor CD36, which has been shown to suppress the effector functions of CD8+ T cells, playing a critical role in tumor immune evasion.786,787 In dendritic cells (DCs), lipid buildup induces ER stress and activates the transcription factor XBP1, potentially leading to failed presentation of tumor-associated antigens and weakening antitumor immune responses.788 Unlike CD8+ T cells, Tregs increase fatty acid utilization through CD36, aiding their function and adaptation in the TME and thereby maintaining survival and immunosuppressive capacities.567,789,790 CD36 is selectively upregulated in Tregs within tumors, enhancing mitochondrial adaptability via PPAR-β signaling. This metabolic reprogramming enables Tregs to thrive in the lactate-rich TME.789 Additionally, the availability and utilization of fatty acids by Tregs in the TME are linked to resistance to anti-PD-1 therapies.791 In addition to Tregs, TAMs also utilize CD36 to support their tumor-promoting activity by engulfing long-chain fatty acids derived from tumor cells.792
Amino acid metabolism
Excessive glutamine uptake by cancer cells results in a shortage of glutamine in the TME, tipping the immune balance toward suppression. This lack of glutamine is essential for inducing Treg cell differentiation.793 Under glutamine starvation conditions, TAMs activate HIF-1α to produce IL-23, which then increases the number of immunosuppressive Tregs.794 These Tregs inhibit cytotoxic T cells through the release of immunosuppressive agents such as IL-10 and TGF-β, aiding in tumor immune evasion.794 Studies have demonstrated that CD8+ cytotoxic T cells generate significantly less IFN-γ and TNF-α when stimulated in glutamine-deprived environments than when stimulated in glutamine-replete conditions.795 This decrease in cytokine production is linked to reduced effector functions of CD8+ T cells, highlighting the adverse effects of glutamine deprivation. In ovarian cancer, a lack of glutamine may induce endoplasmic reticulum stress in cytotoxic T cells, depleting their glutamine transporters and impairing their function.796 This mechanism may enable cancer cells to evade immune attack, contributing to tumor progression.
Cancer cells and other cells within the TME exhibit extensive intercellular crosstalk in energy metabolism. While the phenomenon of energy transfer between cells has been studied, the intricate connections between these metabolic and nutrient-sensing signaling pathways require further exploration. Additionally, the specific roles played by intermediate products of energy metabolism, such as succinate, fumarate, in carcinogenesis in certain tissues, and their deeper connections with epigenetic modifications, demand further investigation.
Although research on the mechanisms of tumor energy metabolism has been ongoing for several decades and efforts have been made to treat tumors through targeting energy metabolism, the current challenges persist, leading to limited therapeutic outcomes. One aspect of this challenge stems from the inherent characteristics of cancer cells, which exhibit high metabolic flexibility and plasticity. This allows cancer cells to rapidly adapt to unfavorable local environments797 and acquire nutrients from the environment and other cells to fuel their growth.798 On the other hand, the diverse metabolic products and complex intercellular interactions within the TME lead to significant crosstalk among signaling pathways, making it difficult to predict the efficacy of targeting a specific pathway.799 Additionally, due to variations among patients in genetics, environment, diet, and lifestyle, the heterogeneity of cancer microenvironments makes it challenging to identify universal metabolic targets. This shift towards personalized medicine is becoming increasingly emphasized in current research. Despite the numerous challenges, advancements in technology allow for a more comprehensive analysis of energy metabolism changes in the microenvironment through integrating transcriptomics, proteomics, metabolomics, and other omics approaches. Future developments combining molecular probes and biosensors for real-time monitoring of dynamic changes in energy metabolism may help identify appropriate therapeutic windows.
Integrated interactions among diverse metabolic pathways
The intracellular metabolic network is responsible for maintaining metabolic homeostasis through intricate and precise interactions, thereby ensuring a stable energy supply. In various pathological conditions, such as neurodegenerative and metabolic diseases, the interplay among these metabolic pathways becomes increasingly complex. This complexity involves alterations in molecular regulatory mechanisms, the fine-tuning of enzyme activities, the reorganization of metabolite supply and demand, and substantial restructuring of cellular signaling pathways. These changes are crucial for cellular adaptation to pathological environments, enabling survival and the maintenance of physiological functions. For example, cancer cells exhibit significant metabolic plasticity, switching between glycolysis and OXPHOS via complex metabolic networks mediated by HIF-1 and AMPK.800 This metabolic flexibility not only supports their proliferation and survival in diverse environments but also contributes to drug resistance, emphasizing the value of understanding and potentially targeting these metabolic pathways in cancer therapy.
To elucidate these sophisticated metabolic regulatory networks and signaling mechanisms, the concept of “metabolic checkpoints” has emerged.801 Metabolic checkpoints are molecular mechanisms that sense cellular metabolic states and modulate cellular function in response. This concept advances our comprehension of how cells recalibrate their metabolic pathways to maintain homeostasis across various disease contexts. AMPK serves as a prototypical metabolic checkpoint; in response to decreased energy levels from inhibited glucose and amino acid metabolism, it activates lipolysis and suppresses fatty acid synthesis, thus regulating the balance of glucose and lipid metabolism.802 AMPK influences lipid metabolism through several pathways, including the AMPK-PPAR-α-CPT1A axis.803,804 Furthermore, PRMT6 has been identified as a novel metabolic checkpoint, facilitating the metabolic shift between FAO and glycolysis. The absence of Prmt6 enhances FAO-related gene expression via reduced H3R2 asymmetric dimethylation on promoters.805 Similarly, GCN2 functions as a metabolic checkpoint by phosphorylating eIF2α during amino acid deprivation, inhibiting protein synthesis while promoting the expression of genes involved in amino acid biosynthesis, thereby modulating T-cell metabolism and immune response.801
Furthermore, elucidating the metabolic adaptability of bacteria in various environments remains a critical focus for future research. Examining the interactions among diverse metabolic pathways can deepen our understanding of metabolic adaptation mechanisms under pathological conditions and inform the development of innovative therapeutic strategies. This approach holds promise for the design of personalized treatment regimens, offering significant potential in addressing complex metabolic disorders and cancer.
Advancements for disease detection on the basis of energy metabolism
Cellular energy conversion is an intricate, multidimensional process characterized by the dynamic interplay of ions, metabolites, and biochemical products. Disruptions in energy metabolism, or imbalances and dysfunctions in these metabolites, are frequently associated with the pathogenesis of diseases such as diabetes, malignancies, neurodegenerative disorders, and cardiovascular conditions.806,807 Alterations in key metabolic intermediates, particularly those involved in glycolysis and mitochondrial oxidative pathways, such as glucose, lactate, NADH, ROS, glutamate, and ATP, hold potential as biomarkers for disease diagnosis.806 A range of advanced technologies are employed to investigate energy metabolism for diagnostic purposes, including blood and urine biochemical assays, spectrometry, bioprobes, magnetic resonance imaging (MRI) and positron emission tomography (PET) imaging, nanotechnology, metabolomics, and techniques such as electrochemistry and sensing (Fig. 9). These methods play a vital role in identifying metabolic abnormalities and elucidating disease mechanisms.
Fig. 9
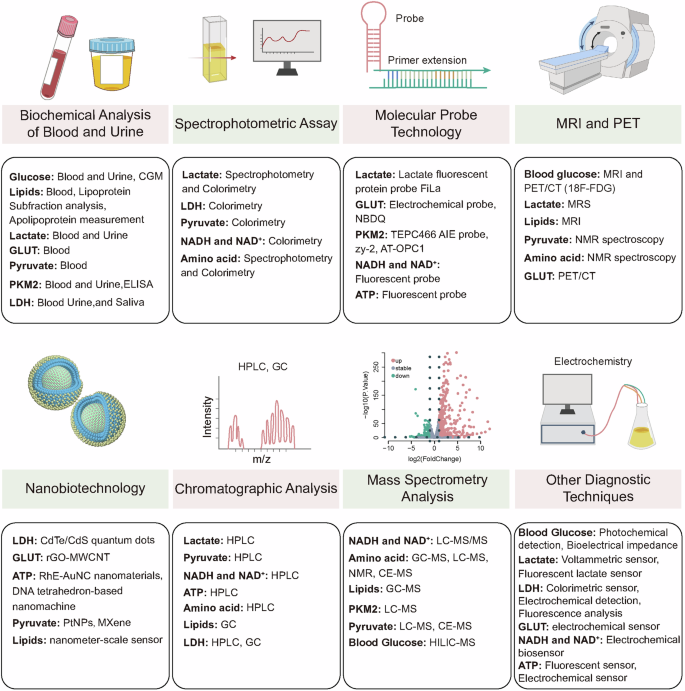
Methods for detecting energy metabolism. MRI magnetic resonance imaging, PET positron emission tomography, HPLC high-performance liquid chromatography, GC gas chromatography, MS mass spectrometry, CE-MS capillary electrophoresis-mass spectrometry
However, each of these techniques has its own advantages and limitations (Table 1). NMR and MS are the most commonly used techniques in metabolomics research. NMR is non-destructive for sample or disease detection, with better reproducibility of results. However, it requires higher concentrations of metabolites, affecting sensitivity.808 On the other hand, mass spectrometry has the advantage of detecting very low concentrations of metabolites but often lacks reproducibility due to the influence of various ions or proteins in the samples and cannot be directly used for in vivo detection.808 Mass spectrometry focuses on metabolite levels, and stable isotope labeling can non-destructively provide dynamic information about metabolites and can be used for in vivo monitoring, but safety issues regarding some radioactive isotopes must be considered.809 Compared with mass spectrometry techniques, fluorescence analysis offers the advantages of convenience, sensitivity, and selectivity. Despite these benefits, the use of basic fluorescence analysis remains limited. This is because fluorescence probes are employed mainly for individual metabolic reactions and metabolites, limiting the ability to conduct flux analysis across entire metabolic pathways.810 On the other hand, research on biologically nanostructured materials such as biosensors is currently a hot topic, as they can continuously and non-destructively monitor specific changes in various circulating metabolites, with improved sensitivity and accuracy, enabling personalized prevention, diagnosis, and treatment.811 Spatial metabolomics based on mass spectrometry imaging provides subcellular resolution (2 μm), offering precise spatial distribution information of metabolites in tissues. However, it can only be used for ex vivo tissue detection, and challenges in slicing adipose and bone tissues may reduce accuracy, while the complexity of the data adds to the difficulty of integration and analysis. Therefore, integrating these techniques in current research will be beneficial for improving the accuracy and sensitivity of studies. The following section introduces the applications of these techniques in detecting specific energy metabolic pathways.
Table 1 Principles, advantages, and disadvantages of different detection techniques
Glucose and lipid detection
Glucose is a fundamental energy source for cells, and its regulation is intricately linked to numerous pathological states, most notably diabetes. Conventional methods for glucose monitoring include capillary blood glucose testing, venous blood glucose analysis, and urine glucose testing. Each method presents limitations: venous blood testing demands frequent sampling and lacks immediacy, whereas urine tests are heavily influenced by renal function, rendering them unsuitable for severe cases. In light of these limitations, innovative glucose monitoring technologies have been developed. Continuous glucose monitoring (CGM) systems have emerged as formidable tools that deliver real-time, continuous glucose data, which enables proactive management of diabetes and the prevention of complications by detecting glucose fluctuations promptly.812 Recent technological advances have introduced noninvasive glucose monitoring techniques such as photochemical, microwave, and bioimpedance methods. The photochemical approach measures glucose changes on the surface of the skin, whereas bioimpedance assesses the electrical resistivity of biological tissues. Despite their noninvasive nature and potential for real-time accuracy, factors such as skin thickness and moisture can affect precision. As technology evolves, these novel glucose detection methods promise more comprehensive, timely glucose data, offering increased support for managing diabetes.
Abnormal lipid metabolism, including dyslipidemia, is a significant risk factor for cardiovascular diseases. Traditional lipid assessments measure total cholesterol, LDL, HDL, and triglycerides and are extensively employed to evaluate cardiovascular risk and inform treatment strategies.813 Advances in the analysis of lipoprotein subfraction and apolipoprotein levels have provided nuanced insights into lipoprotein particle size and concentration, enabling the identification of individuals at increased risk of atherosclerosis.814 These advancements pave the way for individualized approaches to lipid management, aiding in the prevention of cardiovascular complications.
