Nat Immunol
. Author manuscript; available in PMC: 2021 Feb 14.
Published in final edited form as: Nat Immunol. 2018 Jun 20;19(7):665–673. doi: 10.1038/s41590-018-0120-4
Regulatory T cells in autoimmune diseases
Margarita Dominguez-Villar 1,*, David A Hafler 2,*
- Author information
- Article notes
- Copyright and License information
PMCID: PMC7882196 NIHMSID: NIHMS1660995 PMID: 29925983
The publisher's version of this article is available at Nat Immunol
Abstract
During the last years, our understanding of regulatory T cell (Treg cell) biology has greatly expanded. Key observations have challenged the traditional definition of Treg cell and have provided insight into the underlying mechanisms responsible for the development of autoimmune diseases with new therapeutic strategies that improve disease outcome. This review summarizes the newer concepts of Treg cell instability, plasticity and tissue-specific Treg cells and their relation to autoimmunity. These three major concepts have changed our understanding of Treg cell biology: how they interact with other immune and non-immune cells, their functions in specific tissues, and the implications for the pathogenesis of autoimmune diseases.
초록
최근 몇 년간 조절 T 세포(Treg 세포) 생물학에 대한 우리의 이해는
크게 확장되었습니다.
주요 관찰 결과들은 Treg 세포의 전통적 정의를 재고하게 했으며,
자가면역 질환 발병의 근본적 메커니즘에 대한 통찰력을 제공하여
질병 예후를 개선하는 새로운 치료 전략을 제시했습니다.
본 리뷰는
Treg 세포의 불안정성, 가소성, 조직 특이적 Treg 세포라는
새로운 개념과 이들이 자가면역과 맺는 관계를 요약한다.
이 세 가지 주요 개념은
Treg 세포 생물학에 대한 우리의 이해를 변화시켰다:
다른 면역 세포 및 비면역 세포와의 상호작용 방식, 특정 조직에서의 기능,
그리고 자가면역 질환 발병 기전에 대한 함의 등이다.
More than 20 years after their ‘re-discovery’, regulatory T cells (Treg cells) have emerged as an important component in our understanding of the immune response to pathogens and the mechanisms of peripheral tolerance that control the development of allergies and autoimmune diseases. In mice and humans, Treg cells are characterized by the high expression of the IL-2 receptor alpha chain (IL-2Rα, CD25) and the expression of the transcription factor Foxp3, which is required for their development, function and stability1, 2, 3, 4. In humans, as CD25 is also expressed by activated CD4+ T cells, the absence of the IL-7 receptor alpha chain (IL-7Rα, CD127) is used as a complementary marker to CD25 expression to more precisely identify human Treg cells5. Moreover, numerous surface receptors have been described that are variably specific for defined Treg cell subsets, arguing for the heterogeneity of this population6. Treg cells can be broadly classified into two groups based on their developmental origin. Thymic Treg cells (tTregs) – also known as natural Treg (nTreg) cells – are generated in the thymus as a separate lineage at the stage of CD4 single-positive thymocytes, and are thought to be enriched for T cell receptors (TCR) with high affinity for self-peptides7. Although their detailed mechanisms of suppression are still not completely understood and are most probably dependent on the microenvironment and the target population to be controlled, in general they perform their function by both cell-contact mechanisms that involve specific cell surface receptors, and the secretion of inhibitory cytokines such as IL-10, TGF-β and IL-357. Induced Treg cells (iTregs) develop from conventional CD4+ T cells in the periphery after antigen encounter and in the presence of specific factors such as TGF-β and IL-28. To date, we lack a definitive protein marker(s) that distinguishes between these two Treg cell populations in vitro or in vivo, although there are important differences in their epigenetic signature, and particularly in the Foxp3 locus, making iTreg cells intrinsically unstable to inflammatory and/or stress conditions8, 9.
From a historical perspective, the initial observations made in the 1970s that led to the definition of Treg cells in the 1990s are tightly linked to autoimmune diseases. Seminal works by Nishizuka and Sakakura back in 1969 demonstrated that neonatal thymectomy in healthy mice led to inflammation and severe organ-specific autoimmune pathology, implicating a thymic-derived population in control of self-tolerance10. This observation was further confirmed in adult rats that were thymectomized and subjected to sublethal irradiation11. Most importantly, inoculation of CD4+ T cells from healthy syngeneic mice inhibited disease in both systems, and once autoimmunity was established, CD4+ T cells isolated from these mice and adoptively transferred to T cell deficient mice were able to induce disease12. These experiments demonstrated that T cells were able to not only serve as inducers of autoimmune diseases, but also to inhibit them. This led to the hypothesis that in the periphery of normal mice there existed two populations of CD4+ T cells, i.e., one potentially capable of inducing autoimmunity, and a second population of ‘suppressor’ cells, different from helper T cells, that would inhibit autoreactive T cells. Treg cell research during those years floundered, in part due to the failure in finding specific cell markers that could define this population and the ambiguity observed in their mechanisms of suppression. The identification of a population of CD4+ T cells as responsible for controlling autoreactive responses13, and the search for a cell surface marker that would define this population, indicated that a subset of CD4+ T cells present in the periphery of normal mice expressing CD25 was responsible for the inhibition of autoimmunity. Transfer of cell suspensions from spleen of BALB/c mice depleted in CD25+ cells into athymic nude mice induced autoimmunity which afflicted several organs, and co-transfer of CD25+CD4+ T cells inhibited disease4. In 2001, human Treg cells were identified in the thymus and peripheral blood of healthy individuals as those CD4+ T cells expressing very high CD25 and being very similar in phenotype and function to their rodent counterpart14, 15. Although we still lack a definitive marker for human Treg cell isolation, these studies, together with the discovery of Foxp3 as the master transcription factor of Treg cells1, 3, 16, laid the groundwork for the beginning of in depth analysis of Treg cell biology in health and disease (Box 1).
'재발견'된 지 20년이 넘은 지금, 조절 T 세포(Treg 세포)는
병원체에 대한 면역 반응과 알레르기 및 자가면역 질환의 발병을 제어하는
말초 내성 메커니즘을 이해하는 데 중요한 요소로 부상했습니다.
마우스와 인간에서 Treg 세포는
IL-2 수용체 알파 사슬(IL-2Rα, CD25)의 높은 발현과 발달,
기능 및 안정성에 필수적인 전사 인자 Foxp3의 발현으로 특징지어집니다1, 2, 3, 4.
인간에서는 활성화된 CD4+ T 세포도 CD25를 발현하므로,
IL-7 수용체 알파 사슬(IL-7Rα, CD127)의 부재를 CD25 발현에 대한
보완적 표지자로 사용하여
인간 Treg 세포를 보다 정확하게 식별합니다5.
