Re:The significant role of interleukin-6 and its signaling pathway in the immunopathogenesis and treatment of breast cancer
작성자문형철작성시간20.01.29조회수1,166 목록 댓글 0BEYOND REASON
암환자가 설탕(음료수, 아이스크림, 빵)을 먹으면 인슐린이 증가하고
인슐린은 암세포의 염증환경을 지속적으로 자극함.
CRP 수치가 높은 것으로 판단 가능함.
IL 6 암세포의 염증 관여에 관한 연구논문!!
ReviewThe significant role of interleukin-6 and its signaling pathway in the immunopathogenesis and treatment of breast cancer
Keywords
1. Introduction
Breast cancer, one of the most abundant malignant solid tumors, has been considered as the main cause of cancer-related death in women globally [1]. Despite the presence of extensive attempts and various therapeutic approaches, the way to reach a complete remission in breast cancer is yet complex [2]. Our previous approaches for modulation of tumor microenvironment with various immunotherapeutic methods had some hopeful outcomes [[3], [4], [5]]. However, it seems a long road remains to walk through breast cancer therapy. The relation of inflammation and various types of cancer has been suggested by several studies. As we recently showed [6], anti-inflammatory agents can attenuate tumor growth in the breast cancer-bearing mice implying the importance of inflammatory microenvironment in tumor growth. Malignant cells exhibit a high proliferation, which can be enhanced by inflammation. The inflammatory molecules in the tumor microenvironment are mainly secreted by tumor cells themselves and/or other stromal cells [7]. Interleukin-6 (IL-6) is a pro-inflammatory cytokine released by various cells in the tumor microenvironment including the cancerous cells. IL-6 plays a critical role in the expansion and differentiation of tumor cells [8,9]. Increased levels of IL-6 in the serum and tumor site has been demonstrated in several cancers including breast cancer [10]. While this increase is usually accompanied with poor prognosis and lower survival in breast cancer patients, downregulation of IL-6 is related to the better response to treatment [11,12]. IL-6 can affect all aspects of tumorigenesis process by regulating proliferation, apoptosis, metabolism, survival, angiogenesis, and metastasis [13]. IL-6 can also modulate a tumor therapeutic resistance such as multidrug resistance (MDR) [14].
While the obesity is considered as an important risk factor for breast cancer [15], it has been shown that the adipose tissue generates about the 30% of circulating IL-6. On the other hand, cancer-associated adipocytes trigger radio-resistance in breast cancer by secreting IL-6 [16]. Gyamfi and coworkers demonstrated that matured human adipocytes could intensify the aggressive behavior of breast cancer cells and induce an epithelial-mesenchymal transition (EMT)-phenotype through paracrine IL-6 [17]. These facts declare the importance of obesity/IL-6 axis for promoting cancer and therapeutic resistance [18].
Therefore, it seems that blocking IL-6 and/or its receptor can be considered as a potent therapeutic approach for cancers associated with high levels of IL-6 such as breast cancer. This review will try to clarify the role of IL-6 and its receptor in the immunopathogenesis and treatment of breast cancer.
2. IL-6/IL-6R
IL-6 (which is also known as IFN-β2, hepatocyte-stimulating factor, and hybridoma/plasmacytoma growth factor) was identified in 1986 for the first time as B cell stimulation factor which enhances the differentiation of effector B cells toward antibody-producing cells [19,20]. It is a small glycopeptide (185 amino acid, 25 kDa) [19], and secreted by the wide variety of immune and non-immune cells, including T and B cells, monocytes, endothelial cells, fibroblasts, keratinocytes, mesangial cells, and adipocytes. Interestingly, it has been shown that several tumor cells including breast cancer [21], colorectal cancer [22], lung cancer [23], prostate cancer [24], ovarian carcinoma [25], pancreatic cancer [26], and multiple myeloma [27] can also generate this cytokine. IL-6 can modulate several immune and physiological processes in the body, such as the generation of acute-phase proteins, inflammation, antigen-specific immune responses, hematopoiesisand cellular metabolism [28].
