
one-carbon(1C) 대사는
엽산(THF)을 보조인자로 사용해
단일 탄소 단위를 이동·가공하면서
핵산, 메티오닌/SAM, 글리신·세린, NADPH 등을 공급하고,
특히 미토콘드리아–세포질 간 구획화를 통해 에너지·적색/에피게놈 조절까지 관장하는 대사 네트워크입니다.
| Methyl (CH₃) and formyl (CHO) groups are crucial one-carbon units in one-carbon metabolism, a central pathway using tetrahydrofolate (THF) and Vitamin B12 to transfer single carbon atoms for synthesizing DNA (nucleotides), amino acids, and epigenetic regulators like SAM. Formyl groups (from serine, histidine) and methyl groups (from methionine) are vital for purine/thymidylate synthesis and DNA methylation, linking nutrition (folate, B12) to cell growth, replication, and health, with disruptions implicated in cancer and aging. Key Components & Roles
|

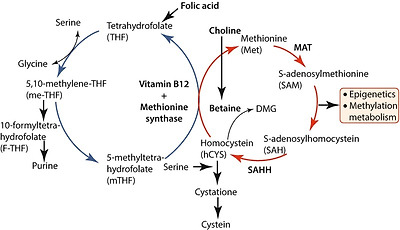

참고) 3 탄소 구조 피루브산
--> 피루브산 산화반응을 거쳐 2탄소 구조 Acetyl-CoA가 되는 과정

참고) 아세틸-CoA(Acetyl-CoA)가 1탄소 구조가 되는 과정
아세틸-CoA(Acetyl-CoA)가 1탄소 구조가 되는 과정.
이는 생화학에서 매우 중요하면서도 흥미로운 주제.
아세틸-CoA는
두 개의 탄소 원자를 포함하는 아세틸기(CH₃CO-)를 가지고 있으며,
주로 탄수화물, 지방, 단백질 대사의 중심에서 에너지 생산을 위해 시트르산 회로(TCA 회로)로 들어가는 역할.
아세틸-CoA의 탄소가 궁극적으로 1탄소(C1) 구조,
즉 이산화탄소(CO₂) 형태로 분해되는 과정은
주로 **시트르산 회로(TCA 회로)**를 통해 일어납니다.
아세틸-CoA의 탄소가 1탄소 구조(CO₂)로 전환되는 과정: 시트르산 회로
아세틸-CoA 자체가
직접 1탄소 분자인 CO₂로 분해되는 것은 아닙니다.
대신,
아세틸-CoA는 미토콘드리아 기질에서 일어나는 일련의 복잡하고 정교한 순환 반응인
시트르산 회로에 진입하여 단계적으로 산화됩니다.
1. 회로 진입 및 결합
아세틸-CoA는
옥살아세트산(oxaloacetate)이라는 4탄소 분자와 결합.
이 반응은
시트르산 생성 효소(citrate synthase)에 의해 촉매되며,
그 결과 6탄소 분자인 시트르산(citrate)이 생성.
이 시점에서
아세틸-CoA에서 유래한 2개의 탄소는 옥살아세트산의 탄소 골격에 통합.
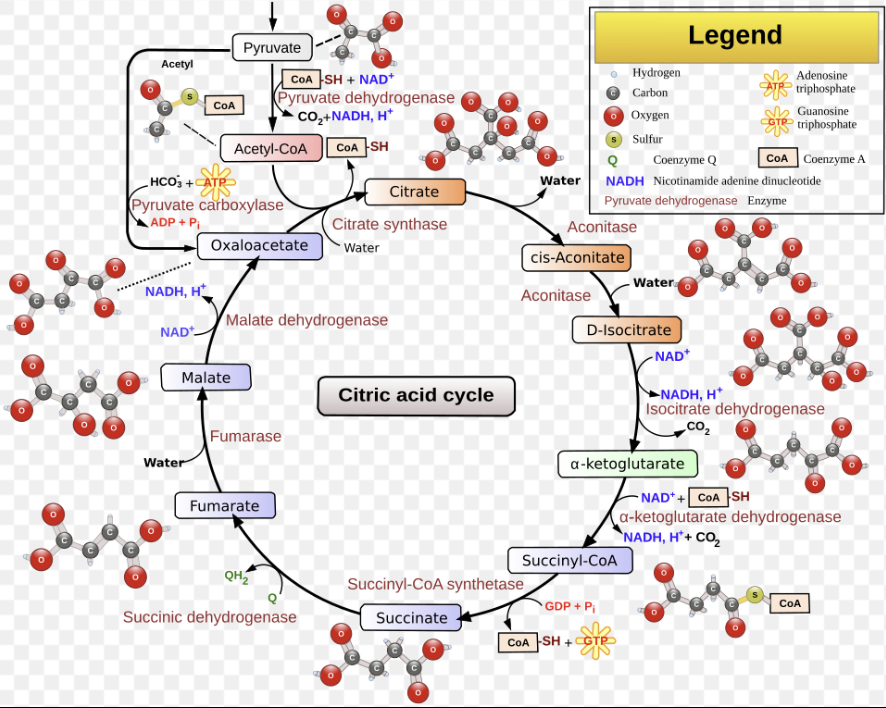
2. 단계적 산화 및 CO₂ 방출
시트르산은 일련의 효소 반응을 거치며 점진적으로 산화됩니다. 이 과정에서 두 번의 산화적 탈카르복실화(oxidative decarboxylation) 반응이 발생합니다.
첫 번째 CO₂ 방출:
6탄소 분자인 아이소시트르산(isocitrate)이 알파-케토글루타르산(alpha-ketoglutarate, 5탄소)으로 전환될 때,
첫 번째 CO₂ 분자가 방출.
두 번째 CO₂ 방출:
알파-케토글루타르산이 석시닐-CoA(succinyl-CoA, 4탄소)로 전환될 때,
두 번째 CO₂ 분자가 방출.
이 두 반응은
모두 탈수소효소 복합체(dehydrogenase complex)에 의해 촉매되며,
이 과정에서 고에너지 전자인 운반체(NADH)도 생성.
3. 탄소의 운명 (중요한 사실)
여기서 흥미로운 점은
아세틸-CoA가 회로에 진입할 때 기여한 두 개의 탄소가
회로의 첫 번째 순환에서 즉시 CO₂로 방출되는 것은 아니라는 사실.
아세틸-CoA의 탄소는
옥살아세트산의 탄소 골격에 합쳐진 후, 여러 단계를 거치며 분자의 대칭성이 바뀌기 때문에,
실제로 첫 번째 회전에서 방출되는 CO₂의 탄소는 원래 옥살아세트산에서 유래한 탄소들.
아세틸-CoA에서 유래한 탄소가 CO₂로 방출되기 위해서는
시트르산 회로가 몇 번 더 회전해야.
4. 회로의 재생성
나머지 4탄소 분자(석시닐-CoA부터 시작)는
최종적으로 옥살아세트산으로 재생성되며,
이 옥살아세트산은 새로운 아세틸-CoA 분자를 받아들여 회로를 다시 시작할 준비를 마칩니다.
결론적으로,
아세틸-CoA는 시트르산 회로라는 복잡하고 아름다운 생화학적 경로를 통해
궁극적으로 1탄소 구조인 이산화탄소(CO₂)로 완전히 산화.
이 과정에서 방출된 에너지는
NADH, FADH₂, GTP(또는 ATP) 같은 고에너지 운반체에 저장되며,
이들은 이후 전자 전달계를 통해 세포 활동에 필요한 막대한 양의 ATP를 생성하는 데 사용.
이처럼 아세틸-CoA의 탄소는
세포 호흡이라는 거대한 여정을 통해 생명체가 에너지를 얻는 핵심적인 원료로 활용되며,
그 마지막은 1탄소 폐기물인 CO₂ 형태로 배출되는 것
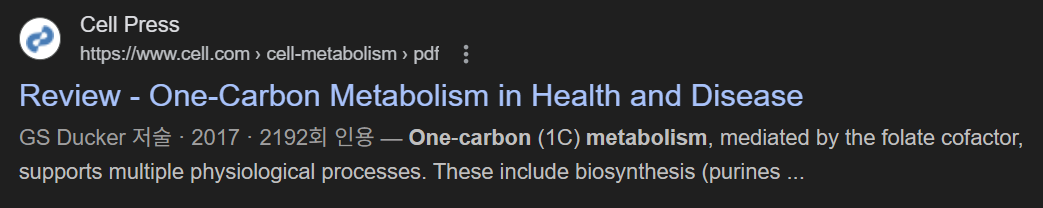
기본 구조: 폴레이트·메티오닌 사이클
폴레이트(THF)는
세린·글리신·콜린 유래 1C 단위를 받아
5,10-methylene‑THF, 5‑methyl‑THF, 10‑formyl‑THF 등으로 산화 상태를 바꾸며
핵산 합성·메티오닌 재메틸화·폼산 생산에 쓰입니다.
메티오닌 사이클에서 5‑methyl‑THF는
호모시스테인을 메틸화해 메티오닌을 만들고,
이는 SAM으로 활성화되어
DNA/히스톤/지질 메틸화 등 에피제네틱 및 생합성 반응의 메틸 공여체로 작동합니다.
| 카르복실기(-COOH)는 중심 탄소(C) 원자에 산소(O) 원자가 이중 결합(C=O)으로 연결되고, 또 다른 산소 원자에는 수소(H) 원자가 단일 결합(C-OH)으로 연결된 형태의 유기 작용기로, 카보닐기(C=O)와 하이드록실기(-OH)가 결합된 구조이며 산성을 띠는 특징이 있습니다. 카복실기의 구조는 그림에서처럼 중심의 탄소 원자에 하나의 산소 원자가 이중 결합으로 연결되어 있고, 하나의 하이드록실기가 단일 결합으로 연결. 분자식에서 보면, 카복실기는 –C=O–의 형태의 카보닐기에 하이드록실기가 붙어있는 형태. 산소와 탄소 사이의 전기음성도의 차이 때문에 탄소 원자는 부분적으로 양전하를 띤다 카복실기는 중심 탄소 원자에 하나의 최외각 전자를 지니고 있기 때문에, 보다 큰 분자와 결합. 하지만, 기본적으로 3개의 결합을 필요로 하는 카복실기는 탄소 사슬의 끝부분에만 위치할 수 있다. 또한 아세트산이 수소 이온을 방출해서 아세테이트가 되는 것처럼 수소이온(H+)을 내놓고 산으로 작용할 수 있다. 용액 및 결정 형태에서 카복실산은 종종 이합체로 존재하며, 여기서 두 개의 서로 다른 분자의 카보닐 산소와 하이드록실 산소는 하이드록실 양성자와 수소 결합을 형성. 쌍의 형성은 이합체당 2개의 수소 결합 형성을 허용. 카복실산과 아미노산에 존재하는 카복실기는 자연에서 가장 자주 생성되는 작용기 중 하나 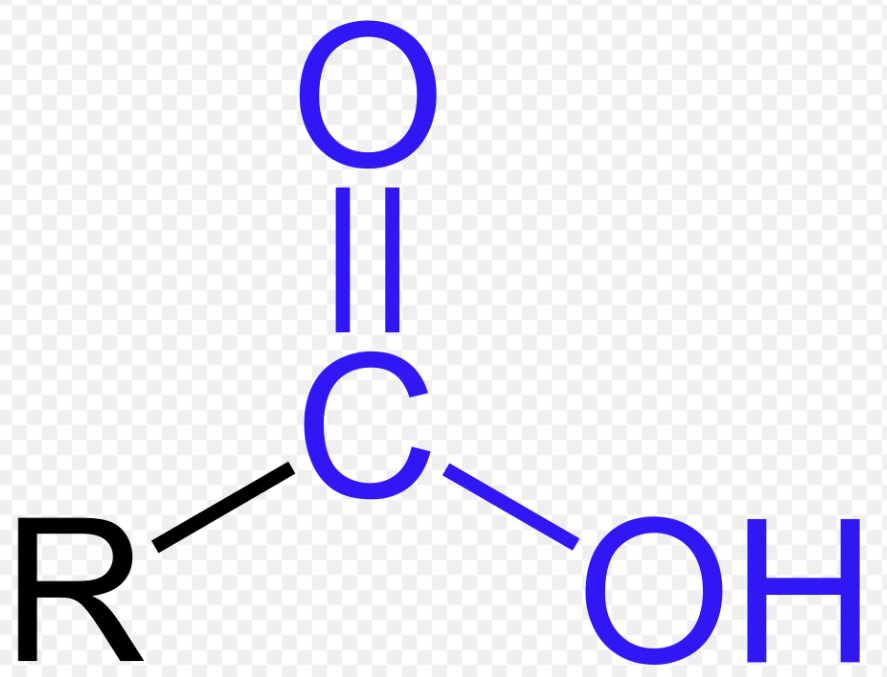 구조적 특징 구성 원자: 탄소(C), 산소(O), 수소(H). 결합 형태: 탄소에 이중 결합한 산소(카보닐기)와 단일 결합한 수산화기(-OH)가 함께 존재합니다. 화학식: R-COOH (R은 다른 탄소 사슬 또는 원자). 공명 구조: 탄소-산소 이중 결합과 단일 결합의 전자가 비편재화되어 공명 구조를 가지며, 이는 카르복실기의 산성 특성에 중요한 역할. 극성:  주요 역할 및 성질 산성: 물에 녹아 수소 이온(H+)을 내놓으며 산성을 띱니다 (예: 아세트산, 포름산). 수소 결합: -OH 그룹 때문에 분자 간 수소 결합이 가능하여 끓는점이 비교적 높습니다. 반응성: 에스터, 아마이드, 무수물 등 다양한 카르복실산 유도체로 쉽게 변환됩니다. |
구획화: 미토콘드리아 vs 세포질
세포질 MTHFD1, SHMT1는
세린·히스티딘·폼산에서 5,10‑methylene‑THF와 10‑formyl‑THF를 만들며,
주로 퓨린·티미딘 합성과 메티오닌 재메틸화를 담당합니다.
