EMBO Rep
. 2014 Oct 13;15(11):1139–1153. doi: 10.15252/embr.201439245
Senescence and apoptosis: dueling or complementary cell fates?
Bennett G Childs 1,†, Darren J Baker 2,3,*,†, James L Kirkland 3, Judith Campisi 4, Jan M van Deursen 1,3,**
- Author information
- Article notes
- Copyright and License information
PMCID: PMC4253488 PMID: 25312810
Abstract
In response to a variety of stresses, mammalian cells undergo a persistent proliferative arrest known as cellular senescence. Many senescence-inducing stressors are potentially oncogenic, strengthening the notion that senescence evolved alongside apoptosis to suppress tumorigenesis. In contrast to apoptosis, senescent cells are stably viable and have the potential to influence neighboring cells through secreted soluble factors, which are collectively known as the senescence-associated secretory phenotype (SASP). However, the SASP has been associated with structural and functional tissue and organ deterioration and may even have tumor-promoting effects, raising the interesting evolutionary question of why apoptosis failed to outcompete senescence as a superior cell fate option. Here, we discuss the advantages that the senescence program may have over apoptosis as a tumor protective mechanism, as well as non-neoplastic functions that may have contributed to its evolution. We also review emerging evidence for the idea that senescent cells are present transiently early in life and are largely beneficial for development, regeneration and homeostasis, and only in advanced age do senescent cells accumulate to an organism’s detriment.
초록
다양한 스트레스에 대응하여 포유류 세포는
세포 노화라고 알려진 지속적인 증식 정지 상태를 겪습니다.
persistent proliferative arrest known as
cellular senescence
많은 노화 유도 스트레스 요인은 잠재적으로 발암성이 있어,
노화가 아포토시스(세포 사멸)와 함께 진화하여
종양 형성을 억제하기 위해 발달했다는 가설을 강화합니다.
Many senescence-inducing stressors are
potentially oncogenic, strengthening the notion that
senescence evolved alongside apoptosis to suppress tumorigenesis.
세포 사멸과 달리,
노화 세포는 안정적으로 생존 가능하며 분비되는 용해성 인자를 통해
주변 세포에 영향을 미칠 수 있습니다.
이러한 인자들은
노화 관련 분비 형질(SASP)로 통칭됩니다.
senescence-associated secretory phenotype (SASP)
그러나
SASP는 조직 및 장기 구조와 기능의 퇴화와의 연관성이 보고되었으며,
심지어 종양 촉진 효과를 가질 수 있어,
왜 세포 사멸이 노화보다 우월한 세포 운명 옵션으로 진화하지 못했는지에 대한
흥미로운 진화적 질문을 제기합니다.
본 연구에서는
세포 노화 프로그램이 암 보호 메커니즘으로서 아포토시스보다 가질 수 있는 장점,
그리고 그 진화에 기여했을 수 있는 비종양성 기능을 논의합니다.
또한,
노화 세포가 생애 초기 단계에서 일시적으로 존재하며
발달, 재생 및 항상성에 크게 기여하며,
노년기에 이르러서야 유기체에 해로운 수준으로 축적된다는 아이디어를 뒷받침하는
최신 증거를 검토합니다.
Keywords: aging, apoptosis, cancer, embryogenesis, senescence
Introduction
Over half a century ago, Hayflick and Moorhead demonstrated that primary human cells in culture have a limited capacity for replication 1. After undergoing a finite number of divisions, these cells entered into a permanent cell cycle arrest, subsequently termed replicative or cellular senescence. They hypothesized that cellular senescence was a model-in-miniature of processes leading to organismal aging. They also noted that cancer cells divided indefinitely in culture, suggesting a role for replicative senescence in preventing cancer.
The intracellular signals that drive senescence remained obscure until the discovery of telomere erosion and telomerase. Telomeres are repetitive DNA sequences that comprise the ends of many linear chromosomes and protect them from degradation and recombination.
Telomeres erode with each cell division due to the biochemical nature of DNA replication: the use of RNA-based priming of the lagging strand and unidirectionality of DNA polymerases. Thus, telomeres have been proposed to be the “molecular clock” that determines the number of divisions a cell can undergo before reaching replicative senescence 2. Telomerase—an enzyme expressed in many human stem and cancer cells 3, as well as broadly in the mouse 4—adds telomeric DNA repeats to the telomere and is capable of conferring an indefinite division potential to several types of primary cells in culture, including fibroblasts 5. Without telomerase, telomeres become critically short and lose their protective function 6, which elicits a DNA damage response (DDR) that upregulates inhibitors of cell cycle progression to effect and enforce the senescence growth arrest 7.
소개
반세기 이상 전,
Hayflick과 Moorhead는
배양된 인간 원시 세포가 복제 능력에 한계가 있음을 입증했습니다 1.
이 세포들은 유한한 분열 횟수를 거친 후
영구적인 세포 주기 정지 상태에 진입했으며,
이는 이후 복제성 또는 세포 노화라고 명명되었습니다.
permanent cell cycle arrest,
replicative or cellular senescence
그들은
세포 노화가 유기체 노화로 이어지는 과정의 축소 모델이라고
가설을 제기했습니다.
또한
암 세포가 배양에서 무한히 분열한다는 점을 지적하며,
복제 노화가 암 예방에 역할을 할 수 있음을 시사했습니다.
They also noted that cancer cells divided indefinitely in culture,
suggesting a role for replicative senescence
in preventing cancer.
복제적 노화가 암 예방에 기여하는 진화적 관점은 흥미로운 주제. 이 관점에 따르면, 복제적 노화는 세포가 무한히 분열하는 것을 막아 암 발생 위험을 줄이는 진화적 메커니즘으로 볼 수 있어요. 즉, 세포가 일정 횟수 이상 분열하지 못하게 함으로써, 암세포로 변이될 가능성을 낮추는 거죠.마치 우리 몸에 '안전 장치'가 있는 것과 비슷해요. 이 안전 장치가 없다면, 세포는 계속 분열하면서 암세포로 변할 위험이 커지겠죠
세포 내 노화를 유발하는 신호는
텔로미어 침식 및 텔로머레이즈의 발견까지 불분명했습니다.
텔로미어는
많은 선형 염색체의 끝을 구성하는 반복적인 DNA 서열로,
염색체의 분해와 재조합으로부터 보호합니다.
The intracellular signals that drive senescence remained obscure until the discovery of telomere erosion and telomerase. Telomeres are repetitive DNA sequences that comprise the ends of many linear chromosomes and protect them from degradation and recombination
텔로미어는 염색체 끝부분을 보호하는 역할을 하는 반복적인 DNA 서열.
마치 신발끈 끝의 플라스틱 팁처럼, 텔로미어는 염색체가 손상되지 않도록 보호하고,
세포 분열 과정에서 유전 정보가 손실되지 않도록 막아주죠
텔로미어는
DNA 복제의 생화학적 특성—지연된 가닥의 RNA 기반 프라이밍과 DNA 폴리메라제의 단방향성—으로 인해
세포 분열 때마다 훼손됩니다.
Telomeres erode with each cell division due to the biochemical nature of DNA replication:
the use of RNA-based priming of the lagging strand and unidirectionality of DNA polymerases.
DNA 복제 과정에서 텔로미어는 복제될 때마다 조금씩 짧아져요. 이건 지연 가닥의 RNA 기반 프라이밍과 DNA 중합 효소의 단방향성 때문이죠. 세포 분열이 일어날 때마다 텔로미어가 점점 짧아지다가, 어느 순간 더 이상 짧아질 수 없게 되면 세포 분열이 멈추고 세포는 노화하게 돼요
지연 가닥의 RNA 기반 프라이밍은 DNA 복제 과정에서 RNA 프라이머가 DNA 가닥에 결합하여 DNA 중합 효소가 새로운 DNA 가닥을 합성할 수 있도록 시작점을 제공하는 걸 말해요. 이 과정은 DNA 복제의 정확성과 효율성을 높이는 데 중요하지만, 텔로미어 복제에는 한계가 있죠.
DNA 중합 효소의 단방향성은 DNA가 항상 5'에서 3' 방향으로만 합성된다는 걸 의미해요. 이 때문에 DNA 복제 과정에서 지연 가닥은 오카자키 절편이라는 짧은 조각들로 나뉘어 복제되고, 이 과정에서 텔로미어가 완전히 복제되지 못하고 짧아지게 되죠
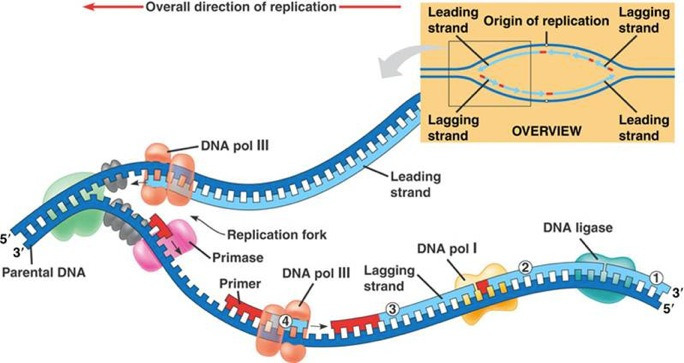
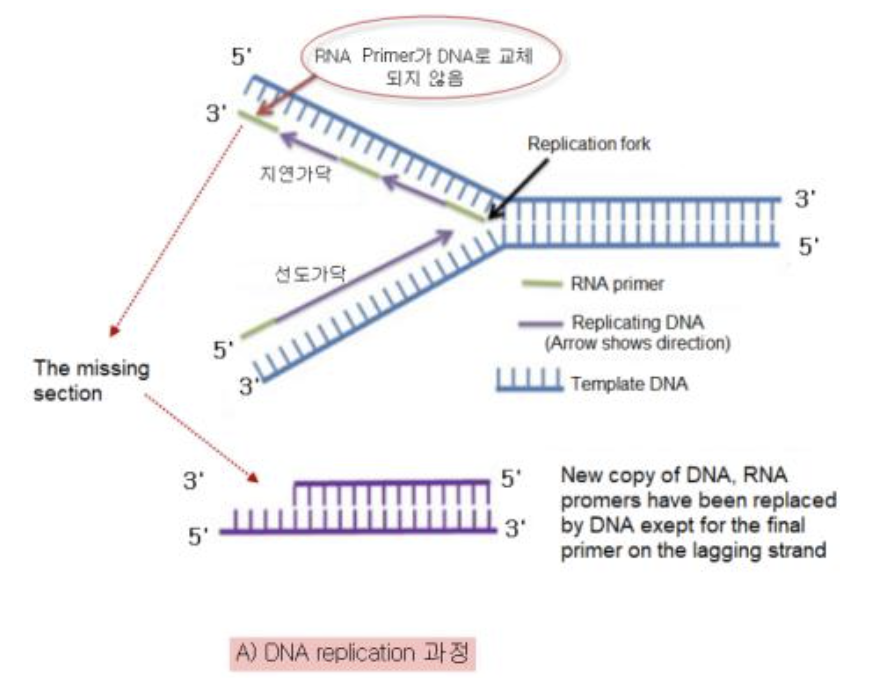
따라서 텔로미어는
세포가 복제 노화 전에 겪을 수 있는 분열 횟수를 결정하는
'분자 시계'로 제안되었습니다 2.
Thus, telomeres have been proposed to be the “molecular clock”
that determines the number of divisions a cell can undergo
before reaching replicative senescence
텔로머레이스—인간 줄기세포와 암세포에서 광범위하게 발현되는 효소 3, 그리고 쥐에서도 널리 발현됩니다 4—는
텔로미어에 텔로미어 DNA 반복 서열을 추가하며,
배양된 여러 유형의 원시 세포(섬유아세포 포함)에
무한 분열 가능성을 부여할 수 있습니다 5.
