NADPH homeostasis in cancer: functions, mechanisms and therapeutic implications
- Review Article
- Open access
- Published: 07 October 2020
NADPH homeostasis in cancer: functions, mechanisms and therapeutic implications
Signal Transduction and Targeted Therapy volume 5, Article number: 231 (2020) Cite this article
52k Accesses
9 Altmetric
Abstract
Nicotinamide adenine dinucleotide phosphate (NADPH) is an essential electron donor in all organisms, and provides the reducing power for anabolic reactions and redox balance. NADPH homeostasis is regulated by varied signaling pathways and several metabolic enzymes that undergo adaptive alteration in cancer cells. The metabolic reprogramming of NADPH renders cancer cells both highly dependent on this metabolic network for antioxidant capacity and more susceptible to oxidative stress. Modulating the unique NADPH homeostasis of cancer cells might be an effective strategy to eliminate these cells. In this review, we summarize the current existing literatures on NADPH homeostasis, including its biological functions, regulatory mechanisms and the corresponding therapeutic interventions in human cancers, providing insights into therapeutic implications of targeting NADPH metabolism and the associated mechanism for cancer therapy.
니코틴아미드 아데닌 디뉴클레오티드 인산염 (NADPH)은
모든 생물체에서 필수적인 전자 공급원으로,
생합성 반응과 산화환원 균형에 필요한 환원력을 제공합니다.
NADPH의 균형은
다양한 신호 전달 경로와 여러 대사 효소에 의해 조절되며,
암 세포에서는 적응적 변화를 겪습니다.
NADPH의 대사 재프로그래밍은
암 세포가 이 대사 네트워크에 대한 항산화 능력에 크게 의존하게 만들고,
동시에 산화 스트레스에 더 취약하게 만듭니다.
암 세포의 독특한 NADPH 균형을 조절하는 것은
이러한 세포를 제거하는 효과적인 전략이 될 수 있습니다.
본 리뷰에서는
인간 암에서 NADPH 균형의 생물학적 기능,
조절 메커니즘 및 이에 대응하는 치료적 개입을 포함한 현재까지의 문헌을 요약하며,
NADPH 대사 표적화와 관련된 암 치료 메커니즘에 대한 통찰을 제공합니다.
Similar content being viewed by others
NAD+ metabolism: pathophysiologic mechanisms and therapeutic potential
Article Open access07 October 2020
NAD+ metabolism, stemness, the immune response, and cancer
Article Open access01 January 2021
Oxidative cell death in cancer: mechanisms and therapeutic opportunities
Article Open access01 August 2024
Background
In cancer cells, the appropriate levels of intracellular reactive oxygen species (ROS) are essential for signal transduction and cellular processes.1,2 However, the overproduction of ROS can induce cytotoxicity and lead to DNA damage and cell apoptosis.3 To prevent excessive oxidative stress and maintain favorable redox homeostasis, tumor cells have evolved a complex antioxidant defense system that strategically adjusts multiple antioxidant enzymes such as catalase, glutathione reductase, and antioxidant molecules. The latter are dependent on the generation of nicotinamide adenine dinucleotide phosphate (NADPH), which is used to maintain reduced glutathione (GSH) and thioredoxin (TRX).4,5,6 NADPH is also well known as an essential electron donor and an indispensable cofactor that is used for transferring and reserving reduction potential for numerous anabolic reactions.7
NADP(H) is predominantly bound to intracellular proteins with different affinities.8 The intracellular content of NADP(H) differs markedly among tissues and cell types. For instance, the total NADP(H) is about 420 nmol/g wet weight in rat liver and 59% of total NADP(H) is found in mitochondria, and 30 nmol/g wet weight in skeletal muscle,5,8 and the NADPH concentration in the cytosol is 3.1 ± 0.3 and 37 ± 2 µM in the mitochondrial matrix in HeLa cells.9 In addition, the redox potentials of the mitochondrial and cytosolic NADP(H) systems are the same around—400 mV in the liver.8
A growing body of evidence has shown that regeneration and maintenance of the cellular NADP(H) content is strongly implicated in a variety of pathological conditions, such as diabetes, cardiovascular disease, neurodegenerative diseases, aging,4,5 especially in tumorigenesis and cancer progression.10 Compared with non-tumor cells, tumor cells usually maintain high levels of NADPH, not only to power redox defense but also to use for biosynthetic reactions to sustain their rapid growth.5,11 This realization has prompted molecular studies of NADPH metabolism and its exploitation for the development of anticancer agents. Recent advances have revealed that therapeutic modulation based on NADPH metabolism has been widely viewed as a novel and effective anticancer strategy.
In this review, we summarize the current existing literatures on NADPH metabolism, including its biological functions, regulatory mechanisms, and the corresponding therapeutic interventions directly or indirectly targeting NADPH metabolism in cancer.
배경
암 세포에서
세포 내 활성 산소 종(ROS)의 적절한 수준은
그러나
ROS의 과도한 생성으로 세포 독성이 유발되어
DNA 손상 및 세포 사멸로 이어질 수 있습니다.3
과도한 산화 스트레스를 방지하고 유리한 환원-산화 균형을 유지하기 위해
암 세포는
카탈라아제, 글루타티온 환원효소, 항산화 분자 등
다양한 항산화 효소를 전략적으로 조절하는 복잡한 항산화 방어 시스템을 진화시켰습니다.
후자는
니코틴아미드 아데닌 디뉴클레오티드 인산(NADPH)의 생성에 의존하며,
이는 환원형 글루타티온(GSH)과 티오레독신(TRX)을 유지하는 데 사용됩니다.4,5,6
NADPH는
또한 수많은 동화 반응에서 환원 잠재력을 전달하고 저장하는 데
필수적인 전자 공급원 및 필수 보조인자로 잘 알려져 있습니다.7
NADP(H)는
세포 내 단백질에 다양한 친화도로 결합되어 있습니다.8
세포 내 NADP(H)의 함량은 조직과 세포 유형에 따라 크게 다릅니다.
예를 들어, 쥐 간에서의 총 NADP(H)는 약 420 nmol/g 습중량이며, 이 중 59%가 미토콘드리아에 존재하며, 골격근에서는 30 nmol/g 습중량입니다.5,8 또한 HeLa 세포의 미토콘드리아 매트릭스에서의 NADPH 농도는 3.1 ± 0.3 및 37 ± 2 µM입니다. 9 또한, 간에서 미토콘드리아와 세포질 NADP(H) 시스템의 환원-산화 전위는 약 -400 mV로 동일합니다.8
점차 증가하는 연구 결과는
세포 내 NADP(H) 함량의 재생 및 유지가
당뇨병, 심혈관 질환, 신경퇴행성 질환, 노화,4,5 특히 종양 발생 및 암 진행과 같은
다양한 병리적 상태와 밀접하게 연관되어 있음을 보여주고 있습니다.10
종양 세포는
비종양 세포에 비해 일반적으로 높은 수준의 NADPH를 유지하며,
이는 산화환원 방어 메커니즘을 활성화하는 것 외에도
빠른 성장 유지에 필요한 생합성 반응에 사용됩니다. 5,11
이 인식은 NADPH 대사 연구와 이를 활용한 항암제 개발에 대한 분자적 연구를 촉진했습니다.
최근 연구 결과는
NADPH 대사 조절을 기반으로 한 치료적 접근이
새로운 효과적인 항암 전략으로 널리 인식되고 있음을 보여주었습니다.
이 리뷰에서는
암에서 NADPH 대사, 그 생물학적 기능, 조절 메커니즘, 그리고
NADPH 대사를 직접 또는 간접적으로 표적으로 하는
치료적 개입에 대한 현재까지의 문헌을 요약합니다.
NADPH-dependent biological functions in cancer
Both NAD(H) and NADP(H) are cofactors that are used for transferring and reserving reduction potential.7,9 Although the structures are closely related, NAD(H) and NADP(H) are recognized by unique compartmentalized enzymes and exert different functions. NAD(H) is mainly involved in catabolic reactions,5,12,13 whereas NADP(H) is primarily involved in cellular antioxidative effects and anabolic reactions as shown in Fig. 1.
NADPH에 의존하는 암의 생물학적 기능
NAD(H)와 NADP(H)는
환원 잠재력을 전달하고 저장하는 데 사용되는
구조는 유사하지만,
NAD(H)와 NADP(H)는 고유한 분획화 효소에 의해 인식되며
서로 다른 기능을 수행합니다.
NAD(H)는 주로 분해 반응에 관여하며,5,12,13
반면 NADP(H)는 세포 내 항산화 효과와 합성 반응에 주로 관여하며,
이는 그림 1에 표시되어 있습니다.
Fig. 1
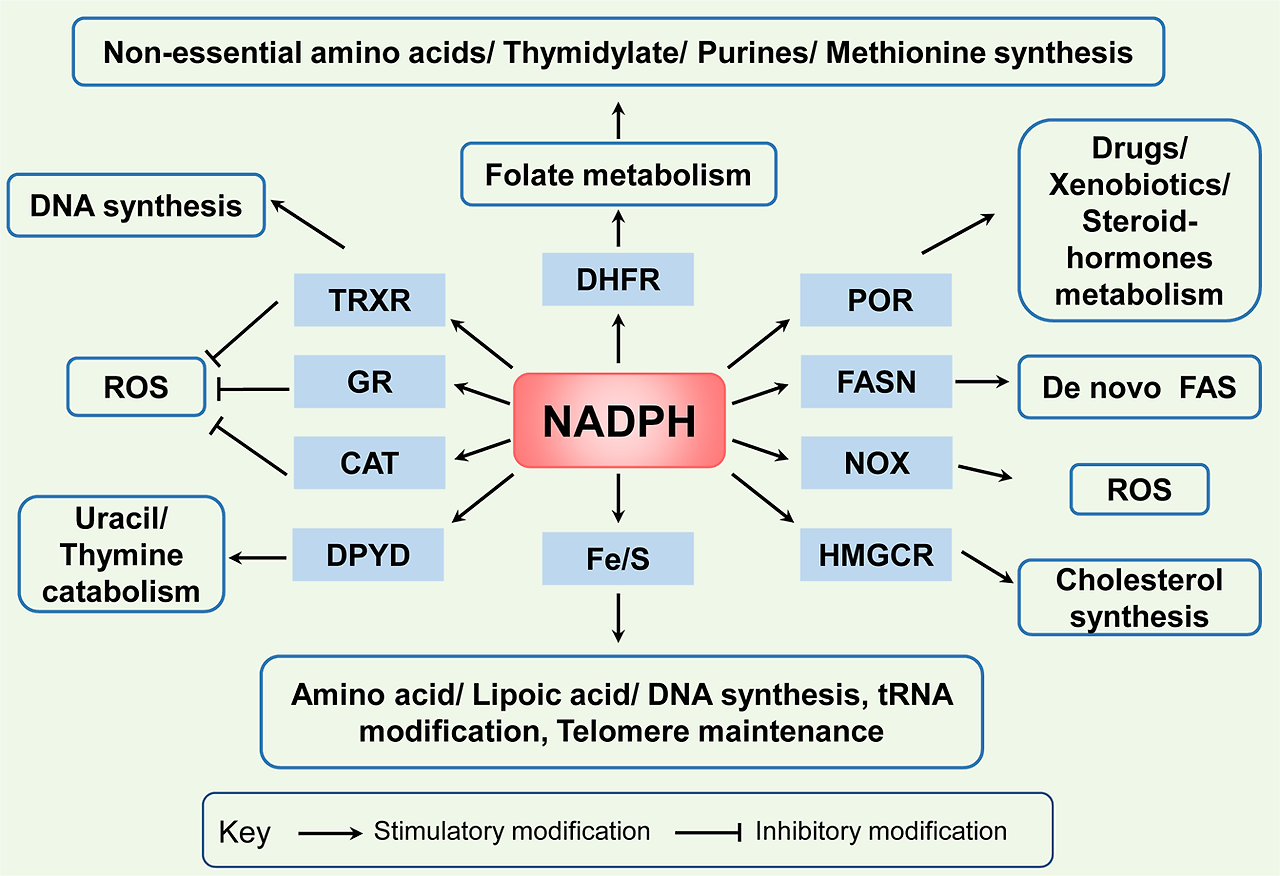
NADPH-dependent biological functions in cancer. There are three principal ways in which NADPH is used. First, NADPH is an essential cofactor of glutathione reductase (GR) and TRXR in GSH and TRX-peroxiredoxin (PRX) system, respectively, and reactivates catalase (CAT) to deactivate ROS for antioxidation; Second, NADPH is a crucial electron source for DHFR, Fe/S, POR, FANS, HMGCR, DPYD contributing to several reductive synthesis reactions, such as FAS, non-essential amino acids, nucleotides, and steroids synthesis; Third, NADPH is a substrate for NOXs to generate ROS
NADPH에 의존하는 암의 생물학적 기능.