미토콘드리아 SHMT2·GCS·MTHFD2/2L·MTHFD1L 축은
세린·글리신·콜린 유래 메틸글리신을 5,10‑methylene‑THF→10‑formyl‑THF→폼산으로 산화하면서
NADH/NADPH를 생성하고,
폼산을 세포질로 수출해 1C 풀을 공급합니다.
주요 산물:
핵산, 아미노산, SAM, NADPH
10‑formyl‑THF는 퓨린 고리 2개의 탄소, 5,10‑methylene‑THF는
dUMP→dTMP 반응에 필요해 DNA·RNA 합성의 1C 수요를 충족합니다.
5‑methyl‑THF는
메티오닌·SAM을 통해 메틸화 반응과 인지질(포스파티딜콜린), 크레아틴, 폴리아민 합성에 1C를 제공하며,
SAM/SAH 비 조절을 통해 에피게놈을 세밀하게 제어합니다.
NADH·NADPH와 산화환원 조절
10‑formyl‑THF를 CO₂로 완전 산화하는 미토콘드리아 ALDH1L2 등은 NADPH를 생성해
미토콘드리아 ROS 방어와 지질 합성에 기여합니다.
미토콘드리아 MTHFD2계의 NADH 생성은
호흡사슬 상태와 연결되어 있으며,
호흡 사슬 이상 시 세린 합성·transsulfuration 경로 재프로그래밍과 1C 리모델링이 일어나
새로운 대사 취약점을 형성합니다.
발달·면역·성체 조직에서의 역할
배아·신경관 형성기에는
미토콘드리아 1C 경로(MTHFD1L, GCS 등)가 필수이며,
결함 시 neural tube defect와 치명적 발달장애가 발생하고
일부는 폼산·메티오닌 보충으로 부분 구제가 가능합니다.
조혈·T세포 활성 등 고증식 면역세포에서는
MTHFD2·SHMT2 축이 강하게 유도되어 퓨린·티미딘 공급 및 NADPH 기반 항산화 방어를 동시에 담당하며,
특정 억제로 선택적 면역 조절·항암 타깃 가능성이 제기됩니다.
암·대사질환에서의 1C 리와이어링
암세포는
세린/글리신 섭취와 de novo 합성을 크게 늘리고,
미토콘드리아 1C 효소(SHMT2, MTHFD2)의 발현을 높여 핵산·SAM·NADPH 공급을 극대화하며,
이 경로는 종양 예후 불량 및 약제 내성과 연관됩니다.
최근 리뷰들은
1C 대사가 종양 내 NADPH 항상성·ROS 저항·에피게놈 재프로그램에 핵심 역할을 하며,
MTHFD2·SHMT2 특이 억제제 등이
새로운 고표적성 항암 전략으로 개발 중임을 강조합니다.
전신 대사·신경퇴행에서의 최신 연구
폼산은
미토콘드리아 1C에서 생성되어 혈액을 통해 전신에 공급되는 순환 1C 캐리어로,
숙주–미생물 상호작용·영양상태·산증(메탄올 중독) 등과 연결되는 시스템 수준의
메타볼라이트로 부상하고 있습니다.
한편,
SAM/1C 사이클의 활성은
미토콘드리아 복합체 I 단백질 메틸화와 Fe‑S 클러스터 생합성을 통해 호흡능을 직접 제어하고,
최근 연구에서는
미토콘드리아 1C 플럭스 강화가
알츠하이머병 모델에서 미토콘드리아 기능 개선 및 신경 보호 효과를 보인다는 보고도 나왔습니다.
Cell Metab
. Author manuscript; available in PMC: 2018 Jan 10.
Published in final edited form as: Cell Metab. 2016 Sep 15;25(1):27–42. doi: 10.1016/j.cmet.2016.08.009
One-Carbon Metabolism in Health and Disease
Gregory S Ducker 1,2, Joshua D Rabinowitz 1,2,*
- Author information
- Article notes
- Copyright and License information
PMCID: PMC5353360 NIHMSID: NIHMS852041 PMID: 27641100
The publisher's version of this article is available at Cell Metab
Abstract
One-carbon (1C) metabolism, mediated by the folate cofactor, supports multiple physiological processes. These include biosynthesis (purines and thymidine), amino acid homeostasis (glycine, serine, and methionine), epigenetic maintenance, and redox defense. Both within eukaryotic cells and across organs, 1C metabolic reactions are compartmentalized. Here we review the fundamentals of mammalian 1C metabolism, including the pathways active in different compartments, cell types, and biological states. Emphasis is given to recent discoveries enabled by modern genetics, analytical chemistry, and isotope tracing. An emerging theme is the biological importance of mitochondrial 1C reactions, both for producing 1C units that are exported to the cytosol and for making additional products, including glycine and NADPH. Increased clarity regarding differential folate pathway usage in cancer, stem cells, development, and adult physiology is reviewed and highlights new opportunities for selective therapeutic intervention.
초록
엽산 보조인자에 의해 매개되는 일탄소(1C) 대사는
다양한 생리적 과정들을 지탱한다.
여기에는
생합성(퓨린 및 티미딘),
아미노산 항상성(글리신, 세린, 메티오닌),
후성유전적 유지, 그리고
산화환원 방어가 포함된다.
biosynthesis (purines and thymidine),
amino acid homeostasis (glycine, serine, and methionine),
epigenetic maintenance, and
redox defense
진핵세포 내부뿐만 아니라 기관 간에서도
1C 대사 반응은 구획화되어 있다.
본 종설에서는 포유류 1C 대사의 기초를 다루며,
서로 다른 세포 소기관, 세포 유형, 생물학적 상태에서 활성화되는 경로들을 포함한다.
특히
현대 유전학, 분석화학, 동위원소 추적 기법을 통해
가능해진 최근의 발견들에 초점을 맞춘다.
새롭게 부각되는 주제는
미토콘드리아 1C 반응의 생물학적 중요성으로,
이는 세포질로 수출되는 1C 단위를 생산할 뿐 아니라
글리신과 NADPH를 포함한 추가적인 산물들을 생성한다.
암, 줄기세포, 발생 과정, 성체 생리에서의
차별적인 엽산 경로 사용에 대한 이해가 더욱 명확해지고 있으며,
이는 선택적 치료 개입을 위한 새로운 기회를 강조한다.
Introduction
Folate metabolism, which supports a broader set of transformations known as one-carbon (1C) metabolism, is a universal metabolic process that serves to activate and transfer 1C units for biosynthetic processes including purine and thymidine synthesis and homocysteine remethylation. Whereas most bacteria, yeast, and plants can synthesize folate, animals require dietary folate intake. In adults, insufficient dietary folate leads to anemia. In developing fetuses, it creates a disposition to birth defects known as neural tube defects, which involve failure of neural tube closure early in pregnancy. Outcomes range in severity from anencephaly, causing fetal loss, to spina bifida with partial leg paralysis (Copp et al., 2015).
Due to its essential role in nucleic acid synthesis, inhibition of folate metabolism blocks cellular proliferation, and inhibitors of bacterial folate synthesis and transformations (sulfamethoxazole and trimethoprim) or mammalian folate transformations (methotrexate and pemetrexed) are widely used antibiotics and chemo-therapeutics (Chattopadhyay et al., 2007). Folate metabolism also contributes to homocysteine remethylation, impacting epigenetics and possibly also cardiovascular health.
서론
일탄소(1C) 대사로 알려진 보다 광범위한 전환 반응들을 지탱하는 엽산 대사는
퓨린 및 티미딘 합성과
호모시스테인 재메틸화를 포함한 생합성 과정에서
1C 단위를 활성화하고 전달하는 보편적인 대사 과정이다.
대부분의 박테리아, 효모, 식물은 엽산을 합성할 수 있는 반면,
동물은 식이를 통해 엽산을 섭취해야 한다.
성인에서 식이 엽산이 부족하면 빈혈이 발생한다.
발생 중인 태아에서는
엽산 결핍이 신경관 결손이라 불리는 선천성 기형의 소인을 형성하는데,
이는 임신 초기의 신경관 폐쇄 실패와 관련된다.
그 결과는
무뇌 태아 소실을 초래하는 중증 형태부터,
하반신 부분 마비를 동반한 이분척추증에 이르기까지 다양하다(Copp et al., 2015).
핵산 합성에서의 필수적 역할 때문에,
엽산 대사를 억제하면 세포 증식이 차단된다.
박테리아의 엽산 합성 및 전환을 억제하는 약물(설파메톡사졸, 트리메토프림)이나
포유류 엽산 전환을 억제하는 약물(메토트렉세이트, 페메트렉시드)은
항생제 및 항암 화학요법제로 널리 사용되고 있다(Chattopadhyay et al., 2007).

엽산 대사는 또한 호모시스테인 재메틸화에 기여하며,
이는 후성유전학적 조절 및 심혈관 건강에도 영향을 미칠 가능성이 있다.
Folate Chemistry
The term folate encompasses a complex set of molecules that share a common core structure involving three chemical moieties: a pteridine ring that can be reduced or oxidized, a para-aminobenzoic acid (PABA) linker that together with the pteridine ring binds 1C units, and a variable chain length polyglutamate tail that serves to localize the molecule within the cell (Figure 1). The biologically active form of folate is the reduced pteridine species, tetrahydrofolate (THF). Nearly all natural folate species in diet and in the body are present in the reduced form, typically 5-methyl-THF in humans (Wright et al., 2007). Folic acid (vitamin B9), a common synthetic food additive used to prevent neural tube defects, must be sequentially reduced to first dihydrofolate (DHF) and then THF before it can enter the folate cycle (Figure 1).
엽산의 화학적 특성
엽산이라는 용어는
공통된 핵심 구조를 공유하는 복합적인 분자군을 포괄한다.
이 구조는 세 가지 화학적 구성 요소로 이루어져 있다.
첫째는 환원 또는 산화될 수 있는 프테리딘 고리,
둘째는 프테리딘 고리와 함께 1C 단위를 결합하는 파라-아미노벤조산(PABA) 연결체,
셋째는 세포 내에서 분자의 위치를 결정하는 가변적인 길이의 폴리글루탐산 꼬리이다(그림 1).
생물학적으로 활성인 엽산 형태는
환원된 프테리딘 종인 테트라하이드로폴레이트(THF)이다.
식이와 체내에 존재하는 거의 모든 자연 엽산 종은 환원형으로 존재하며,
인간에서는 주로 5-메틸-THF 형태이다(Wright et al., 2007).
신경관 결손 예방을 위해 흔히 사용되는 합성 식품 첨가물인 엽산(비타민 B9)은
엽산 회로에 들어가기 전에 먼저 디하이드로폴레이트(DHF),
이어서 THF로 순차적으로 환원되어야 한다(그림 1).
Figure 1. Chemical Transformations of Folates.