텔로머레이스는 텔로미어의 길이를 유지하는 효소예요. 텔로미어가 짧아지는 걸 막아줘서 세포가 더 오래 분열할 수 있게 하죠. 텔로머레이즈는 암세포에서 활발하게 작용해서 암세포가 계속 분열하도록 만들기도 해요.
텔로머레이스는 텔로미어 DNA에 상보적인 RNA 서열을 가지고 있어서, 이 RNA를 주형으로 사용해서 텔로미어 DNA 서열을 합성할 수 있어요. 텔로머레이스가 텔로미어 DNA에 결합하면, RNA 주형이 텔로미어 DNA 말단에 결합하고, 텔로머레이스의 효소 활성에 의해 텔로미어 DNA가 연장되죠. 이렇게 해서 텔로미어의 길이가 유지되는 거랍니다
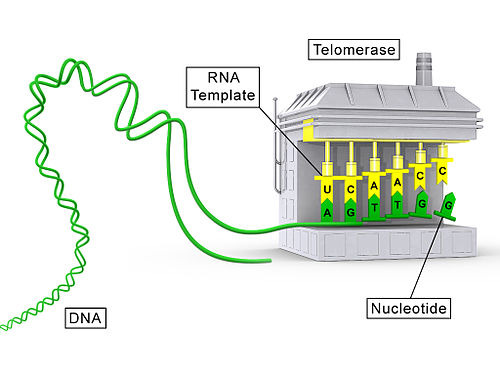
텔로머라제가 결여되면
텔로미어가 극도로 짧아져 보호 기능을 상실합니다 6,
이는 DNA 손상 반응(DDR)을 유발하여
세포 주기 진행 억제 인자를 활성화시켜 노화 성장 정지를 유발하고 강제합니다 7.
DNA damage response (DDR)
https://pmc.ncbi.nlm.nih.gov/articles/PMC8985209/

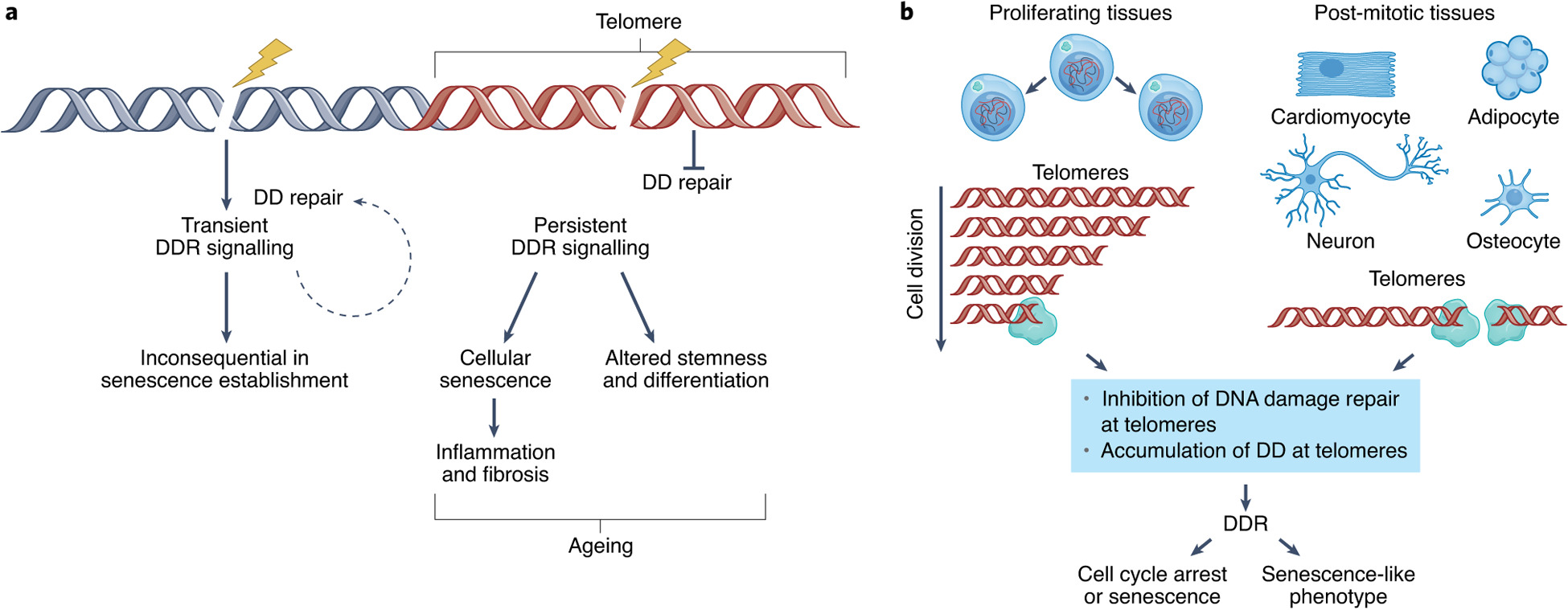
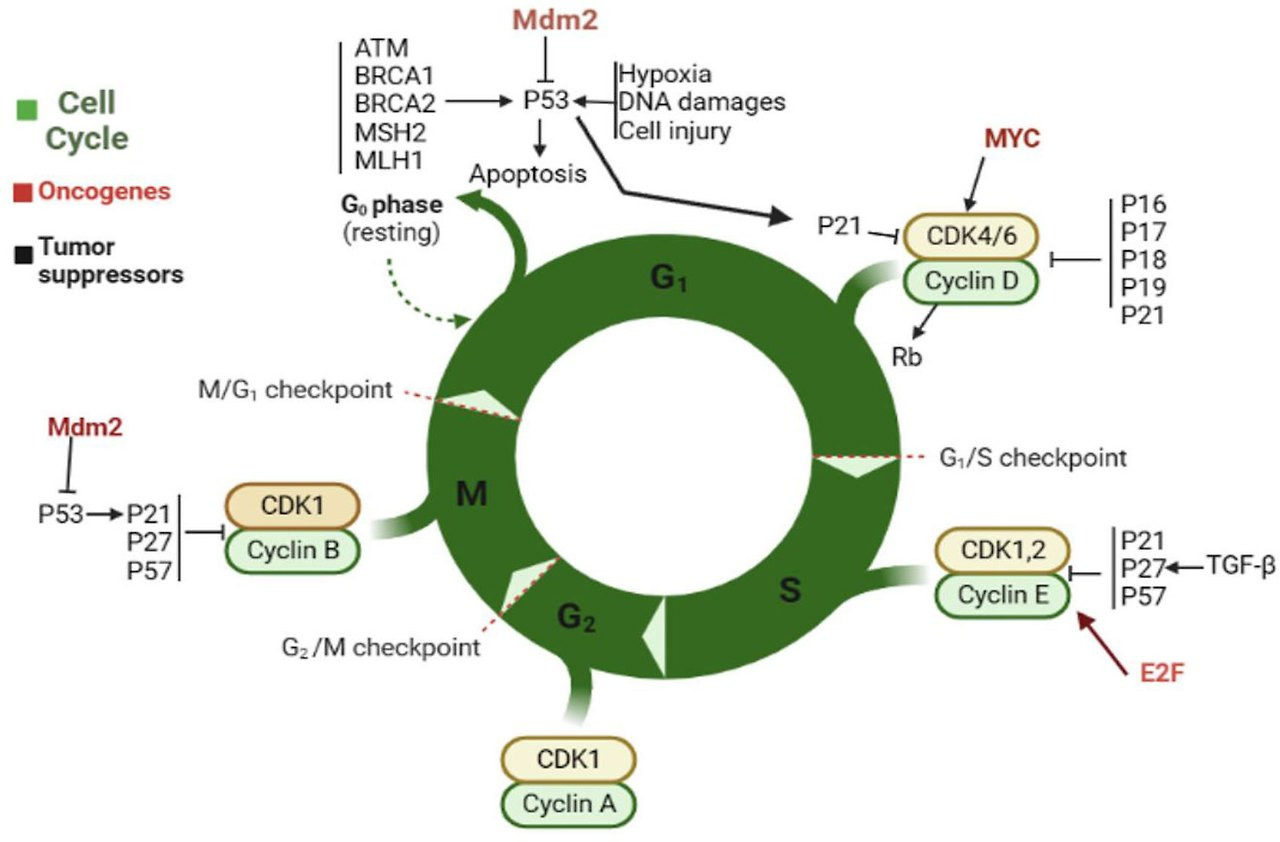
The concept of replicative senescence established a framework for understanding the signaling pathways that drive senescence. Damage sensor proteins, such as ATM in the replicative senescence of human cells, recognize a stress—for example short telomeres—and activate a master regulator (generally p53 through ATM-dependent phosphorylation), which in turn upregulates effectors of cell cycle arrest 8. p53 can also be stabilized through the action of p19Arf (p14Arf in human cells), an inhibitor of the ubiquitin ligase MDM2 that targets p53 for degradation. However, ATM can suppress ARF in some cancer cells, so the final effect on cell cycle progression depends on the balance between upstream signals 9.
p21, a cyclin-dependent kinase inhibitor (CDKi) responsible for the initial cell cycle arrest, is one of the most important targets of p53 transcriptional activity in senescent cells. The p21-mediated cell cycle arrest can act as a temporary respite for cells with low to moderate amounts of damage, preventing S-phase entry under unfavorable conditions for DNA replication 10. If the damage is successfully repaired, cells may resume the transiently interrupted cell cycle. However, prolonged arrest leads to upregulation of the CDKi p16Ink4a 11, which activates the transcriptional regulator Rb. Prolonged p16Ink4a expression results in a permanent cell cycle arrest, often with a 2N G1 DNA content. Using the FUCCI reporter system to monitor cycle stages in live human cells, Nakanishi and colleagues recently found that senescence stimuli leading to a DDR can also result in “skipped” mitosis and a G1 arrest with 4N DNA content 12.
복제 노화 개념은
노화를 유도하는 신호 전달 경로를 이해하는 틀을 제공했습니다.
인간 세포의 복제 노화에서 ATM과 같은 손상 감지 단백질은
스트레스(예: 짧은 텔로미어)를 인식하고
마스터 조절자(일반적으로 ATM에 의존하는 인산화 과정을 통해 p53)를 활성화합니다.
이 마스터 조절자는
차례로 세포 주기 정지 효과기를 활성화합니다 8.
p53은
MDM2라는 유비퀴틴 리가아제의 억제제인 p19Arf(인간 세포에서는 p14Arf)의 작용을 통해
안정화될 수 있습니다.

그러나
ATM은 일부 암 세포에서 ARF를 억제할 수 있으므로,
세포 주기 진행에 대한 최종 효과는 상위 신호 간의 균형에 달려 있습니다 9.
p21은
세포 주기 초기 정지를 유발하는 사이클린 의존성 키나제 억제제(CDKi)로,
노화 세포에서 p53의 전사 활성의 주요 표적 중 하나입니다.
p21에 의한 세포 주기 정지는
DNA 복제 조건이 불리한 상황에서 S상 진입을 방지하여
저~중등도의 손상을 입은 세포에 일시적인 휴식 역할을 할 수 있습니다 10.
손상이 성공적으로 복구되면
세포는 일시적으로 중단된 세포 주기를 재개할 수 있습니다.
그러나
장기간의 정지는
CDKi인 p16Ink4a의 발현을 증가시킵니다 11,
이는 전사 조절인자 Rb를 활성화합니다.
p16Ink4a의 장기간 발현은
2N G1 DNA 함량을 동반한 영구적인 세포 주기 정지로 이어집니다.