NADPH는 세 가지 주요 방식으로 사용됩니다.
첫째, NADPH는 글루타티온 환원효소(GR)와 TRXR의 필수적인 보조인자로 각각 GSH 및 TRX-퍼옥시레독신(PRX) 시스템에서 작용하며, 카탈라아제(CAT)를 재활성화하여 ROS를 비활성화시켜 항산화 작용을 수행합니다.
둘째, NADPH는 DHFR, Fe/S, POR, FANS, HMGCR, DPYD의 중요한 전자 공급원으로 작용하여 FAS, 비필수 아미노산, 뉴클레오티드, 스테로이드 합성과 같은 여러 환원 합성 반응에 기여합니다;
세 번째로, NADPH는 NOXs의 기질로 작용하여 ROS를 생성합니다.
Antioxidative effects
In cancer cells, overcoming oxidative stress is a critical step for tumor progression. NADPH plays a key role in cellular antioxidation systems by providing reducing equivalents to generate reduced forms of antioxidant molecules, which are highly corrected with cancer cell biological behaviors.14 On the one hand, GSH reductase converts GSSG to GSH using NADPH as an important cofactor, then GSH acts as a cosubstrate for GSH peroxidase (GPX) that reduces hydrogen peroxide (H2O2) and other peroxides to H2O or alcohol to deactivate ROS.15,16 On the other hand, TRX reductase (TRXR) utilizes NADPH as an electron donor to maintain the reduced form of TRX, which contributes to scavenge H2O2 and reduce ribonucleotide reductase (RNR) for DNA synthesis.17,18 In addition, in some cell types, NADPH binds to the important H2O2-disposing enzyme: catalase, and reactivates it when it has been inactivated by H2O2.19
항산화 효과
암 세포에서 산화 스트레스를 극복하는 것은 종양 진행의 중요한 단계입니다.
NADPH는
항산화 분자의 환원 형태를 생성하기 위해 환원 등가물을 공급함으로써
세포 내 항산화 시스템에서 핵심 역할을 합니다.
이는 암 세포의 생물학적 행동과 밀접하게 연관되어 있습니다. 14
한편, GSH 환원효소는
NADPH를 중요한 보조인자로 사용하여 GSSG를 GSH로 전환하며,
GSH는 과산화수소(H₂O₂) 및 기타 과산화물을
H₂O 또는 알코올로 환원하여 활성산소종(ROS)을 무해화시키는
GSH 과산화효소(GPX)의 보조기질로 작용합니다. 15,16
다른 한편으로,
TRX 환원효소(TRXR)는
NADPH를 전자 공여체로 사용하여 TRX의 환원형을 유지하며,
이는 H₂O₂를 제거하고 DNA 합성에 필요한 리보뉴클레오티드 환원효소(RNR)를 환원시키는 데 기여합니다. 17,18
또한 일부 세포 유형에서는
NADPH가 중요한 H₂O₂ 제거 효소인 카탈라제에 결합하여
H₂O₂에 의해 비활성화된 후 재활성화시킵니다.19
Reductive synthesis
NADPH is also a crucial electron source for several reductive synthesis reactions, including fatty acids, amino acids, nucleotides, and steroids synthesis to sustain rapid tumor cell growth.20 Primarily, NADPH provides reducing equivalents for fatty acid synthase (FASN), the main rate-limiting enzyme, to synthesize fatty acids with acetyl-CoA serving as a primer and malonyl-CoA as a two-carbon donor,21,22 and provides the needed electrons for iron–sulfur (Fe/S) protein assembly that participate in non-essential amino acid biosynthesis and lipoic acid synthesis, tRNA modification, DNA replication and repair, and telomere maintenance.23 NADPH is also needed for dihydrofolate reductase (DHFR) enzyme to catalyze the reduction of dihydrofolate to tetrahydrofolate (THF) in folate metabolism, which is required for de novo biosynthesis of thymidylate, purines, methionine, and some amino acids.24 Besides, NADPH acts as the reducing reagent for 3-hydroxy-3-methylglutaryl-coenzyme A reductase (HMGCR), the rate-limiting enzyme of the mevalonate pathway, which leads to the synthesis of cholesterol and nonsterol isoprenoids.25 NADPH also acts as a cosubstrate for dihydropyrimidine dehydrogenase (DPYD), which catalyzes the reduction of uracil and thymine to 5,6-dihydrouracil and 5,6-dihydrothymine, respectively.26 In addition, the activity of the cytochrome P450 reductase (POR) also requires NADPH, which has a major role in the metabolism of drugs, xenobiotics, and steroid hormones.27
환원 합성
NADPH는
지방산, 아미노산, 뉴클레오티드, 스테로이드 합성과 같은
여러 환원 합성 반응의 중요한 전자 공급원으로,
빠른 종양 세포 성장 유지에 필수적입니다. 20
주로 NADPH는
지방산 합성 효소(FASN)의 주요 속도 제한 효소에 환원 등가물을 제공하여
아세틸-CoA를 초시제로, 말론일-CoA를 2탄소 기증체로 사용하여
철-황(Fe/S) 단백질 조립에 필요한 전자를 공급합니다.
이 단백질은
필수 아미노산이 아닌 아미노산 생합성,
리포산 합성,
tRNA 변형,
DNA 복제 및 수리,
텔로미어 유지에 필요합니다.23
NADPH는 또한 엽산 대사에서
디히드로포레이트를 테트라히드로포레이트(THF)로 환원하는
디히드로포레이트 환원효소(DHFR) 효소의 촉매 작용에 필요하며,
이는 티미딜산, 푸린, 메티오닌 및 일부 아미노산의 신규 합성에 필수적입니다. 24
또한 NADPH는
메발론산 경로의 속도 제한 효소인 3-하이드록시-3-메틸글루타릴-코엔자임 A 환원효소(HMGCR)의 환원제로 작용하며,
이는 콜레스테롤과 비스테롤 이소프렌오이드의 합성을 유도합니다. 25
NADPH는 또한
디히드로피리미딘 탈수소효소(DPYD)의 보조 기질로 작용하여
우라실과 티민을 각각 5,6-디히드로우라실과 5,6-디히드로티민으로 환원시키는 반응을 촉매합니다. 26
또한 사이토크롬 P450 환원효소(POR)의 활성도
NADPH를 필요로 하며,
이는 약물, 엑스노바이오틱스, 스테로이드 호르몬의 대사에서 중요한 역할을 합니다.27
Free radical generation
In addition, NADPH is also responsible for the generation of free radicals by NADPH oxidases (NOX) as a substrate. NOXs (NOX1–5 and dual oxidases (DUOX) 1 and 2) catalyze the superoxide anions or H2O2 from NADPH and oxygen.28,29,30 NOX-mediated ROS broadly and specifically regulate various redox-sensitive signaling pathways involved in cancer progression via stimulating oncogenes, such as Src and Ras, and inactivating tumor suppressor proteins, such as TP53 and PTEN.31
자유 라디칼 생성
또한
NADPH는
NADPH 산화효소(NOX)를 기질로 삼아
자유 라디칼을 생성하는 데도 관여합니다.
NOX(NOX1–5 및 이중 산화효소(DUOX) 1과 2)는
NADPH와 산소로부터 슈퍼옥사이드 음이온 또는 H₂O₂를 생성하는 반응을 촉매합니다. 28,29,30
NOX에 의해 매개되는 ROS는
암 진행에 관여하는 다양한 산화환원 민감성 신호 전달 경로를 광범위하고
특이적으로 조절합니다.
이는 Src 및 Ras와 같은 발암 유전자의 활성화를 촉진하고
TP53 및 PTEN과 같은 종양 억제 단백질을 비활성화함으로써 이루어집니다.31
Molecular mechanisms of NADPH homeostasis in cancer
Understanding NADPH production and consumption routes is essential to a global understanding of cancer metabolism. As shown in Fig. 2, the NADPH homeostasis is mainly regulated by several metabolic pathways and enzymes including NAD kinase (NADK), the pentose phosphate pathway (PPP), the folate-mediated one-carbon metabolism, malic enzymes (ME), the nicotinamide nucleotide transhydrogenase (NNT), cytosolic or mitochondrial NADP-dependent isocitrate dehydrogenase (IDH1 and IDH2), the glutamine metabolism, and the fatty acid oxidation (FAO). However, for the general NADPH generation in cells, the relative contribution of these pathways and enzymes to NADPH production remains elusive. Recent study show that cellular NADPH could be largely generated by PPP, the folate-mediated one-carbon metabolism and ME in cancer and proliferation cells.32,33 Also, mounting evidence suggests that these different processes and enzymes have functional connections for NADPH homeostasis in cancer. For instance, FAO accelerates the TCA cycle to produce citrate, which is exported to the cytosol to engage in NADPH production through ME1 and IDH1.34 Here we review current knowledge of the underlying mechanisms of NADPH homeostasis following its de novo synthesis, relative contribution of related enzymes and pathways in cancer.
암에서의 NADPH 항상성의 분자적 메커니즘
NADPH의 생성 및 소비 경로를 이해하는 것은
암 대사 전반을 이해하는 데 필수적입니다.
그림 2에 표시된 바와 같이,
NADPH 균형은
NAD 키나아제 (NADK),
펜토스 인산 경로 (PPP),
엽산 매개 1탄소 대사,
말산 효소 (ME),
니코틴아미드 뉴클레오티드 트랜스히드라제 (NNT),
세포질 또는 미토콘드리아 NADP 의존성 이소시트르산 탈수소효소 (IDH1 및 IDH2),
글루타민 대사, 및 지방산 산화(FAO)에 의해 조절됩니다.
그러나
세포 내 일반적인 NADPH 생성에 있어 이러한 경로와 효소가
NADPH 생산에 기여하는 상대적 비율은 여전히 명확하지 않습니다.
최근 연구는
암 세포와 증식 세포에서 세포 내 NADPH가
주로 PPP, 엽산 매개 1탄소 대사 및 ME에 의해 생성될 수 있음을 보여주었습니다.32,33
또한,
이러한 다양한 과정과 효소가
암에서의 NADPH 균형에 기능적 연결성을 가질 수 있다는 증거가 점점 더 쌓이고 있습니다.
예를 들어,
FAO는 시트르산을 생성하기 위해 TCA 회로를 가속화하며,
이 시트르산은 세포질로 수출되어 ME1과 IDH1을 통해 NADPH 생성에 참여합니다.34
본 연구에서는
NADPH의 신규 합성 이후 NADPH 균형 유지의 근본적 메커니즘,
암에서 관련 효소 및 경로의 상대적 기여도에 대한 현재의 지식을 검토합니다.