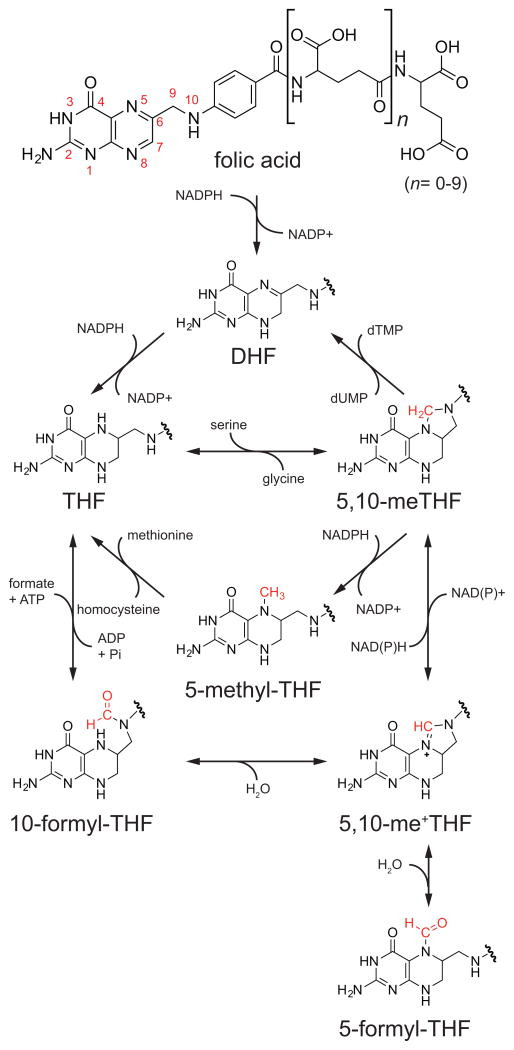
Folic acid is reduced to THF, which can then accept a 1C unit and undergo a series of oxidative/reductive transformations. DHF, dihydrofolate; THF, tetrahydrofolate; 5,10-meTHF, 5,10-methylene-THF; 5,10-me+THF, 5,10-methenyl-THF.
Folate molecules function as carriers for 1C units, allowing them to be manipulated and assembled in support of metabolic processes. To this end, 1C units are covalently bound to the 5-position nitrogen atom on the pterdine ring moiety and the 10-position nitrogen atom on the PABA moiety of THF (Figure 1). Through different covalent bonds to these nitrogen atoms, 1C units can be held in three different carbon oxidation states, each of which plays specific biosynthetic roles (Figure 1). New 1C units primarily enter the system as 5,10-methylene-THF, which can be made from the amino acids serine and glycine and the choline degradation products dimethylglycine and methylglycine (sarcosine). As 1C-loaded folates are not known to transfer across intracellular membranes, 5,10-methylene-THF must be generated in both the mitochondria and cytosol (Anderson et al., 2011). Once bound to THF, 1C units can be interconverted between different oxidation states, with 5,10-methylene-THF, 5-methyl-THF, and 10-formyl-THF each supporting distinct biosynthetic functions (Figure 1). Another formyl-THF species, 5-formyl-THF, does not play a direct biosynthetic role, but serves as a 1C reserve.
엽산과 테트라하이드로폴레이트(THF) 대사 개요
엽산(folic acid)은 환원되어 테트라하이드로폴레이트(THF)가 되며,
이후 1탄소 단위(1C unit)를 받아 일련의 산화·환원 변환을 거친다.
DHF는
디하이드로폴레이트, THF는 테트라하이드로폴레이트, 5,10-meTHF는 5,10-메틸렌-THF, 5,10-me⁺THF는 5,10-메테닐-THF를 의미한다.
엽산 분자는
1탄소 단위를 운반하는 역할을 하여,
다양한 대사 과정에 활용될 수 있도록 한다.
이를 위해 1탄소 단위는
THF의 프테리딘 고리의 5번 질소와 PABA 부분의 10번 질소에 공유 결합된다(그림 1).
이 두 질소 원자와의 결합 방식에 따라,
1탄소 단위는 서로 다른 세 가지 산화 상태로 존재할 수 있으며, 각각 고유한 생합성 기능을 수행한다.
새로운 1탄소 단위는 주로 5,10-메틸렌-THF 형태로 시스템에 유입되며,
이는 세린, 글리신, 그리고 **콜린 분해 산물인 디메틸글리신과 메틸글리신(사르코신)**에서 생성된다.
1탄소를 적재한 엽산은 세포 내 막을 자유롭게 통과하지 못하므로,
5,10-메틸렌-THF는 미토콘드리아와 세포질 모두에서 생성되어야 한다.
THF에 결합된 1탄소 단위는 서로 다른 산화 상태로 상호 전환될 수 있으며,
5,10-메틸렌-THF,
5-메틸-THF,
10-포밀-THF
는 각각 서로 다른 생합성 기능을 지원한다.
한편 5-포밀-THF는
직접적인 생합성 역할은 없지만, 1탄소 저장고(reserve) 역할을 한다.
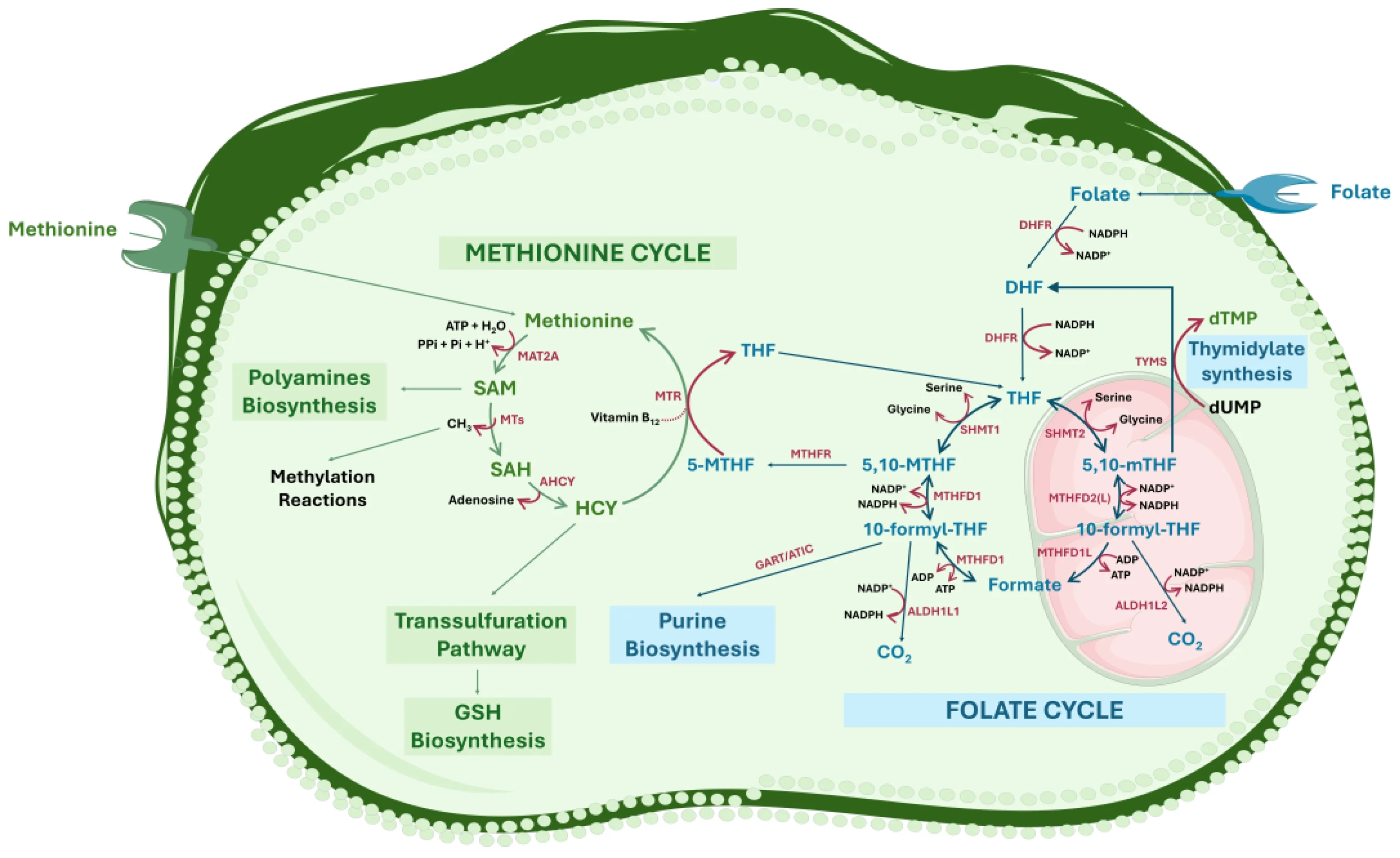
Outputs of Folate Metabolism
Products of 5,10-Methylene-THF: Thymidine and Serine
Thymidylate synthase (TYMS) converts deoxyuridine monophosphate (dUMP) to deoxythymidine monophosphate (dTMP) in a 5,10-methylene-THF-dependent reaction and is the target for the suicide inhibitor chemotherapeutic agent 5-fluorouracil (Figure 2).
엽산 대사의 산출물
1. 5,10-메틸렌-THF의 산물: 티미딘과 세린
**티미딜레이트 합성효소(TYMS)**는
5,10-메틸렌-THF 의존적으로 dUMP를 dTMP로 전환한다.
이 효소는 항암제 **5-플루오로유라실(5-FU)**의 표적이다.
세린 하이드록시메틸전이효소(SHMT)는
5,10-메틸렌-THF를 사용해 글리신을 세린으로 전환하며,
이 반응은 가역적이다.
세포는
한 구획에서 세린을 합성하고 다른 구획에서 분해할 수 있으며,
이는 각 구획의 1탄소 수요와 공급에 따라 달라진다.
Figure 2. Products and Compartmentalization of Folate-Mediated 1C Metabolism.
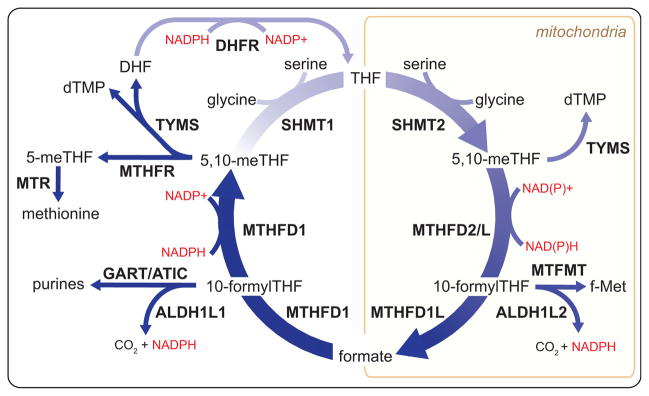
Through an interlinked set of mitochondrial and cytosolic reactions, folate metabolism supports 1C anabolic reactions. All abbreviations are standard gene names. Certain descriptions utilize the common protein name for clarity. SHMT1/2, serine hydroxymethyl transferase, cytosolic(1)/mitochondrial (2); MTHFD1, methylenetetrahydrofolate dehydrogenase, cyclohydrolase, and formyltetrahydrofolate synthetase 1; MTHFD2/L, methylenetetrahydrofolate dehydrogenase 2/2-like; MTHFD1L, monofunctional tetrahydrofolate synthase, mitochondrial; MTFMT, mitochondrial methionyl-tRNA formyltransferase; TYMS, thymidylate synthetase; MTHFR, methylenetetrahydrofolate reductase; MTR, methionine synthase; DHFR, dihydrofolate reductase; GART, phosphoribosylglycinamide formyltransferase; ATIC, 5-aminoimidazole-4-carboxamide ribonucleotide formyltransferase/IMP cyclohydrolase; ALDH1L1/2, cytosolic (1)/mitochondrial (2) 10-formyltetrahydrofolate dehydrogenase.
Serine hydroxymethyltransferase (SHMT) uses 5,10-methylene-THF to convert glycine into serine. The reaction is reversible. Cells can use SHMT to make serine in one compartment and catabolize it in another, with the direction of flow depending upon the supply and demand of 1C units within each compartment (Ducker et al., 2016).
Product of 5-Methyl-THF: Methionine
The most reduced form of folate 1C unit, 5-methyl-THF, has a unique cellular fate, the remethylation of homocysteine to form methionine. 5-methyl-THF is produced by the cytosolic NADPH-dependent activity of methylene tetrahydrofolate reductase (MTHFR) andconsumedbythecobalamin (vitamin B12)-dependent enzyme methionine synthase (MTR) (Figure 2). Mammalian cells can also regenerate methionine from homocysteine in a folate-independent manner using betaine (trimethylglycine, a product of choline degradation) via the enzyme betaine-homocysteine methyltransferase (BHMT). Expression of BHMT is limited to liver and kidney (Pajares and Pérez-Sala, 2006), whereas widespread methionine synthase expression may enable homocysteine remethylation throughout the body.