FUCCI 보고자 시스템을 사용하여
살아있는 인간 세포의 세포 주기 단계를 모니터링한 Nakanishi와 동료들은
DDR을 유발하는 노화 자극이 “건너뛴” 분열과 4N DNA 함량을 동반한
G1 정지를 초래할 수 있음을 최근 발견했습니다 12.
Senescent cells accumulate with organismal aging, but telomere erosion is not the sole cause 13,14. Other stresses that engage the DDR, such as exposure to oxidants, γ-irradiation, UVB light, and DNA damaging chemotherapies, can all induce senescence. Overexpression of oncogenic Ras drives cultured cells into senescence due to the DNA damage induced during the initial period of hyperproliferation, supporting the idea that senescence is a barrier to the proliferation of pre-cancerous or potentially neoplastic cells 15. Other oncogenes, such as BRAFV600E 16, and the loss of tumor suppressors, such as PTEN 17, also promote senescence but through proliferation-independent mechanisms that are quite different from Ras. BRAFV600E-induced senescence involves multiple mechanisms, including suppression of the metabolic enzyme pyruvate dehydrogenase 18, direct activation of p16Ink4a 19,20, and upregulation of IL-6 and IL-8 21, whereas loss of PTEN leads to senescence through mTORC1 17. These results show that different oncogenic stimuli can induce an irreversible senescent state, termed oncogene-induced senescence (OIS), despite acting through different signaling pathways. Further work is needed to establish whether these “stimulus-specific pathways” are distinct or shared by multiple types of senescence (Sidebar A). For example, is pyruvate dehydrogenase suppressed in replicative senescence or PTEN-loss-induced senescence 18? Regardless of differences in signaling pathways, Ras overexpression, BRAF mutation, and Pten deletion ultimately activate common effectors of senescence such as p19Arf and p16Ink4a 22. These cell culture results suggest that senescence is a barrier to transformation and are supported by studies showing that mice lacking p19Arf 23 or p16Ink4a 24 are predisposed to cancer.
Many stimuli leading to a DDR can also induce apoptosis, which is a form of programmed cell death. Apoptosis removes damaged or pre-neoplastic cells, suggesting it should be more capable of restricting tumorigenesis than senescence (Sidebar A). This expectation is especially true in light of the fact that senescent cells actively secrete a suite of cytokines, chemokines, and matrix-remodeling enzymes known as the senescence-associated secretory phenotype (SASP) 25,26 or senescence-messaging secretome (SMS) 27. Thought to be responsible for stimulating the clearance of senescent cells by the innate immune system or to elicit autocrine signaling to maintain the senescent state, many SASP factors also have pro-tumorigenic properties 25. Indeed, senescent cells encourage the growth and invasion of breast cancer 28,29 and mesothelioma cells 30 through their SASP.
Given that senescence seems to be an imperfect tumor-suppressive mechanism, what advantage could it have over apoptosis? How is the choice between senescence and apoptosis determined? Guiding our first inquiries into these questions are two studies demonstrating that senescence is a non-essential but integral part of embryogenesis, a stage in the life of every metazoan that also depends on apoptosis (Sidebar A).
노화 세포는
유기체 노화와 함께 축적되지만,
DDR을 활성화하는 다른 스트레스 요인,
예를 들어 산화제 노출, γ-방사선, UVB 광선, DNA 손상 화학요법 등은
모두 노화를 유도할 수 있습니다.
종양 유발성 Ras의 과발현은
초기 과증식 기간 동안 유발된 DNA 손상으로 인해 배양 세포를 노화로 유도하며,
이는 노화가 전암성 또는 잠재적 종양 세포의 증식을 차단하는 장벽이라는 가설을 지지합니다 15.
다른 종양유발 유전자인 BRAFV600E 16 및 종양 억제 유전자의 상실(예: PTEN 17)도
노화를 촉진하지만,
Ras와 완전히 다른 증식 독립적 메커니즘을 통해 작용합니다.
BRAFV600E에 의한 노화는
피루vate dehydrogenase 18의 억제, p16Ink4a 19,20의 직접 활성화,
IL-6 및 IL-8 21의 발현 증가 등 다중 메커니즘을 포함하며,
반면 PTEN의 상실은 mTORC1 17을 통해 노화를 유발합니다.
이 결과는
서로 다른 종양 유발 자극이 서로 다른 신호 전달 경로를 통해 작용함에도 불구하고,
종양 유전자 유발 노화(OIS)라고 불리는
불가역적인 노화 상태를 유도할 수 있음을 보여줍니다.
추가 연구가 필요하며,
이러한 '자극 특이적 경로'가 다양한 유형의 노화(사이드바 A)에서
서로 다른지 또는 공유되는지 확인해야 합니다.
예를 들어,
피루vate dehydrogenase는 복제 노화나 PTEN 상실 유도 노화에서 억제되나요? 18
신호전달 경로의 차이와 무관하게,
Ras 과발현, BRAF 돌연변이, 및 Pten 결손은
결국 p19Arf 및 p16Ink4a와 같은 노화의 공통 효과기를 활성화합니다 22.
이 세포 배양 결과는 노화가 변환의 장벽임을 시사하며,
p19Arf 23 또는 p16Ink4a 24가 결손된 마우스가 암에 취약하다는 연구 결과로 뒷받침됩니다.
DDR로 이어지는 많은 자극은
세포 사멸의 한 형태인 아포토시스도 유발할 수 있습니다.
아포토시스는
손상된 또는 전암성 세포를 제거하며,
이는 노화보다 종양 형성을 억제하는 데 더 효과적일 것으로 예상됩니다(부록 A).
이 기대는 노화 세포가
또는 노화 메시징 분비체(SMS) 27로 알려진 사이토킨, 케모킨,
매트릭스 재구성 효소 복합체를 적극적으로 분비한다는 사실에
특히 적용됩니다.
SASP 요인들은
선천성 면역 체계에 의해 노화 세포의 제거를 자극하거나
자가분비 신호를 유발하여 노화 상태를 유지하는 것으로 알려져 있지만,
많은 SASP 요인들은 종양 발생 촉진 성질을 가지고 있습니다 25.
실제로
노화 세포는
SASP를 통해 유방암 28,29 및 중피종 세포 30의 성장과 침투를 촉진합니다.
노화가 불완전한 종양 억제 메커니즘으로 보인다면,
노화는 아포토시스보다 어떤 장점을 가질 수 있을까요?
노화와 아포토시스 사이의 선택은 어떻게 결정될까요?
이러한 질문에 대한 첫 번째 탐구를 안내하는 두 가지 연구는 노화가 모든 다세포 생물의 생명 주기 단계인 배아 발생의 필수적이지만 필수적이지 않은 부분이며, 이 단계는 아포토시스에도 의존한다는 것을 보여줍니다(부록 A).
Developmental versus stress-induced senescence and apoptosis
Apoptosis in vivo was originally associated with pathology and identified as a form of non-necrotic cell death during liver injury 31. Sulston and colleagues were the first to identify apoptosis in a non-pathologic process during the embryonic development of the nematode Caenorhabditis elegans. This organism undergoes a fixed, genetically determined period of embryogenesis in which each developing hermaphrodite loses exactly 131 cells, mostly neurons, through apoptosis 32. Many of the molecular effectors of apoptosis, including caspases, were discovered through mutagenic screens that disrupted this process 33. Although differing in details—for example, macrophages engulf apoptotic debris in mammals, whereas non-specialized neighboring cells have this role in nematodes—most factors identified in C. elegans screens have human and mouse homologs 34.
Apoptosis is also functionally conserved during development. Many cells produced in abundance in the embryo are subsequently eliminated by apoptosis. Such cells include mammary tissue in males 35 and the interdigital webbing 36. Likewise, peripheral afferent neurons extend from the spinal ganglia in numbers far exceeding their targets, so only those that successfully contact muscle or skin avoid apoptotic death 37. Thus, apoptosis regulates patterning in the embryo by altering cellularity in the most direct way possible: cell death (Sidebar A).
발달성 노화 대 스트레스 유발 노화 및 아포토시스
아포토시스는
원래 병리와 연관되어 간 손상 시 비괴사성 세포 사멸의 한 형태로 처음 식별되었습니다. 31.
Sulston과 동료들은
선충 Caenorhabditis elegans의 배아 발달 과정에서 병리적 과정이 아닌 과정에서
아포토시스를 처음 식별했습니다.
이 생물은 유전적으로 결정된 고정된 배아 발생 기간을 거치며,
이 기간 동안 각 발달 중인 양성 생식체는
주로 신경세포인 131개의 세포를 아포토시스 통해 정확히 잃습니다 32.
아포토시스의 분자적 효소인 카스파제 등을 포함한 많은 분자적 효소들은 이 과정을 방해하는 돌연변이 스크린을 통해 발견되었습니다 33. 세부적인 차이점—예를 들어, 포유류에서는 대식세포가 아포토시스 잔여물을 삼키지만, 선충에서는 비특이적 이웃 세포가 이 역할을 수행합니다—에도 불구하고, C. elegans 스크린에서 식별된 대부분의 인자는 인간과 쥐의 동형체를 가지고 있습니다 34.
아포토시스는
발달 과정에서도 기능적으로 보존되어 있습니다.
배아에서 대량으로 생성된 많은 세포는
이후 아포토시스 통해 제거됩니다.
이러한 세포에는
남성의 유방 조직35 및 발가락 사이의 막36이 포함됩니다.
마찬가지로,
말초 구심성 뉴런은
척추 신경절에서 목표 지점을 훨씬 초과하는 수로 확장되므로,
근육이나 피부에 성공적으로 접촉한 뉴런만 세포 사멸을 피할 수 있습니다37.

따라서
세포 사멸은 가장 직접적인 방법인 세포 사멸을 통해
배아의 패턴을 조절합니다 (사이드바 A).
Three groups have recently identified cellular senescence during development. Rajagopalan and Long found that HLA-G secreted by trophoblast cells in the extra-embryonic placenta induces senescence of nearby NK cells by binding the receptor CD158d 38. The SASP from these senescent cells promotes vascular tube formation in culture and is hypothesized to drive vascularization of the placenta in vivo. In addition, two groups independently reported the existence of senescence in embryos. Serrano and colleagues identified senescence in the endolymphatic sac and mesonephros of mouse and human embryos and determined it has a morphogenetic role analogous to that of apoptosis 39. They proposed that cellular senescence, followed by macrophage-mediated clearance of senescent cells or by the overgrowth of nearby cells, alters cellularity, resulting in tissue patterning. Keyes and colleagues found evidence of senescence in the apical ectodermal ridge and neural floorplate and proposed that the SASP of these cells induces tissue remodeling 40. All three groups found that p21, and not p16Ink4a, is the key enforcer of the cell cycle arrest, in combination with the p16Ink4a-related protein p15Ink4b. p15Ink4b-positive cells were identified in the mesonephros and endolymphatic sac. Macrophages clear senescent cells in the embryo 39,40, whereas the cell type responsible for eliminating senescent NK cells in the placenta is unknown. However, immune clearance is not necessary in the endolymphatic sac, where senescent cells are overgrown by their dividing neighbors 39.
최근 세 그룹이 발달 과정에서
세포 노화를 식별했습니다.
Rajagopalan과 Long은
태아 외 태반의 영양막 세포에서 분비되는 HLA-G가 수용체 CD158d에 결합하여
근처 NK 세포의 노화를 유도한다는 것을 발견했습니다 38.
이러한 노화된 세포에서 분비되는 SASP는
배양에서 혈관 관 형성을 촉진하며,
이는 태반의 혈관 형성을 유도하는 것으로 추정됩니다.
또한 두 연구 그룹은
독립적으로 배아에서의 노화를 보고했습니다.