Several immune and non-immune cells including T cells, activated B cells, neutrophils, monocytes, and hepatocytes express the IL-6 receptor (IL-6R) [29]. IL-6 has two types of receptors including the transmembrane IL-6 receptor (mIL-6R) expressed on the cell surface and soluble IL-6 receptor (sIL-6R) present in the circulation [30]. The mIL-6R contains a short cytoplasmic domain that is not involved in ligation-derived signaling. The mIL-6R is also known as IL-6Rα, gp80 or CD126. There are two pathways for the generation of sIL-6R including the alternative splicing of mIL-6R mRNA(10%) and the spatial effect of proteinases ADAM10 and ADAM17 (90%) [30]. The sIL-6R lacks the cytoplasmic and transmembrane domains, but the essential domains involved in binding to IL-6 are present which mediate ligation with IL-6 with comparable affinity to mIL-6R.
The mIL-6R and sIL-6R cannot transduce intracellular signaling themselves, and require another transmembrane protein which is known as glycoprotein130 (gp130, IL6ST, IL6Rβ or CD130). The gp130 can form a low-affinity complex with IL-6R and mediate intracellular cell signaling. It is expressed in various cells and participates in the development, growth, cell survival and tissue homeostasis [31]. The cytoplasmic domain of gp130 contains the critical area such as SHP-2 domain and YXXQ motif which is required to start intracellular signaling. Following ligation of gp130 to IL-6/IL-6R, gp130 forms a homodimerized structure and activates cytoplasmic tyrosine kinasesleading to phosphorylation of different transcription factors. The soluble form of gp130 (sgp130) is mainly produced through alternative splicing. The sgp130 is present in blood circulation and its concentration has a direct effect on the inflammation and cancer. It has been demonstrated that sgp130 may form a complex with the IL-6/sIL-6R in the circulation and prevent trans-signaling [32,33].
3. IL-6 signaling pathways
There are various signaling pathways for IL-6 [34] (Fig. 1). Ligation of IL-6 with IL-6R activates Janus kinase (JAK) tyrosine kinases leading to phosphorylation of signal transducer and activator of transcription 3 (STAT3). Phosphorylation induces homodimerization and entrance of STAT3 into the nucleus [35]. Stimulation of IL-6/JAK/STAT3 pathway in cancer cells modulates the expression of several genes involved in the proliferation, survival, and transformation. The suppressors of cytokine signaling (SOCS) molecules and protein inhibitors of activated STAT (PIAS) proteins are the main modulators of the STAT3 pathway. Activated STAT3 usually enhances the expression of these inhibitors under physiological conditions.
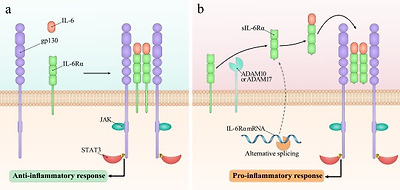
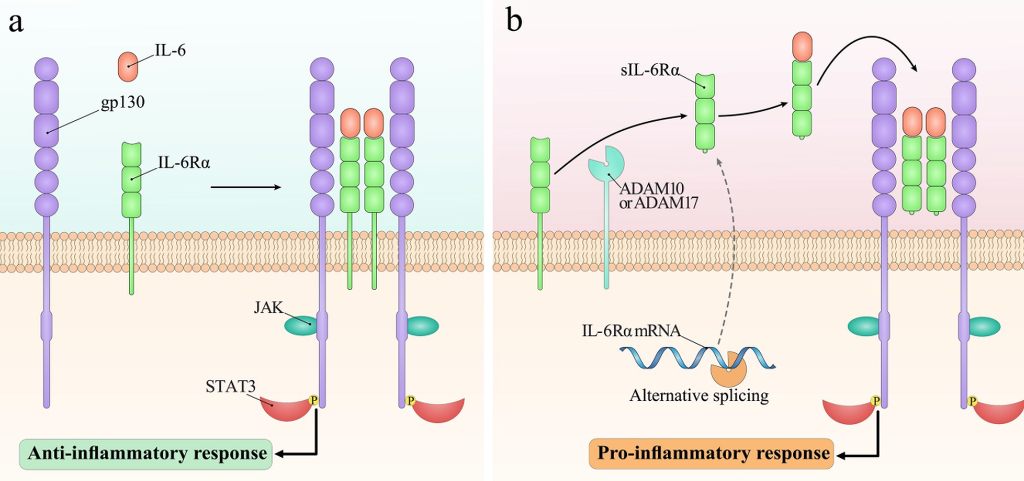
Fig. 1. Different signaling pathways stimulated by IL-6. (a)Classic signaling, which is mainly driven in leukocytes and liver cells, develops through binding of IL-6 to the mIL-6Rα and forming the complex with gp130. (b)On the other hand, trans-signaling can be driven by IL-6 in all gp130-expressing cells. While the stimulation of classic signaling promotes the anti-inflammatory responses, induction of trans-signaling leads to the development of pro-inflammatory responses.