Fig. 2
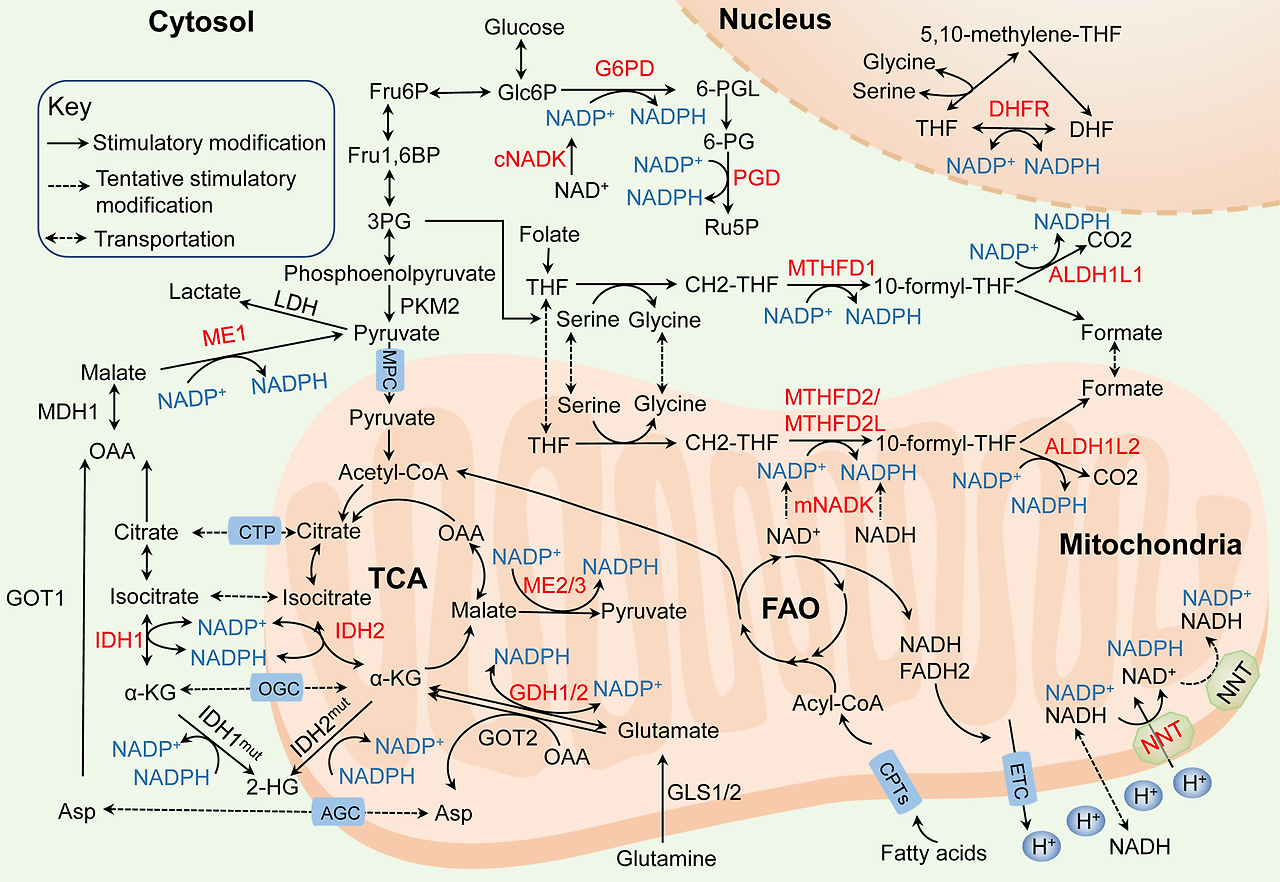
Molecular mechanisms of NADPH homeostasis in cancer. The principal generation of NADPH (blue) with dysregulated pathways and enzymes (red) in cancer: (i) NADKs catalyze the phosphorylation of NAD(H) to form NADP(H) via the de novo synthesis (cNADK in the cytosol and mNADK in mitochondria). (ii) the pentose phosphate pathway (PPP) utilizes G6PD and PGD to maintain the cytosolic NADPH. (iii) the folate-mediated one-carbon metabolism reduces NADP+ to NADPH by MTHFD1/ALDH1L1 in the cytosol, MTHFD2/MTHFD2L/ALDH1L2 in mitochondria and DHFR in the nucleus. (iv) IDH1 located in the cytosol and IDH2 located in mitochondria generate NADPH, but mutant IDHs consume NADPH. (v) ME1 located in the cytosol and ME2/3 located in mitochondria convert NADP+ into NADPH; (vi) the glutamine metabolism generates NADPH by GDH1/2 directly in mitochondria and generates aspartate that is transported into the cytosol for NADPH production depending on ME1. (vii) NNT catalyzes the transfer of hydride ions from NADH to NADP+ and produces NADPH to maintain the mitochondrial NADPH and the reverse-mode NNT that consumes NADPH may exist in cancer cells. (viii) The CPT1/2-mediated FAO generates acetyl CoA that enters the TCA cycle and contributes to NADPH production depending on IDHs and MEs. MPC mitochondirial pyruvate carrier, CTP citrate transport protein, OGC α-ketoglutarate-malate carrier, AGC aspartate–glutamate carrier
암에서의 NADPH 항상성의 분자적 메커니즘.
암에서 NADPH(파란색)의 주요 생성 경로와 조절 장애를 보이는 경로 및 효소(빨간색):
(i) NADKs는 NAD(H)의 인산화 반응을 촉매하여 NADP(H)를 생성하며, 이는 신생합성 경로를 통해 이루어집니다(세포질 내 cNADK와 미토콘드리아 내 mNADK).
(ii) 펜토스 인산 경로(PPP)는 G6PD와 PGD를 활용하여 세포질 내 NADPH를 유지합니다.
(iii) 엽산 매개 1탄소 대사 경로는 MTHFD1/ALDH1L1(세포질), MTHFD2/MTHFD2L/ALDH1L2(미토콘드리아), DHFR(핵)에서 NADP+를 NADPH로 환원합니다.
(iv) 세포질에 위치한 IDH1과 미토콘드리아에 위치한 IDH2는 NADPH를 생성하지만, 돌연변이 IDH는 NADPH를 소비합니다. (v) 세포질에 위치한 ME1과 미토콘드리아에 위치한 ME2/3는 NADP+를 NADPH로 전환합니다;
(vi) 글루타민 대사 경로는 미토콘드리아에서 GDH1/2를 통해 직접 NADPH를 생성하며, ME1에 따라 세포질로 운반되어 NADPH 생성에 사용되는 아스파르트산을 생성합니다.
(vii) NNT는 NADH로부터 NADP+로 수소 이온을 전달하여 NADPH를 생성하며, 미토콘드리아 내 NADPH를 유지합니다. 암 세포에서는 NADPH를 소비하는 역방향 모드의 NNT가 존재할 수 있습니다.
(viii) CPT1/2에 의해 매개되는 지방산 산화(FAO)는 아세틸 CoA를 생성하며, 이는 TCA 회로에 들어가 IDH와 ME에 따라 NADPH 생성에 기여합니다. MPC 미토콘드리아 피루vate 운반체, CTP 시트르산 운반 단백질, OGC α-케토글루타르산-말산 운반체, AGC 아스파르트산-글루타메이트 운반체
NAD kinase
NADPH de novo synthesis is catalyzed by NADKs, which catalyze the phosphorylation of NAD+ to form NADP+. Subsequently, the dehydrogenases/reductases in various metabolic pathways convert NADP+ into NADPH.10,12 NADKs are found in almost all human organs except skeletal muscle, and localized in both cytosol and mitochondria. Compared to cytosolic NADK (cNADK), mitochondrial NADK (mNADK) has a distinctive feature that it can directly phosphorylate nicotinamide adenine dinucleotide (NADH) to generate NADPH to alleviate oxidative stress in mitochondria.35
The Cancer Genome Atlas (TCGA) database indicates both cNADK overexpression and the presence of several cNADK mutants in multiple tumor types.10 Notably, a novel cNADK mutant, NADK-I90F, is found in pancreatic ductal adenocarcinoma cancer (PDAC) patients. CNADK-I90F has a lower Km and higher Vmax for NAD+ compared to wild-type cNADK, which indicates increased enzyme activity. Consistently, compared with cNADK wild-type cells, cells expressing cNADK-I90F have elevated NADPH levels and reduced ROS levels.36,37 In addition, in diffuse large B-cell lymphoma (DLBCL) and colon cancer, silencing cNADK with shRNA impairs the pool of NADPH and suppresses cancer cell growth.38 In terms of NADKs activities, cNADK phosphorylated at S44, S46, and S48, which may be mediated by the phosphoinositide 3-kinase (PI3K)–Akt signaling, has enhanced activity in breast cancer and lung cancer cells, thereby increasing NADPH production.39 Based on its recent discovery, the relevant role of mNADK in human cancers still need to be clarified, but the wild-type and mutant cNADK are potential clinical targets for cancer therapy.
NAD 키나아제
NADPH의 신규 합성은
NADKs에 의해 촉매되며,
이는 NAD+의 인산화 반응을 촉매하여 NADP+를 형성합니다.
이후 다양한 대사 경로의 탈수소효소/환원효소는
NADKs는
골격근을 제외한 거의 모든 인간 장기에서 발견되며,
세포질과 미토콘드리아에 모두 존재합니다.
세포질 NADK(cNADK)와 비교할 때
미토콘드리아 NADK(mNADK)는
미토콘드리아 내 산화 스트레스를 완화하기 위해
니코틴아미드 아데닌 디뉴클레오티드(NADH)를 직접 인산화하여
NADPH를 생성하는 독특한 특성을 가지고 있습니다.35
암 유전체 아틀라스(TCGA) 데이터베이스는 다양한 암 유형에서 cNADK 과발현 및 여러 cNADK 변이체의 존재를 보여줍니다.10 특히, 새로운 cNADK 변이체인 NADK-I90F가 췌관 선암(PDAC) 환자에서 발견되었습니다. CNADK-I90F는 야생형 cNADK에 비해 NAD+에 대한 Km 값이 낮고 Vmax 값이 높아 효소 활성이 증가했음을 나타냅니다. 일관되게, cNADK 야생형 세포와 비교했을 때 cNADK-I90F를 발현하는 세포는 NADPH 수준이 증가하고 ROS 수준이 감소했습니다.36,37 또한 확산성 대세포 B세포 림프종(DLBCL)과 대장암에서 shRNA로 cNADK를 침묵시키면 NADPH 풀이 감소하고 암 세포 성장 억제가 관찰되었습니다. 38 NADK의 활성 측면에서, S44, S46, S48에서 인산화된 cNADK는 인산인오시타이드 3-키나제(PI3K)-Akt 신호전달 경로를 통해 매개될 수 있으며, 유방암 및 폐암 세포에서 활성이 증가하여 NADPH 생산을 증가시킵니다. 39 최근 발견에 따라 인간 암에서의 mNADK의 관련 역할은 여전히 명확히 규명되어야 하지만, 야생형 및 돌연변이형 cNADK는 암 치료를 위한 잠재적 임상 표적이 될 수 있습니다.