Serrano와 동료들은
마우스와 인간 배아의 내림프낭과 중간신장에서 노화를 확인하고,
이는 아포토시스 39와 유사한 형태 발생적 역할을 한다고 밝혔습니다.
그들은
세포 노화 후 대식세포에 의한 노화 세포의 제거 또는 주변 세포의 과도한 증식으로 인해
세포 밀도가 변화되어 조직 패턴 형성이 발생한다고 제안했습니다.
Keyes와 동료들은
아피칼 외배엽 능선과 신경 바닥판에서 노화를 발견하고,
이 세포의 SASP가 조직 재모델링을 유도한다고 제안했습니다 40.
세 그룹 모두 p21이 p16Ink4a가 아닌 세포 주기 정지의 핵심 조절자이며,
p16Ink4a 관련 단백질 p15Ink4b와 결합하여 작용함을 발견했습니다.
p15Ink4b 양성 세포는 메소네프로스와 내림프낭에서 식별되었습니다.
대식세포는 배아에서 노화 세포를 제거합니다 39,40,
반면
태반에서 노화 NK 세포를 제거하는 세포 유형은
알려지지 않았습니다.
그러나
내림프낭에서는 노화 세포가 분열하는
이웃 세포에 의해 덮여지기 때문에 면역 제거가 필요하지 않습니다 39.
Together, these studies suggest that cellular senescence during embryogenesis is a programmed, transient phenomenon that contributes to tissue remodeling through the SASP or to altered cellularity through clearance (Sidebar A).
이러한 연구 결과는 배아 발생 중 세포 노화가 SASP를 통해
조직 재모델링에 기여하거나 제거를 통해 세포 수의 변화를 초래하는
프로그램된 일시적 현상임을 시사합니다(부록 A).
In the young adult, transient senescent cells also exist. This is not to suggest that the senescence cell cycle arrest is reversible, but that these cells serve a similar transient purpose to those in the embryo: the SASP directs tissue repair and regeneration 41,42. Like senescence in development, immunosurveillance clears these cells after their programmed function is performed. Transient senescent cells in the embryo and the adult can be termed “acute senescent cells”, although the molecular pathways involved are slightly different (Fig 1). In advanced age, senescent cells may accumulate due to several factors: declining immune function 43, decreased ability to stabilize p53 to levels required to cause apoptotic death 44, or slow accumulation of macromolecular damage that does not reach the threshold for cell death 14. These “chronic senescent cells” may act to the detriment of the organism by promoting tumorigenesis and tissue dysfunction through the SASP (Sidebar A).
젊은 성인에서도
일시적 노화 세포가 존재합니다.
이는 노화 세포 주기 정지가 가역적임을 의미하는 것은 아니지만,
이러한 세포는 배아에서의 역할과 유사한 일시적 기능을 수행합니다:
SASP는
https://pmc.ncbi.nlm.nih.gov/articles/PMC2984611/

https://pmc.ncbi.nlm.nih.gov/articles/PMC3394879/

발달에서의 노화와 마찬가지로
면역 감시는 이러한 세포가 프로그램된 기능을 수행한 후 제거합니다.
배아와 성인에서의 일시적 노화 세포는
분자 경로가 약간 다르지만 “급성 노화 세포”라고 명명될 수 있습니다(그림 1).
고령화 과정에서 노화 세포는
여러 요인으로 인해 축적될 수 있습니다:
면역 기능의 감소 43,
p53을 세포 사멸을 유발하는 수준으로 안정화하는 능력의 감소 44,
또는 세포 사멸 임계치를 넘지 않는 거대 분자 손상의 서서히 축적 14.
이러한 “만성 노화 세포”는
SASP를 통해 종양 발생과 조직 기능 장애를 촉진함으로써
유기체에 해로운 영향을 미칠 수 있습니다(부록 A).
Figure 1. Senescence in development and in the adult.
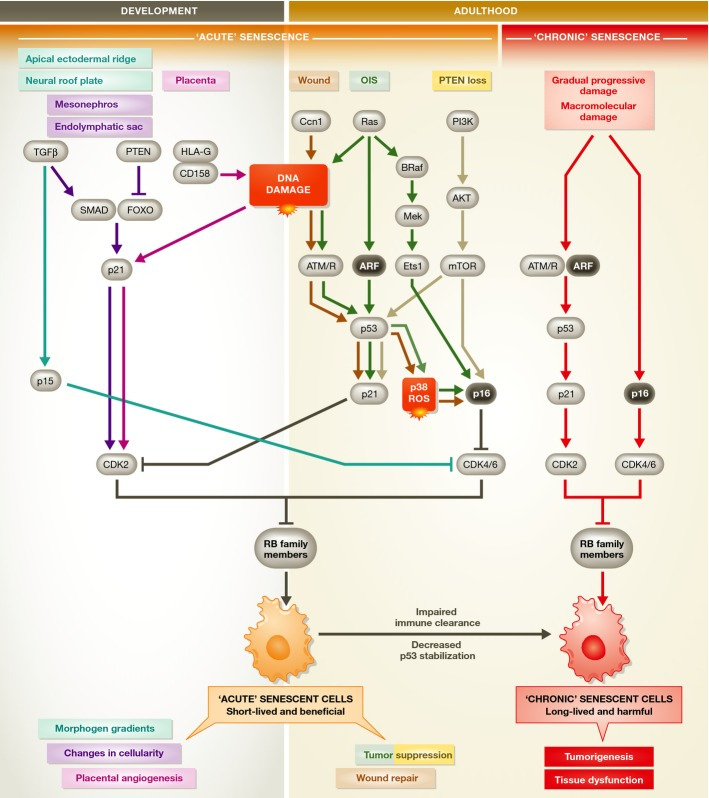
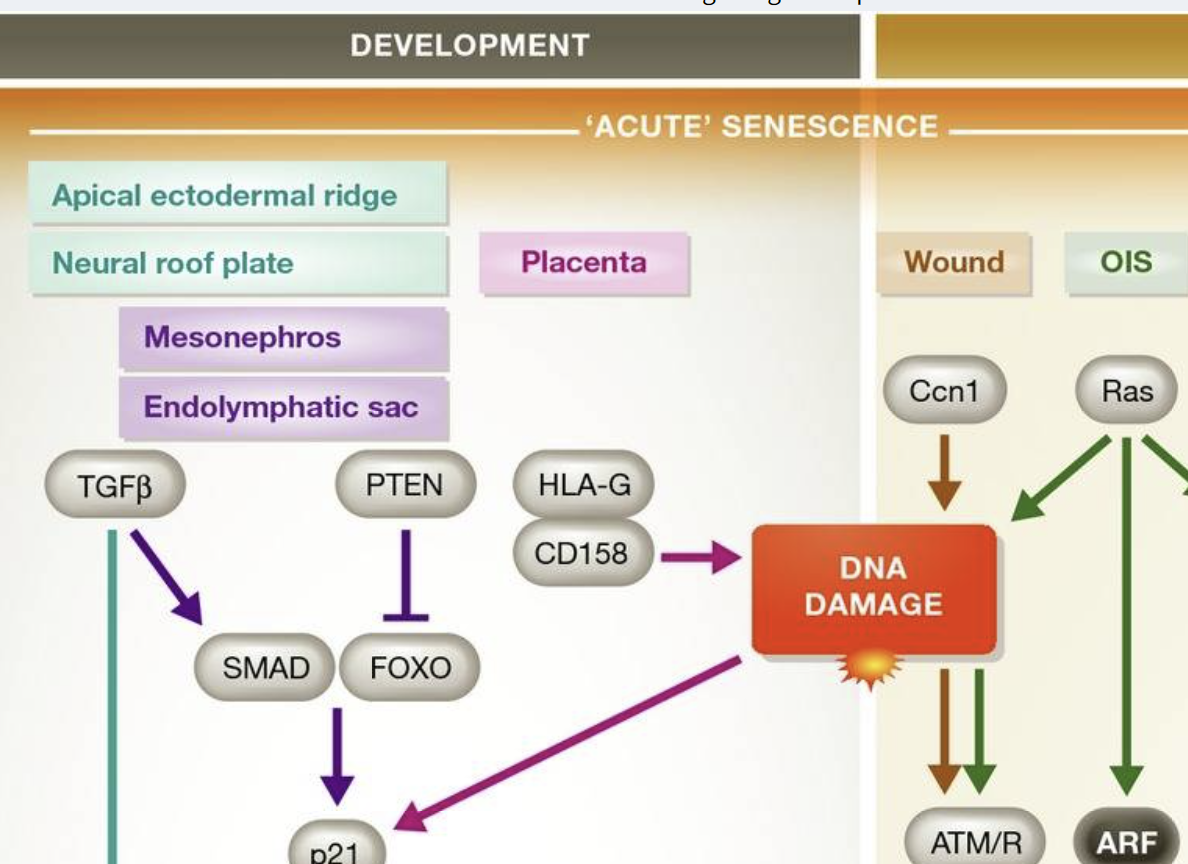
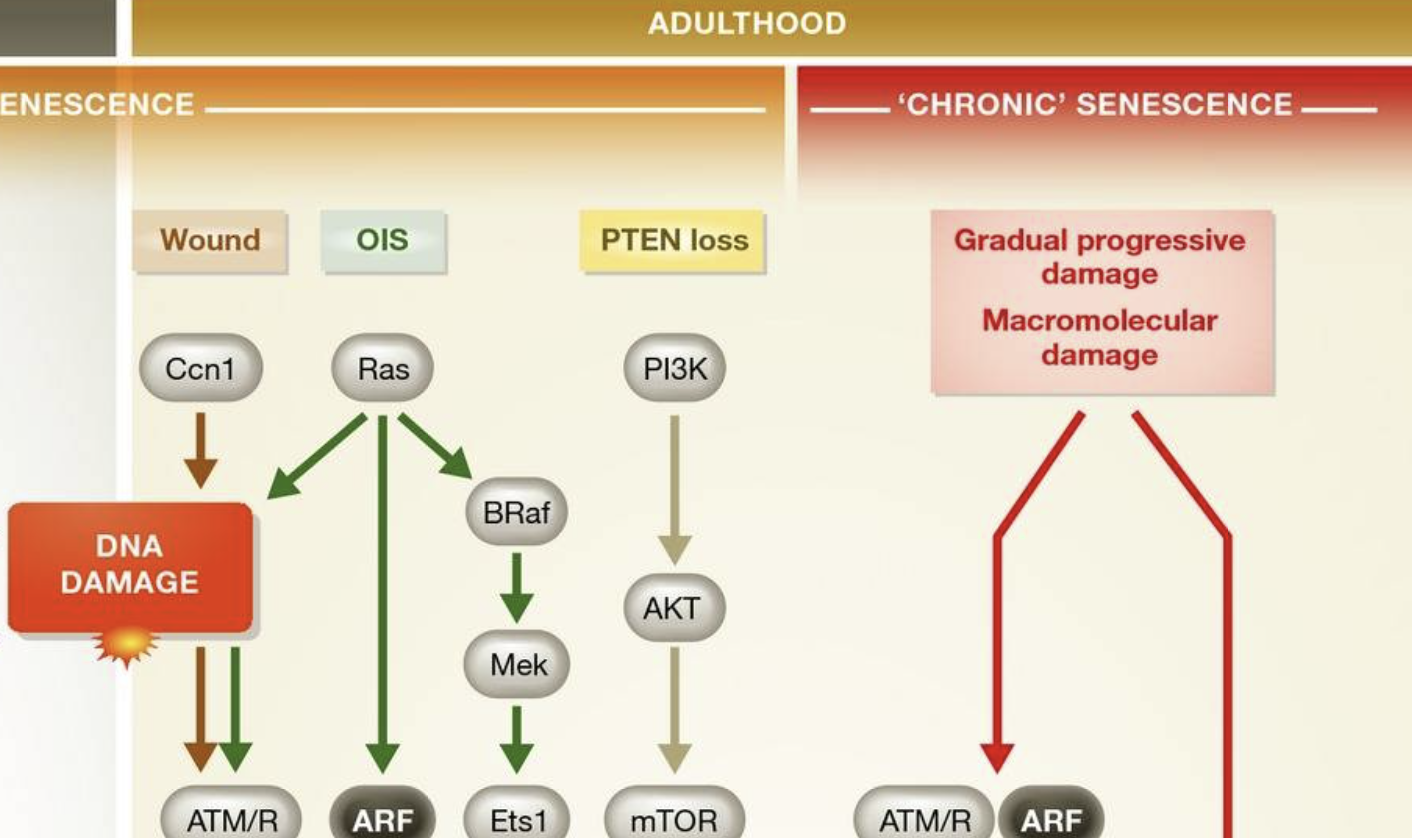
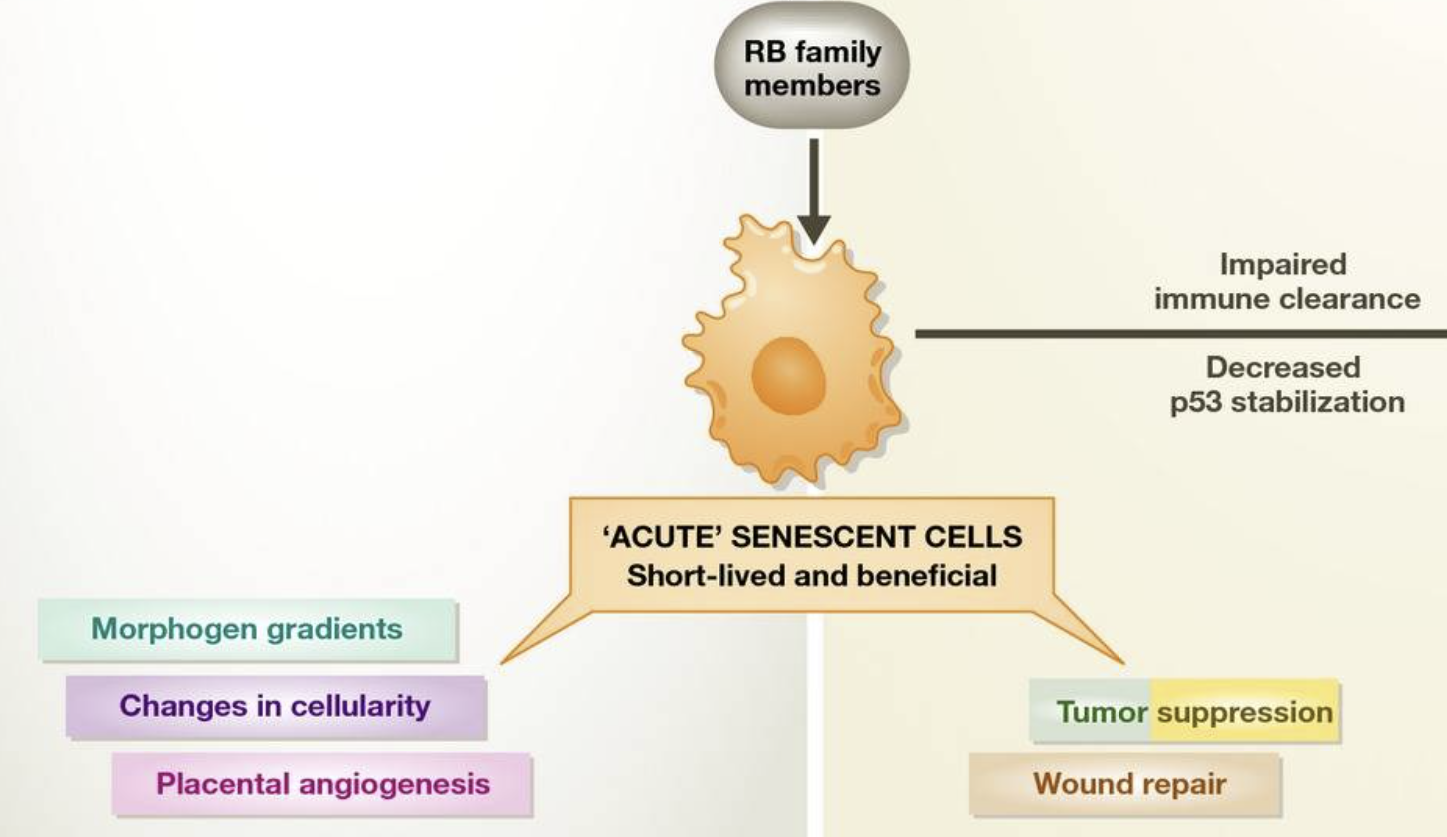
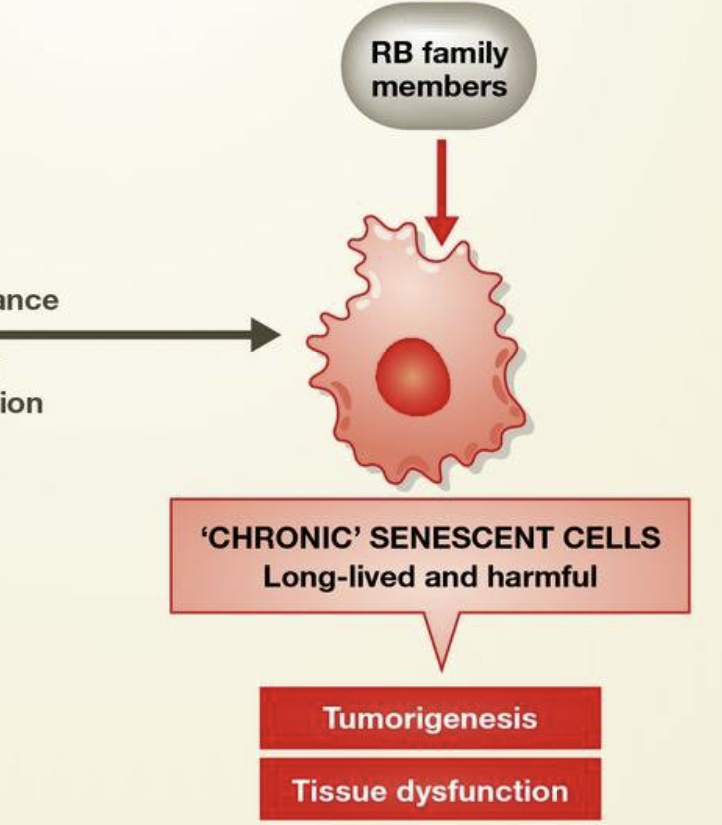