Lactate detection
Lactate is essential for numerous cellular functions and plays a significant role in regulating energy metabolism and signaling. Its buildup in tissue microenvironments is characteristic of many inflammatory and cancerous conditions.778 The Warburg effect, often linked to cancer, is also observed in noncancer diseases such as pulmonary hypertension, fibrosis, heart failure, atherosclerosis, and polycystic kidney disease.815,816 Under stress conditions such as trauma or infection, the body produces lactate through aerobic glycolysis. Thus, lactate serves as a biomarker for various conditions: urinary lactate indicates diabetic nephropathy,817 whereas cerebrospinal fluid lactate provides prognostic insights into cryptococcal meningitis.818
In diagnostics, lactate levels are typically measured via enzyme-based spectrophotometric and colorimetric methods. In clinical settings, especially intensive care, lactate is quantified via LDH-catalyzed reactions, producing NADH, which is measurable by spectrophotometry at 340 nm and correlates with plasma lactate levels.819 Other techniques include voltammetric sensors using ferrocene boronic acid820 and NADH-based fluorescence methods.821 Moreover, high-performance liquid chromatography (HPLC) with fluorescence or mass spectrometry detection is used to analyze lactate in biological samples such as saliva, plasma, and urine.822,823
Traditional detection methods, such as blood, breath, and urine tests, lack the capability for in vivo and live-cell spatiotemporal monitoring, leaving several aspects of lactate metabolism unexamined. Recent innovations include FiLa probes developed by Zhao and collaborators. These advanced fluorescent probes allow high-resolution, real-time lactate monitoring at the subcellular level, surpassing traditional methods such as Seahorse analysis.754,824,825 FiLa technology offers in situ, quantitative tracking of lactate dynamics, an enhanced understanding of lactate distribution, regulatory networks, drug screening, and clinical diagnostics. The team also pioneered dynamic imaging with cell encapsulation systems and portable, high-throughput techniques for rapid lactate detection in small fluid samples, providing a versatile solution.826 Additionally, magnetic resonance spectroscopy (MRS) offers a valuable alternative for assessing lactate metabolism, capturing data unattainable by blood tests alone, as shown in studies on the impact of dichloroacetate in neurological and cancer situations.827
LDH detection
LDH is key in converting lactate to pyruvate.828,829 Subtypes such as LDHA convert pyruvate to lactate, whereas LDHB prefers lactate to pyruvate.828,830,831 Normal LDH levels are under 200 U/L; higher levels can indicate conditions such as malaria,832,833,834 myocardial infarction, and more.835 Persistently high levels postsurgery may suggest residual tumors, whereas decreases indicate successful removal.836 LDH serves as a biomarker for cancers, including colorectal837 and lung cancer,838 and it aids in pleural effusion and chemotherapy guidance.839 This underscores the demand for reliable, simple LDH detection methods.
Colorimetric assays are popular because of their affordability and simplicity, allowing easy concentration measurement.840,841,842 Researchers have crafted a high-stability colorimetric biosensor for LDH, detecting levels as low as 13 U/L in under 5 min, using minimal reagents and imaging technology for quantification.843 A paper-based sensor employing a magnetic immunoassay reached a 0.39 U/L detection limit within 20 min.844 Moreover, a rapid, cost-effective paper-based prototype offers LDH level assessments in less than 4 min, which is ideal for resource-constrained environments, utilizing minimal blood volume and smartphone technology for analysis.843,845
Despite the simplicity of colorimetric methods, their limited sensitivity can hinder broader application.841,846 To address this, a straightforward spectrophotometric assay for salivary LDH detection has been developed, employing NADH oxidation measured at 340 nm. Additionally, a microfluidic microplate-based immunoassay offers rapid detection with reduced antibody use compared with traditional ELISA, reaching limits as low as 6.25 × 10−3 U/L. The challenge of external influences on UV spectrophotometry, such as sample coloration, is mitigated by electrochemical methods, which rely on electrical signal changes and offer advantages in cost and speed via techniques such as cyclic voltammetry, differential pulse voltammetry, and square wave voltammetry.847,848
Widely used in detecting LDH, immunoassays have increased the use of gold nanoparticle-based sensors, which are crucial for detecting elevated LDH in malaria patients.849 These sensors offer heightened sensitivity and reproducibility over commercial kits and are suitable for integration with mobile technology.850 Fluorescent analysis, which uses approaches such as CdTe/CdS quantum dots and SiQDs, provides high sensitivity and a broad linear range.851,852
While current LDH detection methods meet basic needs, innovations are essential for better early screening, with a focus on sensitivity, cost reduction, and improved point-of-care solutions.
GLUT detection
GLUTs are specialized proteins embedded in cell membranes that facilitate glucose movement through diffusion or secondary active transport.853,854 Humans have 14 identified GLUTs categorized into three classes (I, II, III) on the basis of their sequence similarity.855 Research has linked GLUTs with various diseases, suggesting their potential as biomarkers. For example, lower serum levels of GLUT1 and GLUT4 are found in patients with both hypothyroidism and heart failure.856 Neurodegenerative diseases involve decreased GLUT1 and GLUT3, impairing glucose metabolism.857 Furthermore, low GLUT1 expression in the blood is a predictor of severe COVID-19 outcomes.858 The number of cases of gestational diabetes is increasing, with studies reporting higher GLUT1 and GLUT3 levels in the placenta than in healthy individuals.859 In T2DM, increased intestinal GLUT2 expression is observed.860 Early AD is characterized by reduced neuronal GLUT1 and GLUT3, leading to lower D-glucose levels.281 GLUTs also play roles in cancer, where high GLUT1 is linked to poor prognosis in lung, bladder, and oral cancers.861,862,863 Elevated GLUT3 is related to unfavorable outcomes in cancers of the lung, larynx, and oral cavity;861,863,864 thus, GLUTs could serve as diagnostic biomarkers.
Advancements in detecting GLUTs show promise. A study used an rGO-MWCNT composite with a TBO-graphene-gold nanoparticle-GLUT1 antibody as an electrochemical probe, creating a detection platform for live cells. This sensor provides a linear range of 10^5 cells/mL and shows stability and selectivity in detecting GLUT1 across tumor cell types, matching conventional methods such as flow cytometry and Western blotting. It can assess tumor malignancy and differentiate glucose uptake paths, offering cost-effective healthcare solutions.865 Another innovation, is that the NBDQ probe acts as a nonantibody GLUT1 inhibitor and is 30 times more sensitive than traditional tracers in cancer imaging because of the Warburg effect. It shows superior tumor selectivity and biocompatibility in vivo, especially in triple-negative breast cancer models.866
Pyruvate detection
Pyruvate, a key glycolysis product, is essential in cellular energy processes. It is generated in the cytoplasm and fuels the Krebs cycle and OXPHOS in mitochondria.867 Under aerobic conditions, pyruvate is oxidized to produce ATP. Under anaerobic conditions, it is converted to lactate. Interestingly, some cells opt for pyruvate reduction even with oxygen, a process known as the Warburg effect, which maintains energy equilibrium.868 Irregular pyruvate levels are associated with conditions such as diabetes, liver cirrhosis, cardiovascular issues, and neurological disorders.819,869 Its measurement is gaining importance in cancer diagnostics.870,871
Despite its diagnostic potential, pyruvate detection in emergency settings is limited by selectivity issues, as its levels are lower than those of lactate. Proton nuclear magnetic resonance (NMR) facilitates quantitative and qualitative pyruvate analysis.872,873 HPLC combined with fluorescence or mass spectrometry is used to measure pyruvate in body fluids such as saliva and plasma.822,823
Traditional methods, such as ELISA, face challenges such as time consumption and specificity issues. In contrast, biosensors offer rapid, sensitive, and simple detection. A recent biosensor employing platinum nanoparticles on 2D MXenes was developed to detect pyruvate in serum efficiently, with a wide detection range and a low detection limit of 0.7 μM, which was validated in human serum analysis.874 The potential of MXenes as biosensing materials is promising for diverse sensor applications. Additionally, sensors that use Lewis acid/base interactions with diphenylboronic esters for pyruvate detection are being explored for bioimaging purposes.875
PKM2 detection
PKM2, a key glycolytic enzyme, is a prominent PK isoform in mammalian cells because of its vital role in metabolic reprogramming in cancer and active immune cells.876 It serves as a promising marker and therapeutic target in various conditions. Notably, elevated PKM2 levels in the urine of diabetic patients, which are absent in healthy individuals, suggest its potential as a biomarker for early diabetic nephropathy.877 PKM2 is also an early marker for acute kidney injury.878 Its utility as a rapid, noninvasive biomarker for the early detection of structural colon diseases could reduce unnecessary endoscopies.879 PKM2 overexpression in cancers aids in tumor growth and spread,880 reinforcing its role as both a diagnostic marker and a therapeutic target, with significant diagnostic implications.
Recent advancements in PKM2 detection include the TEPP-46-based AIE probe TEPC466, which shows high selectivity and sensitivity for PKM2 via the AIE effect and is particularly useful in imaging colorectal cancer cells for diagnosis and treatment.881 Additionally, the fluorescent probe zy-2 was developed for specific imaging of PKM2 and is able to track it in real time on the basis of concentration and time in PKM2-positive cells, making it ideal for cancer detection.882 Furthermore, the AT-OPC1 probe is designed to label PKM2 at the Lys305 site and uses electrophilic reactivity for precise protein detection through gel-based proteome imaging and real-time cell imaging, offering significant potential in cancer diagnostics.883
NADH and NAD+ detection
NADH and NAD+ play pivotal roles in metabolic processes such as OXPHOS, the TCA cycle, and glycolysis.884 They are integral to cellular oxidation, signal transduction, and safeguarding DNA repair mechanisms.885 Imbalances in the NAD+/NADH ratio are closely associated with conditions such as PD and are associated with elevated NADH concentrations in breast cancer cells relative to those in normal cells. Thus, sensitive and selective real-time monitoring of NADH is essential for diagnosing diverse pathological states and monitoring therapeutic efficacy.
In recent advancements, electrochemical biosensors have emerged as promising alternatives to conventional optical assays such as absorbance or fluorescence-based measurements. Researchers have developed sophisticated sensing technologies, including the synthesis of silver nanoparticles with various morphologies, such as nanorods, nanoprisms, and nanospheres. These nanoparticles are utilized in NADH sensors that are both simple and highly sensitive.
Further innovations involving genetically engineered biosensors such as SoNar, which are designed to monitor NAD+/NADH ratios in live cells, specifically targeting mitochondrial SoNar or the cytosolic SoNar, have been reported. These biosensors provide fluorescence signals that linearly correlate with in situ physiological NAD+/NADH ratios. The differing responses of the cytosolic and mitochondrial NAD+/NADH ratios to acute metabolic perturbations highlight distinct NAD pools. These ratios are modulated by NAD+ precursor availability and are significantly altered under pathophysiological conditions. The deployment of compartment-targeted biosensors alongside real-time imaging offers profound insights into subcellular NAD+/NADH redox dynamics, enhancing future research into the mechanistic roles of NAD+/NADH redox in cellular physiology and disease progression.886
ATP detection
ATP is the principal energy currency in human cells and is critical for various physiological and pathological processes.887 It supports energy balance, metabolic regulation, and cellular communication, playing a key role in conditions such as neurodegenerative and cardiovascular diseases, immune disorders, diabetes, cancer, and obesity.888,889,890,891,892,893,894,895 Precise ATP measurement is essential for understanding disease mechanisms and enhancing diagnostic capabilities.
Despite its importance, efficient and equipment-free ATP monitoring is challenging. Researchers have developed innovative hydrogel microneedles embedded with ATP-specific dual-emission gold nanoclusters. These microneedles allow quick ATP sampling and detection, providing visual identification with high sensitivity, marking a significant advancement in ATP-sensing technologies.896 Additionally, a biosensor with a two-dimensional DNA structure and multiple ATP aptamers enables rapid and sensitive ATP detection. This system capitalizes on aptamer-induced conformational changes, cutting the detection time to 30 min and achieving a detection limit as low as 0.3 pM.897
Multiplexed detection systems have significant implications for disease diagnostics. A comprehensive sensor platform using enzyme-coupled reactions simultaneously detects ATP and lactate. It features dual electrodes on a microcontroller-driven potentiostat chip, incorporating enzymes such as adenylate kinase and PK to generate hydrogen peroxide, resulting in resistance to interference from blood components such as ascorbate and urate.898
Emerging ATP detection technologies, including ATP-targeted fluorescent probes899 and nanomaterial-based signal amplification methods,900 continue to progress. These advancements offer unprecedented sensitivity and speed, enhancing biomedical applications and disease diagnostics.901
Amino acid metabolism detection
Detection methods for amino acid metabolism are crucial for diagnosing and monitoring diseases such as diabetes, neurological disorders, cardiovascular conditions, and cancer, as disruptions in this metabolism are common across these ailments. In diabetes, abnormal amino acid metabolism is linked to insulin resistance, with increased blood amino acid levels. Neurological disorders often involve altered amino acid metabolism, which affects neurotransmitter processes. In cancer, amino acid metabolism dysregulation is tied to tumor growth and metabolic changes. Therapeutically, targeting amino acids, such as asparaginase, in leukemia treatment has demonstrated clinical potential through the disruption of specific amino acid pathways.