또한, 정의된 Treg 세포 하위 집합에 대해
가변적으로 특이적인 수많은 표면 수용체가 보고되어 이 집단의 이질성을 뒷받침합니다6.
Treg 세포는
발달 기원에 따라 크게 두 그룹으로 분류될 수 있다.
흉선 Treg 세포(tTregs) – 자연 Treg(nTreg) 세포라고도 함 – 는
흉선에서 CD4 단일 양성 흉선 세포 단계에서 별도의 계통으로 생성되며,
자가 펩타이드에 대한 높은 친화력을 가진 T 세포 수용체(TCR)가 풍부한 것으로 여겨진다7.
그들의 억제 메커니즘의 세부 사항은 아직 완전히 이해되지 않았으며,
통제 대상인 미세환경과 표적 집단에 따라 달라질 가능성이 높지만,
일반적으로 그들은
특정 세포 표면 수용체를 포함하는 세포 접촉 메커니즘과
IL-10, TGF-β 및 IL-35와 같은 억제성 사이토카인의 분비를 통해
기능을 수행합니다.
유도성 Treg 세포(iTregs)는
항원 접촉 후 말초에서 일반적인 CD4+ T 세포로부터 발달하며,
TGF-β 및 IL-2와 같은 특정 인자의 존재 하에서 발생합니다8.
현재까지 이들 두 Treg 세포 집단을 체외 또는 생체 내에서 구분하는
결정적인 단백질 표지자는 존재하지 않으나,
특히 Foxp3 유전자좌에서 관찰되는 후생유전학적 서명(epigenetic signature)의 중요한 차이점들로 인해
iTreg 세포는 염증 및/또는 스트레스 조건에 본질적으로 불안정합니다8, 9.
역사적 관점에서 볼 때, 1970년대에 이루어진 초기 관찰은 1990년대에 Treg 세포의 정의를 이끌어낸 것으로 자가면역 질환과 밀접하게 연관되어 있습니다. 1969년 니시즈카(Nishizuka)와 사카쿠라(Sakakura)의 선구적 연구는 건강한 생쥐에서 신생아기 흉선 적출이 염증과 심각한 장기 특이적 자가면역 병리를 유발함을 보여주었으며, 이는 자가 내성 조절에 흉선 유래 세포 집단이 관여함을 시사했다10. 이 관찰은 흉선을 적출하고 치사량 미만의 방사선 조사(sublethal irradiation)를 받은 성인 쥐에서도 추가로 확인되었다11. 가장 중요한 것은, 건강한 동종 마우스의 CD4+ T 세포를 접종하면 두 시스템 모두에서 질병이 억제되었으며, 일단 자가면역이 확립된 후에는 이 마우스에서 분리된 CD4+ T 세포를 T 세포 결핍 마우스에 이식하면 질병을 유발할 수 있었다는 점이다12. 이러한 실험들은 T 세포가 자가면역 질환의 유발자 역할을 할 뿐만 아니라 이를 억제할 수도 있음을 보여주었다. 이로 인해 정상 생쥐의 말초에는 두 종류의 CD4+ T 세포 집단이 존재한다는 가설이 제기되었습니다.
즉, 하나는 자가면역 반응을 유발할 가능성이 있는 집단이고,
다른 하나는 헬퍼 T 세포와는 다른 ‘억제’ 세포 집단으로 자가반응성 T 세포를 억제하는 집단이라는 것입니다.
당시 Treg 세포 연구는 이 집단을 정의할 수 있는 특이적 세포 표지자를 찾지 못한 점과 억제 기전의 모호성으로 인해 어려움을 겪었다. 자가반응성 반응을 제어하는 역할을 하는 CD4+ T 세포 집단의 확인13과 이 집단을 정의할 세포 표면 마커에 대한 탐색은 정상 마우스 말초에 존재하는 CD25를 발현하는 CD4+ T 세포의 하위 집합이 자가면역 억제를 담당한다는 것을 시사했다. CD25+ 세포가 제거된 BALB/c 생쥐의 비장 세포 현탁액을 무흉선 누드 생쥐에 이식했을 때 여러 장기를 침범하는 자가면역이 유발되었으며, CD25+CD4+ T 세포를 함께 이식하면 질병이 억제되었다4. 2001년에는 건강한 개인의 흉선과 말초 혈액에서 매우 높은 수준의 CD25를 발현하고, 형상 및 기능 면에서 설치류의 해당 세포와 매우 유사한 CD4+ T 세포로서 인간 Treg 세포가 확인되었다14, 15. 인간 Treg 세포 분리용 결정적 표지자는 아직 부족하지만, 이러한 연구들과 Treg 세포의 핵심 전사 인자로서 Foxp3의 발견1, 3, 16은 건강 및 질병 상태에서의 Treg 세포 생물학 심층 분석의 초석을 마련했다(Box 1).
Box 1. How are Treg cells studied in autoimmunity?
Treg cell studies in autoimmunity have been facilitated by the existence of mouse models for many autoimmune diseases130. While these models of autoimmune disease allow fate mapping and gene modification experiments that cannot be performed in humans, they do not generally reflect the genetic architecture that underlies human autoimmune diseases. That is, the models often reflect the efferent aspects of disease pathogenesis but not the underlying mechanisms that cause the activation of autoreactive T cells.
Human Treg cell studies are hindered by the lack of a definitive surface marker that uniquely isolates Treg cells from other T cell populations. Thus, many early works examining the frequency of Treg cells in peripheral blood of patients need to be considered with caution, as Treg cells were defined solely by the positive expression of CD25 and the results can be misleading as human conventional T cells can also express CD25 upon activation. Moreover, human studies are limited to ex vivo characterizations or short in vitro experiments and depend on the availability of samples from patients with autoimmunity.
Recent advances in high throughput technologies that require small cell numbers will considerably impact human Treg cell autoimmunity research, allowing the analysis of rare Treg cell populations, from small tissue samples, etc. Current available technologies are able to determine the epigenetic and transcriptional signature at the single cell level. The combination of these datasets and system biology approaches to analyze serial samples obtained from individuals at different time points, before and after therapeutic intervention and from different anatomical locations is delivering enormous amounts of information about response to treatment in patients, disease course, and mechanistic insights.