During development and in the healthy adult, cells can undergo acute senescence, a permanent cell cycle arrest that is physiologically normal. In the embryo and placenta, these cells secrete signaling molecules as part of their SASP to promote morphogenesis. Cell death through immune clearance also complements cell death through apoptosis to change cellularity in developing tissues. In the adult, acute senescent cells function to suppress tumorigenesis and promote wound repair, using different molecular mechanisms than in the embryo. Upon immune dysfunction, acute senescent cells that would normally be cleared by immune surveillance may be chronically present. As they also have a decreased ability to stabilize p53 to the levels required for apoptosis, senescent cells not killed by the immune system may contribute to tumorigenesis and tissue dysfunction. See Glossary for definitions and the text for details.
발달 과정과 건강한 성인에서 세포는
급성 노화(acute senescence)를 겪을 수 있으며,
이는 생리적으로 정상적인 영구적인 세포 주기 정지입니다.
배아와 태반에서 이러한 세포는
SASP(senescence-associated secretory protein)의 일환으로
신호 분자를 분비하여 형태 형성을 촉진합니다.
면역 제거를 통한 세포 사멸은
발달 중인 조직의 세포 수를 변화시키기 위해 아포토시스(apoptosis)를 통한 세포 사멸과
보완적으로 작용합니다.
성인에서 급성 노화 세포는
종양 발생을 억제하고 상처 치유를 촉진하는 역할을 하며,
배아와 다른 분자 메커니즘을 사용합니다.
면역 기능 장애 시,
일반적으로 면역 감시 의해 제거되어야 할 급성 노화 세포가 만성적으로 존재할 수 있습니다.
또한 p53을 아포토시스 요구 수준으로 안정화시키는 능력이 감소되어
면역 체계에 의해 제거되지 않은 노화 세포는
종양 발생과 조직 기능 장애에 기여할 수 있습니다.
정의는 용어집을, 자세한 내용은 본문을 참고하시기 바랍니다.
Apoptosis versus senescence in the adult
Whether an individual cell in an embryo is faced with apoptosis and senescence as alternative fates is unknown, as is how it might decide between them, although recent work demonstrates that in p21-knockout animals, embryonic senescence is partially replaced by compensatory apoptosis 39,40. This observation raises the possibility that senescence and apoptosis pathways are simultaneously engaged in certain processes or stress responses and that it is the particular wiring of each cell type that decides which outcome—senescence or apoptosis—will occur first (Sidebar A). Here, we focus on the cell-autonomous features of the choice between senescence and apoptosis in the adult animal, as well as in cultured somatic cells. The emerging molecular factors in this fate choice are the activity of the p53-p21 axis, the role of signaling through PTEN-PI3K-AKT-mTOR, and the degree of macromolecular damage (Fig 2).
성인의 세포 사멸과 노화
배아의 개별 세포가 세포 사멸과 노화 중 어느 쪽을 선택하는지,
그리고 그 선택이 어떻게 이루어지는지는 아직 알려지지 않았지만,
최근 연구에 따르면 p21 결핍 동물에서는 배아의 노화가 부분적으로
보상적 세포 사멸로 대체된다는 것이 밝혀졌습니다 39,40.
이 관찰은
노화와 아포토시스 경로가 특정 과정이나 스트레스 반응에서 동시에 활성화될 수 있으며,
각 세포 유형의 특이적인 회로가 노화나 아포토시스 중
어느 결과가 먼저 발생할지 결정한다는 가능성을 제기합니다(부록 A).
본 연구에서는
성인 동물 및 배양된 체세포에서
노화와 아포토시스 사이의 선택에 대한 세포 자율적 특성에 초점을 맞춥니다.
이 운명 선택에 관여하는 새로운 분자 요인으로는
p53-p21 축의 활성, PTEN-PI3K-AKT-mTOR 신호전달 경로의 역할,
그리고 대분자 손상의 정도가 있습니다(그림 2).
Figure 2. Signaling pathways that enforce the choice between cell cycle arrest, senescence, and apoptosis.