Pentose phosphate pathway
The PPP diverges at the first step of glycolysis, which serves as the largest contributor of cytosolic NADPH and NADPH generation undergoes three irreversible reactions in the PPP oxidative branch.40,41,42 Studies have proved that NADPH production is dramatically increased by enhancing the flux of glucose into the PPP oxidative branch in various cancers.43,44 Glucose-6-phosphate dehydrogenase (G6PD) that exists as either an active dimer or an inactive monomer dehydrogenates G6P to yield 6-phosphogluconolactone (6-PGL) and NADPH in the first reaction. Then, 6-phosphogluconate dehydrogenase (PGD) that often functions as a homodimer catalyzes the oxidative decarboxylation of 6-phosphogluconate (6-PG) to synthesize ribulose-5-phosphate (Ru5P) and a second NADPH in the third reaction.45,46
Increasingly, more studies have shown that G6PD activity is increased in several types of cancers, including bladder, breast, prostate, gastric cancers compared with normal tissues, and the high expression of G6PD predicts poor clinical outcome in various cancer patients and plays critical roles in tumorigenesis and chemoresistance.47,48 PGD is also hyperactive and plays a fundamental role in tumor growth.49,50 G6PD or PGD depletion significantly decrease NADPH levels and enhance chemotherapeutic drugs-induced cell apoptosis by redox modulation.51,52 For what concerns activity regulation, NADP+ is required for G6PD enzymatic activity, whereas NADPH negatively regulates its activity. Hence, tumor cells with higher NADPH consumption exhibit higher levels of active G6PD.45 Interestingly, a study also shows that NADPH level is not changed by silencing PGD expression, which is possible that a temporally increased NADP+/NADPH ratio compensatory increased G6PD activity, thus generating NADPH.45
The NADPH homeostasis is also regulated by the rate-limiting enzyme activity affected by the posttranslational modification. Studies indicate that the glycosylation, SIRT5-mediated deglutarylation and SIRT2-mediated deacetylation all enhance G6PD activity and maintain cellular NADPH homeostasis.53,54,55 Both the phosphorylation of PGD at Y481 upon EGFR activation and acetylation of PGD at K76 and K294 by acetyltransferases enhance its activation for producing NADPH in cancer cells.56,57 Conversely, protein kinase A (PKA) inhibits G6PD activity by directly phosphorylating it on serine and threonine residues.58 Additionally, G6PD activity can be regulated by several signaling pathways in tumors, such as the PI3K/AKT, Ras, Src, Nrf2, mTORC1, PETEN, ATM, and TP53 pathways, in a direct or indirect manner (reviewed in refs. 45,47). For instance, the PTEN protein and cytosolic TP53 bind to G6PD to prevent the assembly of G6PD monomers into active dimers and thus decease the PPP flux.59,60
펜토스 인산 경로
PPP는 글리코lysis의 첫 번째 단계에서 분기되며, 세포질 NADPH의 가장 큰 공급원 역할을 합니다. NADPH 생성은 PPP 산화 분지에서 세 가지 불가역적 반응을 거칩니다.40,41,42 다양한 암에서 글루코스 유입을 증가시켜 PPP 산화 분지로의 유동을 강화하면 NADPH 생산이 급격히 증가한다는 것이 입증되었습니다. 43,44 글루코스-6-인산 탈수소효소(G6PD)는 활성 이량체 또는 비활성 단량체 형태로 존재하며, 첫 번째 반응에서 G6P를 탈수소화하여 6-포스포글루코노락톤(6-PGL)과 NADPH를 생성합니다. 그 다음, 종종 동형 이량체로 기능하는 6-포스포글루코네이트 탈수소효소(PGD)는 6-포스포글루코네이트(6-PG)의 산화 탈카복실화 반응을 촉매하여 리불로즈-5-포스페이트(Ru5P)와 두 번째 NADPH를 생성합니다.45,46
최근 연구에서 G6PD 활성이 방광암, 유방암, 전립선암, 위암 등 여러 종류의 암에서 정상 조직에 비해 증가했으며, G6PD의 고발현은 다양한 암 환자의 예후가 불량함을 예측하며 종양 발생과 화학요법 저항성에 중요한 역할을 한다는 것이 밝혀졌습니다.47,48 PGD도 과활성 상태이며 종양 성장에 근본적인 역할을 합니다. 49,50 G6PD 또는 PGD의 고갈은 NADPH 수준을 유의미하게 감소시키고 환원-산화 조절을 통해 화학요법 약물 유발 세포 사멸을 촉진합니다.51,52 활성 조절 측면에서 NADP+는 G6PD 효소 활성에 필수적이며, NADPH는 그 활성을 음성적으로 조절합니다. 따라서 NADPH 소비량이 높은 종양 세포는 활성 G6PD 수준이 높습니다.45 흥미롭게도, 한 연구에서는 PGD 발현을 억제해도 NADPH 수준이 변화하지 않는 것으로 나타났으며, 이는 일시적으로 증가한 NADP+/NADPH 비율이 G6PD 활성을 보상적으로 증가시켜 NADPH를 생성했기 때문일 수 있습니다.45
NADPH 균형은 후전사적 변형에 의해 영향을 받는 속도 제한 효소 활성에 의해 조절됩니다. 연구 결과, 글리코실화, SIRT5에 의한 deglutarylation 및 SIRT2에 의한 deacetylation은 모두 G6PD 활성을 증강시키고 세포 내 NADPH 균형을 유지합니다. 53,54,55 EGFR 활성화 시 PGD의 Y481 위치에서의 인산화 및 아세틸전달효소에 의한 PGD의 K76 및 K294 위치에서의 아세틸화는 암 세포에서 NADPH 생성을 위해 PGD 활성을 증가시킵니다.56,57 반면, 단백질 키나제 A (PKA)는 세린 및 트레오닌 잔기에서 직접 인산화하여 G6PD 활성을 억제합니다. 58 또한 G6PD 활성은 PI3K/AKT, Ras, Src, Nrf2, mTORC1, PETEN, ATM, TP53 경로와 같은 종양 내 여러 신호전달 경로를 통해 직접적 또는 간접적으로 조절될 수 있습니다(참조 문헌 45,47에서 검토됨). 예를 들어, PTEN 단백질과 세포질 TP53은 G6PD에 결합하여 G6PD 단량체가 활성 이량체로 조립되는 것을 방지함으로써 PPP 유동을 감소시킵니다.59,60
Folate-mediated one-carbon metabolism
Folate-mediated one-carbon metabolism has been long recognized and attributed to its function of producing one-carbon units for nucleic acid and methionine synthesis, another crucial function of this pathway is generating reducing power NADPH.61,62 Serine and glycine are the major carbon sources of this pathway. The activation of serine biosynthesis pathway enhances NADPH generation in cancer cells.63 Conversely, eliminating serine from the medium decreases the NADPH/NADP+ ratio and impairs cancer cell growth.64 Methylene tetrahydrofolate dehydrogenases (MTHFD1 in cytosol and MTHFD2 or MTHFD2L in mitochondria) catalyze the oxidation of 5,10-methylene-THF (CH2-THF) to form 10-formyl-THF, and 10-formyl-THF dehydrogenases (ALDH1L1 in cytosol and ALDH1L2 in mitochondria) catalyze the oxidization of 10-formyl-THF to generate CO2 with concomitant NADPH production. In the nucleus, the THF carrier is oxidized to DHF in an NADPH-generating reaction with electrons used to reduce one-carbon units to the methyl level.65,66,67
MTHFD2 is postulated to be the “main switch” that produces additional one-carbon units in mitochondria to enable rapid growth.63 The expression of MTHFD2 is closely related to the response of the folate antagonist methotrexate (MTX) and the thymidylate synthase inhibitor pemetrexed.68,69 Both MTHFD2 and MTHFD1 are markedly elevated and correlated with poor survival across human cancers.70,71,72 Moreover, study indicates that combining serum AFP with MTHFD1 enhances the prognostic prediction accuracy in hepatocellular carcinoma (HCC).73 Quantitative flux analysis reveals depletion of either MTHFD2 or MTHFD1 results in decreased cellular NADPH/NADP+ and GSH/GSSG ratios and increased cell sensitivity to oxidative stress.32 Suppression of MTHFD2 disturbs redox homeostasis, accelerates cell death in both colorectal cancer (CRC),74,75 and acute myeloid leukemia (AML).64 MTHFD2 is also critical for cancer stem-like properties and chemoresistance, suggesting that disturbing NAPDH homeostasis may prevent recurrence and eradicate tumors.76 And, MTHFD1 depletion reduces both the frequencies of circulating melanoma cells in the blood and metastatic disease burden in mice bearing melanoma,77 suggesting that NAPDH homeostasis represents therapeutic targets to impede distant metastasis. However, the association between MTHFD2L, which can use either NAD+ or NADP+ for dehydrogenase activity, and tumors remains to be investigated.
Cytosolic ALDH1L1 mainly regulates reduced folate pools and purine biosynthesis, while mitochondrial ALDH1L2 produces NADPH in response to oxidative stress.78 Although ALDH1L1 is overexpressed in NSCLC and GC cancer,79,80 ALDH1L1 is reported profoundly downregulated or silenced in cancers, rendering it a candidate tumor suppressor.81,82 Nevertheless, ALDH1L2 is highly expressed and presents as an independent prognostic factor for overall survival in melanoma, PDAC, and CRC.77,78,83 Depletion of ALDH1L2 markedly decreases the NADPH/NADP+ and GSH/GSSG ratios, reduces the circulating tumor cells in blood and alleviates the metastatic burden.77,83,84 In addition, the expression of ALDH1L2 is upregulated by some certain drugs, such as thapsigargin and tunicamycin, endoplasmic reticulum stress inducers in immortalized human B cells,85 mitotane, an adjuvant monotherapy used for treating adrenocortical carcinoma,86 and the indomethacin, an anti-inflammatory agent in breast cancer cells.87 Thus, further exploration of the association between the effects of these drugs on the ALDH1L2 expression and the cellular response to redox stress is needed.
엽산 매개 1탄소 대사
엽산 매개 1탄소 대사는 핵산 및 메티오닌 합성에 필요한 1탄소 단위를 생성하는 기능으로 오랫동안 인정받아 왔으며, 이 경로의 또 다른 중요한 기능은 환원력 NADPH를 생성하는 것입니다.61,62 세린과 글리신은 이 경로의 주요 탄소 공급원입니다. 세린 생합성 경로의 활성화는 암 세포에서 NADPH 생성을 증가시킵니다. 63 반면, 배지로부터 세린을 제거하면 NADPH/NADP+ 비율이 감소하고 암 세포의 성장에 장애를 초래합니다.64 메틸렌 테트라하이드로폴산 탈수소효소(MTHFD1은 세포질에, MTHFD2 또는 MTHFD2L은 미토콘드리아에 존재)는 5,10-메틸렌-THF(CH2-THF)의 산화를 촉매하여 10-포르밀-THF를 형성하며, 10-포르밀-THF 탈수소효소(세포질의 ALDH1L1과 미토콘드리아의 ALDH1L2)는 10-포르밀-THF의 산화를 촉매하여 이산화탄소(CO₂)를 생성하며 동시에 NADPH를 생성합니다. 핵 내에서는 THF 운반체가 NADPH 생성 반응을 통해 DHF로 산화되며, 이 과정에서 전자(electrons)는 일탄소 단위(one-carbon units)를 메틸 수준으로 환원하는 데 사용됩니다.65,66,67
MTHFD2는 미토콘드리아에서 추가적인 1탄소 단위를 생성하여 빠른 성장을 가능하게 하는 '주요 스위치'로 추정됩니다.63 MTHFD2의 발현은 엽산 억제제 메토트렉세이트(MTX)와 티미딜산 합성효소 억제제 페메트렉세드에 대한 반응과 밀접하게 관련되어 있습니다.68,69 MTHFD2와 MTHFD1은 인간 암에서 생존율이 낮은 경우 모두 현저히 증가하며 서로 관련이 있습니다. 70,71,72 또한 연구 결과, 혈청 AFP와 MTHFD1을 결합하면 간세포암(HCC)의 예후 예측 정확도가 향상됩니다.73 정량적 유동 분석 결과, MTHFD2 또는 MTHFD1의 감소는 세포 내 NADPH/NADP+ 및 GSH/GSSG 비율 감소와 산화 스트레스에 대한 세포 감수성 증가를 초래합니다. 32 MTHFD2 억제는 대장암(CRC)74,75 및 급성 골수성 백혈병(AML)에서 산화환원 균형을 교란시키고 세포 사멸을 가속화합니다.64 MTHFD2는 암 줄기세포 유사 특성 및 화학요법 저항성에 필수적이며, 이는 NADPH 균형 교란이 재발을 예방하고 종양을 근절할 수 있음을 시사합니다. 76 또한, MTHFD1 결핍은 멜라노마를 가진 쥐에서 혈중 순환 멜라노마 세포의 빈도와 전이성 질환 부담을 감소시킵니다.77 이는 NAPDH 균형이 원격 전이를 차단하기 위한 치료 표적이 될 수 있음을 시사합니다. 그러나 NAD+ 또는 NADP+를 모두 사용해 탈수소효소 활성을 발휘하는 MTHFD2L과 종양 간의 연관성은 추가 연구가 필요합니다.
세포질 내 ALDH1L1은 주로 감소된 엽산 풀과 푸린 생합성을 조절하며, 미토콘드리아 내 ALDH1L2는 산화 스트레스에 반응하여 NADPH를 생성합니다.78 ALDH1L1은 NSCLC와 위암에서 과발현되지만,79,80 암에서 심하게 하향 조절되거나 침묵화되어 종양 억제 후보로 제시되었습니다. 81,82 그럼에도 불구하고 ALDH1L2는 멜라노마, PDAC, CRC에서 전체 생존율의 독립적인 예후 인자로 고발현됩니다.77,78,83 ALDH1L2의 고갈은 NADPH/NADP+ 및 GSH/GSSG 비율을 현저히 감소시키며, 혈중 순환 종양 세포를 감소시키고 전이 부담을 완화합니다. 77,83,84 또한 ALDH1L2의 발현은 타프시가린과 투니카마이신과 같은 내소체 스트레스 유도제, 불멸화 인간 B 세포에서,85 부신피질 암 치료에 사용되는 보조 요법인 미토탄,86 유방암 세포에서 항염증제인 인도메타신에 의해 상향 조절됩니다. 87 따라서, 이러한 약물이 ALDH1L2 발현에 미치는 영향과 세포의 산화환원 스트레스 반응 간의 연관성을 추가로 탐구하는 것이 필요합니다.