With regards to Treg cell function in human autoimmune diseases, early studies with patients revealed that in a number of autoimmune disorders there is either a defect in the numbers or in the function of Treg cells isolated from peripheral blood, with these data being supported by in vivo models of disease (for a comprehensive list of autoimmune diseases, 17). Although some of these early data are confounded due to the identification of human Treg cells based on the positive expression of CD25 as the only marker for Treg cell identification, subsequent works have clearly demonstrated that most autoimmune diseases display defects in either the number and/or the function of tTreg cells measured in peripheral blood, for example Type 1 diabetes18, 19, 20, multiple sclerosis21, 22, systemic lupus erythematosus (SLE)23, myasthenia gravis24, rheumatoid arthritis25 and others17.
Investigations during the past two decades have advanced our knowledge regarding the mechanisms that underlie tTreg cell development in the thymus, their gene expression signature, their role in controlling both immune and non-immune cell responses, their impact in immune-mediated diseases (reviewed in 7, 26); and finally identifying the tissues where they exert functions beyond suppression27, 28. Moreover, the traditional view that Treg cells are a terminally differentiated population not capable of secreting pro-inflammatory cytokines and whose only function is to suppress T cell responses has been challenged based on more recent data. Specifically, Treg cells possess some degree of plasticity and instability, although we still do not understand the molecular mechanisms that drive these two states in depth or the relationship between each other in disease settings.
This review will provide an update on recent discoveries in Treg cell biology in relation to autoimmune diseases. From our perspective, three major observations have changed our understanding of what Treg cells are, how they function in peripheral lymphoid organs and non-immune tissues, how they relate to other immune and non-immune cells, and how their phenotype and function can be modulated, with clear consequences for the development of new therapeutic strategies for a number of autoimmune diseases. These are: 1) instability of Treg cells and acquisition of an effector phenotype after losing Foxp3 expression under inflammatory conditions, 2) plasticity of the Treg cell phenotype, with acquisition of effector-like properties while maintaining Foxp3 expression, and 3) the discovery of tissue-specific Treg cells, demonstrating again that Treg cells, as other immune cells, are influenced by their environment. These observations arise at a time where the development of high throughput genomic, epigenetic and proteomic technologies allow the analysis of rare cell populations to a single cell level, which will undoubtedly improve our knowledge of basic Treg cell biology in general, and human Treg cell biology in particular.
Foxp3 as a master regulator of Treg cell phenotype and function.
The discovery of Foxp3 as the master regulator of Treg cell development and function was critical for our understanding of Treg cell biology1, 2, 3. Inactivating mutations in the Foxp3 gene causes spontaneous development of severe autoimmunity with a scurfy phenotype in mice29 and Immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) syndrome in humans30, 31. Foxp3 is necessary for tTreg cell development, maintenance and function1, 2, 3, although alone it is not sufficient to fully recapitulate the Treg cell phenotype9, 32. Besides Foxp3, the second requirement necessary for the Treg cell functional program to be established is the generation of a specific epigenetic signature acquired during development and finalized in the periphery9, 33. Both play essential roles in maintaining Treg cell function, and alteration of Foxp3 or epigenetic modifications during auto-inflammatory conditions are likely the cause of Treg cell instability and aberrant plasticity observed in several autoimmune settings (Fig. 1). Due to the importance of Foxp3 in Treg cell maintenance and prevention of autoimmunity, regulation of Foxp3 expression is a matter of active research. Foxp3 is subjected to two major layers of regulation, i.e. transcriptional and post-translational, both of which are responsive to positive and negative regulation by factors in the tissue environment, including cytokines, metabolic mediators and inflammatory factors.
Figure 1. Treg cell functional program in health and autoimmunity.
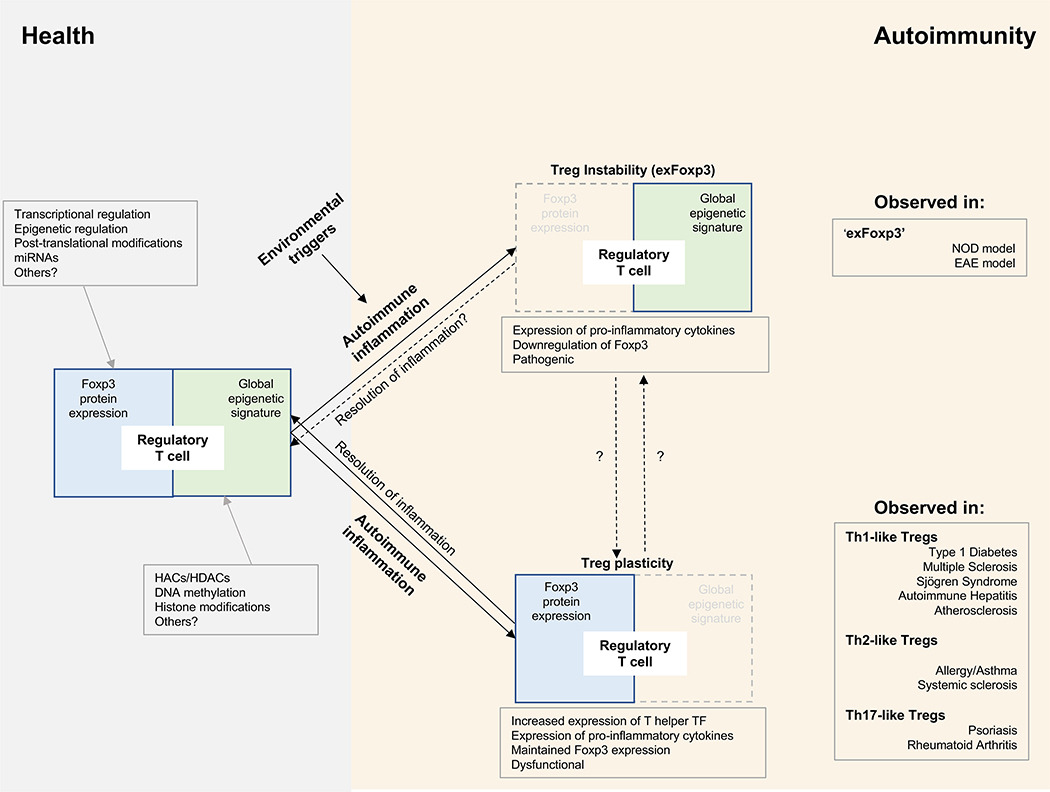
The Treg cell functional program depends on two major axes: Foxp3 expression and a global epigenetic signature, both of which are regulated by a number of factors (gray boxes) and maintained under healthy conditions. In genetically susceptible individuals, environmental triggers induce inflammation, and a percentage of Treg cells can lose Foxp3 expression and become unstable (Treg cell instability, exFoxp3), or can maintain Foxp3 expression but alter their global epigenetic signature (Treg cell plasticity), secrete pro-inflammatory cytokines and display reduced function. Treg cell instability leads to the so-called ‘exFoxp3’ cells, which have been observed in mouse models of diabetes (NOD) and multiple sclerosis (EAE). Treg cell plasticity favors the acquisition of TH1-like, TH2-like or TH17-like properties by the Treg cells, each of which have been observed in a number of autoimmune diseases in mice and humans. While the mechanistic connection between Treg cell instability and plasticity is not known, plastic Treg cells can become ‘normal’ Treg cells after resolution of inflammation. It remains to be determined whether this is also the case for Treg cell instability. Abbreviations: miRNA, microRNA; HACs, histone acetylases; HDACs, histone deacetylases; TF, transcription factors; NOD, non-obese diabetic; EAE, experimental autoimmune encephalomyelitis.