Ras, which is another molecule induced following ligation of IL-6 to its receptor, can hyperphosphorylate and activate mitogen-activated protein kinases (MAPK). MAPK then activates various transcription factors involved in enhancing cell growth, increasing immunoglobulin synthesis, and generating acute phase protein [36].
The phosphoinositol-3 kinase (PI3K)–protein kinase B (PkB)/Akt is another signaling pathway which is induced by IL-6. Following phosphorylation by JAK, PI3K phosphorylates and converts phosphatidylinositol-4,5-bisphosphate (PIP2) to phosphatidylinositol-3,4,5-trisphosphate (PIP3). PIP3 subsequently phosphorylates PkB/Akt serine/threonine kinase [37]. Afterward, Akt modulates the expression of several genes involved in the cell survival [38].
4. IL-6/IL-6R in breast cancer
Local and systemic overexpression of IL-6 has been reported in several cancer types such as breast cancer [39]. Upregulation of IL-6 serum levels is generally associated with poor prognosis and low survival rate in patients with breast cancer [11] (Fig. 2). On the other hand, it has been shown that STAT3 is highly active in more than 50% of breast cancers and this is an important issue, because we know IL-6 is its primary activator [40]. Several cell types such as cancer cells, tumor-associated macrophages (TAMs), helper T (Th) cells, myeloid-derived suppressor cells (MDSCs) and fibroblasts are considered as the primary sources of IL-6 in the tumor microenvironment [41,42]. Thus, it seems we can suppose a new tumor promoting mechanism for these cells in addition to their conventional previously described tumor promoting mechanisms [43]. Cancerous cells usually use IL-6 as a growth factor in an autocrine manner, and paracrine release of IL-6 by other cells has less importance in survival and progression [44]. Nevertheless, both autocrine and paracrine release of IL-6 affects tumor progression via IL-6 trans-signaling [9,45].
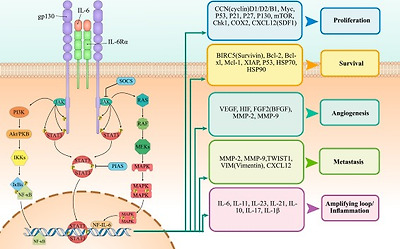
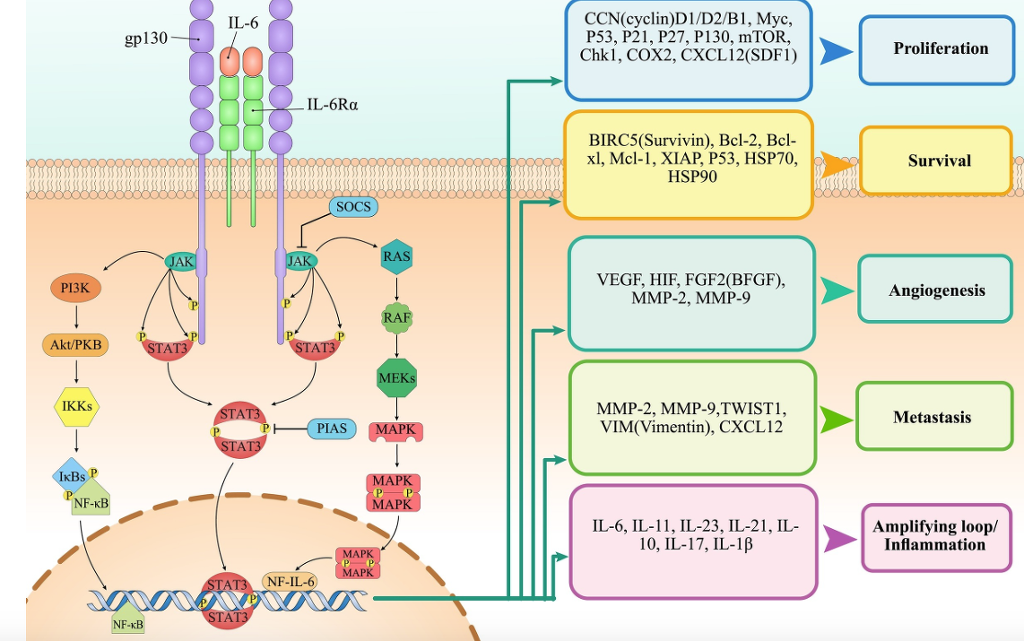
Fig. 2. The role of IL-6 in tumor development. Ligation of IL-6 with its receptor on cancer cells can induce various tumor-promoting pathways including JAK/STAT3, PI3K/AKT and Ras/MAPK which leads to expression of molecules involved in survival, proliferation, invasion, migration, and angiogenesis.