Homocysteine remethylation is of particular physiological importance because methionine is the substrate for S-adenosylmethionine (SAM) synthetase. SAM, the reactive methyl carrier, is the second most common enzymatic cofactor after ATP and plays a major role in epigenetics; biosynthetic processes including phosphatidylcholine, creatine, and polyamine synthesis; and sulfur metabolism (Finkelstein, 1990; Mudd et al., 2007; Su et al., 2016). In fact, phosphatidylcholine synthesis from phosphoethanolamine is likely the largest 1C sink in adult mammals and consequently the largest source of S-adenosylhomocysteine (SAH) (Stead et al., 2006). Changes in methionine concentrations lead to changes in the ratio of SAM to SAH that impact many methylation reactions including histone methylation (Mentch et al., 2015). The SAM/SAH ratio is tightly regulated by metabolic mechanisms including inhibition of MTHFR by SAM (Kutzbach and Stokstad, 1971), inhibition of the SAM-consuming enzyme glycine-N-methyltransferase (GNMT) by 5-methyl-THF (Yeo and Wagner, 1992), and sequestration of 5-methyl-THF by cytosolic serine hydroxymethyltransferase (SHMT1) (Herbig et al., 2002; Stover and Schirch, 1991). In mice, SHMT1 loss increases, and SHMT1 overexpression decreases SAM levels (MacFarlane et al., 2008, 2011).
Products of 10-Formyl-THF: Purines, Formate, and CO2
The most oxidized form of folate carbon, 10-formyl-THF, is required for de novo synthesis of purines. In proliferating mammalian cells in culture, purine synthesis is the largest demand for folate 1C units. For every newly synthesized DNA or RNA base, an average of one 10-formyl-THF is required. (In comparison, 5,10-methylene-THF is needed only for DNA, at a rate of only one equivalent per four bases.) Purine synthesis is an 11-reaction process incorporating two 10-formyl-THF units into the purine backbone. The enzymes that perform this chemistry have been shown to co-localize in the single complex on the outside of mitochondria, termed the purinosome (An et al., 2008; French et al., 2016). Neither this complex nor its constituent enzymes have been identified within the mitochondrial matrix. However, there is a requirement for mitochondrial 10-formyl-THF. Like in bacteria, mitochondrial initiator methionine tRNAs are formylated. This formylation requires 10-formyl-THF and is required for translation of mitochondrially encoded proteins (Tucker et al., 2011).
To enable transfer of 1C units between compartments, 10-formyl-THF can be hydrolyzed to formate. Hydrolysis of 10-formyl-THF to free formate is coupled to ATP production and reversible. Finally, 10-formyl-THF 1C units can be fully oxidized to CO2 in an NADPH-generating reaction, enabling their terminal elimination (Krupenko et al., 2010).
Side Products of 1C Metabolism
The above sections describe the products made from the 1C unit itself. However, the reactions can also generate other important metabolic products. Glycine is a required precursor for many biosynthetic pathways including glutathione, purine, creatine, and heme synthesis. Cells can acquire glycine from the serum directly or by catabolism of choline, serine, or (in most mammals, but not humans) threonine. When exogenous glycine is unavailable, its synthesis from serine is essential to both purine and glutathione synthesis (Ducker et al., 2016). Serine can be synthesized from glucose, and the combination of the serine biosynthetic pathway and the SHMT reaction provides a route from carbohydrates to glycine.
Additionally, 1C transformations produce and consume redox equivalents. While many of these reactions are reversible, the oxidation of 10-formyl-THF to CO2 is not and generates an NADPH equivalent (Figure 2), with recent evidence suggesting that folate-mediated NADPH production may be particularly important in mitochondrial redox homeostasis (Fan et al., 2014; Piskounova et al., 2015).
Metabolic Cycles and CompartmentalizationDihydrofolate-Tetrahydrofolate Cycle
Many folate enzymes alter the redox state of the 1C unit (Figure 2). Only a single one, however, alters THF itself: TYMS. Using 5,10-methylene-THF as the substrate, TYMS transfers a methyl unit to dUMP to make dTMP, abstracting from THF the two electrons required to reduce the 1C unit and producing DHF as the product. THF is regenerated from DHF by the NADPH-dependent activity of dihydrofolate reductase (DHFR), the canonical target of the antibacterial trimethoprim and the anticancer agent methotrexate. Each of these drugs leads to cellular toxicity in part by trapping folate as DHF and thereby depleting the cellular levels of THF required for other folate reactions.
Mitochondrial-Cytosolic 1C Cycle
A complete set of enzymes linking serine to formate exists in both the cytosol and mitochondria. This allows for not only parallel metabolic processes, but also for a complete oxidative/reductive cycle, which catabolizes serine in the mitochondria and synthesizes serine in the cytosol.
The subcellular compartmentalization of folate reactions has been thoroughly reviewed by Tibbetts and Appling (Tibbetts and Appling, 2010). Both compartments encode enzymes with 5,10-methylene-THF dehydrogenase activity, cyclohydrolase activity to interconvert the resulting 5,10-methenyl-THF (an intermediate with no biosynthetic role) with 10-formyl-THF, and formate-THF ligase activity that can release or reassimilate formate in an ATP-dependent manner. In the cytosol, each of these activities is localized to a single trifunctional protein, MTHFD1. In contrast, in mitochondria there are two enzymes with bifunctional dehydrogenase/cyclohydrolase activity (MTHFD2 and MTHFD2L) and a separate formate-THF ligase enzyme (MTHFD1L, which structurally resembles MTHFD1 but with loss of catalytic activity in both the dehydrogenase and cyclohydrolase domains).
The cycle that catabolizes serine in mitochondria and synthesizes serine in the cytosol is thermodynamically driven by the difference in electrochemical potential between mitochondrial NADH (and also NADPH) and cytosolic NADPH (Yang and MacKenzie, 1993). MTHFD1 uses NADPH and MTHFD2/L uses NADH, whereas MTHFD2L may use both substantially under physiological conditions, although cellular tracing studies are needed to confirm this (Shin et al., 2014). The high cytosolic NADPH/NADP+ ratio favors flux from formate toward serine in the cytosol.
Tracing Mitochondrial and Cytosolic Serine Catabolism
The introduction of deuterated serine ([2,3,3-2H]serine) as a tracer has enabled compartment-specific analysis of the flux of 1C units from serine, their main source, by mass spectrometry. When serine is metabolized within the mitochondria to formate, the oxidation of the 1C unit removes one of the deuterons; reductive incorporation of this 1C unit into thymidine or methionine in the cytosol results in their being singly labeled (M+1) (Figures 3A and 3B). Conversely, cytosolic metabolism of serine directly to 5,10-methylene-THF by SHMT1 involves no loss of deuterium and results accordingly in M+2 labeling of thymidine and methionine. This technique was developed by the Gregory and Stover groups, who first used it to trace the contribution of mitochondrial-derived serine to homocysteine remethylation in cell culture and in human volunteers (Gregory et al., 2000; Herbig et al., 2002). Both found that the majority of methionine 1C units originated from the mitochondria. When applied to MTHFD1L deletion mouse embryonic fibroblasts, this labeling technique demonstrates that in the absence of functional mitochondrial folate metabolism, cytosolic SHMT1 makes 5,10-methylene-THF from serine, and M+2 is now observed on methionine (Pike et al., 2010). Across a diverse panel of proliferating cell lines in culture, Ducker et al. found that most make 1C units originate solely in the mitochondria; however, several engaged in both cytosolic and mitochondrial serine catabolism (Ducker et al., 2016). Moreover, upon genetic ablation of the mitochondrial pathway, even cells that normally use only the mitochondrial pathway to catabolize serine will switch to making 1C units from serine in the cytosol.
Figure 3. [2,3,3-2H]Serine Is a Tracer for Mitochondrial Folate Pathway Activity.
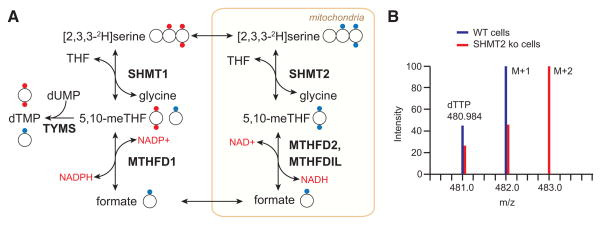
(A) Compartment-specific metabolism of serine can be determined by following the incorporation of 2H from [2,3,3-2H]serine into terminal products of 1C metabolism such as thymidine. If serine is metabolized in the mitochondria, one proton attached to the 3-position carbon will be lost during oxidation to formate, as represented by the blue dots. Reductive incorporation of the formate into the cytosolic folate pool will result in singly deuterated thymidine monophosphate (TMP M+1). In contrast, a serine catabolized in the cytosol by SHMT1 will transfer both protons onto 5,10-methylene-THF and generate a doubly labeled TMP M+2, as represented by the red dots.
(B) Representative mass spectra of WT and SHMT2 deletion cells demonstrate the application of this tracing technique. Adapted from Ducker et al. (2016).
The deuterium from [2,3,3-2H]serine can be traced into NAD(P)H and from there into other metabolites (Fan et al., 2014). Lewis et al. demonstrated using a clever mutant isocitrate dehydrogenase (IDH) reporter strategy that folate metabolism selectively generates NADPH in mitochondria (Lewis et al., 2014). The authors transfected cells with IDH1 (cytosolic) or IDH2 (mitochondrial) constructs that carried active site mutations, leading to the production of the oncometabolite R(−)-2-hydroxyglutarate (2HG) from the NADPH catalyzed reduction of α-ketoglutarate (Dang et al., 2009). 2H-labeled 2HG was produced from [2,3,3-2H]serine in cells with the mutant mitochondrial IDH2, but not cytosolic IDH1. Thus, cytosolic folate metabolism proceeds in the reductive, NADPH-consuming direction.
This cofactor-mediated deuterium tracing can also be accomplished by direct monitoring of cellular metabolites without the introduction of a mutant enzyme. In cell lines with mutations that block mitochondrial folate metabolism, cytosolic folate-mediated NADPH production results in fatty acid labeling from 2H-serine. In cells with intact mitochondrial 1C metabolism, mitochondrial folate metabolism makes both NADH and NADPH, as evidenced by NADH-mediated labeling of malate and NADPH-mediated labeling of proline from 2H-serine (Ducker et al., 2016). Using clustered regularly interspaced short palindromic repeat (CRISPR) deletion mutants, this technique was able to isolate the source of most mitochondrial folate-derived NADPH to the activity of the formyl-THF oxidizing enzyme mitochondrial 10-formyltetrahydrofolate dehydrogenase (ALDH1L2).
Impact of NADH and NADPH Levels
Production of NADH by mitochondrial 5,10-methylene-THF dehydrogenase activity links 1C metabolism to the respiratory state of the cell. Mitochondrial NAD+/NADH ratios are maintained by oxidative phosphorylation, and early experiments in isolated mitochondria showed that formate production from serine was respiration dependent (García-Martínez and Appling, 1993). Conversely, in ρ0 cells, which are depleted for mitochondrial DNA, no alterations in folate metabolism were observed (Patel et al., 2003); however, these cells require exogenous pyruvate to grow, which functions in part to restore the NAD+/NADH ratio (Birsoy et al., 2015; Sullivan et al., 2015; Titov et al., 2016). The commonly used anti-diabetic agent metformin targets mitochondrial complex I and thus decreases the NAD+/NADH ratio. Metformin also inhibits cancer cell growth, in part through inhibition of biosynthetic metabolism (Griss et al., 2015). Metformin-induced growth inhibition can be partially rescued by supplementation with hypoxanthine and thymidine, products of 1C metabolism (Corominas-Faja et al., 2012). It remains unclear, however, if the impact of metformin on 1C metabolism is clinically significant at normal therapeutic doses.