Amino acid metabolism detection employs two main methodologies: biochemical analyses and advanced metabolomics. Techniques such as HPLC, gas chromatography‒mass spectrometry (GC‒MS), and liquid chromatography‒tandem mass spectrometry (LC‒MS/MS) are vital for measuring amino acids in samples such as blood and urine.902,903,904 HPLC separates amino acids and applies specific detection methods.905 GC‒MS combines chromatography and mass spectrometry and uses spectral profiles to identify and quantify amino acids.904 LC‒MS/MS combines liquid chromatography with mass spectrometry, which is sensitive, making it effective for analyzing amino acids in biological matrices.906 Nuclear magnetic resonance spectroscopy can be used to analyze several metabolites, including amino acids, simultaneously.907,908
Metabolomics leverages high-throughput sequencing and bioinformatics to offer comprehensive insights into amino acid metabolic pathways.909 These advanced technologies allow the study of global alterations in amino acid metabolism, the identification of metabolic products, and the understanding of their roles in disease progression, providing critical insights into the biochemical foundations of health and disease.
Platelet mitochondrial detection and disease diagnosis on the basis of energy metabolism
Mitochondria are crucial for cellular energy metabolism, and their dysfunction is intricately associated with a variety of diseases.910 Mitochondrial impairment serves as an important biomarker for a range of conditions, including neurological, cardiovascular, infectious, cancerous, and metabolic disorders.911,912,913,914,915,916 Innovative blood-based bioenergetic tests offer a promising noninvasive alternative to traditional tissue biopsies, providing new avenues for assessing mitochondrial function.910,917 Platelets have been validated as indicators of both systemic and tissue-specific bioenergetic shifts in various diseases owing to their abundance, accessibility, and active metabolic role.918,919,920
In neurodegenerative disorders such as AD and PD, mitochondrial dysfunction in platelets may serve as a biomarker for early diagnosis and tracking of therapeutic efficacy, reflecting bioenergetic changes directly linked to these diseases.921,922,923,924,925 In cardiovascular diseases, examining platelet mitochondrial function can provide insights into conditions such as peripheral artery disease, where exercise interventions have been shown to enhance mitochondrial performance, thereby improving patients’ physical capabilities and quality of life.926,927 Moreover, platelet bioenergetics can serve as a prognostic tool in acute infections such as sepsis and chronic infections such as HIV, indicating mitochondrial dysfunction and informing treatment strategies.928,929,930,931,932 In metabolic disorders, such as T2DM and cardiovascular disease, changes in platelet metabolism can mirror the bioenergetic states critical for disease tracking and predicting treatment responses.933,934
Noninvasive metabolic imaging and disease diagnosis
Noninvasive imaging technologies have revolutionized the assessment of metabolic processes within the body. Techniques such as PET and MRI are crucial for visualizing tissue metabolism. PET, particularly with the use of the glucose analog 18F-fluorodeoxyglucose (FDG), is an effective tool for detecting metabolically active tumors, assessing treatment responses, and monitoring disease progression.935 The advent of hybrid PET/MRI systems has advanced metabolic imaging by combining functional and structural data, greatly improving the diagnosis and management of cancer and metabolic disorders.
New metabolic pathways and molecules are increasingly recognized as potential biomarkers. Enzymes and metabolites from the one-carbon metabolism pathway, such as 10-formyltetrahydrofolate dehydrogenase (FDH) and hydroxyprostaglandin dehydrogenase, are often dysregulated in cancers, contributing to tumor development. Additionally, genes related to OXPHOS, such as UQCRQ, NDUFB7, and UQCRC2, are frequently downregulated in gastric cancer, suggesting their potential as prognostic markers.936 Investigating these pathways offers new biomarkers that can enhance early disease detection. Although the discovery and validation of these biomarkers demand extensive collaborative research, their potential impact on diagnostics and therapy is significant.
Advancements in metabolism-focused disease diagnosis have immense clinical importance. Continuous glucose and lipid metabolism monitoring has greatly improved diabetes and cardiovascular disease management, enabling early interventions and reducing complications. Noninvasive imaging techniques have transformed cancer care by allowing precise detection of active tumors and assessing treatment efficacy. Additionally, metabolomics and biomarker discovery offer new insights into disease mechanisms, supporting the development of personalized treatment strategies. Genetic testing has revolutionized the diagnosis of hereditary metabolic disorders, significantly advancing precision medicine. The integration of these diagnostic advancements with emerging technologies such as artificial intelligence and big data analytics holds great promise. The wealth of data from metabolic monitoring and omics methodologies can be used to develop predictive models and decision support systems, fostering early detection and customized treatment plans. Moreover, the discovery of novel metabolic pathways and therapeutic targets could result in innovative treatments specifically designed for metabolic abnormalities, heralding a new era in precision healthcare.
In the field of energy metabolism detection technologies, current challenges primarily include the sensitivity and specificity of the techniques. Owing to significant differences in metabolite concentrations, the detection of low-concentration metabolites demands increased sensitivity from these technologies. Additionally, sample complexity poses a challenge, particularly when dealing with tissue samples or in vivo detection, as the crossover of different cell types and metabolic pathways may affect the accuracy of data interpretation. Moreover, the complexity of data integration and analysis serves as a bottleneck; further research is needed to effectively integrate and accurately interpret data from various technologies.
Advancements for targeting energy metabolism for disease intervention
Energy metabolism is essential for maintaining cell viability, converting nutrients into energy, and ensuring their efficient use within the cell. Balancing this process is vital for cellular health. Disruption of this gene can lead to diseases such as neurodegenerative disorders, cardiovascular issues, and cancer. Therefore, managing and targeting energy metabolism presents significant therapeutic opportunities. Researchers are developing strategies to address metabolic imbalances. One method involves targeting key enzymes or pathways with drugs or gene therapy to restore equilibrium. Another strategy is to adjust patients’ diets and lifestyles, such as adopting low-sugar diets or increasing physical activity, to positively affect metabolic health. Furthermore, innovative therapies, such as pairing metabolism-targeting drugs with standard chemotherapy for improved cancer outcomes, are being explored to improve existing treatments or disease responses.
This section reviews therapeutic targets in energy metabolism disorders, existing drugs, and current clinical trials. These investigations provide fresh insights into disease treatment and steer future medical progress. By understanding and precisely intervening in energy metabolism, we strive to develop more effective and personalized patient treatments.
Targeting energy metabolism for cancer therapy
Metabolic reprogramming, recognized as a fundamental mechanism in cancer progression, provides novel perspectives and strategic avenues for tumor therapy. In-depth exploration of the metabolic characteristics of tumor cells, coupled with the development of targeted therapies that exploit these specific traits, represents a critical and promising trajectory for future cancer research endeavors (Fig. 10 and Table 2).
Fig. 10
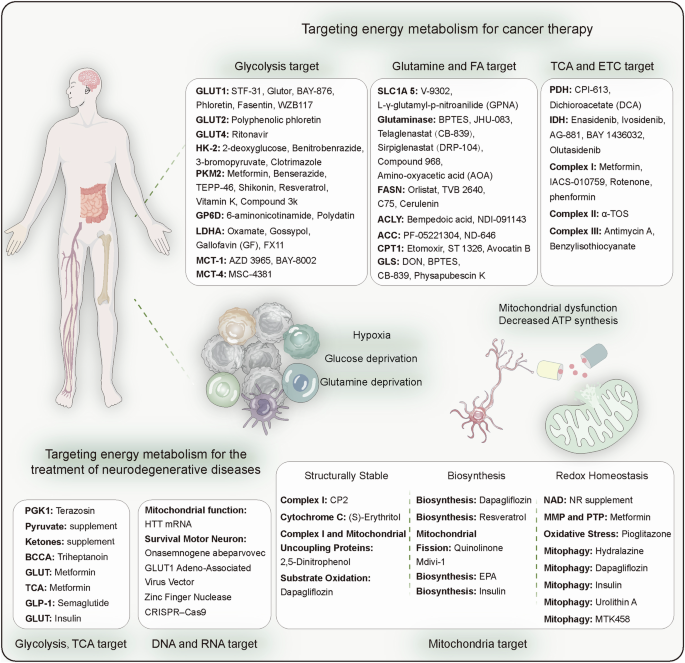
Targeting energy metabolism for cancer and neurodegenerative disease therapy. In cancer, owing to the significant increase in glycolysis, therapeutic drugs are often used to target the glycolytic process. In neurodegenerative diseases, because of mitochondrial dysfunction, therapeutic drugs are often used to target the mitochondrial metabolic process
Table 2 Summary table of selective potential drugs/compounds targeting cancer metabolism pathways
GLUT
GLUTs play crucial roles in the metabolism of cancer cells, with their overexpression linked to increased glucose uptake, which is characteristic of their glycolytic phenotype. This association makes GLUTs attractive targets for cancer therapies.937,938 However, their ubiquitous presence in all cell types complicates their selective inhibition.
GLUT1 inhibitors, including STF-31, GLUTOR, and BAY-876,939 have shown promising anticancer effects by reducing glucose uptake in cancer cells, subsequently suppressing tumor growth. For example, the natural compound WZB117 blocks GLUT1 and inhibits lung and breast cancer cell proliferation while increasing the efficacy of chemotherapies such as cisplatin and paclitaxel.940 Additionally, BAY-876 has been used to increase the efficacy of PD-1 checkpoint inhibitors in models of pancreatic and lung cancer.941 However, GLUT3 overexpression might counter some effects of GLUT1 inhibition, indicating that dual targeting of GLUT1 and GLUT3 could be more effective. Phloretin, an apple-derived compound, acts as a GLUT2 antagonist in triple-negative breast cancer. Studies suggest that phloretin inhibits cancer cell growth in a p53-dependent manner and causes cell cycle arrest.942,943,944 It also increases cancer cell sensitivity to chemotherapy by impeding GLUTs.945 Moreover, GLUT2 activation influences p53 signaling, affecting cell cycle control and apoptosis.946 Ritonavir, an antiviral drug approved by the FDA, has been identified as a GLUT4 inhibitor in multiple myeloma, highlighting its potential for new cancer treatment strategies.947,948
In summary, GLUT inhibitors represent a promising avenue for cancer therapy. By meticulously targeting cancer cell energy pathways, these inhibitors offer the potential for more effective and selective treatments. Future research should aim to further elucidate the mechanisms and applications of GLUT inhibitors in clinical settings to improve cancer treatment outcomes.
HK2
HK2, which is essential for initiating glycolysis, is frequently overexpressed in various cancers. In addition to its glycolytic role, HK2 associates with mitochondrial proteins, sustaining their function and influencing apoptosis.949 This makes it an appealing target for cancer therapies.
HK2 inhibitors present new potential in cancer treatment. Notably, 2-deoxy-D-glucose (2-DG) and 3-bromopyruvate (3-BP) are promising HK2-targeting agents.950 As a glucose analog, 2-DG is incorporated into glycolysis but halts further metabolism due to structural modifications, blocking glucose conversion to G6P, thereby decreasing ATP production and inducing cancer cell death.951,952,953,954 Despite inhibiting growth in multiple cancer cell lines955 and undergoing early clinical trials for certain cancers (NCT 00633087), its development has been curtailed by toxicity concerns.
3-BP has shown robust anticancer activity over the past twenty years by detaching HK2 from mitochondria, thereby reducing ATP levels and promoting cancer cell death. However, clinical trials of 3-BP in cancer therapy have not yet been sanctioned.956,957,958,959 A recent breakthrough introduced benzyl nitrile benzohydrazone (BNBZ) as a new HK2 inhibitor. By targeting HK2, it effectively inhibits pancreatic cancer growth and promotes apoptosis, providing valuable insights into HK2 inhibition. Overall, HK2 inhibitors represent promising avenues for cancer therapy. Overcoming existing challenges and advancing our understanding of HK2 mechanisms could lead to more refined and effective cancer treatment options.