Foxp3 gene expression is a tightly regulated process, and while the mechanisms of Foxp3 transcriptional regulation by transcription factors during tTreg cell development and in mature Treg cells have been clearly established (reviewed in34), data obtained in rodents have shown that Foxp3 gene transcription is also epigenetically controlled. The Foxp3 locus contains several conserved non-coding sequences (CNS) that are critical for the initiation and maintenance of Foxp3 transcription 35. From the three CNS described so far, only CNS2 has been demonstrated to prevent autoimmunity. It is a TCR-responsive enhancer with binding sites for Runx1-CBFβ complexes that is important in maintaining Foxp3 stability. CNS2-deficient mice develop spontaneous autoimmunity, underscoring the importance of CNS2 for Treg cell stability and function36, 37. Furthermore, CNS2 contains a conserved CpG island (also called the TSDR region) that is specifically hypomethylated, and thus, transcriptionally active, in tTregs, but hypermethylated in naive or effector T cells38, 39. This region has been widely used to distinguish bona fide tTreg cells from those conventional T cells transiently upregulating Foxp3 expression and iTregs38, 39.
Three major post-translational modifications have been described for the Foxp3 protein: acetylation, phosphorylation and ubiquitination. These modifications affect Foxp3 protein stability and DNA binding capacities, thus modulating Treg cell function and the development of autoimmunity. Acetylation in specific lysine (Lys) residues by Lys-acetyltransferases globally stabilizes Foxp3 expression and promotes Treg cell function by favoring Foxp3 binding to its transcriptional targets40 and avoiding proteasomal degradation40. Several acetylases and deacetylases have been shown to interact with Foxp3 and control its acetylation, such as TIP6041 and p30040, 42. Other post-translational processes include phosphorylation at Serine (Ser) and Threonine (Thr) residues by several kinases, including PIM-1, PIM-2 and CDK243, 44, 45, 46, and ubiquitination at Lys residues, that targets Foxp3 for proteasomal degradation47. Inflammatory stimuli result in proteasomal-dependent degradation of Foxp3 mediated by the ubiquitinase Stub1, which binds to Foxp3 and promotes its K48-linked ubiquitination48. Overexpression of Stub1 abrogates Treg cell suppressive capacity in vitro and in vivo and confers Treg cells with a TH1-like phenotype, posing the question of whether the T effector-like Treg cell phenotype in this setting is an intermediate stage towards instability and loss of Foxp3 protein expression. In contrast, the deubiquitinating enzyme USP7 is highly expressed in Treg cells and is associated with Foxp3 in the nucleus, regulating Foxp3 turnover. Under inflammatory conditions this enzyme is downregulated, facilitating Foxp3 degradation40. Furthermore, conditional deletion of USP7 on Treg cells leads to lethal autoimmunity, with decreased numbers of Treg cells in the periphery that display an aberrant TH1-like phenotype in vitro and in vivo49. USP7 targeting decreased recruitment of acetyltransferase TIP60 to the CNS2 region of Foxp3. TIP60 promotes acetylation-dependent dimerization of Foxp350, 51 and in the absence of TIP60 there is lethal autoimmunity52. Other factors have also been shown to control Foxp3 expression, such as HIF-1α, which is induced by IL-6 and TCR stimulation and inhibits Foxp3 through ubiquitination53.
Both ubiquitination and acetylation target Lys residues, so they may compete for regulating Foxp3 expression. In fact, hyper-acetylation of Foxp3 prevents polyubiquitination and proteasomal degradation, increasing Foxp3 stability40. These data suggest that post-translational modifications of Foxp3 play a crucial role in modulating Treg cell plasticity or instability, adding a new layer of complexity to the regulation of the Treg cell functional program with potential consequences for the development of autoimmunity.
Foxp3 works in concert with other transcription factors and proteins, forming multiprotein complexes that determine the transcriptional signature and effector functions of Treg cells. Hundreds of protein partners have been described, including transcription factors Gata-3, NFAT, Runx1, Eos and others54. Global analysis of the Foxp3 interaction network suggests a model in which Foxp3 and its binding partners form multi-protein complexes that bind to pre-existing DNA enhancers55 and regulate transcription positively or negatively depending on the recruited interaction proteins56. As previously mentioned, Foxp3 also associates with proteins that mediate epigenetic modifications, such as TIP60, Sirtuin 1, or HDAC741, 57, which alter the acetylation state of partner loci and Foxp3 itself, with consequences for transcription factor binding, and histone modifications. In this regard, disruption of Foxp3 interactions with specific proteins diminishes Treg cell function and leads to autoimmune responses due to increased Foxp3 polyubiquitylation58, 59. Other evidence that supports the importance of Foxp3 interaction partners for Treg cell function comes from the observation that different Foxp3 mutations result in a wide range of IPEX disease severity, reflecting the relative importance of the affected residues in the integrity of the Foxp3 protein and the protein partners that form the DNA-binding complexes. For example, the most common mutation in IPEX, p.A384T, which disrupts the sequence specificity of Foxp3 DNA binding and alters Foxp3 interactions with specific targets genes31, inhibits Treg cell function but preserves the capacity of Treg cells to repress inflammatory cytokine production, due in part to a specific inhibition of Foxp3 interaction with TIP60 (ref. 60). Furthermore, experiments in mice that contained this mutation specifically in Treg cells perturbed Foxp3 binding to specific target genes including Batf, which was partly responsible for the induction of a unique pattern of tissue-restricted inflammation in certain non-lymphoid tissues due to defective function of these Treg cells61. Additionally, a Foxp3 reporter mouse that expresses an NH2-terminal EGFP-Foxp3 fusion protein, which disrupts the interaction of Foxp3 with many cofactors including TIP60, p300 and Eos, does not experience apparent autoimmunity, but its Treg cells display alterations in function in vivo, with autoimmune-prone NOD mice developing diabetes faster than their wild-type counterparts58. Interestingly, these same Treg cells are more potent suppressors of antibody-mediated arthritis due to a preferential interaction of EGFP-Foxp3 with IRF462. These findings demonstrate that not only are certain cofactors crucial for Treg cell-mediated function, but they suggest that Treg cells might be ‘tuned’ to control particular types of inflammation by modulating the constituents of the Foxp3 protein complexes under specific environmental conditions. Furthermore, it is tempting to speculate that the variety of functions that Treg cells perform in different tissue environments could be accompanied by the formation of specific multi-protein complexes with tissue-specific proteins that would cooperate with Foxp3 in performing Treg cell function in specific environments.