The responsiveness of breast cancer cells to IL-6 intimately depends on the expression of estrogen and progesterone receptors (ER and PR). Hormone-sensitive cells exhibit a higher response to IL-6 than hormone insensitive cells, which is associated with the intrinsic generation of higher IL-6 in these cells [46,47]. While the ER-expressing breast cancer cells mostly secrete the sIL-6R, ER-negative cells mainly express the mIL-6R [48]. It has also been shown that IL-6 suppresses ER-negative cells under normal condition in an autocrine manner [49].
IL-6 also increases estrogen levels in circulation and tumor site by activating estrogen-generating enzymes including aromatase, estrone sulfatase, and 17 β-hydroxysteroid dehydrogenase [50]. Estrogen sulfate remains in circulation more than estrogen, so that it is considered as an estrogen reservoir. On the other hand, it is demonstrated that the estrone sulfatase is overexpressed in malignant breast tissues. Accordingly, IL-6 is a crucial modulator of converting estrone to estradiol in MCF-7 ER-positive cells [51], which may imply the cause of high concentration of estradiol in malignant breast tissue.
The IL-6 polymorphisms have also been linked with breast cancer risk. Several studies have performed on the IL-6 promoter single nucleotide polymorphism (SNP) 174 G > C (rs1800795), where IL-6 was upregulated in the samples with the expression of 174 G allele and downregulated in the samples with 174C allele [52]. Another study implied that GG SNP at IL-6 (rs1800795) could enhance metastasis incidence of primary breast cancer [53]. It has also been shown that IL6 rs1800797 AG or AA genotypes are related to a lower disease-free survival [54]. A cohort investigation of 634 primary breast cancer patients in Sweden indicated that regardless of ER-status, chemotherapy-treated patients with C-genotype show a higher risk of early events compared to GG-genotype [55]. Interestingly, polymorphisms of the IL-6R gene can also affect the breast cancer prognosis [56]. These studies suggest that genetic variants of IL-6 and IL-6R can help to the diagnosis and better treatment or prevention, but still we need further investigations to highlight their relation.