PKM2
Pyruvate kinase, particularly PKM2, plays a vital role in cancer metabolism by converting phosphoenolpyruvate to pyruvate and producing ATP. While PKM1 is prevalent in normal tissues, PKM2 is highly expressed in tumor cells, making it an appealing target for cancer therapies.960
Preclinical studies have shown that metformin enhances chemotherapy sensitivity by disrupting the HIF-1α/PKM2 pathway, inducing apoptosis, and preventing epithelial‒mesenchymal transition.961,962,963 The anticancer potential of metformin, mainly through AMPK activation, is under investigation in numerous clinical trials. In addition to metformin, other agents, such as benserazide—used for PD—exhibit antitumor effects linked to PKM2 inhibition in melanoma cells.964 Natural compounds such as shikonin, resveratrol, vitamin K, and gliotoxin inhibit PKM2, exhibiting substantial anticancer activities. Shikonin, derived from Lithospermum, suppresses PKM2 activity and is effective against various cancers, including bladder and lung cancers.963,965,966,967 Resveratrol decreases PKM2 levels, increasing apoptosis by increasing ER stress and mitochondrial dynamics.968 Vitamins K3 and K5 hinder glycolysis by reducing PKM2, thus lowering HeLa cell viability,969 and they inhibit tumors via multiple pathways.970,971,972 Gliotoxin interferes with cancer cell growth by directly targeting PKM2. Compound 3k (C3k) selectively inhibits PKM2, demonstrating significant anticancer effects in cells with high PKM2 expression.973 Other inhibitors, such as TEPP-46 and mitapivat, have further expanded therapeutic options.974 In summary, PKM2 inhibitors and activators present promising avenues for cancer treatment. Understanding the role of PKM2 may lead to the development of targeted therapies that improve patient outcomes.
LDH
LDH plays a vital role in cancer metabolism, especially in advanced stages where elevated lactate levels are common. The isoenzyme LDHA facilitates the conversion of pyruvate to lactate, regenerating NAD+ from NADH. This process sustains glycolysis and creates an acidic TME associated with metastasis, recurrence, and poor prognosis.
Targeting LDHA is becoming a promising cancer treatment approach. Oxamate, a pyruvate analog, competes for the active site of LDH, displaying antitumor activity in diverse cancer cell lines, such as gastric, medulloblastoma, cervical, liver, and non-small cell lung cancers.975,976,977,978,979 Gossypol, a compound from cottonseed, inhibits LDHA by competing with NADH, offering anticancer benefits.980 It also affects Bcl-2 proteins, induces cell cycle arrest, and promotes autophagy. FX11, a small molecule that competes with NADH, has antitumor effects on the gallbladder, prostate, and neuroblastoma, reducing ATP and inducing oxidative stress, effectively inhibiting lymphoma and pancreatic cancer progression.981,982,983,984 Galloflavin targets both LDH isoforms by binding directly to the enzyme, resulting in in vitro antitumor effects across various cancer cell lines.985,986,987,988 When metformin is used, it enhances treatment effects against pancreatic ductal adenocarcinoma, opening new strategies for tackling solid tumors and metastasis.989 Additional LDHA inhibitors, such as GNE-140, NCI-006, and GSK 28387808, are being studied.974
With a deeper understanding of the role of LDH in cancer, LDHA inhibitors are paving the way for innovative cancer therapies. By employing various mechanisms—competing at active sites with cofactors or altering enzyme binding—these inhibitors hold promise for more targeted and effective cancer treatments.
Monocarboxylate transporter (MCT)
Monocarboxylate transporters (MCTs), especially MCT1, play key roles in cellular metabolism and energy balance. High MCT1 levels in tumors are associated with greater invasiveness and a worse prognosis. MCT1 supports lactate transport, helping tumor cells thrive in acidic conditions and thereby fostering growth and metastasis. AZD3965, a selective MCT1 inhibitor, shows antitumor activity in vitro against diffuse large B-cell lymphoma, non-Hodgkin lymphoma, and Burkitt lymphoma cell lines. By blocking lactate transport, AZD3965 increases lactate inside cells, inhibiting their growth. Its effectiveness was proven in vivo with a Raji xenograft Burkitt lymphoma model.990 It also shows strong cytotoxicity in MCT1-positive, MCT4-negative cells and enhances effects when combined with the GLS1 inhibitors doxorubicin and rituximab.990 AZD3965 is currently in phase I trials for solid tumors and lymphomas (NCT01791595). BAY-8002, another MCT1 inhibitor, reduces cell proliferation and tumor size in Daudi and Raji cells but may cause resistance due to increased MCT2 and MCT4 expression.991 Despite the good tolerance of advanced tumors to AZD3965, its development has paused. However, recent studies suggest that combining lactate-targeting methods, including MCT4 inhibitors, with immunotherapy may offer new treatment options.992,993 Future work should explore the roles and clinical uses of MCT1 inhibitors and their potential synergistic combinations with other therapies.
SLC1A5
SLC1A5, or ASCT2, is essential for the uptake of glutamine, a vital nutrient for cancer cell proliferation. Its overexpression in tumors positions SLC1A5 as a promising target for cancer therapies.994,995
Early inhibitors, such as V-9302 and the monoclonal antibodies KM 4008 and KM 4012, aimed to block SLC1A5. L-γ-Glutamyl-p-nitroanilide (GPNA) mimics glutamine to disrupt SLC1A5 and Na+-dependent amino acid transport, inhibiting glutamine absorption and mTOR pathway activation.996 GPNA effectively reduces cancer cell growth in vitro and in vivo, affecting lung cancer, neuroblastoma, prostate cancer, multiple myeloma, breast cancer, and endometrial cancer.997,998,999,1000,1001,1002,1003 Research suggests that V-9302 might influence other transporters, such as SNAT2, SLC38A2, LAT1, and SLC7A5, rather than directly targeting SLC1A5,1004 indicating that further study is needed to clarify its mechanisms and targets. In addition to V-9302 and GPNA, compounds such as benzylserine and benzylcysteine also inhibit SLC1A5 by competing for glutamine binding sites, displaying antitumor activity in breast, endometrial, and gastric cancers.1003,1005,1006,1007 Importantly, V-9302 enhances T-cell-mediated antitumor responses in triple-negative breast cancer models.1008 In lung and colorectal cancer cells, V-9302 is associated with NF-κB-mediated PD-L1 upregulation, potentially affecting tumor immune evasion.1009
Despite some uncertainties, these inhibitors show promise because they target cancer cells in a glutamine-dependent manner. Continued research should clarify their mechanisms, selectivity, and combination potential to increase cancer treatment efficacy.
GLS
GLS converts glutamine into glutamate, fueling tumor cell growth and survival, making GLS inhibitors a key focus in cancer treatment research. Inhibitors such as BPTES, CB-839, and compound 968 act allosterically, each with unique modes of action. BPTES has antitumor effects on several cancers, such as breast cancer, lymphoma, glioma, pancreatic cancer, non-small cell lung cancer, and renal cancer.1010,1011,1012,1013,1014,1015 However, its clinical potential is limited by its low solubility and bioavailability.1016 CB-839, a BPTES derivative, has overcome these limitations and has received FDA approval. It is undergoing clinical trials for both blood-related and solid tumors, alone and with other anticancer drugs.1017,1018 These trials examine combinations such as everolimus for renal cancer (NCT 03163667), talazoparib or palbociclib for solid tumors (NCT 03875313, NCT 03965845), paclitaxel for triple-negative breast cancer (NCT 03057600), azacitidine for myelodysplastic syndromes (NCT 03047993), and cabozantinib for metastatic renal cancer (NCT 03428217). While some trials fell short of expectations, CB-839 is still being studied in combination therapies.
Compound 968, which targets Rho GTPase, disrupts glutamine metabolism, thereby inhibiting tumor cell growth and invasion and shrinking tumors in animal models.1019 It increases the sensitivity of specific cancers, such as non-small cell lung cancer and ovarian cancer, to chemotherapy.1020 Sirpiglenastat (DRP-104) is another glutamine analog under early clinical trials aimed at treating advanced cancers either as a standalone or with immune checkpoint inhibitors (NCT 04471415 and NCT 06027086).68
In addition to GLS inhibitors, other factors related to glutamine metabolism are promising targets for cancer therapy. For example, transaminase inhibitors such as amino-oxyacetic acid (AOA) have demonstrated efficacy in reducing tumor activity in laboratory and animal studies.1021 Overall, targeting GLS and associated metabolic pathways offers new avenues for cancer treatment strategies.
FASN
FASN is a promising target for cancer therapy because it is overexpressed in various cancers. Preclinical research has shown that FASN inhibitors, such as C93 and FAS31, can significantly suppress tumor growth in small lung cancer and melanoma models.1022,1023 Newer inhibitors such as TVB-316 and TVB-2640 have shown reduced toxicity and are currently in trials for non-small cell lung cancer, prostate cancer, and HER2-positive metastatic breast cancer (NCT 03808558, NCT 03179904, and NCT 05743621).1024 Additionally, the novel compounds TVB-3664 and TVB-3166 demonstrated antitumor effects in laboratory and animal studies, but further clinical trial data are needed.
Orlistat, which is known for its ability to treat obesity, inhibits fat absorption by targeting lipases and acts against FASN, indicating that it is effective against a range of cancers.1022,1025,1026,1027,1028,1029,1030 Cerulenin, another FASN inhibitor, interacts with the β-ketoacyl synthase domain, but its reactive epoxy group raises safety issues. This prompted the creation of C75, a more stable alternative that targets multiple FASN sites, despite retaining side effects similar to those of cerulenin.1031,1032,1033,1034 FASN continues to be a critical focus for new cancer therapies, with research efforts geared toward maximizing efficacy while minimizing adverse effects.
ATP citrate lyase and ACC1 and ACC2
ACLY is a vital enzyme in cellular metabolism that is targeted by several inhibitors. Bempedoic acid, a notable ACLY inhibitor, effectively reduces lipid levels and received FDA approval in 2020 as a cholesterol-lowering drug.1035 In ACLY inhibitor research, NDI-091143 stands out for its strong nanomolar inhibitory activity as an allosteric inhibitor. This compound binds specifically to the homotetramer of ACLY, hindering its catalytic function. NDI-091143 competes with citrate and simultaneously inhibits ATP, allowing precise modulation of ACLY activity.1036
ACC1 and ACC2 regulate cellular fatty acid synthesis. The novel oral inhibitor PF-05221304 is under clinical evaluation for treating nonalcoholic fatty liver disease (NAFLD) and fibrosis (NCT 03248882). While promising for liver disease, its efficacy and application in cancer therapy need further exploration. ND-646, an allosteric inhibitor of ACC1 and ACC2, has been shown to suppress tumor growth in non-small cell lung cancer (NSCLC) xenograft models.1018 These findings position ND-646 as a promising candidate for cancer therapy, emphasizing its potential utility in oncology.
Carnitine palmitoyltransferase 1
CPT1, an enzyme on the mitochondrial outer membrane, is key to FAO. Inhibitors such as etomoxir hold promise in cancer treatment, showing antitumor effects in prostate and nasopharyngeal cancers, acute myeloid leukemia, and breast cancer.727,1037,1038,1039 Etomoxir also works synergistically with CD47 antibodies and radiotherapy.1040 ST1326, a selective CPT1A inhibitor, has strong antitumor activity in Burkitt lymphoma and leukemia cell lines, increasing the effectiveness of the Bcl-2 inhibitor ABT-199.1041,1042,1043,1044,1045 A limited number of studies have shown that oxfenicine, another CPT1 inhibitor, has anticancer activity in malignant melanoma cells.1046 Avocatin B, a natural CPT1 inhibitor from avocados, inhibits AML cell growth without harming normal hematopoietic stem cells, suggesting a potential new approach for blood cancer treatment.1047 Perhexiline, initially developed for angina as an inhibitor of CPT1 and CPT2,1048,1049 may also reduce cancer cell viability in chronic lymphocytic leukemia, as supported by animal model studies.1048
Isocitrate dehydrogenase
IDH mutations are pivotal in various cancers. Agios Pharmaceuticals’ ivosidenib (Tibsovo®) targets these mutations and has received FDA approval to treat AML and cholangiocarcinoma with IDH1 mutations.1050 Ivosidenib is being tested in trials for combination therapies with other cancer drugs, including a phase III trial with azacitidine (NCT 03173248), a phase I trial with cytarabine and fludarabine (NCT 04250051), and a phase I/II trial with venetoclax (NCT 03471260). It is also in a phase III trial for advanced cholangiocarcinoma (NCT 02989857).