Further evidence in support of a physiological link between mitochondrial NADH and 1C metabolism was provided by two recent studies of metabolic alterations induced by mitochondrial respiratory chain deficiency (Bao et al., 2016; Nikkanen et al., 2016). Using an inducible dominant-negative mitochondrial DNA polymerase, Bao et al. showed that mitochondrial DNA depletion triggers, via the transcription factor ATF4, expression of serine synthesis genes (PHGDH, PSAT1, and PSPH), thereby increasing serine to feed 1C metabolism. In related work, Nikkanen et al. investigated the outcome of mitochondrial DNA depletion in vivo. In a model of mitochondrial myopathy induced by mutation of the mitochondrial DNA helicase TWINKLE, reductions in tissue formate and 10-formyl-THF co-occurred with increases in expression of mitochondrial folate 1C enzymes (MTHFD2 and MTHFD1L). In the muscle and blood of progressive external ophthalmoplegia patients, increases in 1C-related metabolites, including serine and cystathionine, were observed. Together, these studies are consistent with a model whereby impaired electron chain transport activity lowers mitochondrial formate production, inducing an adaptive response to increase both 1C donors and mitochondrial 1C metabolic enzymes, but these changes are insufficient to regain full wild-type (WT) pathway functionality.
What role does cytosolic NADP+/NADPH play in driving reductive cytosolic folate metabolism? Unlike in the case of the NAD+ in the mitochondria, there is as of yet not an easy way to modulate steady-state cytosolic NADPH levels. However, the case of mitochondrial folate mutant cell lines is instructive. These cells now process 1C units oxidatively in the cytosol in order to produce 10-formyl-THF for purine biosynthesis. In engineered HEK293T cells deficient for MTHFD2, the NADP+/NADPH ratio does not significantly change between WT and mutant. Instead, there is a dramatic decrease in 10-formyl-THF. These results argue that changes in folate pools, as opposed to NADPH, control the thermodynamically favored direction of cytosolic 1C flux (Ducker et al., 2016).
Rationale for Compartmentalization
The differential electrochemical potential of NADH versus NADPH explains why folate metabolism, as constructed, involves mitochondrial formate shuttling to the cytosol. But it does not explain why the mitochondria need be included in the first place. If cytosolic MTHFD1 used NAD+ for the dehydrogenase reaction, it would favor the oxidative direction (West et al., 1996). Mitochondrial-specific 1C demand is small and could be met in theory by transport or a low-activity pathway. We believe that nature has chosen to localize most serine catabolism and 1C unit oxidation to mitochondria to uncouple 1C metabolism from glycolysis. Glucose fermentation to lactate is redox balanced, but this balance can be thrown off by any other NADH-generating cytosolic reaction. If 1C oxidation were to instead occur in the cytosol with concomitant NADH generation, cells could become depleted of cytosolic NAD+, blocking glycolysis. Thus, the mitochondrial localization of oxidative 1C metabolism enhances cellular robustness.
Polyglutamation Encodes Compartmentalization
The mitochondrial/cytosolic division of folate metabolism is encoded both genetically by the different isozymes and chemically by differences in the folates themselves. Folate is distributed by the circulation as THF-monoglutamate species (Wright et al., 2007). These monoglutamates are actively transported across the cell membrane using the anionic reduced folate transporter (RFC, SLC19A1) (Hou and Matherly, 2014). To retain folates within the cell, they are polyglutamated, which reduces their affinity for the transporter and enhances their affinity for folate enzymes (Kim et al., 1996). Folate polyglutamate synthetase (FPGS) activity is essential for cell proliferation (Freemantle et al., 1995). Compared to bacteria, the degree of polyglutamation is higher in eukaryotes (Leung et al., 2013; Lin et al., 1993; Lu et al., 2007). Changes in RFC and FPGS function are mechanisms of acquired resistance to antifolates (Zhao and Goldman, 2003). Mutations that impair RFC transport provide resistance to methotrexate in leukemic cells, whereas loss of FPGS expression leads to pemetrexed resistance, as polyglutamation is essential for its activity (Liani et al., 2003).
A mitochondrial folate transporter (SLC25A32) is responsible for translocation of THF into the mitochondria, and mutants deficient for this six-transmembrane helix protein are glycine auxotrophs, a common phenotype of mitochondrial folate pathway loss (Chasin et al., 1974; Titus and Moran, 2000). Folate polyglutamates are not thought to be substrates for this transporter, necessitating mitochondrial localization of FPGS activity, achieved by a mitochondria splice variant of the gene (Freemantle et al., 1995; Lowe et al., 1993). Mitochondrial folate polyglutamates are of larger average chain length, but this may only reflect the processive activity of FPGS on the trapped slow exchanging mitochondrial folate pool. The compartmental requirements for FPGS were recently elegantly confirmed by Lawrence and colleagues using inducible versions of either the mitochondrial or cytosolic isoforms of FPGS in an FPGS null cell line (Lawrence et al., 2014). When FPGS was expressed only in the cytosol, cells were glycine auxotrophs, whereas when expression was confined to the mitochondria, cells required thymidine and purines to circumvent their now defective cytosolic biosynthetic pathways.
Nuclear Folate Metabolism
Folate metabolism has long been identified in purified nuclei (Prem veer Reddy and Pardee, 1980). Isotope tracer studies in isolated nuclei show that dUMP is converted to dTMP and that formate can function as the 1C source (Anderson and Stover, 2009; Field et al., 2014). No nuclear isoforms of folate genes have been discovered. Rather, it has been proposed that cytosolic folate enzymes perform nuclear functions. SHMT1, SHMT2α (an alternatively spliced isoform of SHMT2), and MTHFD1 show partial nuclear localization as GFP-tagged overexpression constructs, and SHMT1 may associate with the nuclear lamin (Anderson et al., 2012b; Field et al., 2016), and its cell-cycle-specific sumoylation has been proposed to control the formation of a nuclear complex with TYMS (Anderson et al., 2012a). Such a complex, however, is not essential for maintaining nuclear thymidine pools, as HEK293 cells engineered via CRISPR to lack both SHMT1 and 2 (and thus lacking any SHMT-dependent nuclear complex) grow normally when supplemented with formate (Bao et al., 2016). More generally, the lack of nuclear-specific enzymes has hindered efforts to assign function to nuclear folate metabolism.
Sources of 1C Units
Eukaryotic cells mobilize multiple carbon sources to supply 1C metabolism. Serine, glycine, formate, histidine, and the choline-derived methylglycine species (sarcosine and dimethylglycine) can all directly contribute to the THF-bound 1C pool (Figure 4). Quantitatively, choline, serine, and glycine are the most important dietary sources of folate 1C units. Beyond supporting biosynthetic processes, 1C metabolism plays a pivotal role in catabolism of these nutrients.
Figure 4. Major Sources of 1C Units in the Mammalian Cell.
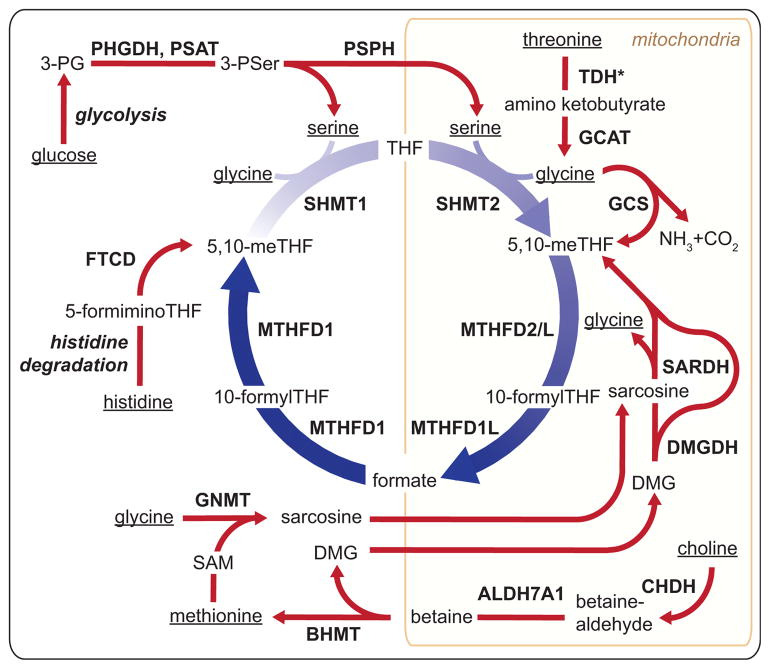
1C sources originate in the diet and are metabolized to generate 1C units. Dietary sources of 1C are underlined and include glucose, threonine, methionine, glycine, serine, histidine, and choline. TDH*, human TDH encodes a pseudogene without functional catalytic activity; 3PG, 3-phoshoglycerate; 3PSer, 3-phosphoserine; DMG, dimethylglycine; TDH, L-threonine dehydrogenase; GCAT, glycine C-acetyltransferase; GCS, glycine cleavage system; SARDH, sarcosine dehydrogenase; DMGDH, dimethylglycine dehydrogenase; CHDH, choline dehydrogenase; ALDH7A1, aldehyde dehydrogenase 7 family member A1; BHMT, betaine-homocysteine S-methyltransferase; GNMT, glycine N-methyltransferase; FTCD, formimidoyltransferase cyclodeaminase; PHGDH, phosphoglycerate dehydrogenase; PSAT, phosphoserine aminotransferase; PSPH, phosphoserine phosphatase.
Among the various 1C donors, only three are capable of directly feeding into the cytosolic folate-bound 1C pool: histidine, serine, and formate (Figure 4). Histidine degradation generates 5-formimino-THF, which is converted into 5,10-methylene-THF in the cytosol by formimidoyltransferase cyclodeaminase (Solans et al., 2000). Serine, which can be synthesized from glucose but under typical conditions in mammals mainly comes from dietary protein intake, can directly feed the cytosolic 1C pool via SHMT1. In rapidly proliferating cultured cells, however, the main direct source of cytosolic 1C units is formate released from their mitochondria.
Within the mitochondria, 1C units can be made from serine, glycine, sarcosine, or dimethylglycine and excreted into the cytosol as formate (Figure 4). SHMT2 converts serine into a 1C unit and glycine that can be further metabolized by the glycine cleavage system (GCS). Thus, SHMT2 and the GCS are poised to work in tandem. GCS is a mitochondrial-localized four-enzyme complex that decarboxylates glycine to form CO2 and deaminates the remaining carbon to form mitochondrial 5,10-methylene-THF (Kikuchi et al., 2008). This reaction is reversible in vitro (Kochi and Kikuchi, 1974). While not functional in humans, in all other mammals (including chimpanzees) glycine can also be produced in the mitochondria from threonine via threonine dehydrogenase (TDH) and glycine C-acetyltransferase (GCAT). While SHMT2 is the dominant 1C source, as determined by isotope tracing in most proliferating cells cultured in vitro, glycine catabolism is a major 1C source in stem cells, embryogenesis, and quiescent adult tissues.
Choline is an important dietary nutrient that supports both folate-independent methionine synthesis and, via the resulting dimethylglycine, folate-bound 1C pools. In the liver, choline can either be used for lipid head group synthesis or catabolized to dimethylglycine (Ueland, 2011). Choline catabolism begins with its two-step mitochondrial oxidation to trimethylglycine (betaine). In the cytosol, betaine remethylates homocysteine in a folate-independent reaction catalyzed by BHMT (Figure 4). After donating a methyl group to homocysteine, betaine becomes dimethylglycine, which can yield additional 1C units via folate-dependent mitochondrial reactions: dimethylglycine dehydrogenase (DMGDH) makes sarcosine and sarcosine dehydrogenase (SARDH) makes glycine.
In addition to being synthesized via the choline degradation pathway, sarcosine can be made in the cytosol by SAM-mediated methylation of glycine catalyzed by the enzyme GNMT (Luka et al., 2009). This provides a potential route from methionine to folate-bound 1C units. Sarcosine levels are elevated in aggressive and metastatic prostate cancer tumors (Sreekumar et al., 2009). This has raised great interest in sarcosine as a biomarker for prostate cancer; however, studies attempting to relate urine or blood sarcosine concentrations to prostate cancer have yielded inconsistent results (Kelly et al., 2016).
Circulating formate is another 1C source. In addition to being a product of folate metabolism, formate may also be produced from folate-independent processes including the kynurenine pathway of tryptophan degradation, the polyamine/methionine cycle, and the methanol detoxification pathway. Free formate concentrations in mammalian serum range from 20 to 50 μM and originate from both dietary sources and internal metabolic processes (Lamarre et al., 2013, 2014). The role for circulating formate in meeting normal 1C demand is unclear. Formate is typically considered a toxin and is responsible for metabolic acidosis resulting from methanol or formaldehyde poisoning. Flux from glycine to serine catalyzed by SHMT1 consumes 1C units and may serve the biological function of formate detoxification. Clearance of formate is known to be folate dependent, and its cross-species variance correlates with susceptibility to methanol poisoning (Sweeting et al., 2010). Thus, the folate system does not just support anabolism, but also has important catabolic and detoxification functions.