Malic enzymes
ME participate in reactions that link the components of catabolic metabolism in glycolysis and the Krebs cycle via the oxidative decarboxylation of malate to pyruvate, thereby inducing the anabolic metabolism with concomitant NADPH production.32,88 A quantitative flux analysis showed that the direct contribution of ME to NADPH generation was estimated to equal the contribution of the PPP.89 ME family consists of three isoforms: ME1 is located in the cytosol and ME2, ME3 are located in mitochondria. ME1 and ME3 require NADP+ and ME2 utilizes either NAD+ or NADP+ for their catalytic activities, thus NADPH can be produced by ME both directly and indirectly through the activity of the NNT that catalyzes the transfer of hydride ions from NADH to NADP+ and produces NADPH in mitochondria.90 However, ME1 and ME2 seem to be the main isoforms because ME3 is hardly negligibly detected in many assessed mammalian cells.91
The overexpression of ME1 is significantly associated with a poor prognosis for people with cancer, including those with gastric cancer, oral squamous cell carcinoma, breast cancer, lung cancer, etc.92,93,94,95 Silencing ME1 markedly reduces NADPH and increases ROS levels, ultimately induces cell apoptosis under oxidative stress, such as glucose starvation or anoikis.96,97 Moreover, the ME1 protein is hypophosphorylated at S336 and hyperacetylated at K337 by PGAM family member 5 and acetyl-CoA acetyltransferase, respectively, resulting in ME1 translocation from mitochondria to the cytosol, dimerization and activation, thus strongly promoting NADPH generation and tumorigenesis.98 ME1 expression is also regulated by well-known tumor suppressors or oncogenes such as TP53 or KRAS.91,99 Intriguingly, there is a direct crosstalk between ME1 and PPP components, and ME1 increases the ability of PGD to bind to 6-PG, enhancing NADPH generation.100
ME2 is also overexpressed in several cancers according to recent investigations, and is closely associated with cancer growth, metastasis, and poor outcomes.101,102 ME2 depletion, accompanied by an increased NADP+/NADPH ratio and ROS levels, impacts PI3K/AKT signaling and enhances the sensitivity of erythroleukemia and NSCLC cells to cisplatin.103,104 Besides, ME2 ablation results in elevated cellular ROS levels, which activates the AMPK pathway and then stimulates TP53 to attenuate melanoma cell proliferation.105,106 ME2 is frequently hemizygously codeleted along with tumor suppressor SMAD4 in human solid tumors including gastric cancer and PDAC.107,108 In ME2-unexpressed gastric cancer cells, its isoenzyme ME1 is upregulated to replenish intracellular NADPH and promotes cell survival under glucose starvation and anoikis.107 ME3 is in lower enzymatic activity than do ME2 in mitochondria. However, in ME2 homozygously deleted PDAC cell lines, its isoenzyme ME3 plays the compensatory roles for intracellular NADPH homeostasis.108,109 These findings provide a prime ‘collateral lethality’ therapeutic strategy for the treatment of a substantial fraction of GC or PDAC patients.
말산 효소
ME는 말산의 산화적 탈카복실화 반응을 통해 말산을 피루vate로 전환함으로써 글리코lysis와 크렙스 회로의 분해 대사 구성 요소들을 연결하는 반응에 참여하며, 이 과정에서 NADPH 생성과 함께 합성 대사 과정을 유도합니다.32,88 정량적 유동 분석 결과, ME의 NADPH 생성 직접 기여도는 PPP의 기여도와 동일하다고 추정되었습니다.89 ME 가족은 세 가지 이소형으로 구성됩니다: ME1은 세포질에 위치하며, ME2와 ME3는 미토콘드리아에 위치합니다. ME1과 ME3는 NADP+를 필요로 하며, ME2는 NAD+ 또는 NADP+를 촉매 활성에 사용합니다. 따라서 ME는 미토콘드리아에서 NNT의 활성을 통해 NADH에서 NADP+로 수소 이온을 전달하고 NADPH를 생성함으로써 NADPH를 직접 및 간접적으로 생성할 수 있습니다.90 그러나 ME3는 많은 평가된 포유류 세포에서 거의 검출되지 않기 때문에 ME1과 ME2가 주요 이소형으로 보입니다.91
ME1의 과발현은 위암, 구강 편평상피암, 유방암, 폐암 등 암 환자의 예후가 나쁜 것과显著하게 연관되어 있습니다.92,93,94,95 ME1을 침묵시키면 NADPH가 현저히 감소하고 ROS 수준이 증가하며, 결국 글루코스 결핍이나 anoikis와 같은 산화 스트레스 하에서 세포 사멸을 유도합니다. 96,97 또한 ME1 단백질은 PGAM 가족 구성원 5에 의해 S336에서 저인산화되고 아세틸-CoA 아세틸트랜스퍼레이즈에 의해 K337에서 과아세틸화되어 미토콘드리아에서 세포질로 이동하며, 이로 인해 이량체화 및 활성화되어 NADPH 생성과 종양 형성을 강력히 촉진합니다. 98 ME1 발현은 TP53 또는 KRAS와 같은 잘 알려진 종양 억제제나 종양 유전자에 의해 조절됩니다.91,99 흥미롭게도 ME1과 PPP 구성 요소 사이에 직접적인 교차 작용이 존재하며, ME1은 PGD가 6-PG에 결합하는 능력을 증가시켜 NADPH 생성을 촉진합니다.100
ME2는 최근 연구에서 여러 암에서 과발현되며 암 성장, 전이, 및 예후와 밀접하게 연관되어 있습니다.101,102 ME2 결핍은 NADP+/NADPH 비율과 ROS 수준 증가와 동반되어 PI3K/AKT 신호전달을 영향을 미치며, 에리트로레우미아 및 NSCLC 세포의 시스플라틴 감수성을 증가시킵니다. 103,104 또한 ME2 제거는 세포 내 ROS 수준을 증가시켜 AMPK 경로를 활성화하고 TP53을 자극하여 멜라노마 세포 증식을 억제합니다.105,106 ME2는 위암 및 PDAC를 포함한 인간 고형 종양에서 종양 억제인자 SMAD4와 함께 반수체적으로 공동 삭제됩니다. 107,108 ME2가 발현되지 않은 위암 세포에서 그 이소효소 ME1은 세포 내 NADPH를 보충하여 포도당 결핍 및 anoikis 조건 하에서 세포 생존을 촉진합니다.107 ME3는 미토콘드리아에서 ME2보다 효소 활성이 낮습니다. 그러나 ME2가 동형접합적으로 삭제된 PDAC 세포주에서는 그 이소효소 ME3가 세포 내 NADPH 균형 유지에 보상 역할을 합니다.108,109 이러한 결과는 위암 또는 PDAC 환자의 상당 부분에 대한 치료를 위한 주요 '부수적 치사성' 치료 전략을 제공합니다.
Nicotinamide nucleotide transhydrogenase
NNT is an integral mitochondrial inner membrane protein in eukaryotes that catalyzes the transfer of hydride ions from NADH to NADP+ and produces NADPH utilizing the proton motive force generated by the electron transport chain (ETC).110 The process is essential for maintaining the mitochondrial NADPH and NADH pools. NNT activity contributes to 45% of the total NADPH in mitochondrial pool, indicating a significant role of NNT for NADPH pool maintenance,111 and NADPH obtained by NNT is also used for the reductive carboxylation of α-KG to isocitrate mediated by IDH2.112 In contrast to this prevailing view, a fascinating work illustrates that the NNT reverses the direction upon NADPH consumption to support NADH and ATP productions under a pathological workload, at the cost of NADPH-linked antioxidative capacity. The models unexpectedly show that lacking a functional NNT presents with less oxidative damage to the heart compared to mice with active NNT.113 This finding provides potentially fresh insights into pathology and metabolic regulation, but more study about the NNT reversal process in cancer is urgently needed.
In cancer cells, NNT activity is stimulated by hyperpolarized mitochondria. Further, the NADH from increased glycolysis in the cytosol can be transferred to mitochondria to drive NADH-dependent NNT.89 Additionally, NNT is overexpressed in gastric cancer cell, which is associated with lower overall survival and disease-free survival. NNT knockdown shows limited ability to maintain NADPH levels and reduces tumorigenicity under oxidative stress conditions, such as that induced by anoikis, glucose deprivation in vitro, or impairs peritoneal dissemination and lung metastasis in vivo.114 Similar effects are observed in liver cancer,115 pheochromocytoma116 and NSCLC,111 and NNT is likely to be activated by NADPH consumption, such as in IDH-mutant cells.117 Additionally, considered as a key antioxidative enzyme, NNT is critical for inducing macrophage inflammatory responses118 and preventing ROS-induced cytotoxicity in T cells exposed to asbestos that can cause a reduction in antitumor immunity.119 To date, NNT appears to play a key role in tumorigenesis and modification of NNT may regulate immune effects of anti-tumor. Unfortunately, pharmacological inhibitors specific for NNT have not been reported and need to be developed.
니코틴아미드 뉴클레오티드 트랜스히드로게나제
NNT는 진핵생물의 미토콘드리아 내막에 존재하는 필수 단백질로, NADH로부터 NADP+로 수소 이온을 전달하여 전자 전달 사슬(ETC)에 의해 생성된 프로톤 동력에 의해 NADPH를 생성합니다.110 이 과정은 미토콘드리아 내 NADPH 및 NADH 풀의 유지에 필수적입니다. NNT의 활성은 미토콘드리아 풀 내 총 NADPH의 45%를 차지하며, 이는 NNT가 NADPH 풀 유지에 중요한 역할을 함을 보여줍니다.111 또한 NNT를 통해 생성된 NADPH는 IDH2에 의해 매개되는 α-KG의 환원적 카복실화 반응을 통해 이소시트레이트로 전환됩니다. 112 이 일반적인 관점과 달리, 흥미로운 연구는 병리학적 부하 하에서 NADPH 소비 시 NNT가 방향을 역전시켜 NADH 및 ATP 생성을 지원하며, 이는 NADPH 관련 항산화 능력을 희생시키는 것으로 나타났습니다. 모델 결과는 기능적 NNT가 결여된 경우 활성 NNT를 가진 쥐에 비해 심장 산화 손상이 덜 발생함을 의외로 보여주었습니다.113 이 발견은 병리학과 대사 조절에 대한 새로운 통찰을 제공하지만, 암에서의 NNT 역전 과정에 대한 추가 연구가 시급히 필요합니다.
암 세포에서 NNT 활성은 과분극화된 미토콘드리아에 의해 자극됩니다. 또한 세포질에서 증가한 글리코lysis로부터 생성된 NADH는 미토콘드리아로 전달되어 NADH 의존적 NNT를 촉진합니다.89 또한 위암 세포에서 NNT는 과발현되며, 이는 전체 생존율과 질병 무재발 생존율의 감소와 연관되어 있습니다. NNT 발현을 억제하면 산화 스트레스 조건 하에서 NADPH 수준을 유지하는 능력이 제한적이며, anoikis, 체외에서 포도당 결핍으로 유발된 산화 스트레스 조건에서 종양 형성 능력을 감소시키거나, 체내에서 복막 확산 및 폐 전이를 방해합니다.114 유사한 효과는 간암,115 페오크로모시토마,116 및 NSCLC에서 관찰되며, NNT는 IDH 돌연변이 세포에서와 같이 NADPH 소비에 의해 활성화될 가능성이 있습니다. 117 또한 주요 항산화 효소로 간주되는 NNT는 대식세포의 염증 반응을 유도하는 데 필수적이며,118 석면 노출로 인한 ROS 유발 세포 독성으로 항종양 면역력이 감소하는 것을 방지합니다.119 현재까지 NNT는 종양 발생에 핵심적인 역할을 하며, NNT의 조절은 항종양 면역 효과를 조절할 수 있습니다. 불행히도 NNT에 특이적인 약리학적 억제제는 보고되지 않았으며 개발이 필요합니다.