Other IDH1 inhibitors, such as BAY 1436032, olutasidenib (FT-2102), and IDH-305, are under development. BAY 1436032, an allosteric oral inhibitor, penetrates the blood‒brain barrier,1051 and although its AML phase I results have shown limited responses, trials for gliomas continue (NCT 02746081). Olutasidenib is undergoing phase I/II trials for AML and myelodysplastic syndromes (NCT 02719574),1052 including advanced solid tumors and gliomas (NCT 03684811). IDH-305 is in phase I trials for malignancies with IDH R132 mutations (NCT 02381886). Enasidenib (Idhifa®) is an IDH2 inhibitor for the R140Q and R172K subtypes. It lowers serum 2-HG by inhibiting the α-KG conversion of mutant enzymes,1050 which has been FDA-approved since August 1, 2017, for relapsed/refactory AML with IDH2 mutations.1053 AGI-6780, another IDH2 inhibitor, shows in vitro promise for cell differentiation, although in vivo studies of tumors are lacking.1054 AG-881 is currently in clinical trials for AML patients with IDH2 or mixed IDH1/2 mutations (NCT 02492737).
ETC
The ETC, comprising four complexes (CI-IV), is vital for drug development, particularly in cancer therapy. Targeting these complexes provides innovative treatment approaches. Metformin, which is recognized for its ability to inhibit complex I, is used to treat T2DM and has shown anticancer potential in laboratory and clinical settings. In contrast, IACS-010759, a targeted complex I inhibitor, is in clinical trials for acute myeloid leukemia (AML) and advanced cancer.1055 While rotenone and deguelin also inhibit complex I, which has anticancer potential, their neurotoxic effects restrict their clinical adoption.1056,1057 Alpha-tocopheryl succinate (α-TOS) inhibits complex II, inducing cytotoxicity in tumor cells by promoting electron leakage and ROS generation.1058 Similarly, complex III inhibitors, such as benzyl isothiocyanate, trigger ROS accumulation and apoptosis in breast, liver, and lung cancer cells.1059 For complex IV and ATP synthesis, inhibitors such as resveratrol and oligomycin have shown promising antitumor activity in skin cancer, neuroblastoma, and lymphoblastic leukemia models.1057,1060
Dietary interventions in cancer treatment
Specific diets hold significant potential in preventing tumor onset, delaying tumor growth, and enhancing the efficacy of current cancer treatments. Given the heterogeneity of cancer and host metabolism, some diets are tailored to target specific vulnerabilities. Caloric restriction, ketogenic diet, and intermittent fasting are among the dietary interventions predominantly employed in cancer therapy research. Since cancer cells typically rely on glucose as their primary nutrient for energy production, restricting glucose intake is a promising therapeutic approach. Limited glucose intake redirects the body towards the utilization of ketone bodies, resulting in beneficial effects in cancer treatment with the ketogenic diet.1061 Owing to the high demand of cancer cells for protein synthesis, restricting essential amino acid intake in the diet can inhibit tumor growth.1062 However, the role of protein intake in cancer initiation and progression remains debatable. Cancer cells often exhibit high expression of lipid receptors and transport proteins. It has been demonstrated that diets rich in lipids derived from the diet and adipocytes contribute to tumor progression and metastasis.1063,1064 Therefore, limiting lipid elevation may have anti-tumor effects, but conclusive data from more preclinical models are needed. Importantly, dietary manipulations induce systemic responses that are not limited to the tumor itself and influence other stromal behaviors, such as the immune system and overall homeostasis. Therefore, the impact of dietary restrictions should be viewed holistically with the aim of maintaining functional anti-tumor immune responses and avoiding cachexia development. Dietary manipulation in cancer therapy may be short-term and should be combined with other treatment modalities to increase patient compliance.
Metformin in cancer treatment
Metformin demonstrates a significant role in energy regulation in cancer treatment, exhibiting certain inhibitory effects on various tumors. One of its primary mechanisms of anticancer activity involves the inhibition of complex I in the cell’s mitochondrial respiratory chain, thereby activating AMPK.1065 Activated AMPK not only exerts anticancer effects by inhibiting the mTOR signaling pathway but also suppresses fatty acid synthesis, effectively inhibiting cancer cell proliferation.1066 Additionally, metformin may induce intracellular and extracellular acidification by regulating lactate metabolism.1067 This acidification of the TME disrupts NAD+ regeneration, inhibiting cancer cell proliferation.1068 Metformin also reduces insulin-like growth factor (IGF) signaling pathway activity, decreases glucose absorption, and promotes glucose breakdown, thus reducing energy supply and growth of cancer cells.1069 In adjunctive therapy, the combined application of metformin with chemotherapy aids in enhancing the sensitivity of patients to chemotherapy. This effect may be attributed to the blood glucose-lowering effect of metformin, which maintains a glucose-deprived environment unfavorable for cancer cell growth, thus increasing the efficacy of chemotherapy.1070 Overall, the mechanisms of action of metformin in cancer treatment are diverse and complex, with the potential to inhibit tumor growth and metastasis. Comprehensively, the combined application of metformin with other treatment modalities in cancer treatment may yield superior therapeutic outcomes compared with standalone approaches.
Brown and beige adipose tissue in cancer treatment
BAT has recently emerged as a potential target in cancer therapy. Studies have shown that cold-induced brown adipose heat production can inhibit tumor growth.1071 Both BAT and tumor cells rely on glucose as their primary energy source. The activation of BAT under cold conditions increases the demand for glucose, potentially limiting the uptake of glucose by tumor cells, thereby inhibiting their growth.1072 Brown adipocytes can efficiently utilize glucose and other nutrients to produce heat rather than ATP, which aids in suppressing tumor growth.1073 Cancer cells often alter metabolic pathways to support their rapid proliferation. Activating brown adipose tissue may alter the overall metabolic state, influencing the metabolic pathways of tumor cells and thereby inhibiting their growth. This metabolic regulation could offer novel insights and approaches for cancer therapy. Therefore, studying and harnessing the anti-tumor effects of BAT may provide new research avenues for cancer treatment, offering more treatment options and opportunities.
Although metabolic enzymes present attractive therapeutic targets for cancer treatment, various targeted drugs are at different stages of clinical trials for multiple reasons. Only a few have been approved by the FDA for cancer therapy. Nucleoside analogs were among the first chemotherapeutic drugs introduced for cancer treatment; however, they not only affect cancer cells but also impact normal proliferating cells. Similarly, targeting other metabolic complexes or enzymes is restricted by their toxicity to normal tissues. Additionally, the metabolic plasticity of cancer cells, where cells can upregulate alternative pathways or acquire nutrients from the environment to adapt to metabolic changes, poses a challenging task that will require simultaneous targeting of metabolic pathways and nutrient clearance routes.1074 The focus of future studies on immunometabolism will be to enhance the understanding of the multifaceted functions of complex immune metabolic signaling pathways within the TME, develop highly effective targeted drugs that combine specificity and safety, or improve cancer immunotherapy in combination with immune checkpoint inhibitors (ICIs) to enhance resistance. The identification of specific, mutation-dependent metabolic vulnerabilities in particular cancers by targeting them to synergize with radiotherapy, chemotherapy, or immunotherapy to induce cytotoxicity is suggested.
Targeting energy metabolism for neurodegenerative disease therapy
Disruptions in energy metabolism, particularly in glycolysis and mitochondrial processes, are central pathogenic mechanisms in neurodegenerative diseases. Targeting these metabolic pathways has shown therapeutic potential in treating these conditions (Fig. 10 and Table 3). Although a decrease in cerebral glucose metabolism has been widely observed in both clinical and preclinical trials of NDs, this reduction in glucose metabolism appears to be unrelated to circulating glucose levels but rather associated with increased blood sugar, such as in diabetes. Therefore, merely supplementing glucose is not a feasible strategy for treating ND; enhancing mitochondrial energy metabolism and improving impaired cerebral basal metabolism may be a more effective treatment approach.
Table 3 Pharmacological treatment of cardiovascular diseases
Targeting glycolysis and the TCA cycle
In neurodegenerative diseases, the downregulation of PGK1, an essential glycolytic enzyme, is prevalent. Activating PGK1 presents a potential treatment strategy. Terazosin enhances PGK1 activity, increasing glycolysis and ATP levels in the brain to address energy deficits in mice.343,1075 Supplementing with pyruvate, a key link between glycolysis and the TCA cycle, improves brain energy status.1076,1077 Ketone bodies such as β-hydroxybutyrate and caprylic acid increase TCA cycle activity by increasing acetyl-CoA levels, independent of glycolysis.1078,1079 These ketones, which are transported to the brain, increase TCA efficiency.1080 Triheptanoin compensates for low levels of BCAAs in Huntington’s disease (HD), addressing energy metabolism problems.1081,1082 Tributyrin formulations improve mitochondrial function and ketone availability, supporting glucose metabolism in AD.1083 The specific effects of ketone body intervention on AD, and its potential relationship with brain lipid distribution and lipid metabolism, merit further investigation. Furthermore, while ketone supplementation has been effective in experimental animal models, its impact in clinical trials has not been as pronounced,1084 warranting further research on its long-term effects on a broader population of individuals with AD. This variability in response may be attributed to individual metabolic differences, emphasizing the importance of personalized treatment approaches.
Diabetes poses a risk for neurodegenerative diseases, and diabetes treatments may aid in disease management. Metformin increases ATP production via glucose metabolism regulation, potentially easing ND symptoms.1085 Semaglutide, a GLP-1 agonist, offers neuroprotection in PD and AD by increasing autophagy, reducing apoptosis, and reducing α-Syn and Aβ toxicity.1086,1087,1088 The further development of complex multi-target drugs that simultaneously possess neuroprotective potential and target specific shared pathways in T2DM and AD holds promise as potential therapeutic interventions.
Mitochondrial targeting in neurodegenerative diseases
Mitochondrial abnormalities, such as decreased respiratory capacity, increased mitochondrial fragmentation, and imbalanced mitochondrial fission/fusion, have been identified in the brains of individuals with AD prior to the deposition of pathological β-amyloid plaques.1089 Thus, mitochondrial dysfunction plays a critical role in neurodegenerative diseases.1090 Research efforts have aimed to increase mitochondrial function by supporting the ETC, stimulating biogenesis, and reducing oxidative damage.1091 Coenzyme Q10 provides neuroprotective effects by enhancing mitochondrial function, but this is only effective in patients with a mitochondrial type of PD. In future treatments, a potential direction could be to assess the risk of mitochondrial dysfunction more accurately for targeted therapy. CP2, through partial inhibition of complex I, enhances mitochondrial energy efficiency, potentially increasing ATP levels in the brain.1092 It also activates AMPK, increasing neuronal protection against oxidative stress, reducing tau and Aβ accumulation, and improving axonal transport.1092,1093
Therapeutic approaches include targeting estrogen receptor-β with (S)-equol to increase cytochrome c oxidase activity in AD.1094 Uncoupling proteins offer new ways to bolster the cellular defense against oxidative stress. Selective inhibition of complex I and targeting of these proteins may enhance ND treatment outcomes.1094 Low-dose 2,4-dinitrophenol has shown promise in ND experimental models.1095 Dapagliflozin safeguards mitochondria in diabetic mouse models, normalizing their size and reducing oxidative damage.1096
Mitochondrial biogenesis, which is crucial for cellular health, is promoted by resveratrol via the SIRT1-AMPK-PGC-1α pathway, which also enhances autophagy to clear damaged mitochondrial components.132,1097 The inhibition of mitochondrial fragmentation is beneficial in ND contexts.1098 Quinazolinone derivatives such as mdivi-1 inhibit excess division, offering neuroprotection in AD, PD, and brain injury.1099,1100 EPA, an ω-3 fatty acid, also supports mitochondrial integrity by altering lipid composition.1101,1102
Maintaining redox balance is targeted by MitoQ, which reduces oxidative stress and offers neuroprotection in animal models of ND.1103 However, clinical trials have not demonstrated neuroprotective effects in PD patients, further emphasizing the differences between animal models and clinical diseases. Altering the NAD+/NADH ratio enhances brain energy status; NR, a precursor to NAD+, alleviates cognitive issues in AD models.272 Metformin and pioglitazone aid in mitochondrial health; hydralazine increases activity, indicating therapeutic potential.1104 Natural antioxidants such as alpha-tocopherol, curcumin, resveratrol, quercetin, and rosmarinic acid are also highlighted as potential therapeutic strategies to counteract oxidative damage and ROS production in AD.1105 However, the specific protective mechanisms of these drugs still require further investigation, including their impact on mitochondrial complexes, modulation of the calcium ion balance, and stabilization of mtDNA.