1C Metabolism in Development and Proliferative Adult Tissues
Decades of research have confirmed the essential requirement for dietary folate in both development and the adult. The best characterized function of folate metabolism is nucleic acid synthesis to support cellular proliferation, and folate metabolism is essential in proliferating tissues. What has only recently been uncovered, however, is the role specific folate-utilizing enzymes play in supporting growth and development, and which pathways are activated in fetal development, immune proliferation, and tissue homeostasis. Probably the most important insight over the past decade regards the role and importance of mitochondrial folate metabolism. Deletions in mitochondrial folate enzymes lead to neural tube defects and loss of viability. Many of these defects cannot be rescued metabolically, suggesting that mitochondrial folate metabolism is intrinsically required to supply 1C units in highly proliferative states, such as the embryo. This essentiality in development has hindered a rigorous examination of the requirement for mitochondrial folate metabolism in adult tissues, though expression patterns and limited tracing data suggest that it plays an important role in these contexts as well. Development of conditional and tissue-specific folate gene knockouts should eventually illuminate more precisely the roles of folate metabolism in adulthood.
Neural Tube Defects
Demand for 1C units is highest during fetal development, and folate deficiency leads to neural tube and congenital heart defects (Bailey and Berry, 2005; Beaudin and Stover, 2009). As the only enzyme capable of generating the cytosolic 10-formyl-THF required for purine synthesis, deletion of the trifunctional MTHFD1 enzyme is embryonic lethal (MacFarlane et al., 2009). The source of the embryonic 1C units appears to be substantially mitochondrial as SHMT1 deletion mice are viable and fertile (MacFarlane et al., 2008), although these animals do have vulnerabilities: in SHMT1−/− dams fed a standard diet no fetal abnormalities were observed; however, when folate and choline were removed from the diet, a fraction of fetuses developed neural tube defects (Beaudin et al., 2011). Maternal supplementation of the folate-poor diet with the thymidine precursor deoxyuridine completely rescued the neural tube defects, suggesting that in this instance, inadequate DNA synthesis was at least in part responsible for the neural tube defects (Martiniova et al., 2015). Surprisingly, however, thymidine was less effective as a rescue agent, and formate was not tested; thus, further investigation into the underlying mechanism is merited.
Mice defective in mitochondrial 1C metabolism are generally nonviable. MTHFD1L and GCS deletions are embryonic lethal due to neural tube defects (Momb et al., 2013; Narisawa et al., 2012). MTHFD1L−/− defects can be partially ameliorated through supplementation of dams with formate, but no viable offspring are produced (Momb et al., 2013). GCS mutants (deleted for the AMT gene) are nearly universally afflicted with neural tube defects. Maternal supplementation with methionine, but not thymidine, ameliorated some fetal deformities but was insufficient to recover viable pups. Interestingly, using a genetrap allele strategy targeting a different GCS component, GLDC, live mice deficient in GCS activity were created whose neural tube defects were formate rescuable (Pai et al., 2015). A genetrap allele is a selectable marker inserted into an early intron of a gene that contains both a splice acceptor site and stop codon followed by a polyadenylation sequence. Upon transcription, the unnatural splice site is incorporated into the nascent mRNA, resulting in early termination and functional deletion of the targeted gene, but unlike homozygous deletion techniques, some functional transcripts can be created due to missed splicing of the genetrap construct. Mice homozygous for the GLDC genetrap (GLDCGT1/GT1) were recovered at less than Mendelian ratios, and neural tube defects were common among fetuses. The live mice have brain defects including domed head, enlarged brain, and hydrocephalus leading to premature death. These abnormalities were largely rescuable by maternal formate supplementation, suggesting that the underlying neural tube defects are due to a lack of 1C units required to support neuronal proliferation (Pai et al., 2015). In humans, deficient GCS activity due to mutations in the AMT or GLDC genes causes nonketotic hyperglycinemia, which commonly presents as severe neonatal neurological dysfunction (Kure et al., 2006).
Although there has been no published attempt to make an SHMT2−/− mouse, serine appears to be a critical 1C source during development. Serine does not readily pass the blood-brain barrier, and accordingly, the main source of CNS serine is de novo synthesis from glucose. Knockout of the serine biosynthetic enzyme phosphoglycerate dehydrogenase (PHGDH) in mice causes severe neurological abnormalities including absence of the olfactory bulb, ganglionic eminence, and cerebellum (Yoshida et al., 2004). In humans, severe mutations in PHGDH or the downstream serine synthesis genes phosphoserine aminotransferase (PSAT1) and phosphoserine phosphatase (PSPH) result in the fatal Neu-Laxova syndrome, which is characterized by fetal malformation including microcephaly (Acuna-Hidalgo et al., 2014). Although definitive mouse genetic studies are lacking, methylglycine species appear to be less important as 1C sources, as a patient with an inactivating mutation in DMGDH has been identified and is apparently healthy (Binzak et al., 2001). Thus, mitochondrial generation of 1C units from both serine and glycine (but not methylated glycine species), supported by de novo serine synthesis from glucose, is required for normal mammalian development (Figure 5).
Figure 5. 1C Metabolism across Different Cell Types.
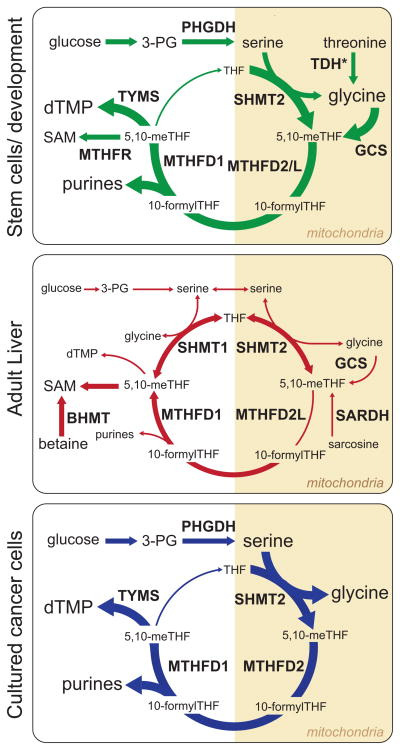
A cartoon representation of the major folate-dependent 1C metabolic pathways in three representative cell types. Variable font size and line widths represent the relative importance of each species or reaction in the indicated cell type. Reactions not drawn for an individual cell type are considered to be negligible in that context. TDH*, TDH activity is important in murine stem cells, but this enzyme is non-functional in humans.
MTHFD2, Hematopoiesis, and the Immune System
Hematopoiesis and immune response are among the most proliferative processes in the body and thus highly folate sensitive. In adults, dietary folate deficiency manifests as macrocytic anemia. Immune cells have long been noted to be sensitive to conventional antifolate therapies, limiting the effective doses that can be given for cancer therapy. Moreover, low-dose methotrexate is a mainstay in the treatment of certain autoimmune disorders including rheumatoid arthritis and lupus (Pincus et al., 2003). The downside of such therapy includes side effects in other proliferative tissues, including the gastrointestinal tract, and also in the liver. Thus, more selective inhibition of immune cell folate metabolism would be clinically useful.
Mouse genetics has uncovered a hematopoietic lineage-specific folate isozyme, MTHFD2. In contrast to mice lacking the closely related pathway genes MTHFD1L and AMT, those deleted for MTHFD2 do not show a neural tube defect but rather normal body development with a failure of the liver tissue to support hematopoiesis (Di Pietro et al., 2002). MTHFD2−/− embryos are smaller and paler than their WT littermates and lack normal liver redness. No rescue was observed when dams were supplemented with formate or a glycine-rich diet, and, in fact, supplemental glycine appeared to accelerate fetal death (Patel et al., 2003). One possible explanation for the lack of neural tube defect in this model is the presence of MTHFD2L, which is expressed throughout the embryo beginning at neural tube closure (Shin et al., 2014). This result suggests that MTHFD2 is of greatest functional importance in the hematopoietic lineage, including immune cells. The lineage specificity of MTHFD2 may be a function of the demand for 1C units for rapid proliferative growth; the reported Kcat/KM for MTHFD2 is 50-fold higher than for MTHFD2L (Shin et al., 2014).
Consistent with this, new work in T cell activation shows a requirement for rapid 1C metabolism. This is met by large changes in gene expression that serve both to mobilize existing stored 1C from 5-formyl-THF by 5-formyltetrahydrofolate cycloligase, and to produce new 1C units from serine via the SHMT2-MTHFD2 pathway (Ron-Harel et al., 2016). Interestingly, inhibition of T cell activation by SHMT2 short hairpin RNA (shRNA) was fully rescued by a combination of formate and the general antioxidant N-acetylcysteine, but not either alone, implicating the mitochondrial 1C pathway in T cells in both 1C unit generation and redox defense, as previously observed in HEK293T cells (Ducker et al., 2016). Given the widespread expression of MTHFD2L outside of the immune system, it seems likely that existing side effects of antifolate therapies could be mitigated by specific MTHFD2 inhibition. Accordingly, there is great promise in the development of MTHFD2-specific antifolates.
1C Metabolism in Stem Cells
Mouse embryonic stem cells (ESCs) divide at rates (4–5 hr doubling) far in excess of standard cultured cell lines, leading to unique metabolic stresses. Interestingly, murine ESCs selectively utilize glycine derived from TDH to generate 1C units for both nucleotide and methionine synthesis. Without threonine, mouse ESCs cannot synthesize DNA (Wang et al., 2009). The TDH reaction also generates acetyl-CoA, and threonine withdrawal alters histone modification by modulating both SAM and acetyl-CoA levels (Shyh-Chang et al., 2013). Threonine withdrawal can be rescued by a combination of pyruvate (as an acetyl-CoA source) and other 1C donors including glycine, dimethylglycine, and betaine.
The requirement of mouse ESCs for threonine raises two questions: Why the need for threonine as a 1C source in the mitochondria? And how is this demand met in humans given our lack of a functional TDH? Among all animals studied to date, humans appear to have uniquely lost the activity of the TDH gene (Edgar, 2002; Wang et al., 2009). In vitro studies have found that SHMT can break threonine into glycine via a retroaldol mechanism (Schirch and Szebenyi, 2005). Might such activity play a role in human ESCs? Or might murine, but not human, ESCs be defective in direct glycine uptake into mitochondria and/or mitochondrial serine catabolism, and therefore require threonine as a glycine source? Further research is needed to address these questions.
Folate Metabolism in Non-proliferative Adult Tissues
In development and in proliferative tissues, folate metabolism has an anabolic role in generating purines and thymidine, and in some cases also methionine, glycine, or serine. Among non-proliferative adult tissues, anabolic 1C reactions are largely localized to the liver and kidney, where they support the synthesis of the energy molecule creatine and the lipid head group choline, major organismal sinks for SAM-bound 1C units. In other non-proliferative tissues, the role of folate metabolism shifts to maintaining homeostasis of nucleotides and amino acids, and utilization of dietary choline, serine, and glycine as substrates for ATP and NADPH generation.
Folate metabolism is a major contributor to amino acid homeostasis (Brosnan et al., 2015). Isotopic tracer infusion studies in humans illustrate the high total body turnover in 1C-related amino acids. In one study, 13C-glycine was infused into healthy volunteers and GCS activity assayed by mass spectrometry. A large fraction of glycine was catabolized by the GCS and generated 1C units at over 20× the demand for homocysteine remethylation (Lamers et al., 2007). Similarly, in a different study to quantify the serine contribution to homocysteine remethylation, a small fraction of serine metabolic flux (~3%) could account for all of the homocysteine remethylation (Davis et al., 2004). These studies show that glycine and serine metabolism are dynamic but do not reveal the tissue specificity or function of the underlying reactions.
Amino Acid Homeostasis
1C metabolism directly controls the levels of three amino acids: methionine, serine, and glycine. Folate metabolism is the only direct route to catabolize glycine into the terminal waste products ammonia and CO2. 1C metabolism also indirectly controls cysteine levels via its effect on the transsulfuration pathway that transfers sulfur from homocysteine to serine to make cysteine.