During metastasis, the major cause of breast cancer-related deaths, malignant cells migrate toward distant organs by invading surrounding local tissues and acquiring a mesenchymal phenotype. The mesenchymal-to-epithelial transition and epithelial-to-mesenchymal transition (EMT) are two phenotype switching processes of metastatic cancer cells [57]. It is well-known that inflammation plays a crucial role in EMT promotion [57,58]. Accordingly, it has been demonstrated that IL-6 participates in EMT and enhances the recruitment of mesenchymal stem cells (MSC) toward human breast cancer cells [59,60]. IL-6 enhances the metastasis of many cancers to the bone by upregulating CXCR4 through STAT3 and c-Jun [61,62]. It has been shown that autocrine release of IL-6 by MCF-7 cells enhances their invasion capacity to the extracellular matrix [63].
The loss of function in the E-cadherin, a membrane adhesion molecule, which leads to mobilize fixed tumor cells is one of the key events in the EMT process [64]. Treatment of MCF-7 cells with IL-6 induced the EMT phenotype through upregulating molecules such as N-cadherin, Snail, Vimentin, and Twist, and downregulating E-cadherin [65].
Furthermore, IL-6/STAT3 axis facilitates angiogenesis in several cancers by upregulating VEGF, MMP9, and bFGF in tumor-associated endothelial cells, tumor-associated macrophages and MDSCs [63,66,67]. It is also reported that IL-6 enhances Notch-3-mediated ERK activation which leads to induction of CA-IX (a hypoxia survival gene) and JAG-1 (Notch ligand) in breast cancer. IL-6-induced CA-IX enhances the invasive potential of breast cancer cells and mammospheres [63].
5. IL-6/IL-6R as a therapeutic target for cancer therapy
As discussed earlier, IL-6 has a critical function in cancer development and resistance to current anti-cancer-therapeutics. IL-6 exerts these functions in cooperation with various factors; however, STAT3 is the most important player among them [8,68]. Accordingly, targeting IL-6/JAK/STAT3 pathway was associated with promising outcome in several cancer types. Therefore, blockage of this pathway can be considered as a potent therapeutic approach for cancers associated with overexpression of IL-6 including breast cancer. Various therapeutics including the anti- IL-6/IL-6R or anti-sIL-6R monoclonal antibodies (mAb) [12,69,70], selective inhibitors of IL-6 downstream signaling factors such as STAT3 or kinase inhibitors (like JAK inhibitor) [71] have been used for blockage of IL-6/JAK/STAT3 pathway, as discussed below(Fig. 3).
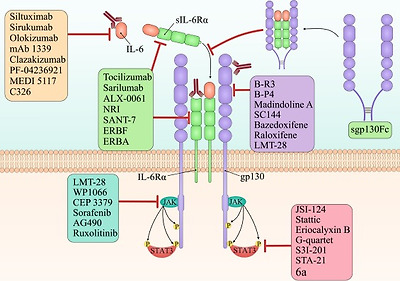