Clearing dysfunctional mitochondria is key to maintaining cellular metabolism. Insulin is crucial for regulating glucose metabolism and mitochondrial dynamics.1106 Urolithin A promotes mitophagy, reduces inflammation, and improves cognition.1107,1108 PINK1 activators such as MTK458 facilitate mitophagy, suggesting therapeutic avenues for PD.1109
RNA and DNA therapeutics
Recent advancements in oligonucleotide therapies have shown significant clinical success,1110,1111 particularly with the development of antagomirs and locked nucleic acids, which enhance the relevance and efficacy of oligonucleotides in treating neurodegenerative diseases.1112,1113 In Huntington’s disease, antisense oligonucleotides target mutant HTT mRNA to prevent disruptions in mitochondrial transport and function.1114 Allele-specific approaches reduce mutant HTT while preserving normal HTT expression, offering new treatment avenues. Similar strategies are being explored for PD and amyotrophic lateral sclerosis (ALS) with frontotemporal dementia (FTD), addressing mitochondrial gene mutations.1115,1116
Oligonucleotides and small interfering RNAs (siRNAs) modulate pre-mRNA splicing or neutralize microRNAs, increasing the expression of energy-producing genes that are often downregulated in neurodegenerative conditions.1111,1117 Direct gene therapies involve the delivery of full gene copies to restore deficient expression, as observed with the use of AAV vectors for spinal muscular atrophy.1118 Strategies targeting dysfunctional PGC-1α may restore mitochondrial functions in DA pathways in PD models.1119 DNA and RNA editing technologies such as zinc finger nucleases and CRISPR–Cas9 show therapeutic promise. In AD, converting ApoE4 alleles to ApoE3 through gene editing has demonstrated potential in improving brain energetics.1117,1120 Although in vivo APOE editing is nascent, rapid advancements are underway to neutralize ApoE4 and improve AD-related glucose metabolism.1121 HD-linked mitochondrial dysfunction due to excessive mRNA translation highlights a new therapeutic target, with complexes that inhibit translation offering a path to restore mitochondrial energy integrity.1122 Autophagy-targeting chimeric molecules eliminate dysfunctional mitochondria, restoring mitochondrial function and ATP levels in Down syndrome fibroblasts, potentially preventing AD progression.1123 However, the aforementioned studies were primarily conducted in mouse models, and the clinical outcomes have been consistently unsatisfactory. This discrepancy may stem from the fact that disease progression in clinical patients is more severe than that in experimental models, rendering treatments less effective. Nevertheless, further research on mitochondrial gene therapy remains crucial.
Dietary interventions
Diet plays a crucial role in enhancing energy metabolism in neurodegenerative diseases. As mentioned earlier, ketones can serve as a vital energy source for neurons, with dietary control being an effective strategy to intervene in ketone metabolism. A ketogenic diet is crucial for improving neuroinflammation by inhibiting glycolysis and promoting ketone body production. Ketone bodies regulate insulin secretion by inhibiting glycolysis, enhancing insulin sensitivity, and improving glucose tolerance.1124 They also ameliorate mitochondrial dysfunction in neurons and glial cells.1125 The antioxidative effects of ketone bodies on mitochondrial function occur primarily by regulating mitochondrial respiratory complexes, reducing ROS levels, and enhancing antioxidant capacity.1126 Following ketone supplementation, an increase in glutathione levels in hippocampal mitochondria of rats can be observed, which helps protect neurons from damage.1127 The supplementation of ketone bodies in AD and PD patients results in improvements in cognitive and motor functions.1128,1129 However, further research is needed to understand how to enhance the brain’s absorption and utilization of ketone bodies. The consumption of foods rich in antioxidants, such as vitamin C, vitamin E, and polyphenols, assists in scavenging free radicals, reducing oxidative stress-related damage to neurons. Certain nutrients in the diet, such as tyrosine and tryptophan, serve as precursors for neurotransmitter synthesis, enhancing neurotransmitter release and neural signal transmission. Foods containing polyunsaturated fatty acids and omega-3 fatty acids contribute to improving lipid metabolism and alleviating neuroinflammatory responses. The Mediterranean diet and the Jiangnan diet are rich in fiber, antioxidants, polyunsaturated fatty acids, phenolic compounds, and other beneficial substances, which have positive protective effects on neurons.1130
Identifying new vectors, novel therapeutic targets, and reliable transgenic delivery pathways remains a primary focus in research on gene therapy for NDs. While clinical trials related to gene therapy are ongoing, satisfactory outcomes have not yet been achieved. Future research should focus on the cellular and molecular mechanisms associated with neurodegenerative diseases to identify more effective diagnostic and therapeutic targets. Despite promising therapeutic avenues for ND treatment indicated by recent studies, the majority of new treatments have failed in clinical trials. The development of new drugs remains a constant area of research aimed at delaying disease progression and preventing cell death. Personalized treatments focusing on comprehensive metabolomics and genetics are essential for addressing neurodegenerative diseases, considering their prevalence in aging populations and co-occurrence with multiple conditions.
Targeting molecular pathways for populations with metabolic disturbances may be more effective, such as the use of PPAR agonists like pioglitazone, which has shown therapeutic efficacy in clinical trials. Statin drugs, on the other hand, may be more beneficial for populations with comorbidities like hyperlipidemia. Recognizing the limitations of single-target drugs such as antioxidants and neuroprotective agents in improving ND symptoms, there is a continued emphasis on developing new drugs or combination therapies to address multifactorial causes, potentially offering more effective treatments for this disease.
Inflammation and energy metabolism collectively influence disease progression, yet the efficacy of anti-inflammatory drugs in alleviating ND symptoms is limited. Further exploration is needed to understand the role and therapeutic potential of inflammation at different stages of the disease. Understanding the unique nature of the blood-brain barrier is crucial for improving drug permeability and targeting to avoid peripheral organ side effects. Additionally, in addition to targeting neuronal cells, targeting immunometabolic reprogramming in astrocytes to prevent neuroinflammation may present a new avenue for ND treatment, necessitating the differentiation of various cell types within the substance.1131,1132,1133
Targeting energy metabolism for cardiovascular disease therapy
Harnessing the modulation of energy metabolism represents a novel therapeutic approach that shows promise for cardiovascular disease management. The study of metabolic interventions has increasingly concentrated on the dynamics of cardiac fatty acid and glucose metabolism, along with mitochondrial oxidative capacity, as pivotal areas impacting cardiovascular health. Strategically adjusting metabolic processes within cardiomyocytes—including the enhancement of glucose oxidation and the reduction of FAO—offers a pathway to bolster the energy provision for heart cells, consequently increasing cardiac pump efficiency (Fig. 11).
Fig. 11
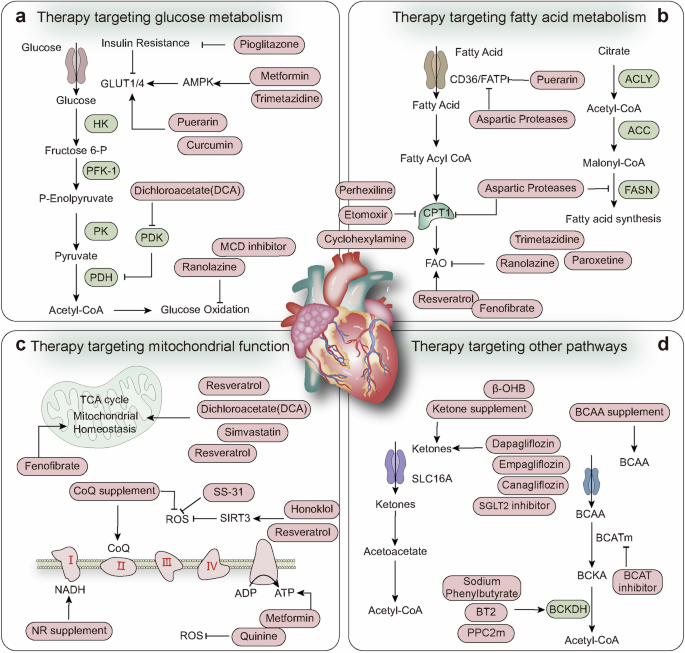
Targeting energy metabolism for cardiovascular disease therapy. a Various drugs, by targeting the expression of glucose transport proteins and glycolytic enzymes, can increase energy production in the heart and inhibit the process of oxidative stress. b Medications that target FAO inhibit the highly oxygen-consuming process of FAO and synthesis, shifting towards glycolysis, which can maintain the stability of heart function under hypoxic conditions. c By targeting mitochondria to maintain the stability of the TCA cycle and the electron respiratory chain, the generation of ROS can be reduced, ATP production can be promoted, and the stability of the heart can be maintained. d By supplementing with external energy substances, including ketone bodies and branched-chain amino acids, the energy synthesis and functional stability of the heart can be promoted
Targeting glycolysis
Glycolysis has emerged as a pivotal therapeutic target in cardiovascular diseases, serving as a critical energy source for cardiomyocytes adapting to hypoxic conditions. The enhancement of glycolytic pathways holds promise for early intervention. Metformin, by activating AMPK, restores glucose uptake in insulin-resistant myocardial cells, showing efficacy in diabetes-related cardiac issues.1134 Pioglitazone improves insulin sensitivity and glucose uptake while preventing mitochondrial dysfunction, mitigating damage during ischemia‒reperfusion.1135,1136 Trimetazidine activates AMPK, enhancing mitochondrial function, insulin sensitivity, and GLUT4 translocation and increasing glucose uptake.1137 Ranolazine significantly augments glucose oxidation in ischemic and reperfused rat hearts,1138 whereas malonyl-CoA decarboxylase inhibitors increase glucose oxidation and improve ischemic cardiac function.1139
Notably, trimetazidine shifts cardiac metabolism from FAO to glucose utilization by inhibiting mitochondrial long-chain 3-ketoacyl CoA thiolase.1140 Carvedilol optimizes substrate preference from fatty acid to glucose oxidation, reducing oxygen consumption and enhancing efficiency.1141 Neuregulin modulates PACC, CPT1, CD36, and PGC-1α expression while increasing GLUT4 transport, optimizing cardiac energy metabolism.1142
Natural compounds such as puerarin, curcumin, and resveratrol show therapeutic potential. Puerarin improves postinfarction function in diabetic mice by modulating GLUT4 and CD36 expression and translocation.1143 Curcumin activates the SIRT1-Fox1 and PI3K/Akt pathways in diabetic rat hearts, promoting glucose uptake and utilization and inhibiting oxidative stress and apoptosis.1144 Resveratrol normalizes FAO, enhances glucose use, and mitigates oxidative stress.428 These findings underscore the importance of targeting glycolysis and metabolic modulation in cardiovascular disease management.