Amino acid metabolism is constrained by that fact that any amino acids consumed in excess of immediate protein synthesis demand must be catabolized. The majority of amino acid catabolism is thought to occur in the liver and kidney where carbon can contribute to gluconeogenesis and nitrogen can be converted to urea for excretion (Brosnan, 2003). Quantitative measurements of the contributions of different organs to amino acid homeostasis are consistent with a major role for liver and kidney in 1C metabolism. These measurements are based on regional arterial-venous differences in serum amino acid concentrations, which reflect metabolic activity of the intervening organ. The liver is fed by both the arterial circulation and the portal vein, which runs from the small intestine and carries nutrients absorbed there. While sampling from the portal vein of humans is too invasive, the metabolic contribution of the liver can be determined in fasted individuals, where amino acid uptake from the intestines is minimal. During fasting, comparison of hepatic venous blood to arterial blood has revealed liver uptake of methionine and glycine that roughly matches their production from muscle (Felig, 1975). Thus, liver appears to be the major site of methionine and glycine utilization. In contrast, serine is consumed by many peripheral organs (Felig, 1975) and produced substantially from the kidney, which can make it from glucose, protein catabolism, and/or 1C-mediated conversion of glycine (Brosnan, 1987).
Liver Control of Systemic Methylation Potential
In parallel to consuming methionine, the liver also makes methionine. Folate-dependent homocysteine remethylation occurs throughout the body, but is likely most important in liver. Murine knockout of MTHFR, which produces the 5-methyl-THF required for homocysteine remethylation, results in homocystinuria (pathological elevation of homocysteine in the blood and urine), with early postnatal death and defects in brain development. Feeding of betaine to dams during pregnancy, which provides an alternative substrate for liver (but not brain) homocysteine remethylation, ameliorates the brain development defects and prevents death of the pups (Schwahn et al., 2004). MTHFR variants with severely reduced catalytic activity occur rarely in the human population and show similar phenotypes to the knockout mouse (Goyette et al., 1995). These observations suggest that brain development is dependent on liver 1C metabolism to maintain SAM homeostasis.
In contrast to the rarity of MTHFR alleles with severely reduced catalytic activity, alleles with moderately reduced catalytic activity are common in the human population and result in an elevation in circulating homocysteine levels without any developmental brain abnormalities. These low-activity alleles are associated with a modestly increased risk of cardiovascular disorders (Casas et al., 2005; Wald et al., 2002). Despite effectively lowering homocysteine levels, folate and cobalamin supplementation do not reduce the incidence of cardiovascular events in the general population, arguing against high homocysteine levels directly causing cardiovascular disease (Bazzano et al., 2006; Lonn et al., 2006; Zhou et al., 2011).
Increased homocysteine also occurs in liver disease, including non-alcoholic fatty liver disease (NAFLD) (Dai et al., 2016). In animals fed high-fat diets to induce NAFLD, liver 1C metabolism is dysregulated, as evidenced by intrahepatic increases in SAH and free homocysteine and decreases in methionine and the GNMT enzyme (Pacana et al., 2015). At the same time, deletions of 1C enzymes lead to development of liver disease. Mice lacking BHMT have elevated liver betaine and homocysteine and lower SAM, and develop hepatocellular carcinoma by 1 year of age (Teng et al., 2011).
Homocysteine concentrations are also regulated by the activity of the transsulfuration pathway, which is the only route to degrade homocysteine and thus irreversibly consume the methionine backbone. In this pathway, the homocysteine sulfur atom is transferred to serine to make cysteine. Transsulfuration occurs exclusively in the liver, brain, and pancreas; in the brain it is required to support glutathione synthesis (Vitvitsky et al., 2006). Cystathionine beta synthetase (CBS) is the first enzyme in this pathway and has an SAM regulatory site that activates the enzyme. Mutation of the site results in homocystinuria (Kluijtmans et al., 1996). Through the synthesis of reduced cysteine, this pathway contributes to glutathione synthesis and thus cellular redox homeostasis. By generating a reduced cysteine, it conserves an NADPH equivalent that would otherwise be required to reduce extracellular cystine taken in from the circulation (Eriksson et al., 2015).
Several mechanisms protect against excess buildup of methionine relative to homocysteine. These include SAM inhibition of MTHFR and disposal of excess SAM 1C units by GNMT. Mice deleted for GNMT develop progressive liver disease marked by steatosis and fibrosis, leading to hepatocellular carcinoma (Martínez-Chantar et al., 2008). Interestingly, while GNMT and BHMT deletions both cause progressive liver disease resulting in hepatocellular carcinoma, they have opposite metabolic effects: GNMT deletion causes high SAM and methionine and low homocysteine, consistent with human patients with GNMT deficiency (Liu et al., 2007; Luka et al., 2006). Apparently, either too high or too low liver SAM/SAH ratios can alter epigenetics and thereby promote cancer.
Mitochondria-to-Cytosol 1C Shuttling in Liver and Kidney
By combining expression data of 1C metabolic enzymes with flux constraints from arterial-venous amino acid measurements, it is possible to infer an approximate map of the active 1C pathways in the kidney and liver (Figure 5). BHMT is unique to the liver and kidney and together with methionine synthase supports the high liver demand for SAM, with liver estimated to carry out 80% of body total methylation (Stead et al., 2006). Enzymes catalyzing 1C unit generation from serine, glycine, and dimethylglycine, as well as the multifunctional cytosolic enzyme MTHFD1, show abundant expression in liver and kidney, with SHMT1 nearly exclusive to these two organs (Girgis et al., 1998) (Kure et al., 2001; Lang et al., 1994). Although mitochondria isolated from rat liver make formate, indicating a complete 1C pathway (Barlowe and Appling, 1988), rodent kidney and especially liver show very low expression of MTHFD2(L) and MTHFD1L (Bolusani et al., 2011; Shin et al., 2014). This raises the possibility that in these organs, mitochondrial 1C units are not completely oxidized and excreted as formate, but rather used to synthesize serine from glycine, with the serine exported to the cytosol and catabolized there via SHMT1. For supporting homocysteine remethylation, compared to 1C unit transfer via formate, transfer of 1C units from mitochondrial to cytosol via serine reduces the required number of enzymes and conserves cytosolic NADPH. Infusion of [2,3,3-2H]serine in human subjects produces serum methionine and liver-synthesized apoprotein B100 in both the M+1 and M+2 labeling states, indicating at least some cytosolic 1C production from serine (Gregory et al., 2000). Thus, liver and kidney may engage in an alternative 1C shuttle in which SHMT1 makes cytosolic 1C units.
Most Adult Tissues Express Both Mitochondrial and Cytosolic 1C Enzymes
The function of 1C metabolism in other adult non-proliferative tissues is unclear. Their modest demands for 1C units could potentially be met in a variety of ways, including circulating formate. Based on enzyme expression data, however, all adult tissues appear to have the ability to generate 1C units de novo from mitochondrial folate metabolism, transfer formate to the cytosol, synthesize thymidine and purines, and remethylate homocysteine. Excess folate-bound 1C units can be disposed of by the universally expressed enzyme cytosolic 10-formyltetrahydrofolate dehydrogenase (ALDH1L1), while the mitochondrial ALDH1L2 is largely confined to heart, pancreas, brain, and muscle (Krupenko et al., 2010). Beyond these housekeeping functions, little is known about relative fluxes and regulation. Given the roles of glycine and serine in neurotransmission, it seems likely the brain is an active site of folate metabolism. Expression data show high GCS transcripts in brain. SHMT1 is expressed selectively in the hippocampus, including the dentate gyrus and cornu ammonis, regions associated with cell proliferation (Abarinov et al., 2013). A polymorphism in MTHFD1L was recently associated with late-onset Alzheimer’s disease (Naj et al., 2010). Interestingly, elevated circulating homocysteine levels are associated with increased cognitive decline and progression to Alzheimer’s in multiple prospective trials, although both mechanistic understanding of this association and high-quality intervention studies to test the disease-modifying potential of lowering homocysteine are lacking (Smith and Refsum, 2016). In light of the confirmed fetal neurological defects associated with defective folate metabolism, and strong associations between 1C metabolism and cognition, detailed study using isotopic tracers in the brain is warranted.
Dietary Folate Intake and Adult Disease
While folate supplementation is recommended for pregnant women to prevent birth defects, the benefit of supplementation in other adults remains unclear. As noted above, elevated homocysteine levels are correlated with diseases of aging including cardiovascular disease and cognitive decline; however, clinical trials have not shown a benefit in supplemental vitamin B intake for these conditions. Across many studies using both experimental and epidemiological methods, gastrointestinal and in particular colorectal cancer incidence is inversely correlated with folate consumption (Giovannucci, 2002). Similar results have been reported in population-based studies examining breast and pancreatic cancer (Ericson et al., 2007; Larsson et al., 2006). However, in a clinical trial to test if supplemental folic acid would suppress the recurrence of colon polyps in patients recently diagnosed with at least one adenoma, no significant difference was observed between placebo and treatment groups, with a trend toward both more polyps and carcinoma in the folate group (Chae and Yun, 2007). These results suggest that adequate dietary folate over the long term may support genome integrity, but added folate may fuel the growth of existing lesions. The impact of the intake of the 1C donor vitamin choline is less studied, with a recent review not finding any well-proven associations between choline consumption and disease (Leermakers et al., 2015).
1C Metabolism Is Upregulated in Cancer
Targeting 1C metabolism led to the first selective chemotherapeutic agents, the drug aminopterin and its closely related analog, methotrexate, which is still used in treatment of leukemias today. 1C metabolism is also targeted by 5-fluorouracil, a suicide inhibitor of TYMS, and by the new antifolate pemetrexed, approved a decade ago for treatment of mesothelioma and now also a first-line treatment for lung cancer. Unlike methotrexate, which is a near perfect structural analog of folic acid, pemetrexed is more distinct, no longer containing the pterdine ring. Both methotrexate and pemetrexed interact with a wide diversity of folate enzymes. Methotrexate most potently blocks DHFR, while pemetrexed more potently inhibits TYMS and 5-aminoimidazole-4-carboxamide ribonucleotide (AICAR) transformylase (Chattopadhyay et al., 2007; Racanelli et al., 2009). Overexpression of DHFR and TYMS (as well loss of FPGS and RFC function, mentioned above) are mechanisms of acquired resistance to these agents. While pemetrexed is argued to have superior activity in solid tumors, neither fully fulfills the potential of 1C inhibition for anticancer therapy due to toxicities caused by inhibition of 1C metabolism in non-transformed cells, including those of the intestinal epithelium and bone marrow, resulting in gastric complications, anemia, and immune deficiency. While rescue therapy with 5-formyl-THF (folinic acid, marketed as leucovorin) or folate is commonly given in conjunction with these treatments, their therapeutic index remains modest.
Serine Synthesis and Mitochondrial Folate Genes Are Overexpressed in Cancer
New genomics and metabolomics approaches have highlighted distinctive aspects of 1C metabolism in cancer and rekindled interest in targeting this pathway with more selective antifolates (Amelio et al., 2014; Locasale, 2013). Multiple studies have identified increased expression of two 1C pathways in cancer: the de novo serine pathway and the mitochondrial 1C pathway. Expression of these pathways is controlled by common master transcription factors, as well as by genomic amplifications in the first enzyme of the serine pathway, PHGDH.
Consistent with the increased demand for DNA synthesis required for the proliferative cancer phenotype, multiple 1C metabolic enzymes are upregulated in cancer (Mehrmohamadi et al., 2014). Not surprisingly given their importance as drug targets, among all genes, TYMS and DHFR are some of the most overexpressed in cancer. Unexpectedly, mitochondrial folate enzymes are equally upregulated. A recent meta-analysis of tumor gene expression revealed MTHFD2 as the most consistently overexpressed metabolic gene in cancer (Nilsson et al., 2014). The gene for the mitochondrial enzyme directly upstream in the pathway, SHMT2, was identified as recurrently amplified and important for tumor growth (Lee et al., 2014). These two genes are co-overexpressed with TYMS in a large variety of human cancers, linking the DNA synthesis demands of tumors with the mitochondrial folate system. Intriguingly, a different meta-analysis found that across cell lines, expression of these and other mitochondrial folate enzymes was better correlated with sensitivity to methotrexate than expression of the cytosolic enzymes considered to be their primary targets (Vazquez et al., 2013). Total MTHFD2 expression correlates with invasiveness and poor prognosis in breast cancer (Lehtinen et al., 2013; Liu et al., 2014). In acute myeloid leukemia (AML), 1C genes such as MTHFD2 stand out as being among the most strongly repressed by differentiating agents such as all-trans retinoic acid (Pikman et al., 2016). Thus, cancer cells activate the immune/early embryogenesis-specific mitochondrial dehydrogenase, MTHFD2. Selective targeting of this enzyme may restrict antifolate associate toxicities to the immune system and improve therapeutic indices.