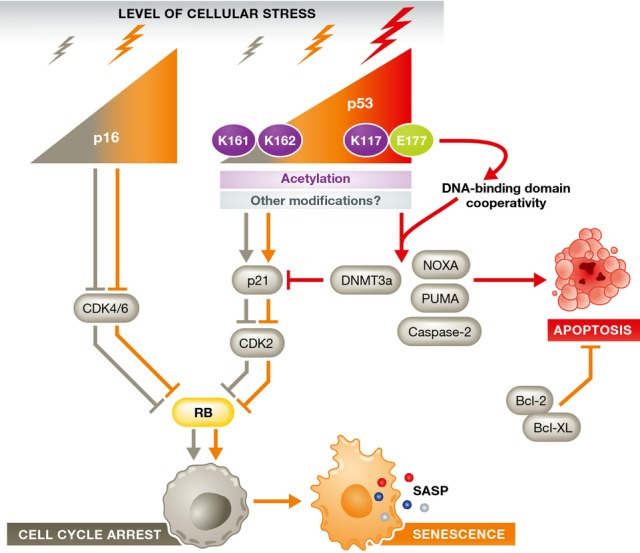
Pathways are colored as follows: leading to arrest in gray, leading to senescence in orange, and leading to apoptosis in red. The weight of the arrow reflects the level of stress. The p16-RB and p53-p21 pathways are known to be important for cellular response to stress. The decision to activate p16, p53, or both is determined by the stress level and cell type. Low levels of p16 promote a transient arrest, whereas high levels lead to senescence. Low levels of p53, with transient kinetics and K161/K162 acetylation, promote cell cycle arrest and senescence. High levels of p53, K117 acetylation, and cooperativity of DNA binding domains within the p53 tetramer lead to the transcription of apoptotic genes and ensuing apoptosis, both directly and by blocking pro-senescence signals. See Glossary for definitions and the text for details.
Stress level
In some circumstances, apoptosis is a response to overwhelming stress, whereas senescence is a consequence of less severe damage 45. For example, doxorubicin leads to senescence at low doses and apoptosis at high doses in MCF7 breast cancer cells 46. A similar dose-dependent response to doxorubicin is seen in neonatal rat cardiomyocytes 47,48. Other stresses that lead to DNA damage induce senescence at low doses and apoptosis at higher ones, such as fibroblast exposure to etoposide 49 or UVB 50, and keratinocyte exposure to UVB 51. Oxidative damage also induces a dose–response effect: high-dose H2O2 causes apoptosis, whereas lower doses of H2O2 induce senescence in F65 and IMR90 human diploid fibroblasts 52,53. However, some DNA damaging agents that produce bulky adducts, such as busulfan 49, cause senescence but not apoptosis regardless of the dose. This suggests that the nature of the DNA damage, in addition to its severity, can determine the cellular response. Finally, the cell type determines the response to a given stress. Human stromal fibroblasts senesce in response to up to 50 Gy ionizing radiation (IR), whereas T lymphocytes undergo apoptosis in response to only 2 Gy IR (J. Campisi, unpublished data). It is unclear whether these different outcomes are the result of differences in DNA repair efficiency or downstream “preferences” for apoptosis or senescence. Thus, the crucial determinants of whether a cell responds to damage by undergoing senescence or apoptosis are the cell type and the nature and intensity of the damage.
The p53-p21 axis
In addition to the nature and degree of stress, the balance between pro-senescent and pro-apoptotic pathways also decides cell fate. One such pathway is controlled by the tumor suppressor protein p53. p53 was first shown to trigger apoptosis in response to cellular stress, but is now known to, depending on the stress and cell type, modulate genes involved in homeostasis, transient cell cycle arrest, and senescence 54. p53 levels, kinetics, and transcriptional activity are all key determinants of how cells respond to various stressors (Fig 2). For example, MEFs expressing the hypomorphic R172P p53 mutation senesce rather than apoptose in response to UVB, fail to upregulate the pro-apoptotic factors PUMA and NOXA and express high levels of the pro-survival gene BCL-2 55. Conversely, human diploid fibroblasts treated with a dose of H2O2 sufficient to induce a mixture of apoptosis and senescence lead to p53 induction in both cases, but twice as much p53 is expressed in cells destined for apoptosis 53. Intriguingly, PKC family members are upregulated in MRC5 human lung fibroblasts undergoing IR-induced senescence. Knockdown of PKCζ or PKCλ reduces the levels of BCL-2, phospho-BAD and phospho-CREB, leading to a marked p53 induction and apoptosis 56. p53-dependent apoptosis relies on a combination of p53 expression level and post-translational modifications, which regulate its activity and localization. The observation that p53 expression is necessary, but not sufficient, for oncogene- and DNA damage-induced senescence suggests that, likewise, changes in p53 post-translational modifications are important for senescence 57–59.
The kinetics of expression is an additional way through which p53 is regulated to control cell fate. Low levels of γ-irradiation—such as 2.5–5 Gy—usually induce a transient rise in p53 levels 60, leading to a transient cell cycle arrest followed by recovery 61. However, when p53 degradation is prevented with the MDM-2 inhibitor Nutlin-3a, the ensuing higher, stable p53 levels lead to cellular senescence 61. Furthermore, p53 stabilization by Nutlin-3a treatment promotes senescence, with no evidence of apoptosis, in cultured MEFs upon oxidative stress 62. Finally, stabilization of p53 is impaired with age in splenocytes 44. If this is a general phenomenon, attenuated p53 signaling may prevent severely damaged cells from undergoing apoptosis and contribute to senescent and neoplastic cell accumulation with age.
Preferential p53-mediated transactivation of apoptosis- or senescence-specific target genes can also direct cell fate (Fig 2). p53 transactivation activity is fine-tuned by both post-translational modifications 63 and cooperativity between the DNA binding domains of the p53 tetramer. For instance, separation-of-function mutants show that individual acetylation sites on p53 can control the choice to senesce or apoptose 64. Thus, cells from p53K117R/ K117R mutant mice—in which the acetyl acceptor lysine is replaced by arginine—cannot upregulate PUMA and NOXA to induce apoptosis, but can still undergo cell cycle arrest and senescence through p21 upregulation. Ablation of K161 and K162, in addition to K117, also eliminates the arrest and senescence responses. Despite the fact that p53 has been shown to be phosphorylated at different positions after replicative senescence or DNA damage 65, it is unclear whether mutation of these residues would lead to “preference” for an apoptotic fate.
The quaternary structure of the p53 complex also appears to be important for the cellular response to stress. Disrupting cooperation between p53 DNA binding domains with the point mutation E177R interferes with activation of pro-apoptotic genes, but not other p53 targets involved in senescence and metabolism 66,67.
One important p53 target gene is p21, which enforces the initial cell cycle arrest in cells undergoing senescence and apoptosis 68. p21 expression has been proposed to negatively regulate p53-dependent apoptosis 69. Low concentrations of doxorubicin promote senescence in SKN-SH neuroblastoma 70 and colorectal carcinoma cells 71, associated with high p21 expression, whereas high doses of doxorubicin result in low p21 expression and apoptosis. These findings show the inverse relationship between p21 and apoptosis sensitivity expected for an anti-apoptotic protein and suggest the possibility that p21 is actively suppressed in apoptosis. Zhang and colleagues demonstrated that, in apoptotic colorectal carcinoma cells, the p53-target DNMT3a is responsible for suppressing p21. When this p21 antagonism is relieved by DNMT3a knockdown, the high-dose doxorubicin that would normally cause apoptosis leads to senescence instead. Similarly, as mentioned above, developmental senescence does not occur in p21-knockout animals and is partially compensated by apoptosis 39,40. In addition, p21 disruption tips the balance from senescence to apoptosis in colon cancer cells treated with the topoisomerase inhibitors irinotecan or camptothecin 72. Whether this is because p21 knockdown allows cell cycle reentry, with Topo1 inhibition leading to aberrant replication and a pro-apoptotic DDR, is unclear.
PTEN-AKT signaling
PTEN converts the lipid second messenger PIP3 to PIP2, thereby suppressing the activity PI3K and AKT, which are kinases that control pathways important for cell cycle progression 73, size 74, and metabolism 75. The PTEN/PI3K/AKT axis is also important for the choice between apoptosis and senescence. Complete loss of PTEN induces senescence in certain mouse and human cells independently of hyperproliferation, subsequent DNA damage, and ATM kinase activation 17. This is in striking contrast to the senescence caused by overexpression of Ras, which requires hyperproliferation and a DDR 15. However, some cell types proliferate in the absence of PTEN, although the response to stress is altered by PTEN status. For instance, in response to irradiation, human glioma cells with wild-type PTEN undergo apoptosis, whereas PTEN-null glioma cells undergo ROS/p53/p21-dependent senescence 76. Conversely, AKT deficiency confers resistance to replicative- and Ras-induced senescence and promotes apoptosis under conditions of oxidative stress 77. The nuclear functions of PTEN, including DNA damage repair, have been recently shown to depend on SUMOylation at K254 78. Whether selectively altering the nuclear functions of PTEN in the DDR instead of disrupting its cytosolic phosphatase activity would also lead to a pro-senescent, anti-apoptotic phenotype is unknown, and a promising area of research.
Cellular senescence arising from PTEN deficiency or AKT activation occurs through activation of mTORC1, requires p53 activity, and engages p21 79. Consistent with these findings, rapamycin—which inhibits mTORC1—delays replicative senescence and the senescence induced by progerin, the truncated form of Lamin A found in patients with Hutchinson-Gilford progeria syndrome 80,81. If mTOR is the signaling center of the PTEN-PI3K/AKT pathway relevant to a senescence or apoptosis decision, rapamycin should promote apoptosis under conditions of stress. Indeed, rapamycin enhances apoptosis in human and mouse cells treated with cisplatin, although under basal conditions—such as in untreated cultured keratinocytes—rapamycin does not promote apoptosis 82,83.
Effectors and inhibitors of apoptosis
Blocking the ability of damaged cells to execute the apoptotic program can also switch cell fate toward senescence (Sidebar A). Although the extrinsic apoptotic pathway has an arm that is completely caspase independent 84, the intrinsic pathway relies on caspases at multiple stages 85. For example, DNA damage stabilizes p53 to activate the intermediary caspase-2, which triggers mitochondrial outer membrane permeabilization (MOMP) 86. MOMP allows the release of mitochondrial cytochrome c activating the caspase-9-containing apoptosome 87. The apoptosome then triggers the executioner caspases 3, 6, and 9, which degrade protein targets to effect the morphological changes of apoptosis and cause cell death. Inhibition of caspases therefore blocks intrinsic apoptosis at many steps of the pathway, leaving senescence as an alternative cell fate. For instance, treating SKN-SH neuroblastoma cells with doxorubicin and a pan-caspase inhibitor prevents apoptosis and promotes senescence 70. Similarly, FANC-deficient hematopoietic progenitor cells usually undergo apoptosis when damaged by oxidative stress, but blocking apoptosis by caspase inhibition allows cells to survive and become senescent 88.
Manipulating upstream mediators of apoptosis, such as the anti-apoptotic Bcl-2 family proteins, can also influence the choice between senescence and apoptosis. Overexpression of Bcl-2, which prevents cytochrome c release during MOMP 89, forces senescence in fibroblasts treated with an otherwise lethal dose of doxorubicin 90. This also occurs in cancer cells, in which chemotherapy-induced senescence is an important alternative cell fate to apoptosis (Sidebar A). One of the first studies to report an apoptosis-independent function to p53 in cancer showed that murine lymphomas overexpressing Bcl-2 undergo p53/p16-dependent senescence rather than apoptosis in response to cyclophosphamide 91. Conversely, knockdown of the anti-apoptotic BCL-XL protein in colon cancer cells subject to DNA damage induced by irinotecan switches senescence to apoptosis 72.
Apoptosis resistance or sensitivity in senescence
If senescence and apoptosis are truly alternative cell fates, one hypothesis would be that cellular changes that are pro-senescent are actively anti-apoptotic and that senescent cells are resistant to apoptosis (Fig 3). Seluanov and colleagues provided strong evidence for the existence of apoptosis resistance in replicatively senescent HDFs, which they showed occurs through p53 signaling. They found that early-passage WI-38 cells undergo p53-dependent apoptosis in response to actinomycin D, low-dose cisplatin, or UVB irradiation, whereas p53-independent apoptosis occurred with high-dose cisplatin and etoposide. When senescent cells were challenged with p53-dependent apoptotic stimuli, they underwent necrosis instead 92. Exogenous expression of p53 in senescent cells restored their ability to undergo p53-dependent apoptosis. The response of senescent cells to the p53-independent agents was similar to that of early-passage cells, suggesting that apoptosis resistance in senescent cells may be mediated by changes in p53 signaling. Supporting this idea, exposure to Fas ligand induces p53-independent apoptosis to the same extent in senescent and non-senescent WI-38 cells 93.