Targeting mitochondria
Mitochondria, as principal energy sources, play indispensable roles in cardiovascular health by regulating intracellular calcium levels, oxidative reactions, and apoptosis.1145 However, their dysfunction is a catalyst for cardiovascular disease progression, as evidenced by significant mitochondrial disruptions during the early stages of these diseases.1146 The pathogenesis of cardiovascular conditions is closely linked to oxidative stress, which arises from an imbalance in ROS and is coupled with decreased endogenous antioxidant enzymes.1147
Enhanced lipid peroxidation, which leads to vascular membrane damage, is central to cardiovascular disease mechanisms.1148 To address such challenges, several mitochondria-protective drugs have been identified. For example, coenzyme Q10 supports mitochondrial integrity by increasing ATP levels, enhancing radical scavenging, and promoting electron transport in the respiratory chain.1149,1150 Metformin mitigates oxidative stress, reduces mitochondrial HSCoA depletion, and elevates cardiac HSSA and ATP levels.1151
Similarly, simvastatin sustains mitochondrial function by inducing lipid droplet accumulation, facilitating mitochondrial autophagy, and preventing mitochondrial damage.422 MCD inhibitors enhance mitochondrial antioxidative capacity by modulating malonyl-CoA levels, consequently reducing FAO and proton generation.430 Dichloroacetate (DCA), a PDK inhibitor, enhances Krebs cycle flux, preventing myocardial hypertrophy and restoring contractile reserve without altering the expression of key metabolic regulators.1152 Pioglitazone, a PPAR-γ agonist, has the potential to prevent mitochondrial disruption and intramyocellular lipid imbalance, which are crucial for reversing severe pulmonary arterial hypertension and vascular remodeling.1135 Nicotinamide riboside (NR), an NAD+ precursor, has been demonstrated to protect mitochondrial function in peripheral blood mononuclear cells and reduce inflammation in advanced heart failure patients.1153 Fenofibrate contributes uniquely to cardiovascular treatment by enhancing mitochondrial function and FAO and increasing SDH activity and the expression of genes such as PPAR-α and SIRT3.441
In addition to synthetic drugs, natural compounds have shown remarkable mitochondrial protective effects. Antioxidants reduce the risk of heart disease through their ability to scavenge free radicals, which inhibits the generation of ROS and enhances endogenous antioxidant enzymes.1154 Anthocyanins counteract mitochondrial dysfunction through multiple pathways, including the inhibition of cytochrome C reduction and caspase activation.1155 Resveratrol preserves the mitochondrial membrane potential in cardiomyocytes, inhibits apoptosis, increases mitochondrial respiratory enzyme activity, and curtails ROS production.428 Increased lipid peroxidation severely impacts the mitochondrial function of cardiac cells by inducing ferroptosis, making the inhibition of ferroptosis a current focus in the treatment of cardiovascular diseases. Treatment strategies primarily involve reducing the extent of lipid peroxidation, decreasing iron ion concentrations, disrupting iron transport, and regulating the expression of ferroptosis-related proteins.1156
Targeting FAO
Although FAO is a primary energy source, is relatively inefficient because of its high reliance on oxygen molecules during metabolism.432 Enhancing metabolic efficiency in the heart is paramount, especially in cardiac diseases where the oxygen supply may be compromised. CPT1 is a key player in mitochondrial fatty acid uptake, and modulating its ability to inhibit fatty acid absorption could redirect cardiac metabolism toward more energy-efficient pathways under pathological conditions. Inhibiting CPT1 with compounds such as perhexiline and etomoxir has been shown to reduce FAO, thereby mitigating heart failure symptoms.433 Etomoxir, in particular, inhibits CPT1 and promotes glucose oxidation, conferring cardioprotection against ischemic injury.433 In patients with chronic heart failure, CPT1 inhibitors such as perhexiline and etomoxir have been shown to improve the left ventricular ejection fraction.1157,1158 These inhibitors optimize cardiac energy metabolism by curtailing long-chain fatty acid uptake and activating glucose dehydrogenase to increase carbohydrate oxidation.1159 Notably, etomoxir effectively prevents contractile dysfunction in animal models, likely by enhancing myocardial performance. Perhexiline, a CPT1 inhibitor, induces a metabolic shift from fatty acid to glucose oxidation, triggering a complex rebalancing of carbon and nucleotide phosphate fluxes.1159 This metabolic redirection not only optimizes energy production but also enhances metabolic flexibility by increasing lactate and amino acid uptake.1159 Furthermore, perhexiline may exert additional effects beyond metabolic redirection, such as reducing ROS production throughout the cardiovascular system by altering the NADH/NADPH ratio.1160 Given that ROS are key contributors to cardiovascular damage, this property of perhexiline could have significant implications for the prevention and treatment of cardiovascular diseases.
In addition to limiting lipid absorption, directly inhibiting FAO represents a promising therapeutic approach.1158 Trimetazidine, a drug that restricts FAO by blocking the enzyme 3-ketoacyl-CoA thiolase while simultaneously promoting glucose oxidation,1161 not only improves the cardiac energy supply but also may regulate cardiac function by reducing systemic resting energy expenditure.1162 In patients with dilated cardiomyopathy and myocardial ischemia, trimetazidine has been shown to increase the left ventricular ejection fraction,1163 indicating a direct positive impact on cardiac function. Moreover, trimetazidine treatment has been found to attenuate insulin resistance and improve insulin sensitivity in patients with idiopathic dilated cardiomyopathy.1163 Improving insulin sensitivity is particularly crucial for diabetic patients, as insulin resistance underlies the pathogenesis of T2DM and is associated with various metabolic disorders. By enhancing insulin sensitivity, trimetazidine may contribute to improved metabolic states in the heart. Abnormal lipid metabolism is closely associated with heart diseases, exacerbating cardiomyocyte death and decreasing bioenergetic production through mechanisms such as the induction of ferroptosis. Ferroptosis is closely associated with other types of cell death, including apoptosis, necroptosis, and necrosis. Future research should focus on understanding how the regulation of this mixed cell death affects cardiac energy metabolism. Past studies have highlighted the roles of ZBP1, AIM2, and other regulators of mixed cell death in heart injury; however, research on how these molecules specifically impact the cardiac energy metabolism process is limited. Investigating the functions of these proteins and identifying additional common regulatory molecules as potential intervention targets are crucial.
Dietary interventions
Dietary adjustments play a crucial role in the treatment of cardiovascular diseases. The proportions of fats, proteins, and carbohydrates in the diet directly influence the energy metabolism of individuals with cardiovascular diseases. The intake of antioxidants and unsaturated fatty acids in the Mediterranean diet contributes to improving lipid metabolism, benefiting cardiovascular health.1164 Diets high in fiber and low in saturated fats help control chronic inflammation, improve lipid and glucose metabolism, thereby reducing the risk of cardiovascular diseases. Components of the diet, such as fiber and probiotics, play crucial regulatory roles in balancing the gut microbiota. The Jiangnan dietary pattern shares similarities with the Mediterranean diet in terms of nutritional components and plays a positive role in controlling cardiovascular diseases. However, Western HFDs rich in choline can be converted by gut bacteria, leading to elevated levels of trimethylamine N-oxide, consequently promoting the development of cardiovascular diseases.1165 Foods rich in antioxidants such as vitamin C, vitamin E, and flavonoids in the diet aid in scavenging free radicals, reducing oxidative stress damage and thereby promoting mitochondrial energy production. A ketogenic diet has a beneficial effect on cardiovascular diseases, primarily because of its anti-inflammatory and antioxidant effects and the provision of alternative fuel for cardiac metabolism.91 Moreover, the role of intermittent fasting in improving cardiovascular diseases also relies on ketone generation and the enhancement of risk factors such as obesity and lipid imbalances.91
Cardiovascular diseases often involve alterations in lipid and glucose metabolism. Shifting the balance from fatty acid β-oxidation to glucose oxidation to optimize energy substrate metabolism has been suggested as a therapeutic strategy for treating (ischemic) heart disease. However, increased glycolysis may lead to myocardial hypertrophy, necessitating the prioritization of FAO to alleviate cardiac hypertrophy. This underscores the dynamic transitions in heart disease, demanding corresponding changes in treatment approaches. Recent research on cardiovascular disease treatment has focused on various natural or synthetic active compounds that protect mitochondrial function and maintain energy production. These compounds act by reducing ROS, inhibiting abnormal lipid peroxidation, and stabilizing the ETC, with promising results from clinical trials of these active substances.1166,1167
Despite significant efforts, there are currently no approved treatments specifically targeting cardiac metabolism. This is partly due to differences in energy metabolism between animal models and human physiology and a lack of reliable non-invasive detection methods. With the increasing prevalence of cardiovascular diseases, more significant challenges are posed for effective preventive and therapeutic strategies, necessitating continued exploration and validation of new biomarkers to enhance early diagnosis of cardiovascular diseases. While advances in metabolomics have led to the discovery of numerous biomarkers, translating metabolomics research findings into diagnostic and therapeutic methods in clinical practice still faces various challenges, including accuracy, cost, and practicality. The development of gene editing technologies like CRISPR/Cas9 offers opportunities to screen potential pathogenic genes and target treatments for certain genetic cardiovascular diseases. However, substantial variations in metabolic levels exist among individuals, highlighting the challenge of studying and understanding the differences in cardiovascular disease metabolic characteristics among various individuals for the personalized treatment and prevention of cardiovascular diseases.
Targeting energy metabolism for autoimmune disease therapy
Energy metabolism is a critical factor in the pathogenesis of autoimmune diseases, particularly in supporting the proliferation and function of activated T and B cells, which have high energy demands. These metabolic perturbations are not isolated events but are interconnected and interact within the body. The consequence of this interaction is the breakdown of immune tolerance, manifested by the proliferation of follicular helper T (Tfh) and Th17 cells, impaired function of Tregs, aberrant B-cell activation leading to excessive autoantibody production, elevated levels of inflammatory mediators, multiorgan inflammation, and tissue damage, ultimately culminating in the development of autoimmune disorders. Thus, modulating energy metabolism to ensure proper differentiation and function of immune cells is paramount for maintaining immune homeostasis and treating autoimmune conditions.
Current therapeutic approaches for autoimmune diseases include the use of nonsteroidal anti-inflammatory drugs (NSAIDs) to manage pain and inflammation, glucocorticoids (GCSs) to suppress an overactive immune system, and disease-modifying antirheumatic drugs (DMARDs) to inhibit the release of inflammatory mediators and immune cell proliferation (Table 4). Patients diagnosed with autoimmune diseases in the early stages may benefit from these interventions, as they help mitigate inflammatory symptoms and prevent disease progression.1168 As research has advanced, the significant connection between these therapeutic agents and the modulation of energy metabolism has become increasingly evident. Ongoing studies targeting energy metabolism are providing novel insights and strategies for the treatment of autoimmune diseases, offering promising avenues for future therapeutic interventions.
Table 4 Pharmacological treatment of autoimmune diseases
Targeting glycolysis
Imbalances in immune cell homeostasis are pivotal in the progression of autoimmune diseases, driving their continuous progression. Adaptations in the local microenvironment lead to a reprogramming of energy metabolism to accommodate these changes. Immune cells exhibit distinct metabolic preferences; for example, in RA, increased glycolysis is evident through elevated GLUT1 activity, yet this pattern differs between CD8+ and Tregs.567,1169 These cells offset GLUT1 deficits by upregulating GLUT3 or GLUT6,1169 whereas Treg cells rely on FAO instead of glucose uptake.567 Given these metabolic variations, managing glucose influx via GLUT1 regulation has emerged as a promising therapeutic avenue. The investigative use of CG-5, a broad-spectrum glucose transport inhibitor, highlights its potential in autoimmune therapy by curbing Th1 and Th17 differentiation and diminishing T-cell proliferation in mixed lymphocyte reactions.1170
HK, the enzyme that initiates glycolysis through glucose phosphorylation, is competitively inhibited by 2-deoxyglucose (2-DG), which structurally resembles glucose and binds to HK2, thereby hindering its activity through the accumulation of phosphorylated 2-DG.1171 Preclinical trials have demonstrated the efficacy of 2-DG in decelerating arthritis in K/BxN mice and suppressing immune cell activation.1171 Metformin, a staple in diabetes management, inhibits FLSs via the Akt/mTOR pathway, alleviating arthritis symptoms.1172 When used alongside 2-DG, metformin enhances IL-2 production in CD4+ T cells and mitigates disease symptoms in lupus-prone mice.1173 Inhibiting HK1 and HK2 with lonidamine has been shown to alleviate joint damage in collagen-induced arthritis models1174 while reducing IL-1β and TNF-α levels and restoring macrophage anti-inflammatory functions in RA models. 3-Bromopyruvate (3-Br-PA) not only affects SDH but also inhibits HK2, showing promise as a novel treatment for inflammatory arthritis.582 Its application, alongside FX11, an LDHA inhibitor, effectively decreases lactate production in stimulated synovial fibroblasts, attenuating inflammation in mice.579,1175 Rapamycin, which targets mTOR, has been confirmed to aid in Treg development and function in SLE while inhibiting Th17 cell maturation.1176,1177 Through dual disruption of glycolysis and glutaminolysis, bioactive compounds from plants, such as C28MS, significantly alleviate arthritis severity, suggesting their potential as RA treatments.1178
The metabolic processes underlying anti-inflammatory drug actions in autoimmune disease treatments often involve glycolysis modulation. Among nonsteroidal anti-inflammatory drugs, aspirin dissociates mitochondrial-bound HK2 by disrupting its interaction with VDAC1,1179 whereas diclofenac suppresses GLUT1 and HK2 activity, thus reducing glycolysis.1180 Methotrexate, a csDMARD, diminishes HK2 and GLUT expression in RA FLSs,1181 and iguratimod disrupts the HIF-1α-HK2 axis, compromising Tfh cell function.1182 Tofacitinib, a csDMARD, impairs glycolysis in RA-FLSs, decreasing the expression of HK2, GSK3A, LDHA, and HIF1A1183 and promoting the dissociation of HK2 from the mitochondria.1184
Targeting mitochondria
In autoimmune disorders, immune cells such as activated T and B cells often increase their mitochondrial OXPHOS to fulfill the substantial energy requirements for proliferation and immune responses. However, excessive production of ROS can damage mtDNA, proteins, and lipids, leading to cellular dysfunction and intensified tissue inflammation. Thus, targeting mitochondrial processes represents a promising therapeutic strategy.