Treg cell instability.
Both the expression and stability of Foxp3 play crucial roles in the maintenance of Treg cell function63. Thus, genetic deletion of a conditional Foxp3 allele in mature Treg cells results in effector T cells that are capable of causing inflammatory tissue lesions64. Although the instability of Foxp3 in iTreg cells has been widely observed and it is intrinsic to their developmental origin8, 9, tTreg cells have been investigated to determine how instability of Foxp3 expression under basal or inflammatory conditions in specific tissues impacts the development and resolution of autoimmunity65, 66, 67. Loss of Foxp3 expression by tTreg cells had been previously observed in vitro68, 69, in adoptive transfers into lymphopenic hosts70, in infection settings71 and graft-versus-host disease72. A fate-mapping mouse model where the YFP reporter marks all cells that at any time expressed Foxp3 in both homeostatic and autoimmune inflammatory conditions has been generated. A small percentage of apparently stable Treg cells lost Foxp3 expression and acquired an effector-memory phenotype with secretion of different levels of the pro-inflammatory cytokines IFN-γ and IL-1767. These ‘exFoxp3’ cells were able to induce autoimmunity in an adoptive transfer model in the NOD background and consisted of a mixed population based on the level of TSDR demethylation, suggesting that not all exFoxp3 in this model might have once been de facto tTregs. More recent data suggests that this fate-mapping mouse model was detecting a percentage of Foxp3 tTreg cells that were either upregulating Foxp3 transiently, or were not fully committed to the tTreg cell lineage73. Further data supporting tTreg cell instability shows that Foxp3 expression is lost on MOG38–49-specific tTreg cells during experimental autoimmune encephalitis (EAE) development, with increased frequency of exFoxp3 in the central nervous system at the preclinical and peak stages of EAE that decreases during EAE resolution. These exFoxp3 Treg cells expressed IFN-γ and were able to transfer EAE74. It remains to be determined whether the decrease in exFoxp3 during disease resolution was due to re-acquisition of Foxp3 expression by exFoxp3 cells.
Another genetic fate-mapping mouse model provided evidence that the majority of mature tTreg cells in spleen and lymph nodes are relatively stable under homeostatic conditions66. This model, based on inducible labeling of Foxp3+ cells upon tamoxifen treatment, marks all those cells that express Foxp3 at the moment of tamoxifen administration, and, in contrast to continuous labeling67, prevents cells transiently expressing Foxp3 to be detected. Although stable under homeostatic conditions, with growth factor deprivation or IL-2 receptor blockade which induces autoimmunity75, mature tTreg cells significantly decreased the level of Foxp3 expression per cell, and a small population lost Foxp3 expression completely. However, they did not produce pro-inflammatory cytokines66, suggesting some degree of instability under specific environmental settings. This apparent discrepancy might arise from the different fate-mapping mouse models and the type of labeling of Foxp3 cells, which could lead to the labeling of non-committed Treg cells67, or the absence of labelling of exFoxp3 appearing before tamoxifen administration66. Discordant results could also depend on the inflammatory stimuli utilized to test Foxp3 stability. Regardless, in both fate-mapping models there is a small population of tTreg cells that loses Foxp3 expression, and those Treg cells that remain ‘stable’ display diminished Foxp3 expression at the single cell level66. As decreased levels of Foxp3 in Treg cells isolated from inflammatory sites have been observed in mouse models of autoimmunity63, 76 and in patients with autoimmune diseases24, 77, 78, 79, 80, 81, further works with these models are warranted in order to examine the mechanisms and consequences of long-term decreases in Treg cell Foxp3 expression. Further data has confirmed the observation that most mature tTreg cells are stable under steady-state conditions with a new fate-mapping mouse model where Foxp3 lacks CNS1, but they become unstable when stimulated in vitro and in vivo in a model of EAE, losing Foxp3 expression and acquiring TH1- and TFH cell-like features73. While epigenetic changes such as re-methylation of the CNS2 region could account for the loss of Foxp3 expression in these settings67, 73, the molecular mechanisms that are responsible for the decrease in Foxp3 protein and the potential contribution of post-translational modifications of Foxp3 protein on exFoxp3 cell generation remain to be explored.
Finally, depletion of specific Foxp3 protein partners can also precipitate the appearance of exFoxp3 cells. For instance, Treg cell-specific deletion of the chaperone GP96 in the NOD background led to lethal autoimmunity due to defective Treg cell suppressive capacities in models of diabetes and colitis. In this system Treg cells progressively lose Foxp3 expression and gain IFN-γ secretion, although they maintain their specific TSDR demethylation pattern59. Mice with a Treg cell-specific deletion of the transcription factor Helios develop systemic autoimmune pathology, characterized by increased germinal center formation, lymphocytic infiltration into non-lymphoid organs and glomerulonephritis82. Although Helios does not form protein complexes with Foxp3 nor does it bind to the Foxp3 locus82, Helios-deficient Treg cells express increased IFN-γ and IL-17 and are unstable, with decreased expression of Foxp3 and a tendency to completely lose its expression 82, 83. Treg cells deficient in another transcription factor – Eos – exhibit increased expression of IL-2 and IFN-γ along with reduced suppressive capacity, while forced overexpression of Eos in Treg cells prevents Treg cell instability, even in inflammatory environments. EosloFoxp3+ tTreg cells are detectable in vivo and have regulatory function, with the ability to acquire TH-like effector characteristics while maintaining Foxp3 expression, however such cells exhibit specific changes in the global DNA methylation pattern84.
Regulatory T cell plasticity.