Figure 3. The interplay between senescence and apoptosis is cell type specific.
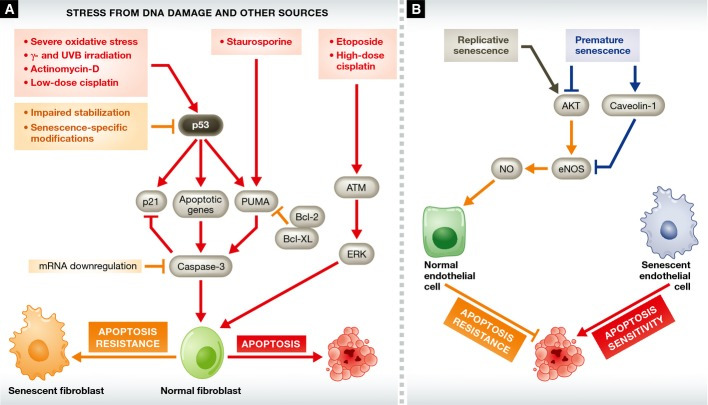
The pathways leading to apoptosis are depicted in red, those leading to apoptosis resistance in orange, and those that sensitize endothelial cells to apoptosis in blue. (A) Senescent fibroblasts are resistant to p53-mediated apoptotic stimuli, such as actinomycin D and low-dose cisplatin, as well as to stimuli—such as staurosporine—that rely on p53 target genes. This resistance can be explained by low p53 levels due to decreased stabilization in senescent cells, as well as the existence of senescence-specific p53 post-translational modifications. Senescent and non-senescent cells have a similar sensitivity to p53-independent apoptotic stimuli. (B) Senescent endothelial cells have increased sensitivity to apoptosis. Senescent endothelial cells lose eNOS expression and express reduced levels of pro-survival NO. This eNOS loss may be due to the loss of the positive regulator AKT, or to the upregulation of negative regulators, such as caveolin-1. However, AKT levels increase during replicative senescence, so the issue of PTEN/PI3K/AKT signaling in the senescent endothelium is unresolved. See Glossary for definitions and the text for details.
The outcome of apoptosis resistance can also be cell survival, rather than necrotic cell death. Senescence induced by mild H2O2 promotes survival rather than apoptosis in response to apoptotic stimuli such as UVB 94 or high-dose H2O2 53, in part by upregulating the anti-apoptotic BCL-2 despite global repressive epigenetic changes 95. Like UVB, high-dose H2O2 upregulates p53 53, leading to p53-dependent apoptosis 96. Similarly, replicative senescent human fibroblasts resist apoptosis in response to serum withdrawal by maintaining high levels of BCL-2 97. This upregulation of anti-apoptotic proteins in senescent cells may explain staurosporine resistance 93,98,99. Staurosporine triggers apoptosis in a p53-independent manner, indicating that alterations in p53 during senescence cannot be directly responsible for this resistance. However, staurosporine requires PUMA to execute apoptosis in MEFs 100 and human colon cancer cells 99. Overexpression of BCL-2 counteracts the pro-apoptotic genes PUMA and NOXA in cancer cells 101, and a similar process occurs during senescence.
Not only fibroblasts can escape apoptotic death. Replicatively senescent primary keratinocytes upregulate NF-κB to resist UVB-induced apoptosis, whereas immortalized HaCat keratinocytes remain sensitive to apoptosis 102. Furthermore, when keratinocytes enter G0 after reaching confluence, they become UVB resistant. Exit from the cell cycle has also been shown to lead to apoptosis resistance in colon cancer cell lines, but this phenomenon has not been widely studied 103. Similar to confluence, cell cycle extension can promote resistance to apoptosis by allowing additional time to repair damage. Consistent with this scenario, late passage, non-senescent human fibroblasts—which have an extended cell cycle time—upregulate BCL-XL and resist UVB-induced apoptosis 104. Modulation of the cell cycle, either by its extension or exit to G0, is therefore a confounding element in studies about the apoptosis resistance of senescent cells that precludes definitive conclusions about the effects of senescence per se.
In contrast to fibroblasts and keratinocytes, senescent endothelial cells are more susceptible to apoptosis than their non-senescent counterparts. For example, porcine pulmonary artery endothelial cells passaged to replicative senescence undergo more spontaneous apoptosis than at early passage, with reduced BCL-2 and increased BAX expression 105. Senescent human umbilical vein endothelial cells (HUVECs) similarly undergo increased apoptotic death in response to exogenous ceramide C2 106. Conversely, senescent human foreskin fibroblasts treated in the same manner are resistant to this compound compared to their early-passage counterparts 106. Apoptosis by exogenous ceramide C2 has been reported to be p53 dependent, although in radiation-induced apoptosis, ceramide C2 production is p53 independent 107.
Fibroblasts show reduced sensitivity to apoptosis as passage number increases, whereas HUVECs are increasingly susceptible to apoptosis with passage 108, showing reduced activity of the anti-apoptotic endothelial nitric oxide synthase (eNOS) 109–112. Hoffman and colleagues attributed the loss of NO production to reduced AKT 108, which phosphorylates and activates eNOS 113. Other mechanisms may also be involved in endothelial cell senescence, such as increased levels of the eNOS negative regulator caveolin-1 112. PTEN/PI3K/AKT pathway activity is reduced during senescence of irradiated endothelial cells 114 and HUVECs exposed to high glucose 115. However, AKT activity rises during replicative senescence of endothelial cells, and its inhibition extends replicative lifespan in vitro 116. Thus, the variations in sensitivity to apoptosis between senescent endothelial cells and fibroblasts may reflect differences in the way these cells modulate pro-survival factors, such as eNOS, or how the PTEN/PI3K/AKT pathway is altered with senescence.
Mice expressing low amounts of the mitotic checkpoint protein BubR1 due to hypomorphic alleles (BubR1H/H) accumulate senescent cells in several tissues early in life 117 and rapidly develop progeroid phenotypes 118. BubR1H/H mice that are prevented from developing senescent cells through genetic ablation of p16Ink4a have a delayed time to onset of these phenotypes; however, they still die early from p16Ink4a-independent effects of BubR1 depletion 117. p16Ink4a-positive cells can be effectively eliminated from BubR1 hypomorphic mice using an INK-ATTAC transgene, in which a fragment of the p16Ink4a promoter is used to drive the expression of a drug-inducible caspase-8/FKBP fusion protein in senescent cells. Administration of the synthetic drug AP20187 causes dimerization of the FKBP domains, forcing caspase-8 activation and apoptotic death.
The clearance of p16Ink4a-positive cells in this manner also delays the development of progeroid phenotypes 119. The cell types undergoing senescence and expressing the INK-ATTAC transgene in BubR1H/H fat and muscle have been defined as adipocyte progenitors/stem cells and fibroadipogenic progenitors, respectively 120. While it is tempting to conclude that these cells are not resistant to apoptotic death because the apoptotic program can be initiated through caspase-8 dimerization, the role of caspase-8 is downstream of any apoptosis-resistance changes described in senescent cells. Nevertheless, these results in BubR1H/H INK-ATTAC mice provide hope for the development of a senescent cell killing therapy, by demonstrating that senescent cells can undergo apoptosis in vivo with the proper stimulus.
Senescence signaling within tissues
Apoptosis leads to a rapid elimination of dysfunctional cells by phagocytes in a manner that does not stimulate inflammation 121. On the other hand, the pro-inflammatory secretion of growth factors and cytokines from senescent cells has the potential to generate prolonged paracrine signaling. In this way, apoptosis can be viewed almost solely as a cell-intrinsic mechanism, as compared to the dual cell autonomous and non-autonomous nature of senescent cells. Emerging data suggest that the presence of senescent cells has an advantage over apoptosis due to this ability to communicate with other cells, raising the possibility that signaling from senescent cells within tissues can be both beneficial and detrimental (Sidebar A).
The senescence program is activated in a variety of benign and pre-malignant lesions in vivo to limit tumor progression in a cell-autonomous manner 16,122–124. Various components of the SASP, however, promote pre-malignant cell growth or invasion through their ability to induce angiogenesis, epithelial–mesenchymal transitions and differentiation within the local microenvironment 25,29,125–127. These effects are clearly pro-neoplastic and thus are detrimental side effects of the SASP.
However, several studies have suggested that the SASP is not always pro-tumorigenic 128. First, the SASP can reinforce and maintain the senescent state in cell culture models of senescence 21,129–131. Second, the SASP attracts the immune system to clear both premalignant and established tumor cells by phagocytosis or cytotoxic-mediated killing, through a “senescence surveillance” process that entails both innate and adaptive immune responses 132–134. Oncogene-induced, pre-malignant hepatocytes present many features of senescent cells, including high levels of p16Ink4a, p21 and senescence-associated (SA)-β–galactosidase activity. It is thought that these cells generate a SASP that initiates a CD4+-T-cell-mediated adaptive immune response to subsequently remove these pre-malignant lesions. Furthermore, reactivation of p53 in a Ras-induced liver-carcinoma mouse model resulted in rapid regression of the existing tumor. Surprisingly, the tumors were not eliminated through apoptosis but through cellular senescence and a SASP, consistent with observations from a sarcoma mouse model 135. The SASP that is generated within the liver tumors triggers the innate immune system to respond to the senescent cells and remove them through the action of macrophages, neutrophils, and NK cells.
With these observations in mind, one could argue that senescence in pre-malignant and established tumor cells has some advantages over apoptosis (Fig 4), although it should be emphasized that apoptosis provides a preferred and effective anti-tumor mechanism in various contexts, including malignancies with Myc mutations 136,137. First, when a cell within an emerging tumor undergoes senescence, it has the potential to negatively impact its neighboring non-senescent tumor cells through the SASP. For instance, it has been shown that senescence and SASP production can trigger senescence in neighboring cells via paracrine signaling, a phenomenon that has been referred to as bystander senescence 138. Second, the mobilization of immune responses to these areas of senescence in pre-malignant and established tumors could have a greater impact on reducing tumor cell burden than apoptosis of single cells. This is in contrast to what is observed with apoptosis of large proportions of neoplastic cells, such as is seen with cytotoxic agents and various mouse models, illustrating that apoptosis is a potent anti-cancer mechanism when it occurs in a coordinated manner in a large percentage of cells. When a limited number of cells must decide between senescence and apoptosis, perhaps senescence has a greater consequence, as there is potential to impact other cells in the microenvironment. However, it is still unclear why certain pre-neoplastic lesions—such as benign nevi that are often caused by oncogenic BRAF mutations—remain for extended periods of time, avoiding immune-mediated senescence surveillance. Whether analogous avoidance processes occur outside the context of tumorigenesis, for example, during senescence in aged tissues, is unknown (Sidebar A).
Figure 4. Consequences of senescence and apoptosis in stressed tissues.