Isocitrate dehydrogenases (IDH)
IDH also facilitates the generation of NADPH from NADP+ by catalyzing the oxidative decarboxylation of isocitrate to α-ketoglutarate (α-KG) for TCA cycle.120 There are three subtypes of IDH: IDH1 is located within the cytosol and peroxisomes, and IDH2/3 are primarily found in mitochondria. IDH1/2 use NADP+ as a cofactor and conduct a reversible reaction, while IDH3 uses NAD+ as a cofactor and conducts irreversible conversion.121,122
Multiple lines of evidences have revealed that IDH1 is overexpressed in numerous cancers and is closely correlated with poor prognoses of patients with non-small cell lung carcinoma (NSCLC),123 PDAC,124 or one of several hematological malignancies.125 Notably, ELISA demonstrate that IDH1 level is also significantly elevated in the plasma of NSCLC patients, suggesting that it can be used as a potential plasma biomarker.126 The upregulation of IDH1 may represent a common metabolic adaptation for diminishing oxidative stress and supporting macromolecular synthesis, consequently promoting tumor growth and therapy resistance.125 Furthermore, IDH1 silencing results in decreased NADPH and α-KG levels, with the increased ROS levels, leading to cancer cell apoptosis in NSCLC.123 Besides, oxidative stress conditions also increase the innately high IDH1 expression, and IDH1 silencing significantly enhances cell sensitivity to cancer chemotherapy, radiotherapy, and photodynamic therapy by reducing NADPH.124,127,128 In addition, IDH1 is hyperacetylated in CRC cells and is significantly correlated with distant metastasis and poor survival. SIRT2-dependent IDH1 deacetylation at K224 impairs its enzymatic activity and represses its malignant behaviors in CRC.129 Specially, studies also found that IDH1 is significantly downregulated in clear cell renal cell carcinoma (ccRCC) compared with normal kidney cells, suggesting that IDH1 may function as a candidate tumor suppressor for ccRCC.130,131
Most studies indicate that IDH2 is also significantly upregulated in ESCC,132 ovarian cancer,133 lung cancer and other types of cancer,134 playing a pro-oncogenic role. Overexpression of IDH2 decreases ROS levels and increases cancer cell growth.121 IDH2 depletion decreases the expression of HIF1α and leads to the attenuation of tumor growth in lung cancer.134 However, because of heterogeneity among cancer cells, other studies have shown that IDH2 expression is decreased in metastatic HCC and gastric cancer tissues compared with paired normal tissues.135,136 The underlying mechanism is that these cells lacking IDH2 show enhanced invasive behavior due to the increase in matrix metalloproteases, which depend on the NF-κB pathway. In addition, NAD+ production by the NNT enhance SIRT3-mediated deacetylation and loss of NAD+-dependent deacetylase SIRT3 increases the acetylation of IDH2 at K413 and decreases its enzymatic activity by reducing dimerization, thus regulates mitochondrial redox status and promotes cell tumorigenesis in luminal B breast cancer,137 and B cell malignancies.138 SIRT5-mediated IDH2 desuccinylation also regulates cellular NADPH homeostasis and redox potential.54
The contribution of IDH to NADPH generation in cancer remains controversial. IDH1 and IDH2 also catalyze the reductive carboxylation and support tumor cells growth with defective mitochondria. Studies show that IDH1/2 syntheses isocitrate/citrate from α-KG with NADPH consumption, then the isocitrate/citrate import into the mitochondria and contribute to suppress mitochondrial ROS.139,140 In addition, recently, IDH1 and IDH2 gene mutations have been prevalent in several diverse malignancies, including glioma, AML, angioimmunoblastic lymphomas, chondrosarcoma, and melanomas.141,142 Recurrent somatic mutation of residues are mainly located at enzymatic active sites that bind to isocitrate, typically at R132 including R132H, R132L, R132S, R132C, and R132G in IDH1, and R140Q or R172K in IDH2.143,144 The mutated IDH1 and IDH2 proteins are endowed with a novel ability to catalyze the reduction of α-KG to generate a rare metabolite, 2-hydroxyglutarate (2-HG), while consuming NADPH.145 Further, the relevance of these mutations and their roles in carcinogenesis and possible therapeutic implications have been extensively reviewed elsewhere.141,146,147
이소시트르산 탈수소효소 (IDH)
IDH는 TCA 회로에서 이소시트르산을 α-케토글루타레이트 (α-KG)로 산화 탈카복실화 반응을 촉매하여 NADP+에서 NADPH를 생성하는 데 기여합니다.120 IDH는 세 가지 하위 유형으로 구분됩니다: IDH1은 세포질과 과산화소체에 존재하며, IDH2/3는 주로 미토콘드리아에 존재합니다. IDH1/2는 NADP+를 보조인자로 사용하며 가역적 반응을 수행하는 반면, IDH3는 NAD+를 보조인자로 사용하며 비가역적 전환을 수행합니다.121,122
다양한 연구 결과는 IDH1이 여러 암에서 과발현되며, 비소세포 폐암(NSCLC),123 췌장암(PDAC),124 또는 여러 혈액암 중 하나와 관련된 환자의 예후와 밀접하게 연관되어 있음을 보여주었습니다.125 특히, ELISA 분석은 NSCLC 환자의 혈장 내 IDH1 수치가 유의미하게 증가함을 보여주어, 잠재적인 혈장 바이오마커로 활용될 수 있음을 시사합니다. 126 IDH1의 발현 증가가 산화 스트레스를 감소시키고 대분자 합성을 지원함으로써 종양 성장과 치료 저항성을 촉진하는 공통된 대사적 적응 메커니즘을 나타낼 수 있습니다.125 또한, IDH1 침묵화는 NADPH 및 α-KG 수치를 감소시키고 ROS 수치를 증가시켜 NSCLC에서 암 세포 사멸을 유발합니다. 123 또한, 산화 스트레스 조건은 본래 높은 IDH1 발현을 증가시키며, IDH1 침묵화는 NADPH 감소로 인해 암 화학요법, 방사선 요법, 광역학 요법에 대한 세포 감수성을 크게 향상시킵니다.124,127,128 또한, IDH1은 CRC 세포에서 과아세틸화되며, 원격 전이와 예후 불량과 유의미하게 관련되어 있습니다. SIRT2에 의존적인 IDH1의 K224 위치에서의 탈아세틸화는 그 효소 활성을 저해하고 CRC에서의 악성 행동을 억제합니다.129 특히, 연구들은 IDH1이 정상 신장 세포와 비교해 투명 세포 신장 세포 암종(ccRCC)에서 유의미하게 하향 조절됨을 발견했으며, 이는 IDH1이 ccRCC의 후보 종양 억제자로 기능할 수 있음을 시사합니다.130,131
대부분의 연구는 IDH2가 ESCC,132 난소암,133 폐암 및 기타 암 유형에서 유의미하게 과발현되며,134 종양 촉진 역할을 한다는 것을 보여줍니다. IDH2 과발현은 ROS 수준을 감소시키고 암 세포 성장 속도를 증가시킵니다.121 IDH2 결핍은 폐암에서 HIF1α 발현을 감소시키고 종양 성장 억제를 유발합니다. 134 그러나 암 세포의 이질성으로 인해 다른 연구에서는 IDH2 발현이 전이성 간세포암(HCC) 및 위암 조직에서 대조군 정상 조직에 비해 감소된 것으로 나타났습니다.135,136 이 메커니즘은 IDH2를 결여한 세포가 NF-κB 경로에 의존하는 매트릭스 메탈로프로테아제(MMP) 증가로 인해 침습성이 강화되기 때문입니다. 또한 NNT에 의한 NAD+ 생산은 SIRT3 매개 탈아세틸화를 촉진하며, NAD+ 의존성 탈아세틸레이즈 SIRT3의 감소는 IDH2의 K413 위치에서의 아세틸화를 증가시키고 이중화 감소로 인해 효소 활성을 감소시켜 미토콘드리아 산화환원 상태를 조절하고 유방암의 luminal B 유형 및 B 세포 악성 종양에서 세포 종양 형성을 촉진합니다.137 138 SIRT5에 의한 IDH2의 데수신일화도 세포 내 NADPH 균형과 환원 잠재력을 조절합니다.54
암에서 IDH가 NADPH 생성에 미치는 기여도는 여전히 논란의 여지가 있습니다. IDH1과 IDH2는 환원성 카복실화 반응을 촉매하며, 미토콘드리아 기능 장애를 가진 종양 세포의 성장에 기여합니다. 연구 결과, IDH1/2는 NADPH 소비를 통해 α-KG로부터 이소시트르산/시트르산을 합성하며, 이 이소시트르산/시트르산은 미토콘드리아로 수입되어 미토콘드리아 ROS 억제에 기여합니다.139,140 또한 최근 IDH1 및 IDH2 유전자 변이가 뇌종양, 급성 골수성 백혈병(AML), 혈관면역성 림프종, 연골육종, 흑색종 등 다양한 악성 종양에서 널리 관찰되었습니다. 141,142 재발성 체세포 돌연변이는 주로 이소시트레이트와 결합하는 효소 활성 부위에 위치하며, IDH1에서는 R132(R132H, R132L, R132S, R132C, R132G)에, IDH2에서는 R140Q 또는 R172K에 주로 발생합니다. 143,144 변이된 IDH1 및 IDH2 단백질은 α-KG의 환원을 촉매하여 희귀 대사산물인 2-하이드록시글루타레이트(2-HG)를 생성하며 NADPH를 소비하는 새로운 능력을 갖추고 있습니다.145 또한 이러한 변이의 관련성, 암 발생에서의 역할 및 잠재적 치료적 함의는 다른 문헌에서 광범위하게 검토되었습니다.141,146,147
Glutamine metabolism
Glutamine metabolism is a major cellular carbon source for the TCA cycle, a nitrogen donor for nucleotide, amino acid, and lipid biosynthesis, it is also critical for maintaining NADPH levels.148,149 Proliferating cancer cells exhibit aerobic glycolysis, leading to a shift in glucose carbon away from the TCA cycle, which results in the increased use of glutamine to fuel anabolic processes to support rapid cell growth with increased NADPH and ammonia generation. Glutaminolysis is the mitochondrial pathway by which glutamine is first deaminated to glutamate by glutaminases (GLS1/2). Then, either NADPH-dependent glutamate dehydrogenases (GDH) or other transaminases, including glutamate oxaloacetate transaminase 2 (GOT2) and glutamate pyruvate transaminase 2 (GPT2), convert glutamate into a-KG to meet the need for corresponding amino acids.89
Conventionally, GDH (coded by the GLUD gene) is the more predominant enzymes vital for the reactions needed to replenish the TCA cycle and yield NADPH than GOT2 and GPT2, which consists of ubiquitously expressed GDH1 and GDH2 mainly existing in neuronal and testicular tissue and having lower activity than GDH1.150 GDH1 is highly expressed in most tumor samples and correlated with tumor progression stage, including breast cancer and lung cancer cells.151,152 GDH1 depletion results in imbalanced redox homeostasis and cell cytotoxicity and attenuates cancer cell proliferation, which as well as the results in erythroleukemia cells, while it negligibly affects normal cell proliferation.151 Additionally, enhanced GDH1 activity has also been reported to be a possible prognostic marker and an indicator of metastasis in patients with CRC or gastric cancer.153,154 Under conditions of insufficient glycolysis caused by glucose deprivation, 2-deoxyglucose treatment or Akt signaling inhibition, glutamine-addicted cells are more sensitive to GDH1 deficiency.155 Furthermore, GDH-derived NADPH is consumed to support the reductive carboxylation of α-KG by IDH2, and the compensatory increase in the expression of GDH1 or GDH2 promote the growth of IDH-mutant glioma cells.156 Besides, with the consumption of extracellular glutamine, GDH can also catalyze ammonia derived from glutaminolysis and α-KG to support the synthesis of glutamate and downstream metabolites by reductive amination in a NADPH consumption manner to meet the cancer cells growth.148,157,158
Specifically, some cancer cells, such as PDAC and CRC cells, depend on a noncanonical glutamine metabolism pathway in the cytosol under the regulation of oncogenic KRAS activation. Glutamine-derived aspartate induced by GOT2 is transported into the cytosol and converted by GOT1 to oxaloacetate, then converted by malate dehydrogenase (MDH1) into malate and subsequently oxidized into pyruvate by ME1 to create NADPH.159,160 GHD1 shRNA has no effect on PDAC cells growth, while knocking down GOT2 elevates ROS levels and leads to cell senescence.161 Further, cytosolic GOT1 inhibition decreases oxaloacetate levels and reduces the cellular NADPH/NADP+ and GSH/GSSG ratios.159 Consistent with these findings, the addition of exogenous malate protects cells from excessive ROS accumulation in MDH1-knockdown cells.162 Consequently, targeting the glutamine metabolism pathway, which is essential for cancer cells but dispensable for normal cells, may lead to novel therapeutic approaches to treat refractory tumors.
글루타민 대사
글루타민 대사는
TCA 회로의 주요 세포 내 탄소 공급원이며,
핵산, 아미노산, 지질 생합성에 필요한 질소 공급원입니다.
또한 NADPH 수준을 유지하는 데
증식 중인 암 세포는 호기성 당분해 과정을 통해
포도당의 탄소 흐름이 TCA 회로에서 벗어나 글루타민의 사용이 증가하여,
NADPH와 암모니아 생성이 증가하며 빠른 세포 성장에 필요한 동화 과정을 지원합니다.
글루타민 분해는
글루타민이 글루타민아제(GLS1/2)에 의해 글루타메이트로 탈아미노화되는
미토콘드리아 경로입니다.