Plasticity is a property inherent to most, if not all, immune cells, which allows them to adapt their phenotype and function to the changing environment and extracellular ‘danger’ signals. Thus it is not surprising that Treg cells possess some degree of plasticity. Treg cells have the capacity to acquire features specific to the type of immune response they control, mostly driven by ‘master’ transcription factors and regulated by environmental signals. Thus, Treg cells acquire T-bet expression to restrain type 1 inflammation during infection85, 86, and utilize IRF4 and STAT3 to inhibit TH2 (Ref.87) and TH17 (Ref. 88) responses, respectively. While this modality of plasticity seems to be advantageous for the host and beneficial for the outcome of the immune response, aberrant plasticity of Treg cells is also observed in several autoimmune diseases, with Treg cells expressing pro-inflammatory cytokines, acquiring T helper-like phenotypes and displaying diminished function in most cases but maintaining Foxp3 expression levels21, 83, 89, 90, 91, 92, 93, 94, 95. Paradoxically, these TH-like Treg cells utilize the same transcription factors used by Treg cells to inhibit specific types of immune responses. Therefore, IFN-γ secretion by TH1-like Treg cells requires T-bet expression21, 91, 92, 93, while IL-6-driven Th17-like Treg cells require STAT3 for IL-17 secretion72, and IL-4-driven TH2-like Treg cells upregulate IRF4 and Gata-395, 96. In most instances, TH-like Treg cells have a demethylated TSDR Foxp3 locus even though they share effector features, suggesting that their phenotype might be reversible21, 90, 93. They display alterations in the epigenetic signature characteristic of Treg cells9, 32, 34, 97, which may be the underlying mechanism that allows for pro-inflammatory cytokine secretion. Current efforts focus on understanding the signaling pathways that drive this plasticity in specific autoimmune diseases, to harness this flexibility on the treatment of human disease92 and the role of Treg cell plasticity in autoimmune-related tissues.
TH1-like Treg cells
Perhaps the best characterized tTreg cell plasticity event is the acquisition of TH1-like features. In mouse models and patients with autoimmune diseases such as type 1 diabetes93, multiple sclerosis21, 91, autoimmune hepatitis98 and Sjogren syndrome99, there is an increased frequency of IFN-γ+Foxp3+ tTreg cells in the periphery that display reduced suppressive capacities as compared to Treg cells from healthy age-matched subjects. TH1-like Treg cells upregulate the transcription factor T-bet and other TH1 markers, such as CCR5 and CXCR3. Furthermore, in the Apoe–/– mouse model of atherosclerosis, which shares a number of pathogenic similarities with autoimmune disorders, there is accumulation of IFN-γ-producing TH1-like Treg cells in the aorta that display altered suppressive capacities in vitro and in vivo90. Using an in vitro model of TH1-like Treg cell generation using IL-12, it has been demonstrated that TH1-like Treg cells possess an activated PI(3)K-Akt-FoxO pathway, which is partly responsible for the secretion of IFN-γ and decreased suppressive capacity91. Interestingly, Treg cells isolated from patients with relapsing-remitting multiple sclerosis (RRMS) also display an activated PI(3)K-AKT-FoxO pathway ex vivo and their suppressive capacity is corrected upon PI(3)K pathway blockade94. In vivo, PI(3)K-Akt activation by Treg cell-specific deletion of the PI(3)K phosphatase PTEN provokes a type 1 autoimmune disorder, with Treg cells downregulating the expression of CD25 and Foxp3 and displaying reduced functionality100, 101. Moreover, FoxO itself has been implicated in the regulation of Treg cell plasticity. Mice with a Treg cell-specific deletion of FoxO succumb to lethal autoimmunity similar to that observed in scurfy mice94, with Treg cells displaying a TH1-like Treg cell phenotype and being unable to prevent disease in a colitis model. IFN-γ seemed to be involved in Treg cell defective function, as Foxo1–/–Ifng–/– mice partially recovered from the wasting syndrome94.
Transcriptomic analysis of IFN-γ+ TH1-like Treg cells at the population91 or single cell90 levels demonstrates that they exhibit reduced expression of immunosuppressive genes as compared to Treg cells and have altered expression of costimulatory molecules, migratory properties and specific signaling pathways. It is unknown whether TH1-like Treg cells play a role in disease pathogenesis/protection in specific tissues. The presence of TH1-like Treg cells has been observed in MOG-specific Treg cells infiltrating the central nervous system of mice during EAE development. These Treg cells were not capable of suppressing central nervous system-infiltrating MOG-specific effector T cells and prevent disease onset, secreting IFN-γ at the onset and peak of disease but with a reduced frequency during the recovery phase102. In contrast, T-bet+Foxp3+ cells in pancreatic tissue seem to be protective in a mouse model of type 1 diabetes103. Lastly, while most work on TH1-like Treg cells suggest that IL-12 and/or type 1 cytokines are inducing the phenotype, the fact that the PI(3)K-Akt is involved in their generation, which is a major pathway integrating diverse environmental signals into cell function, suggests that other environmental cues could have the ability to induce Treg cell plasticity, as it has been observed, for example, with increased dietary salt concentrations104.
TH17-like Treg cells
A small proportion of human peripheral Treg cells produce IL-17 in healthy individuals and upregulate RORC2 (TH17-like Treg cells) ex vivo105 while conserving suppressive capacities105, 106. Considering the well-established developmental relationship between Treg cells and TH17 cells, it remains to be determined whether TH17-like Treg cells are a transient stage in the de-differentiation of tTreg cells into Th17 cells as it has been suggested69, 107. In support of this, conversion of Foxp3+ Treg cells into TH17 cells has a crucial role in the pathogenesis of autoimmune arthritis in a collagen-induced arthritis mouse model. This conversion is driven by IL-1β109 and IL-6, and Foxp3+IL-17+ Treg cells are observed in the synovium of subjects with active rheumatoid arthritis108. Moreover, conversion of Treg cells into TH17 cells has also been reported in the CD18hypo PL/J mouse model of psoriasis109, and TH17-like Treg cells have been observed in skin tissue of psoriatic patients89.
Perhaps the tissue where TH17-like Treg cells have been best identified is the gastrointestinal tract. Lamina propria appears enriched in iTreg cells that express high levels of TH17-defining transcription factor RORγt. These TH17-like Treg cells appear to have a positive function, as their absence exacerbates pathogenesis in several models of mucosal autoimmunity110, 111. Furthermore, recent studies have suggested that Foxp3+RORγt+ Treg cells control glomerulonephritis112, and lack of TH17-like Treg cells results in increased SLE-associated mortality and organ pathology113. Besides the importance of IL-1β and IL-6 on promoting IL-17 secretion by Treg cells114, 115, other environmental factors have been shown to modulate TH17-like Treg cell conversion either indirectly or directly, including indoleamine 2,3-dioxygenase (IDO)116, Toll-like receptor 2 (TLR2) ligation117, and certain infections118.