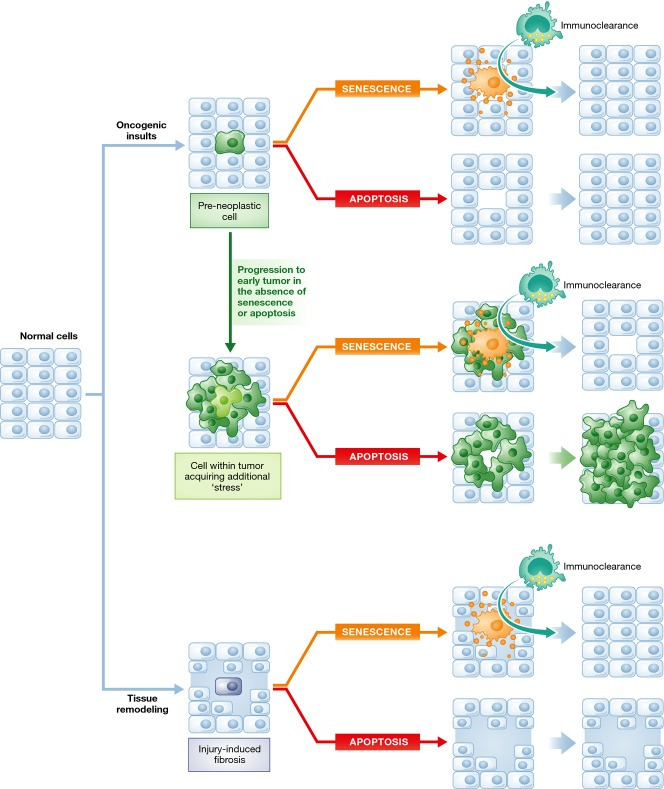
Normal cells are subject to a variety of stressful stimuli, including oncogenic insults (top) and tissue damage (bottom). Cells that have acquired a pre-neoplastic lesion may undergo senescence or apoptosis. The outcome of this decision is largely the same if the senescence surveillance machinery—which ensures that the lesion is efficiently removed—is intact. If pre-neoplastic lesions do not induce senescence or apoptosis, they continue to grow and progress (middle). In this scenario, if senescence is engaged in a fraction of the now established tumor, the SASP and recruitment of the surveillance machinery may be much more effective at removing tumor cells than a single cell that undergoes apoptosis and does not initiate an immune response. Although not illustrated, if a large percentage of tumor cells are coerced into apoptosis, this would also lead to reduction in tumor volume. In response to tissue damage (bottom), senescence would also theoretically be advantageous compared to apoptosis, as the production of the SASP would limit tissue fibrosis and promote tissue remodeling, as long as the SASP-producing cell is ultimately removed by the immune system.
One potential reason why senescent cells become more abundant with age is that the immune system deteriorates over time and becomes less efficient in clearing senescent cells. On the other hand, perhaps mechanisms that maintain tissue/organ size act to suppress the clearance of senescent cells by the immune system to prevent potential tissue dysfunction through cell loss. Future studies to investigate these phenomena and assess whether immune cells target both acute and chronic senescent cells for destruction are clearly needed and warranted. In addition, aging was not selected for during the evolution of most organisms. Thus, the programs that mediate changes with advancing age may be more malleable than previously thought.
Other tissues seem to benefit by initiating senescence upon damage. Hepatic stellate cells of the liver, for example, which promote fibrosis upon activation by injury, undergo senescence upon chronic damage to restrict fibrosis 134. Similarly to what is observed in established liver tumors, NK cells subsequently clear these senescent stellate cells. During the wound-healing response in skin, myofibroblasts proliferate and produce extracellular matrix 139. These cells then senesce and destroy the matrix through the SASP, thereby limiting fibrosis. A number of additional tissues undergo senescence with age and disease (reviewed in 14,128,140,141); however, additional studies are needed to understand the contribution of these senescent cells to disease initiation, progression, and maintenance.
Future directions
Important molecular mechanistic insights about the relationship between senescence and apoptosis have emerged over recent years, although a number of questions about the elimination of senescent cells in vivo remain unresolved. The first concerns the interaction between the immune system and cellular senescence. If, as hypothesized, a decline in immune surveillance underlies the accumulation of senescent cells with age, the rates with which senescent cells are generated could be relatively constant with age and, thus, short-lived senescent cells would exist transiently even earlier in life. One test of this idea would be to compare senescent cell numbers in young normal mice and mice that are deficient in various arms of the immune surveillance network, including mice lacking CD4+ T cells, macrophages or NK cells. An increase in senescent cells in young immunodeficient animals would suggest that immunosurveillance normally limits their presence in healthy young animals.
The question of rates of senescence in youth and old age can be partially addressed by inducible immunodeficiency models. If the rate of senescent cell production increases with age, abrogation of immune surveillance in aged animals should cause greater accumulation of senescent cells than disrupting the immune system in youth. A potential confounder in this experimental design is that immune dysfunction could per se drive de novo formation of senescent cells, rather than sparing existing cells. A complimentary approach would be to label senescent cells during a period of time in young animals and compare them with the number of cells accumulated during an identical interval in old age. An estrogen receptor–Cre fusion protein driven by the p16 promoter, in combination with a floxed cassette inhibiting the expression of GFP, for example, might suffice for this purpose.
Another open question is whether and how apoptosis resistance is altered with the establishment and progression of senescence (Sidebar A). Some features of late senescence, such as the recently described cytoplasmic chromatin processing 142, are reminiscent of blebbing during apoptosis. Whether chromatin blebbing is a hallmark of senescent cells or whether it represents a transition from senescence to apoptosis remains to be determined. Activation of latent transposons occurs late in senescence 143,144, which may cause non-self antigen presentation on the cell surface, leading to cytotoxic T-cell-mediated apoptosis. Thus, additional apoptotic pressures may overwhelm the anti-apoptotic machinery engaged in established senescent cells. However, these hypotheses assume that senescence occurs because it is preferable to apoptosis in certain contexts. One alternative to this model is that pre-neoplastic cells already have critical mutations that inhibit apoptosis, leaving senescence as a fail-safe mechanism. The ability of “single hits”—such as Ras overexpression or BrafV600E mutation—to induce senescence might argue against this possibility, but perhaps these mutants compromise apoptosis and thereby lead to senescence, rather than vice versa.
Whether senescent cells in aged organisms are more detrimental than beneficial to healthy life remains unclear. The lack of any overt downside to senescent cell clearance in the BubR1H/H; INK-ATTAC model suggests that apoptosis of senescent cells might be safe 119. It may therefore be reasonable to develop therapies that kill senescent cells to prolong healthy organismal life. The question then would be to identify the molecular target(s) that constitute an Achilles’ heel of senescent cells, which express a balance of pro-survival and pro-apoptotic signals. Although enhancing p53 levels might tip the balance toward apoptosis, drugs that do so risk having significant off-target effects. Understanding when and how to tip this balance will require more basic research and the ability to translate this research to pre-clinical and eventually clinical settings.
Conclusions
Regardless of the theoretical mechanisms discussed above, the function of senescent cells in vivo, when unrelated to pathologies such as cancer and wounding, remains obscure. Crucial conclusions about senescence are based on cell culture work using non-physiological stresses. Many of the most interesting features of senescent cells observed in vitro, such as G1 arrest with 4N DNA content 12 or cytoplasmic heterochromatin processing 142, have yet to be verified in vivo or in culture by other groups. Given the variability in apoptosis sensitivity observed between senescent endothelial cells and fibroblasts, it is imperative that in vivo senescent cells be identified, isolated, and characterized (Sidebar A).
An even larger challenge to the field is to put what has been learned about senescence and apoptosis, not only in the context of the human or mouse, but into the bigger picture of multicellular life. What other species have cellular programs analogous to senescence? Does C. elegans, the poster child for developmental apoptosis, also use cellular senescence during embryogenesis? If enough data are gathered about the use of these processes in development and aging in vertebrates and invertebrates, it would maybe be possible to ascertain which mechanism is ancestral. The field can also learn whether senescence originated as a developmental force or a tumor suppressor. This question has not been resolved from the apoptosis viewpoint either. While perhaps inappropriate to ask the teleological “Why?” about the choice between apoptosis and senescence, we can ask “How?”—how did they evolve, how do cells choose between them, and, ultimately, how can we make use of this choice to promote a healthy life (Sidebar A).
Sidebar A: In need of answers —
Acknowledgments
We apologize to those whom we were unable to reference due to space limitations. Support was provided by grants from the Paul Glenn, Ellison, and Noaber Foundations and the National Institutes of Health R01CA96985, R01CA166347, and P01AG041122.
GlossaryAKT
Protein kinase B
ARF
Alternate reading protein from CDKN2a locus; p19 in mice and p14 in humans
ATM
Ataxia telangiectasia mutated
BAD
Bcl-2-associated death promoter protein
BAX
Bcl-2-associated X protein
BCL-2
B-cell lymphoma 2, an important anti-apoptotic protein
BCL-XL
B-cell lymphoma extra large
BRAF
Rapidly accelerated fibrosarcoma; a serine–threonine protein kinase in the RAS-RAF-MEK-ERK signaling cascade
CCN1
CCN family member 1; a matrix-associated extracellular signaling protein
CD158
Cluster of differentiation 158; a plasma membrane receptor
CDK
Cyclin-dependent kinase
CDKi
Cyclin-dependent kinase inhibitor
CREB
Cyclic-AMP response element binding protein
DDR
DNA damage response
DNMT3a
DNA methyltransferase 3a
eNOS
Endothelial nitric oxide synthetase
ERK
Extracellular signal-regulated kinase
ETS1
Erythroblastosis homolog 1; a transcription factor
FKBP
FK506 binding protein
FOXO
Forkhead Box O-containing protein a transcription factor
HDF
Human diploid fibroblast; a descriptor of cell karyotype and origin
HLA
Human leukocyte antigen; human equivalent of the major histocompatibility complex genes
HUVEC
Human umbilical vein endothelial cell
IL
Interleukin
INK-ATTAC
Senescent cell killing transgene: apoptosis through targeted activation of caspase (ATTAC) in p16Ink4a (INK)-positive cells
MCF7
Michigan Cancer Foundation-7 human breast cancer cell line
MDM2
Mouse double minute 2
MOMP
Mitochondrial outer membrane permeabilization
MRC5
Human diploid fetal lung fibroblast cell line
mTORC1
Mechanistic target of rapamycin complex 1
NK
Natural killer
NO
Nitric oxide
NOXA
Phorbol-12-myristate-13-acetate-induced protein 1
OIS
Oncogene-induced senescence
p38
MAPK Mitogen-activated protein kinase
PI3K
Phosphoinositide-3 kinase
PIP
Phosphatidylinositol phosphate
PKC
Protein kinase C
PTEN
Phosphatase and tensin deleted in chromosome 10
PUMA
P53 upregulated modulator of apoptosis
Rb
Retinoblastoma
SKN-SH
Bone marrow-derived cell line from a human patient with neuroblastoma
SMAD
Transcription factor downstream of TGF-β signaling
TGF-β
Transforming growth factor-β
UVB
Ultraviolet B light
WI-38
Human diploid fetal lung fibroblast cell line
Conflict of interest
The authors declare that they have no conflict of interest.
References
- Hayflick L, Moorhead PS. The serial cultivation of human diploid cell strains. Exp Cell Res. 1961;25:585–621. doi: 10.1016/0014-4827(61)90192-6. [DOI] [PubMed] [Google Scholar]
- Bodnar AG, Ouellette M, Frolkis M, Holt SE, Chiu CP, Morin GB, Harley CB, Shay JW, Lichtsteiner S, Wright WE, et al. Extension of life-span by introduction of telomerase into normal human cells. Science. 1998;279:349–352. doi: 10.1126/science.279.5349.349. [DOI] [PubMed] [Google Scholar]
- Martinez P, Blasco MA. Telomeric and extra-telomeric roles for telomerase and the telomere-binding proteins. Nat Rev Cancer. 2011;11:161–176. doi: 10.1038/nrc3025. [DOI] [PubMed] [Google Scholar]
- Prowse KR, Greider CW. Developmental and tissue-s