Concurrent with these increases in folate metabolic enzyme expression, cancers increase metabolism of 1C sources, namely serine and glycine (Figure 5). High net consumption of serine and glycine is nearly universal across the NCI-60 cancer panel (Jain et al., 2012). Isotope tracer analysis, however, shows that only serine, and not glycine, feeds into the 1C pool in cancer cells grown in culture (Jain et al., 2012; Labuschagne et al., 2014). Serine-derived 1C units are for nucleotide synthesis, as homocysteine remethylation is not observed in cultured cancer cell lines due to a combination of excess methionine and the lack of cobalamin in standard tissue culture media (Maddocks et al., 2016). p53−/− cells cultured without serine are deficient in growth, as are tumors in mice fed serine/glycine-free diets (Maddocks et al., 2013). In addition to serine uptake, de novo serine synthesis is commonly increased. In both breast cancer and melanoma, PHGDH is overexpressed at the protein level, due in part to genomic amplification of the locus (Locasale et al., 2011; Possemato et al., 2011). Patients with breast cancer tumors that overexpress de novo serine biosynthesis enzymes have worse prognoses, but this may reflect that PHGDH overexpression is highest in the hard-to-treat triple-negative breast cancer subtype (Antonov et al., 2014; Kim et al., 2014).
While genomic amplification has been observed with PHGDH, high expression of most folate pathway enzymes is due to aberrant transcriptional regulation. Recently, ATF4 has been identified as an upstream regulator of both de novo serine biosynthesis and mitochondrial folate enzyme transcription. In lung cancer, ATF4 is commonly activated by NRF2 due to mutations in the NRF2 suppressor KEAP1 (DeNicola et al., 2015; Kansanen et al., 2013). ATF4 can also be activated by mTOR, linking the PI3K signaling axis to overexpression of 1C genes (Ben-Sahra et al., 2016).
GCS activity appears to be important in some tumors. In lung cancer tumor-initiating cells, overexpression of GLDC is required for maintenance of the proliferative phenotype and is itself transforming (Zhang et al., 2012). This required the catalytic activity of GLDC and was associated with changes in 1C metabolites, most notably thymidine, but the authors were unable to prove whether glycine itself was supplying carbon or whether decreasing glycine levels promoted tumor growth through other mechanisms. Kim et al. found that glycine cleavage activity was essential for xenograft growth of the glioma cell line LN229 by preventing the buildup of toxic oxidative glycine byproducts (Kim et al., 2015).
Upregulation of mitochondrial 1C metabolism contributes to cancer independently of providing 1C units for biosynthesis, in part by supporting antioxidant defense. SHMT2 plays a protective role against hypoxic stress. In MYC amplified cancer cell lines grown under hypoxia, SHMT2 is induced and shRNA knockdown leads to decreases in NADPH and impaired survival (Ye et al., 2014). In human gliomas, SHMT2 expression is highest in ischemic tumor regions showing “pseudopalisading necrosis” (Kim et al., 2015). The authors proposed that part of the protective effect of SHMT overexpression was a downregulation of aerobic glycolysis, achieved by SHMT-mediated decreases in the PKM2 activators serine, succinyl-5-aminoimidazole-4-carboxamide-1-ribose-5′-phosphate (SAICAR), and fructose 1,6-bisphophate. In a herculean study of the determinants of metastasis in patient-derived xenografts from melanoma, Piskounova et al. identified a role for the mitochondrial enzyme that consumes the 1C unit of 10-formyl-THF to make NADPH (ALDH1L2) (Piskounova et al., 2015). Expression of this enzyme counteracts oxidative stress and thereby promotes tumor metastasis. In contrast to the cytosolic isoform ALDH1L1, expression of the mitochondrial ALDH1L2 is increased in many tumor types (Krupenko et al., 2010).
New Targets for Cancer Therapy
What then about selectively targeting particular 1C enzymes for the treatment of cancer? Cancer genetics suggest two obvious pathways not targeted by existing therapies, de novo serine synthesis and mitochondrial folate metabolism (Tedeschi et al., 2015). While few good inhibitors have been published, early studies employing genetics suggest that targeting these pathways will not be straightforward.
Nilsson et al. generated constitutive shRNA knockdown cells lines targeting PHGDH and assessed their ability to form tumors as xenografts (Nilsson et al., 2012). In KRAS mutant Colon 26 cell lines, knockdown of PHGDH did not impair xenograft implantation or growth. In the PHGDH amplified MDA-MB-468 breast cancer cells, shRNAs targeting PHGDH led to a significant growth defect that was not serine rescuable and impaired xenograft growth (Possemato et al., 2011). This is likely due to off-target effects of the shRNA, as CRISPR deletions have recently been made and are defective in growth only in the absence of serine. Researchers at Novartis reported that for two out of three separate PHGDH-expressing breast cancer cell lines, induced expression of shRNA constructs targeting PHGDH did not alter growth as xenograft, despite all three cell lines being potently inhibited by knockdown in culture (Chen et al., 2013). The development of small-molecule inhibitors of PHGDH should help further illuminate functional requirements for de novo serine biosynthesis in tumors and in the host (Mullarky et al., 2016; Pacold et al., 2016). It is likely that on-target inhibition of PHGDH will impact cancer growth only in serine-poor environments. The tumor microenvironment is low in serine (Kamphorst et al., 2015). Accordingly, specific PHGDH inhibitors that do not alter tumor cell growth in standard tissue culture media may nevertheless effectively slow tumor growth in vivo due in part to their systemic, rather than tumor-localized, activity.
Several groups have now reported results of experiments genetically targeting mitochondrial 1C genes. Nilsson et al. did not observe a change in growth rate when SHMT2 was knocked down in colon cancer xenografts (Nilsson et al., 2012). In contrast, Woo and colleagues reported robust inhibition of hepatocellular carcinoma xenograft growth using inducible shRNA against SHMT2 (Woo et al., 2016). In a murine model of AML, shRNA knockdown of MTHFD2 extended survival, but animals still succumbed to their disease (Pikman et al., 2016). Ducker et al. generated cancer cell lines with complete deletion for MTHFD2 using CRISPR. When grown as xenografts, only a modest decrease in final tumor volume was observed, with reversal of cytosolic 1C flux meeting 1C demand (Ducker et al., 2016). Simultaneous deletion of the cytosolic enzyme SHMT1 blocked xenograft formation, indicating a requirement for tumor-intrinsic 1C production. When evaluated within the context of relevant experiments from cultured cells, these in vivo studies suggest that although multiple 1C enzymes are highly overexpressed in cancer, no single enzyme’s catalytic activity is likely to be essential for tumorigenesis. Thus, rationally targeting this pathway may require inhibiting multiple enzymes. Alternatively, combined modulation of tumor and systemic metabolism may prove more effective than targeted interventions that hit only the tumor without also altering the availability of circulating nutrients.
Outlook
It has been 70 years since Sidney Farber’s seminal experiments with the antifolate aminopterin in pediatric leukemia proved the importance of folate metabolism to cancer cell growth, launching the modern science of chemotherapy. In parallel, the role of folate in 1C metabolic processes that contribute to fetal growth, bacterial infection, and immune activation were uncovered, all leading to significant medical advances. Nevertheless, our ability to selectively target 1C metabolism therapeutically remains crude. Two major obstacles have prevented the development of more specific 1C therapies. The first has been a lack of understanding of the specific ways in which 1C metabolism is altered in disease. New genetics data highlight unique and potentially actionable nodes of this metabolism in cancer, such as MTHFD2. Excitedly, new data on T cell activation may provide similar insight into the immune system, although overlap in tumor and T cell metabolism may limit therapeutic options for cancer. Second, and relatedly, there has been only a belated understanding of the complete scope of folate-supported reactions and products, and how non-1C “by-products” may contribute to pathogenesis. Whereas most existing 1C drugs have been focused on inhibiting the active transformation of folate 1C into DNA bases, such as inhibiting TYMS by 5-fluorouracil, new work highlights the critical roles 1C metabolism plays in supporting antioxidant defense through glutathione and NADPH. Importantly, these by-products have cell-type and context-dependent importance, potentially allowing for greater selectivity than general inhibition of nucleotide or DNA synthesis.
Metabolism in general and specifically folate metabolism have benefitted tremendously from the development of modern genetic and analytical tools. As these techniques have matured, our understanding of the complexity of these pathways has deepened and much is known beyond the “textbook” description. It is now time to leverage this knowledge for translational benefit, in the form of a new generation of effective folate inhibitors and modulators.
Acknowledgments
G.S.D. is supported by a postdoctoral fellowship from the American Cancer Society (PF-15-190-01-TBE). J.D.R. is supported by NIH grants R01CA163591 and P30DK019525 and Stand Up to Cancer. We thank Li Chen, Jonathan Ghergurovich, and all members of the J.D.R. lab for helpful discussions. J.D.R. is a founder and SAB member of Raze Therapeutics, which targets 1C metabolism.
References
- Abarinov EV, Beaudin AE, Field MS, Perry CA, Allen RH, Stabler SP, Stover PJ. Disruption of shmt1 impairs hippocampal neurogenesis and mnemonic function in mice. J Nutr. 2013;143:1028–1035. doi: 10.3945/jn.113.174417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acuna-Hidalgo R, Schanze D, Kariminejad A, Nordgren A, Kariminejad MH, Conner P, Grigelioniene G, Nilsson D, Nordenskjöld M, Wedell A, et al. Neu-Laxova syndrome is a heterogeneous metabolic disorder caused by defects in enzymes of the L-serine biosynthesis pathway. Am J Hum Genet. 2014;95:285–293. doi: 10.1016/j.ajhg.2014.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amelio I, Cutruzzolá F, Antonov A, Agostini M, Melino G. Serine and glycine metabolism in cancer. Trends Biochem Sci. 2014;39:191–198. doi: 10.1016/j.tibs.2014.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An S, Kumar R, Sheets ED, Benkovic SJ. Reversible compartmentalization of de novo purine biosynthetic complexes in living cells. Science. 2008;320:103–106. doi: 10.1126/science.1152241. [DOI] [PubMed] [Google Scholar]
- Anderson DD, Stover PJ. SHMT1 and SHMT2 are functionally redundant in nuclear de novo thymidylate biosynthesis. PLoS ONE. 2009;4:e5839. doi: 10.1371/journal.pone.0005839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson DD, Quintero CM, Stover PJ. Identification of a de novo thymidylate biosynthesis pathway in mammalian mitochondria. Proc Natl Acad Sci USA. 2011;108:15163–15168. doi: 10.1073/pnas.1103623108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson DD, Eom JY, Stover PJ. Competition between sumoylation and ubiquitination of serine hydroxymethyltransferase 1 determines its nuclear localization and its accumulation in the nucleus. J Biol Chem. 2012a;287:4790–4799. doi: 10.1074/jbc.M111.302174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson DD, Woeller CF, Chiang EP, Shane B, Stover PJ. Serine hydroxymethyltransferase anchors de novo thymidylate synthesis pathway to nuclear lamina for DNA synthesis. J Biol Chem. 2012b;287:7051–7062. doi: 10.1074/jbc.M111.333120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonov A, Agostini M, Morello M, Minieri M, Melino G, Amelio I. Bioinformatics analysis of the serine and glycine pathway in cancer cells. Oncotarget. 2014;5:11004–11013. doi: 10.18632/oncotarget.2668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey LB, Berry RJ. Folic acid supplementation and the occurrence of congenital heart defects, orofacial clefts, multiple births, and miscarriage. Am J Clin Nutr. 2005;81:1213S–1217S. doi: 10.1093/ajcn/81.5.1213. [DOI] [PubMed] [Google Scholar]
- Bao XR, Ong SE, Goldberger O, Peng J, Sharma R, Thompson DA, Vafai SB, Cox AG, Marutani E, Ichinose F, et al. Mitochondrial dysfunction remodels one-carbon metabolism in human cells. eLife. 2016;5:e10575. doi: 10.7554/eLife.10575. Published online June 16, 2016. http://dx.doi.org/10.7554/eLife.10575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barlowe CK, Appling DR. In vitro evidence for the involvement of mitochondrial folate metabolism in the supply of cytoplasmic one-carbon units. Biofactors. 1988;1:171–176. [PubMed] [Google Scholar]