TH2-like Tregs
Patients with systemic sclerosis display an increased frequency of TH2-like Treg cells in skin but not in peripheral blood, characterized by the secretion of IL-4 and IL-13 and upregulation of Gata-3 and IRF-4. Peripheral Treg cells from these patients express high levels of ST2, the receptor for the alarmin IL-33. This cytokine is enriched in the skin, suggesting that it might play a role in the reprogramming of Treg cells into a TH2-like phenotype95. TH2-like Treg cells have also been observed in allergy-susceptible mutant mice (Il4raF709), and in peripheral blood Treg cells from food-allergic patients. TH2-like Treg cells secrete IL-4 and/or IL-13 and upregulate transcription factors IRF-4 and Gata-396. Moreover, mice with Treg cell-restricted deletion of the ubiquitin-ligase Itch show autoimmune features and Treg cells are not able to control TH2 inflammation. This defect is associated with the acquisition of a TH2-like phenotype by the Treg cells, with increased Gata-3 expression, STAT6 activation and secretion of IL-4119.
Many open questions remain to be answered in regards to Treg cell plasticity. Does Treg cell plasticity reflect initial heterogeneity of the Treg cell population, with only a specific subset of Treg cells being able to acquire effector-like properties65? Do TH-like Treg cells represent a transient stage on the path to becoming exFoxp3? Can we harness Treg cell plasticity to design new therapeutic strategies aimed at modulating Treg cell function in diverse disease settings? What is the role of TH-like Treg cells in autoimmune tissues during disease development and progression?
Tissue-resident Treg cells
One of the major recent advances in the Treg cell field is the discovery that Treg cells populate specific tissues in the body during physiological and stress conditions. The characterization of these populations and their mechanisms of action will have important implications to our understanding of the development, maintenance and resolution of autoimmunity in specific organs. Tissue-resident Treg cells actively perform non-immunological functions and work at maintaining tissue homeostasis and wound repair, roles that could be important for tissue homeostasis in autoimmunity settings. A number of studies have begun to define the specific phenotype of tissue-resident Treg cells in muscle, skin, lung, gastrointestinal tract, liver and adipose tissue among others (reviewed in 28). In most cases, these Treg cells appear to be of thymic origin120, 121, 122, with considerable TCR repertoire oligoclonality, indicating that particular antigens in the tissue may be responsible for the accumulation of Treg cells in specific niches120, 122, 123. They are characterized by the expression of tissue-specific transcription factors and mediators that drive the function of other cells in that tissue, supporting the notion that the microenvironment exerts important effects on the phenotype of Treg cells in a given location. The molecular mechanisms by which tissue-resident Treg cells acquire their tissue-specific program have only begun to be explored. Epigenetic analysis of mouse Treg cells isolated from different tissues and compared to lymph node Treg cells have demonstrated that tissue Treg cells undergo extensive epigenetic reprograming that is globally tissue-specific, but there is a common tissue Treg cell population characterized by the expression of KLRG1 and ST2124.
The best studied tissue-specific Treg cell population is that resident in visceral adipose tissue (VAT). VAT Treg cells exert important roles in defending against associated metabolic disorders. In mice, lean fat tissue is populated by T cells with a TH2 phenotype and enriched in Treg cells, that maintain the predominance of resident anti-inflammatory macrophages. Treg cells accumulate over time and acquire a TH2-like phenotype, with expression of Gata-3, BATF, IRF4, CCR4 and IL-10121, and PPARγ, a transcription factor that controls adipocyte differentiation and mediates VAT Treg cell accumulation, phenotype and function121, 125. Treg cell-specific depletion of PPARγ results in a specific loss of VAT Treg cells, demonstrating an important role of this transcription factor in the development and/or maintenance of VAT Treg cells121. VAT Treg cell frequency diminishes with age in obese mice, suggesting their role in modulating obesity-associated fat tissue inflammation123. Given that obesity is an established risk factor in autoimmune diseases, it will be important to define the features and function of VAT Treg cells in the development of autoimmunity.
Similarly, lung and muscle Treg cells express the epidermal growth factor receptor (EGFR) ligand amphiregulin to promote tissue repair. Muscle Treg cells accumulate in acutely injured skeletal muscle in mouse models of muscle injury and muscular dystrophy, where they control muscle inflammation upon injury and promote tissue repair by acting on immune and non-immune cells27, 120. Data in a zebrafish model further supports the dual role of Treg cells in immune regulation and tissue regeneration observed in mice. Zebrafish Treg-like cells126 rapidly migrate to damaged organs in models or spinal cord, heart and retina regeneration and their function is dependent on the secretion of organ-specific regenerative factors. Conditional ablation of Treg cells inhibited organ regeneration127.
Treg cells populate healthy skin and help maintain homeostasis. In healthy individuals and rodents, skin Treg cells possess an effector-memory phenotype, with expression of Treg cell-specific surface markers and higher proliferative capacity ex vivo as compared to their blood counterparts. They express IL-17 and IL-10 and display a demethylated TSDR region, suggesting that they are tTregs122. Interestingly, Treg cells reside in close apposition to hair follicles, where they contribute to hair follicle stem cell activation by promoting their proliferation and differentiation through the Notch pathway. Ablation of Treg cells completely abrogates the ability of the hair follicle to regenerate, implicating Treg cells as a key component of the skin stem cell niche128.
Maintenance of local tissue homeostasis is of particular importance in the intestinal mucosa, where the immune system must be able to effectively discriminate between pathogens and dietary factors or commensal flora. Accordingly, deregulated immune responses against luminal flora in genetically susceptible individuals are generally recognized as key factors in the pathogenesis of Inflammatory Bowel Disease (IBD). While the role of commensal flora in inducing Treg cells has been well characterized, the dynamics and characterization of Treg cells in the mucosal tissue has only begun to be appreciated. Thus, lamina propria appears enriched in Treg cells that express high levels of RORγt110, 129, and although most of them are not tTregs, they seem to have a positive function in maintaining tissue homestasis, and their absence exacerbates pathogenesis in several models of mucosal autoimmunity.
How do Treg cells maintain tissue integrity and what is the involvement of tissue-specific Treg cells in autoimmune diseases? One could speculate that defects in specific tissue-resident Treg cells would be involved in either the development, maintenance of resolution of autoimmune disorders affecting that specific tissue, e.g. Is there a specific role for muscle-resident Treg cells in the pathology of myasthenia gravis, or for VAT Treg cells in autoimmune disorders where obesity is an important risk factor, or for skin Treg cells in psoriasis? Lastly, while most studies performed so far have been done in mice, and although there are intrinsic difficulties with translating these observations to humans, it will be crucial to examine the phenotype and function of tissue-specific Treg cells in healthy individuals and patients with autoimmune diseases, to understand the role of tissue-resident Treg cells on the development and maintenance of human autoimmunity.
Conclusions.