MitoTempo, a mitochondrion-targeted antioxidant that mimics superoxide dismutase, effectively neutralizes mitochondrial oxidative stress. Its administration has been shown to decelerate disease progression in MRL/lpr lupus mouse models, demonstrating potential for broader clinical applications.1185
N-Acetylcysteine (NAC), a robust antioxidant, has shown positive therapeutic effects in SLE patients.1186 NAC treatment significantly curtails the production of anti-double-stranded DNA antibodies and reduces T-cell proliferation, which is vital in managing SLE. By increasing the mitochondrial membrane potential in T cells, NAC facilitates apoptosis, aiding in the control of abnormal T-cell activation and proliferation. In CD4+ T cells, NAC additionally enhances the expression of immune regulatory markers such as Foxp3 and CD25, potentially modulating immune responses and mitigating disease symptoms.
BZ-423, a 1,4-benzodiazepine derivative, exerts its effects by increasing ROS levels and promoting apoptosis. In SLE mouse models, BZ-423 effectively inhibits hyperactive autoreactive T cells with elevated ATP synthase expression.1187 Treatment with BZ-423 converts mitochondrial oxygen to ROS, triggering apoptosis and significantly improving clinical manifestations in SLE models.
Metformin modulates immune responses by activating AMPK and inhibiting mitochondrial complex I activity.1188 This action reduces ROS formation, effectively blocking NETosis and IFNa production in SLE.1189,1190 Inhibitors of the JAK-STAT pathway, such as tofacitinib, possess strong anti-inflammatory properties, significantly decreasing the mitochondrial membrane potential, mass, and ROS generation in RA synovial fibroblasts.1183 These inhibitors affect key mitochondrial genes and increase OXPHOS and ATP production while reducing the expression of genes related to glycolytic pathways and related genes.1183
Itaconate, an SDH inhibitor, modulates succinate and inflammatory cytokine levels in activated macrophages,1191 showing associations with decreased disease activity and enhanced treatment outcomes in animal models of arthritis.1192
Targeting FAO
Elevated levels of BAFF have been observed in various autoimmune diseases.1193,1194 B cells exposed to high levels of BAFF show enhanced metabolic capabilities and can evade tolerance checkpoints.1195,1196 The BAFF-specific monoclonal antibody Belimumab, which inhibits this dysregulated signaling pathway, has been approved as an adjunct therapy for SLE and has demonstrated efficacy in clinical trials, including improvements in B-cell dysfunction.1197 While the primary effects of rapamycin are attributed to alterations in T cells, it also inhibits BAFF-mediated mTORC1 signaling in B cells, thereby limiting their proliferation and survival.1198
Statins, by competitively binding to the active site of HMG-COA reductase, effectively block the biosynthesis of cellular cholesterol. In the treatment of SLE, statins not only regulate lipid metabolism but also exert beneficial immunomodulatory effects, offering therapeutic advantages to SLE patients.1199 Pioglitazone, a PPAR agonist, promotes the functional expansion of dendritic cells in SLE patients by activating AMPK and inhibiting the mTOR1 signaling pathway. These cells express high levels of PPAR receptors, and the effect of pioglitazone has been validated in vitro.1200 Furthermore, by enhancing the expression of CD36 and activating FAO, pioglitazone promotes lipid absorption, offering a new perspective on metabolic regulation in SLE patients.1201
N-butyldeoxynojirimycin (NB-DNJ) is a compound with lipid metabolism-regulating capabilities that acts through the inhibition of glycosphingolipid (GSL) synthesis. NB-DNJ modulates the function of CD4+ T cells from SLE patients by enhancing TCR signaling, as confirmed in vitro.1202 Additionally, NB-DNJ reduces autoantibody production in cocultures of B and T cells, suggesting a novel strategy for immunoregulatory treatment in SLE.
Dietary interventions
Dietary patterns also play a role in altering metabolic processes in autoimmune diseases. Dietary interventions can modulate blood lipids, benefiting both patients with SLE and patients with RA by reducing disease activity scores.1203 Increasing the intake of omega-3 fatty acids in the diet can raise HDL levels and lower triglyceride levels in adolescent-onset SLE patients,1204 whereas in adult SLE patients, it leads to an increase in HDL and a decrease in VLDL. Short-chain fatty acids (SCFAs) consumed in the diet promote the expansion of Tregs in the gut, indicating the multifaceted role of SCFAs in regulating T cell differentiation.1205 Oral lipid supplements may enhance the effectiveness of conventional therapies by increasing essential fatty acid levels to boost the systemic inflammatory response, potentially relieving joint pain and predicting responsiveness to DMARDs in RA patients.1206 The effects of the aforementioned dietary components suggest that the Mediterranean diet and Jiangnan dietary patterns contribute to improving energy metabolism in autoimmune diseases. However, the impact of intermittent fasting on autoimmune diseases remains inconclusive and requires further research,1207 especially concerning the role of the ketogenic diet, for which current studies are still very limited.
Various pathways of energy metabolism have emerged as potential regulators of immune cell differentiation and hold promise for treating autoimmune diseases. Inhibiting different energy metabolic pathways can induce metabolic reprogramming, necessitating a shift towards alternative pathways to maintain cell differentiation and function. Therefore, adopting an integrated approach combining metabolomics and proteomics aids in comprehensively understanding how metabolic enzymes and metabolites influence immune homeostasis under both normal physiological and autoimmune conditions. Importantly, key enzymes in metabolic pathways may exhibit isoforms in different tissues and cells, underscoring the importance of identifying isoform subtypes of enzymes in specific T cell subsets associated with different autoimmune diseases.
Conclusion and perspectives
The precise regulation of energy metabolism is fundamental for maintaining the balance between energy supply and demand within biological systems. This review outlines the well-established roles of energy metabolism in both health and disease, focusing on key processes such as glycolysis, OXPHOS, FAO, and amino acid metabolism. Disruptions in energy metabolism not only drive the abnormal proliferation of cancer cells and synovial fibroblasts but also lead to imbalances in the differentiation of immune cell populations, including Th17 cells, Tfh cells, Treg cells, and macrophages. These disruptions also cause significant changes in the expression of numerous proteins and enzymes involved in energy metabolism. Metabolic signaling pathways, including the mTOR, SIRT, AMPK, HIF, and Myc pathways, play crucial roles in reprogramming cellular metabolism by regulating the balance between anabolic and catabolic processes. These pathways offer valuable targets for developing new therapeutic approaches with reduced side effects and the potential for targeted elimination of pathological cells. However, despite the promising potential of interventions aimed at correcting metabolic dysfunctions, translating these concepts into practical therapies remains challenging.
In neurodegenerative diseases such as AD and PD, abnormalities in energy metabolism result in insufficient glucose uptake and mitochondrial dysfunction, leading to inadequate energy supply and oxidative stress. These disruptions trigger neuronal malnutrition, structural changes, and functional loss. In contrast, cancer cells undergo metabolic reprogramming to enhance glycolysis and glutaminolysis, adapting to hypoxic and nutrient-deprived environments, which promotes tumor proliferation and metastasis. Metabolic alterations in tumor cells also significantly impact immune cells; effector T cells and M1 macrophages increase glycolysis, whereas memory T cells, Tregs, and M2 macrophages primarily rely on FAO. This metabolic shift provides a survival advantage to Tregs in the TME, leading to competition between tumor and immune cells for energy substrates. Modulating energy metabolism presents a new strategy for treating both neurodegenerative diseases and cancer. Improving mitochondrial function and enhancing OXPHOS could help restore the cellular energy balance and slow the progression of neurodegenerative diseases. Conversely, inhibitors targeting key enzymes or transporting proteins in the glycolytic pathway could reduce the energy supply to tumor cells and inhibit tumor growth. Extensive research into drugs that target energy metabolism is ongoing and has shown promising therapeutic effects; however, the efficacy and safety of these approaches require further validation through clinical studies.
Looking ahead, several key areas in energy metabolism research deserve more in-depth exploration. Mechanistically: 1. Focus on the role of inflammation in energy metabolism: The interplay between inflammation and energy metabolism is reciprocal. However, understanding how inflammation either promotes or disrupts energy metabolism processes, as well as how energy metabolism influences inflammation and the regulatory mechanisms required to maintain their balance, necessitates further investigation. 2. Examine energy interactions among different cell types: The exchange of energy between immune cells and the interaction of energy between cancer cells and stromal cells profoundly impact the overall energy balance in the disease environment. Therefore, exploring the mechanisms of this energy transfer is crucial. 3. Emphasizing regulatory mechanisms: The regulation of energy metabolism has been a cornerstone and challenge in research. Future exploration should delve into the dynamic changes in regulatory signals, particularly focusing on the role of epigenetic modifications in metabolic processes. 4. Development of disease models: Many diseases, especially brain tissue disorders, lack reliable experimental models. The emergence of 3D organoids provides an effective means to explore related mechanisms.
Regarding detection approaches: 1. Focus on metabolic heterogeneity among different cells: Current detection methods struggle to accurately differentiate metabolic variances and dynamic changes between different cell types within disease environments. The development of more precise, noninvasive dynamic monitoring techniques is a significant contemporary challenge. 2. Addressing interindividual variability and early diagnosis: Establishing long-term individual health records and tracking systems to identify disease type, stage, and risk prediction markers on the basis of individual characteristics and metabolic changes is essential. 3. Integration of CRISPR-Cas9 gene editing with multi-omics technologies: The integration of CRISPR-Cas9 gene editing technology with mass spectrometry, single-cell metabolomics, spatial transcriptomics, and other multi-omics methods is crucial for identifying metabolic biomarkers and formulating personalized therapeutic algorithms. Recent studies have successfully linked gene mutations to transcriptional phenotypes through CRISPR screening and single-cell transcriptomics via Perturb-seq methods.1208 Additionally, the development of the CRISPR-human Organoids-Single-cell RNA Sequencing (CHOOSE) system offers a comprehensive screening approach for functional loss in organoids, thus providing a detectable pathway for precise metabolic regulation of diseases.1209 These findings indicate that the combination of CRISPR-Cas9 gene editing technology with single-cell metabolomics and human organoids holds promise for accurately monitoring cellular metabolic changes in relevant diseases. By combining gene editing with mass spectrometry technology, the wide analysis of metabolites in CRISPR-edited cell or animal models to detect changes in metabolic pathways is achievable. The integration of gene editing with spatial genomics enables detailed mapping of the relationships between gene expression patterns and metabolites, including their spatial distributions within tissues and organs.
Therapeutic strategies: 1. Develop multitarget drugs that target multiple metabolic processes, such as glucose metabolism, lipid metabolism, and protein metabolism, while simultaneously regulating multiple key enzymes or signaling pathways. Combining this approach with immunotherapy and dietary therapy, among other methods, can address the inefficiency of current single-target therapies. 2. Address discrepancies between laboratory research and clinical settings: Human pathogenesis and metabolic plasticity are often more complex than animal models. Bridging this gap and focusing on personalized treatments constitute critical aspects for successful clinical translation. 3. Ensuring the safety of gene therapy: Therapies based on CRISPR-Cas9 are on the rise. Future emphasis should focus on improving the targeting specificity and effectiveness of gene therapy to minimize damage to healthy tissues, possibly achieved through enhancing delivery vehicles. 4. Focus on dietary interventions: Approaches such as intermittent fasting and starvation-based therapies have shown initial success in disease treatment. Further clarification of disease dynamics through the aforementioned diagnostics, identification of treatment windows, and tailoring of dietary plans can increase patient compliance and treatment efficacy. 5. Other considerations: The presence of key enzymes as isoenzymes in different tissues and cells underscores the versatility and adaptability of metabolic pathways. Furthermore, mitochondrial transfer is also quite common in diseases. Recent studies suggest that transferring mitochondria from BMSCs to CD8+ T cells significantly enhance antitumor efficacy,1210 providing a promising therapeutic avenue.
In essence, the regulation of energy metabolism as a therapeutic strategy shows considerable promise amidst challenges and hope. Successfully integrating these findings into clinical practice will necessitate rigorous and comprehensive efforts in both fundamental and translational research.
References