이후
NADPH 의존성 글루타메이트 탈수소효소(GDH) 또는
글루타메이트 옥살아세테이트 트랜스아미나제 2(GOT2)와
글루타메이트 피루vate 트랜스아미나제 2(GPT2)와 같은
다른 트랜스아미나제가 글루타메이트를 a-KG로 전환하여
해당 아미노산의 요구를 충족시킵니다.89
전통적으로 GDH(GLUD 유전자에 의해 암호화됨)는 TCA 회로를 보충하고 NADPH를 생성하는 반응에 필수적인 효소로, GOT2와 GPT2보다 더 널리 분포되어 있습니다. GOT2와 GPT2는 주로 신경 세포와 고환 조직에 존재하는 GDH1과 GDH2로 구성되며, GDH1보다 활성이 낮습니다. 150 GDH1은 대부분의 종양 샘플에서 고도로 발현되며 유방암 및 폐암 세포를 포함한 종양 진행 단계와 관련이 있습니다.151,152 GDH1 결핍은 산화환원 균형 장애와 세포 독성을 유발하며 암 세포 증식을 억제하며, 이는 적혈구 백혈병 세포에서도 관찰되지만 정상 세포 증식에는 미미한 영향을 미칩니다. 151 또한, GDH1 활성 증가가 대장암 또는 위암 환자의 예후 지표 및 전이 지표로 가능할 수 있다는 보고가 있습니다.153,154 포도당 결핍으로 인한 글리코lysis 부족 조건에서 2-데옥시글루코스 처리 또는 Akt 신호전달 억제 시 글루타민 의존성 세포는 GDH1 결핍에 더 민감합니다. 155 또한 GDH에서 유래한 NADPH는 IDH2에 의한 α-KG의 환원적 카복실화 지원을 위해 소비되며, GDH1 또는 GDH2 발현의 보상적 증가가 IDH 돌연변이 글리오마 세포의 성장을 촉진합니다. 156 또한, 세포외 글루타민의 소비와 함께 GDH는 글루타민 분해에서 유래한 암모니아와 α-KG를 환원 아미노화 반응을 통해 NADPH 소비 방식으로 글루타메이트 및 하류 대사물의 합성을 촉진하여 암 세포의 성장을 지원합니다.148,157,158
특히, PDAC 및 CRC 세포와 같은 일부 암 세포는 종양 유발성 KRAS 활성화의 조절 하에 세포질에서 비전형적인 글루타민 대사 경로에 의존합니다. GOT2에 의해 유도된 글루타민 유래 아스파르테이트는 세포질로 운반되어 GOT1에 의해 옥살아세테이트로 전환된 후 말산 탈수소효소(MDH1)에 의해 말산으로 전환되고, 이후 ME1에 의해 피루vate로 산화되어 NADPH를 생성합니다.159,160 GHD1 shRNA는 PDAC 세포의 성장에 영향을 미치지 않지만, GOT2를 억제하면 ROS 수준이 증가하고 세포 노화가 발생합니다. 161 또한, 세포질 내 GOT1 억제는 옥살아세테이트 수준을 감소시키고 세포 내 NADPH/NADP+ 및 GSH/GSSG 비율을 감소시킵니다. 159 이러한 결과와 일치하게, MDH1 발현을 억제한 세포에서 외인성 말레이트를 추가하면 과도한 ROS 축적으로부터 세포를 보호합니다.162
따라서
암 세포에는 필수적이지만
정상 세포에는 필수적이지 않은 글루타민 대사 경로를 표적으로 삼는 것은
치료가 어려운 종양을 치료하기 위한 새로운 치료 전략으로 이어질 수 있습니다.
Fatty acid oxidation
In addition, FAO pathway is also key for providing NADPH indirectly, which is indispensable in many cancers especially under metabolic stress. FAO generates NADH, FADH2, and acetyl coenzyme A (CoA) in each round,163 and NADH and FADH2 enter the ETC while the acetyl CoA enters the TCA cycle to produce citrate, which is exported to the cytosol to engage in NADPH production through ME1 and IDH1.34 FAO and FAS are both essential for tumor progression and support each other. Acetyl CoA and NADPH accumulated from FAO metabolism in the cytosol are needed to initiate FAS.164 The carnitine palmitoyl transferases (CPT), the rate-limiting enzymes in the FAO pathway, transport long-chain acyl-CoA from the cytosol to mitochondria.165 CPT-mediated FAO activation is reported to play key roles in maintaining NADPH homeostasis and promoting cell metastasis and chemoresistance in gastrointestinal cancer166,167 and melanoma.168 Recent studies also show that knocking down PPAR coactivator 1α (PGC1α), an important transcriptional coactivator regulating CPT1A and CPT1B, obviously decreases the ratio of NADPH/NADP+ and ATP levels, impairing radiation resistance in nasopharyngeal carcinoma (NPC) cells.169 What’s more, AMP-activated protein kinase (AMPK) also regulates the function of FAO in maintaining NADPH homeostasis and promotes tumor cell survival under oxidative stress or metabolic stress.170,171,172,173
지방산 산화
또한, FAO 경로는
NADPH를 간접적으로 공급하는 데도 핵심적인 역할을 하며,
이는 특히 대사 스트레스 하에서 많은 암에서 필수적입니다.
FAO는 각 단계에서
NADH, FADH2, 및 아세틸 코엔자임 A (CoA)를 생성합니다.163
이 중 NADH와 FADH2는
전자 전달 체계(ETC)로 들어가며,
아세틸 CoA는 트리카르펜산 회로(TCA 회로)로 들어가 시트르산을 생성합니다.
시트르산은 세포질로 수출되어 ME1과 IDH1을 통해 NADPH 생성에 참여합니다.34 FAO와 FAS는 모두 종양 진행에 필수적이며 서로를 지원합니다. 세포질에서 FAO 대사로부터 축적된 아세틸 CoA와 NADPH는 FAS를 시작하기 위해 필요합니다. 164 FAO 경로의 속도 제한 효소인 카르니틴 팔미토일 트랜스퍼레이즈(CPT)는 세포질에서 미토콘드리아로 장쇄 아실-CoA를 운반합니다.165 CPT 매개 FAO 활성화는 위장관 암166,167 및 흑색종에서 NADPH 균형 유지, 세포 전이 촉진, 화학 요법 저항성 증진에 핵심 역할을 한다는 보고가 있습니다. 168 최근 연구에서는 CPT1A와 CPT1B를 조절하는 중요한 전사 공활성인자 PPAR 공활성인자 1α(PGC1α)를 억제하면 NADPH/NADP+ 비율과 ATP 수준이 현저히 감소하여 비인두암(NPC) 세포의 방사선 저항성이 저하된다는 것이 밝혀졌습니다. 169 또한 AMP 활성화 단백질 키나제(AMPK)는 NADPH 균형 유지에 관여하는 FAO의 기능을 조절하며, 산화 스트레스나 대사 스트레스 하에서 종양 세포의 생존을 촉진합니다.170,171,172,173
Therapeutic implications for targeting NADPH metabolism
Compared with their normal counterparts, many types of cancer cell have increased oxidative stress and the upregulation of antioxidant capacity. With the metabolic reprogramming of NADPH, cancer cells increase the demand of NADPH for antioxidative effects and anabolic reactions. The specific vulnerability of tumor cells leveraging the aberrant NADPH-synthesis pathways can be exploited to induce cell death under various cellular stresses. Manipulating ROS levels by redox modulation is a way to selectively kill cancer cells without causing significant toxicity to normal cells. This strategy is the basis for many anticancer therapeutics, including chemotherapeutics, radiotherapies, and most small-molecule inhibitor-based therapies, which impair tumor metabolism and induce excessive ROS accumulation, inducing cell toxicity and death.11,174 As illustrated in Fig. 3, the inhibitors targeting NADPH-synthesis enzymes are being extensively developed. The specific target, anti-tumor effect, and clinical progress of these inhibitors targeting NADPH metabolism are also summarized in Tables 1 and 2.
NADPH 대사 표적화의 치료적 함의
정상 세포와 비교했을 때,
많은 종류의 암 세포는 산화 스트레스가 증가하고
항산화 능력이 활성화됩니다.
NADPH의 대사 재프로그래밍을 통해
암 세포는 항산화 효과와 동화 반응을 위해
NADPH의 수요를 증가시킵니다.
종양 세포가 이상적인 NADPH 합성 경로를 활용하는 특정 취약성은
다양한 세포 스트레스 하에서 세포 사멸을 유도하는 데 활용될 수 있습니다.
산화환원 조절을 통해 ROS 수준을 조작하는 것은
정상 세포에 심각한 독성을 유발하지 않고
암 세포를 선택적으로 죽이는 방법입니다.
이 전략은 화학요법, 방사선 요법, 대부분의 소분자 억제제 기반 치료법 등
종양 대사 장애를 유발하고 과도한 ROS 축적을 유도해 세포 독성과 사멸을 유발하는
그림 3에 표시된 바와 같이, NADPH 합성 효소를 표적하는 억제제들이 광범위하게 개발되고 있습니다.
NADPH 대사 표적 억제제의 특정 표적, 항종양 효과, 임상 진행 상황은 표 1과 2에 요약되어 있습니다.
Fig. 3
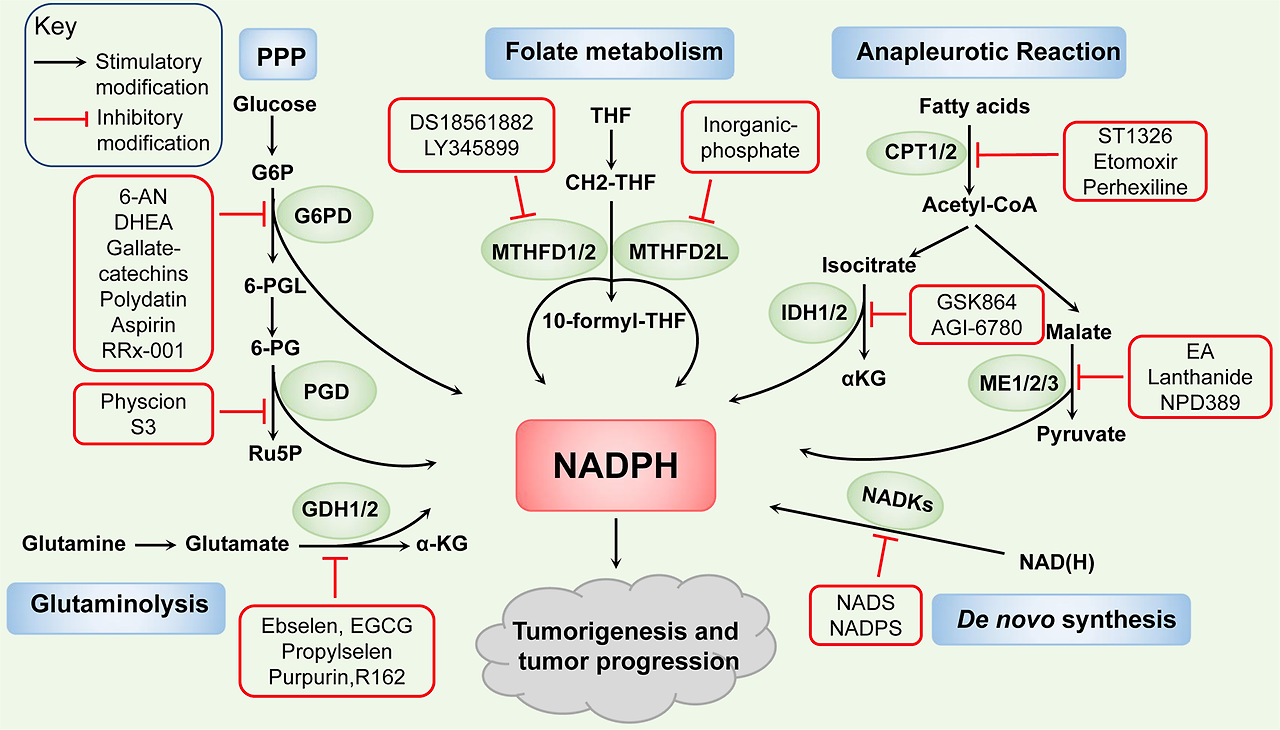
Therapeutic implications for targeting NADPH metabolism. Many inhibitors targeting NADPH-synthesis enzymes have been discovered to impair NADPH pool, thus attenuate tumorigenesis and tumor progression. Such as NADS, NADPS of NADKs. 6-AN, DHEA, gallate-catechins, polydatin, aspirin, RRx-001 of G6PD. Physcion, S3 of PGD. DS18561882, LY345899 of MTHFD1/2. Inorganic phosphate of MTHFD2L. GSK864 of IDH1 and AGI-6780 of IDH2. Lanthanide of ME1 and EA, NPD389 of ME2. ST1326, Etomoxir of CPT1, and perhexiline of CPT2. Ebselen, EGCG, propylselen of GDH1/2 and purpurin, R162 of GDH1
Table 1 The pre-clinical studies with inhibitors targeting NADPH metabolism in cancer
Table 2 The clinical trials with inhibitors targeting NADPH metabolism in cancer
For de novo NADPH synthesis pathway, correlation studies have revealed that thionicotinamide adenine dinucleotide (NADS) and thionicotinamide adenine dinucleotide phosphate (NADPS), converted from the pro-drug thionicotinamide (TN), act as inhibitors of NADKs through targeting the NAD-binding site of NADKs and decreasing the levels of NADPH.37 Combining TN with several chemotherapeutic drugs induces synergistic cell killing, indicating its efficacious antitumor effect in DLBCL and colon cancer.38 Further, reduced NADPH levels induced by NADPS results in accelerated degradation of DHFR and impairment of the folate cycle, which delays cancer cell growth.175
For the PPP enzymes, recent studies have discovered some inhibitors targeting on G6PD, such as NADP+ analogs, the competitive inhibitor 6-aminonicotinamide (6-AN), noncompetitive inhibitors epiandrosterone and dehydroepiandrosterone (DHEA) which reduces the availability of NADPH and inhibits the cell growth.176 The combination of cisplatin and 6-AN optimizes the clinical dose and minimized the side effects.177,178,179 The new small molecule inhibitors are gradually being discovered, such as, gallated catechins (EGCG, GCG, ECG, CG), as the competitive inhibitors of NADP+, repress the activity of G6PD and suppress NADPH production.180 The natural molecule polydatin increases the NADP+/NADPH ratio and decreases the invasion of breast cancer cells by inhibiting G6PD activity.181 Further, the activity of G6PD is also repressed by aspirin casing acetylation of G6PD to decrease the activity of G6PD and the generation of NADPH, and by RRx-001, a novel clinical-stage chemosensitizer and radiosensitizer, which exerts antiproliferative effects in human tumor cells.182,183 Moreover, physcion and its derivative S3, novel small-molecule PGD inhibitors which fits in a pocket of PGD near the binding site of 6-PG to inhibit PGD enzyme activity and then decrease the NADPH level, exhibit excellent anticancer effects and sensitize leukemia cells to antimalarial agent dihydroartemisinin (DHA).184 For the folate metabolism pathway, the NADP+-dependent dehydrogenase activity of MTHFD2 and MTHFD2L can be inhibited by inorganic phosphate.185 Besides, other MTHFD2 inhibitors have been reported, including DS18561882 and LY345899 in a substrate-based manner, and treatments based on them decrease cellular NADPH/NADP+ ratio, increase cellular ROS levels, and impair tumorigenesis and metastasis.74,186 For the glutamine metabolism pathway, ebselen, epigallocatechin-3-Gallate (EGCG), and propylselen are reported to bind to GDH-active sites to abolish NADP+ binding and impair in cancer cell functions.187 A study also shows that purpurin and its analog, R162, acting as mixed model inhibitors of GDH1, inhibit GDH1 activity, elevate ROS levels and thus attenuate cancer cell proliferation.151