Intense research in the Treg cell field has improved our knowledge of their biology and has provided evidence of the existence of instability and plasticity within the Treg cell compartment. Accumulating data have shown the presence of exFoxp3 cells and TH-like Treg cells in a number of autoimmune pathologies over the last years. Current efforts focus on determining the molecular mechanisms responsible for the induction of each of these functional states in the periphery and autoimmune organs as well as the environmental triggers that drive them. The ultimate goal of these studies is to harness Treg cell instability and plasticity mechanisms to modulate Treg cell function not only in autoimmune pathologies, but also in other diseases such as cancer. With regards to tissue-resident Treg cells, although we have begun to understand the mechanisms and factors that control their function in tissues, many questions remain to be answered mostly related to the mechanisms that tissue Treg cells employ to maintain tissue integrity in autoimmune diseases and the characterization of tissue-resident Treg cells in humans (Box 2). Answers to these questions will undoubtedly improve the design of Treg cell-specific therapeutic options for patients with autoimmune diseases.
Box 2. Treg cell-specific therapies for autoimmune diseases.
One of the many therapeutic strategies being investigated in the autoimmunity field is the restoration of peripheral tolerance, and one explored avenue has been the development of Treg cell-based therapeutic strategies. Interesting results have resulted from immune therapies based on the adoptive transfer of polyclonally expanded Treg cells in patients with autoimmune diseases such as type 1 diabetes, cutaneous lupus erythematosus and Crohn’s disease131, 132, 133, 134. Furthermore, with the development of technologies to engineer specific TCR to immune cells such as chimeric antigen-receptors (CARs135), future approaches will potentially achieve optimal numbers of Treg cells with TCR specific for disease-relevant antigens for adoptive transfers to patients with autoimmunity in those cases where the autoantigens are known. Although it is still early to know the long-term consequences of these therapies, the first results are encouraging and promise to adopt these technologies for other autoimmune diseases.
Another potential therapeutic approach to peripheral tolerance restoration is targeting specific molecules and/or pathways that are dysfunctional in the existing pool of Treg cells in patients. For this approach, it will be of importance to determine the physiological relevance and molecular mechanisms driving plasticity and instability of Treg cells in patients, as well as to improve our knowledge on how dysfunction of tissue-specific Treg cells affect the development of certain autoimmune diseases. In this respect, there are many questions still unsolved (see main text and Box 3) that will undoubtedly improve the design of Treg cell-specific therapeutic options for patients with autoimmune diseases.
Box 3. Unanswered questions.
Discovery of markers to distinguish tTreg cells from iTregs.
What are the molecular mechanisms and environmental triggers responsible for exFoxp3 cell generation in autoimmunity?
What are the detailed molecular mechanisms and environmental cues that drive Treg cell plasticity into TH1-, TH22- and TH17-like Treg cell phenotypes in specific autoimmune settings?
What is the contribution of the Treg cell epigenetic signature to Treg cell instability and plasticity in autoimmunity?
What is the epigenetic and phenotypic signature of human Treg cells in specific tissues, and how do they contribute to disease pathology?
Does Treg cell plasticity reflect the initial heterogeneity of the Treg cell population, with only a specific subset of Treg cells being able to acquire effector-like properties?
What is the role of Treg cell plasticity and instability in autoimmune tissues during disease development, progression and resolution of disease?
What are the mechanisms that Treg cells utilize to maintain tissue integrity and what is the involvement of tissue-specific Treg cells in tissue damage in autoimmunity?
References.
- 1.Fontenot JD, Gavin MA & Rudensky AY Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol 4, 330–336 (2003). [DOI] [PubMed] [Google Scholar]
- 2.Hori S, Nomura T & Sakaguchi S Control of regulatory T cell development by the transcription factor Foxp3. Science 299, 1057–1061 (2003). [DOI] [PubMed] [Google Scholar]
- 3.Khattri R, Cox T, Yasayko SA & Ramsdell F An essential role for Scurfin in CD4+CD25+ T regulatory cells. Nat Immunol 4, 337–342 (2003). [DOI] [PubMed] [Google Scholar]
- 4.Sakaguchi S, Sakaguchi N, Asano M, Itoh M & Toda M Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol 155, 1151–1164 (1995). [PubMed] [Google Scholar]
- 5.Liu W et al. CD127 expression inversely correlates with FoxP3 and suppressive function of human CD4+ T reg cells. J Exp Med 203, 1701–1711 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sakaguchi S, Miyara M, Costantino CM & Hafler DA FOXP3+ regulatory T cells in the human immune system. Nat Rev Immunol 10, 490–500 (2010). [DOI] [PubMed] [Google Scholar]
- 7.Josefowicz SZ, Lu LF & Rudensky AY Regulatory T cells: mechanisms of differentiation and function. Annu Rev Immunol 30, 531–564 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kanamori M, Nakatsukasa H, Okada M, Lu Q & Yoshimura A Induced Regulatory T Cells: Their Development, Stability, and Applications. Trends Immunol 37, 803–811 (2016). [DOI] [PubMed] [Google Scholar]
- 9.Ohkura N et al. T cell receptor stimulation-induced epigenetic changes and Foxp3 expression are independent and complementary events required for Treg cell development. Immunity 37, 785–799 (2012). [DOI] [PubMed] [Google Scholar]
- 10.Nishizuka Y & Sakakura T Thymus and reproduction: sex-linked dysgenesia of the gonad after neonatal thymectomy in mice. Science 166, 753–755 (1969). [DOI] [PubMed] [Google Scholar]
- 11.Penhale WJ, Farmer A, McKenna RP & Irvine WJ Spontaneous thyroiditis in thymectomized and irradiated Wistar rats. Clin Exp Immunol 15, 225–236 (1973). [PMC free article] [PubMed] [Google Scholar]
- 12.Sakaguchi S, Takahashi T & Nishizuka Y Study on cellular events in postthymectomy autoimmune oophoritis in mice. I. Requirement of Lyt-1 effector cells for oocytes damage after adoptive transfer. J Exp Med 156, 1565–1576 (1982). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sakaguchi S, Fukuma K, Kuribayashi K & Masuda T Organ-specific autoimmune diseases induced in mice by elimination of T cell subset. I. Evidence for the active participation of T cells in natural self-tolerance; deficit of a T cell subset as a possible cause of autoimmune disease. J Exp Med 161, 72–87 (1985). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Baecher-Allan C, Brown JA, Freeman GJ & Hafler DA CD4+CD25high regulatory cells in human peripheral blood. J Immunol 167, 1245–1253 (2001). [DOI] [PubMed] [Google Scholar]
- 15.Stephens LA, Mottet C, Mason D & Powrie F H