For the NADPH-synthesis enzymes involved in anapleurotic reactions, including IDH1/2, ME1/2/3, and CPT1/2, the targeting inhibitors are also being extensively developed. Study shows that treatment with GSK864 as IDH1 inhibitor binding an allosteric site on IDH1 reduces the NADPH/NADP+ ratio and prolongs the survival of glioblastoma multiforme (GBM) PDXs model.125 AGI-6780 treatment, binding with IDH2 or mutant IDH2 in an allosteric manner at the dimer interface, reduce the IDH2 activity and lead to the repression of cell growth in lung cancer.134 Mutant IDH-targeted therapy and a number of important recent pre-clinical and clinical studies in IDH-mutant solid tumors have been extensively reviewed elsewhere,147 and listed in Table 2. Furthermore, NPD389 binding to ME2 in fast-binding mode impairs its activity,188 and embonic acid (EA) induces the cellular senescence of H1299 cancer cells through its noncompetitive inhibitory activity against ME2.189 Further, ME1 treated with the inhibitor (piperazine-1-pyrrolidine-2,5-dione) has little effect on normal rat intestinal epithelial cells but strongly suppresses human CRC cell growth by targeting ME1 NADP+-binding site and reducing the NADPH level.90 Lanthanide treatment represses cell proliferation and the epithelial–mesenchymal transition (EMT) by inhibiting ME1 in oral squamous cell carcinoma cells.93 In addition, CPTs are also considered to be targeted. Glioma cells with FAO inhibited by etomoxir, a CPT1 inhibitor, exhibits a profound decrease in NADPH levels, reduced GSH content and elevation of intracellular ROS levels. Besides, CPT1A-suppression or etomoxir treatment fails to maintain redox homeostasis in detached CRC cells and induces sensitivity to glucose deprivation in PDAC cells.166,190 Further, in gastrointestinal cancer cells, genetic inhibition or pharmacological treatment of CPT2 with perhexiline disrupts NADPH and promotes cell apoptosis after oxaliplatin treatment. Combining perhexiline with oxaliplatin leads to a significant suppression of cancer progression.167 Other inhibitors of CPTs are also discovered, such as Ro25-0187, ST1326 which are expected to be used for cancer treatment.191
Conclusions
In summary, the essential role of NADPH homeostasis has been increasingly recognized in cancer development and progression through cellular antioxidative effects and anabolic reactions. Pharmacological restriction of cellular NADPH availability by targeting its synthesis pathways to impair NADPH homeostasis is currently recognized as a crucial and potential strategy for cancer treatment.
However, there is an interdependent relationship in which the NADPH pool is simultaneously supported and used by various pathways in cells. For example, pyruvate kinase muscle isoform 2 (PKM2) inactivation can both attenuate the glucose flux to PPP and enhance folate metabolism to mediate NADPH generation.32,43 Moreover, because of the heterogeneous nature of tumors, there are considerable variations in NADPH-related processes in different tumors, for example, the main pathways of glutamine metabolism in the context of PDAC are different from the previous prevailing view as informed by studies of other cancers,159,192 indicating the need for careful analyses of individual characteristics among cancers for establishing individualized precision therapy. Moreover, the special functions of these metabolic enzymes are not fully understood in cancer. For instance, the reverse-mode NNT that consumes NADPH to support NADH and ATP productions in contrast to the conventional view has not been reported with respect to cancer.113 Besides, because of the high plasticity of the metabolic network and metabolite exchange among cancer and stromal cells, a compensatory response can be readily induced to produce limiting metabolites.193 In addition, the relative contribution of these pathways and enzymes to NADPH production can be variable in different cell types and under different conditions. Hence, additional studies are needed to evaluate the entire NADPH metabolome, identify the important interrelationships and determine the main pathway to select more suitable targets. Also, the effects of NADPH metabolism on immune cells in the tumor microenvironment are needed to explore for exploiting novel anticancer opportunities.
As the NADPH metabolism are shared in normal and cancer cells, selectively targeting NADPH synthesis under special circumstances without affecting normal cells is difficult. Therefore, one of the greatest challenges to target cancer metabolism is the induction of toxic effects on noncancerous cells. Further, many reported small-molecule inhibitors target several metabolic enzymes with similar structures, for example, EGCG targets both NADPH-dependent FASN and NADP+-dependent GDH.21,187 The functions can be also markedly different among the isoforms of these enzymes. For instance, cytosolic ALDH1L1 mainly regulates reduced synthesis, while mitochondrial ALDH1L2 produces NADPH to attenuate oxidative stress.78 IDH1/2 use NADP+ as a cofactor while IDH3 needs NAD+121. The development of highly selective or isoform-specific inhibitors will reduce side effects and is an important goal for the near future. Most compounds specifically targeting cancer NADPH metabolism are in preclinical studies, thus there are still challenges to address before these compounds enter the clinic. Collectively, to better understand the therapeutic potential of NADPH metabolism, more preclinical and clinical studies should be implemented to address these difficulties, and combined approaches with immunotherapy and/or chemotherapeutics should be pursued as the best strategies because of their synergistic effects.
결론
요약하자면, NADPH 균형의 필수적인 역할은 세포 내 항산화 효과와 동화 반응을 통해 암의 발생과 진행에 있어 점점 더 중요하게 인식되고 있습니다. 세포 내 NADPH 가용성을 표적화하여 그 합성 경로를 억제함으로써 NADPH 균형을 방해하는 약리학적 접근은 현재 암 치료를 위한 중요한 잠재적 전략으로 인정받고 있습니다.
그러나 NADPH 풀은 세포 내 다양한 경로에 의해 동시에 지원되고 사용되는 상호 의존적인 관계에 있습니다. 예를 들어, 피루vate 키나제 근육 이소형 2(PKM2)의 비활성화는 PPP로의 포도당 유입을 억제하고 엽산 대사 활성을 증가시켜 NADPH 생성을 조절할 수 있습니다. 32,43 또한 종양의 이질적인 특성으로 인해 다양한 종양에서 NADPH 관련 과정에 상당한 차이가 존재합니다. 예를 들어, PDAC(췌장암) 맥락에서 글루타민 대사 주요 경로는 다른 암 연구에서 제시된 기존 관점과 다르며,159,192 이는 암별 특성을 세밀히 분석하여 개인 맞춤형 정밀 치료를 수립하기 위해 주의 깊은 분석이 필요함을 시사합니다. 또한, 이러한 대사 효소의 특수 기능은 암에서 완전히 이해되지 않았습니다. 예를 들어, 전통적인 관점과 달리 NADPH를 소비하여 NADH와 ATP 생성을 지원하는 역방향 NNT는 암에 대해 보고되지 않았습니다.113 또한, 암 세포와 Stromal 세포 간의 대사 네트워크와 대사체 교환의 높은 가소성으로 인해 제한적인 대사체를 생산하기 위한 보상 반응이 쉽게 유도될 수 있습니다. 193 또한 이러한 경로와 효소가 NADPH 생산에 기여하는 상대적 비율은 세포 유형과 조건에 따라 달라질 수 있습니다. 따라서 전체 NADPH 대사체를 평가하고 중요한 상호 관계를 식별하며 주요 경로를 결정하여 더 적합한 표적을 선택하기 위해 추가 연구가 필요합니다. 또한 종양 미세환경 내 면역 세포에 대한 NADPH 대사 영향은 새로운 항암 기회를 탐색하기 위해 연구되어야 합니다.
NADPH 대사는 정상 세포와 암 세포에서 공통적으로 존재하기 때문에, 정상 세포에 영향을 주지 않으면서 특수한 조건 하에서 NADPH 합성을 선택적으로 표적화하는 것은 어렵습니다. 따라서 암 대사 표적화의 가장 큰 도전 과제 중 하나는 비암성 세포에 독성 효과를 유발하지 않으면서 암 대사 경로를 표적화하는 것입니다. 또한, 많은 보고된 소분자 억제제는 유사한 구조를 가진 여러 대사 효소를 표적화합니다. 예를 들어, EGCG는 NADPH 의존성 FASN과 NADP+ 의존성 GDH를 모두 표적화합니다.21,187 이러한 효소의 이소형 간 기능은 현저히 다를 수 있습니다. 예를 들어, 세포질 내 ALDH1L1은 주로 환원 합성을 조절하는 반면, 미토콘드리아 내 ALDH1L2는 NADPH를 생성하여 산화 스트레스를 완화합니다.78 IDH1/2는 NADP+를 보조인자로 사용하지만 IDH3는 NAD+를 필요로 합니다.121 고도로 선택적 또는 이소형 특이적 억제제의 개발은 부작용을 줄일 수 있으며, 가까운 미래의 중요한 목표입니다. 암 NADPH 대사 과정을 표적으로 하는 대부분의 화합물은 전임상 연구 단계에 있으며, 따라서 이러한 화합물이 임상 단계로 진입하기 전에 해결해야 할 과제들이 여전히 존재합니다. 종합적으로, NADPH 대사 과정의 치료적 잠재력을 더 잘 이해하기 위해 이러한 어려움을 해결하기 위한 추가적인 전임상 및 임상 연구가 실시되어야 하며, 면역요법 및/또는 화학요법과의 결합 접근법이 시너지 효과를 고려할 때 최상의 전략으로 추구되어야 합니다.
References
Chiarugi, A., Dolle, C., Felici, R. & Ziegler, M. The NAD metabolome-a key determinant of cancer cell biology. Nat. Rev. Cancer 12, 741–752 (2012).
Chen, Z., Tian, R., She, Z., Cai, J. & Li, H. Role of oxidative stress in the pathogenesis of nonalcoholic fatty liver disease. Free Radic. Biol. Med. 152, 116–141 (2020).
Nogueira, V. & Hay, N. Molecular pathways: reactive oxygen species homeostasis in cancer cells and implications for cancer therapy. Clin. Cancer Res. 19, 4309–4314 (2013).
Ying, W. NAD+/NADH and NADP+/NADPH in cellular functions and cell death: regulation and biological consequences. Antioxid. Redox Signal. 10, 179–206 (2008).
Xiao, W., Wang, R. S., Handy, D. E. & Loscalzo, J. NAD(H) and NADP(H) redox couples and cellular energy metabolism. Antioxid. Redox Signal. 28, 251–272 (2018).
Xu, D. et al. The protein kinase activity of fructokinase A specifies the antioxidant responses of tumor cells by phosphorylating p62. Sci. Adv. 5, eaav4570 (2019).
Cao, X., Wu, L., Zhang, J. & Dolg, M. Density functional studies of coenzyme NADPH and its oxidized form NADP(+): structures, UV–Vis spectra, and the oxidation mechanism of NADPH. J. Comput. Chem. 41, 305–316 (2020).