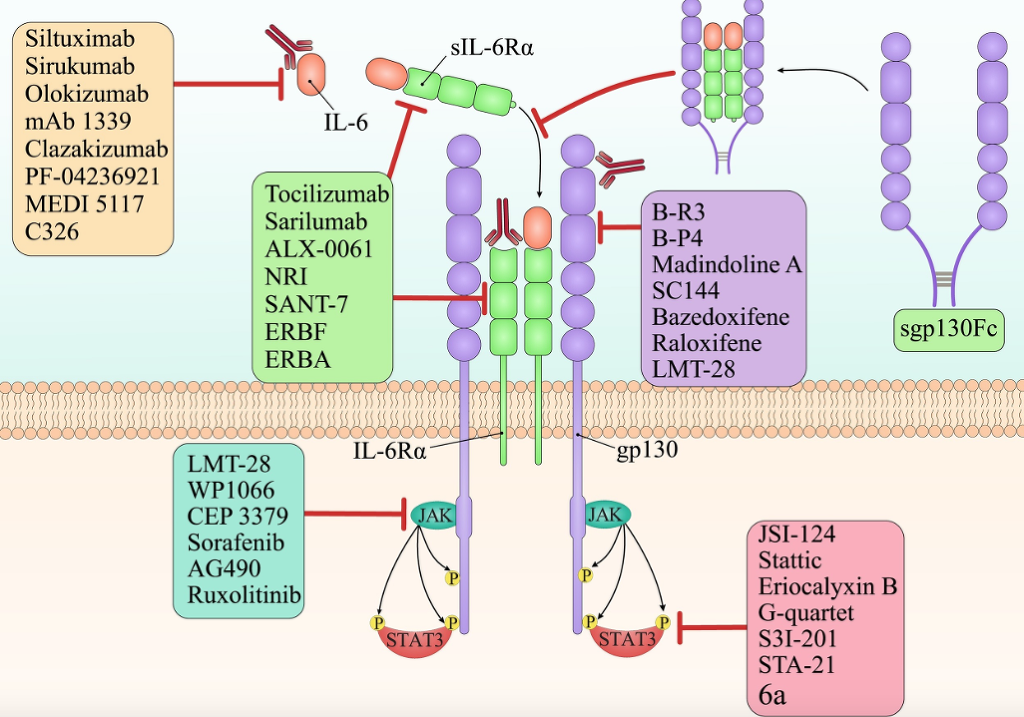
Fig. 3. Targeting IL-6 in cancer therapy. There are various types of targeted agents which inhibit one of the crucial parts of the IL-6 signaling pathway. Siltuximab, sirukumab, olokizumab, mAb 1339, clazakizumab, PF-04236921, MEDI 5117, and C326 are the anti-IL-6 monoclonal antibodies. Tocilizumab(mAb), sarilumab(mAb), ALX-0061(bi-specific nanobody), NRI(anti-IL-6R scFv of tocilizumab fused to IgG1 Fc), SANT-7(super-antagonist of IL-6), ERBF(IL-6R–antagonist), and ERBA(IL-6R–antagonist) are the IL-6R specific inhibitors which inhibit both the classic and trans-signaling of IL-6. However, soluble gp130-Fc fusion protein is a specific inhibitor for trans-signaling without effect on classic-signaling of IL-6. B-R3(mAb), B-P4(mAb), madindoline A(gp130 antagonist), SC144, bazedoxifene(selective ER-modulator), raloxifene (selective ER-modulator), and LMT-28 suppress IL-6 signaling pathway by inhibiting the gp130. LMT-28, WP1066, CEP 3379, sorafenib, AG490, and ruxolitinib are small molecules that inhibit JAKs. Finally, JSI-124, stattic, eriocalyxin B, G-quartet, S3I-201, STA-21, and 6a are STAT3′s inhibitors.
6. IL-6 direct inhibitors
Some investigators have tried to directly block IL-6 for treating cancer which is summarized below (Table 1).
Table 1. Studies related to the blockage of IL-6 for cancer therapy.
| Target | Agent name | Structure | Study type | Models/cell lines | Cancer type | Result(s) | Ref. |
|---|---|---|---|---|---|---|---|
| IL-6 | Siltuximab (CNTO 328) | Chimeric mAb | In vitro, in vivo | SKOV-3 and CAOV-3 cell lines, xenograft mouse modeم | Ovarian cancer | Inhibition of STAT3 and downstream factors which led to cancer cells sensitivity to paclitaxel | [74] |
| In vitro | LNCaP-IL6+ cell line | Prostate cancer | Enhanced cancer cells sensitivity to apoptosis | [82] | |||
| In vitro, in vivo | H1650, H322, H157 cell lines/ mouse xenograft model | Lung cancer | Tumor inhibition | [80] | |||
| Clinical trial | – | Multiple myeloma | Complete remission | [83] | |||
| mAb 1339 (OP-R003) | human mAb | In vitro, in vivo | INA6, XG1cell lines/ mouse xenograft model | Multiple myeloma | Growth inhibition, inhibition of osteoclastogenesis and bone remodeling | [88] | |
| Clazakizumab (BMS945429, ALD518) | Humanized mAb | Clinical trial | Phase I and II | Non-small cell lung cancer | Decreased NSCLC-related anemia and cachexia | [90] |
6.1. Siltuximab (CNTO 328, Sylvant)
Siltuximab (CNTO 328), an anti-IL-6 chimeric mAb, is recently licensed for the treatment of multicentric Castleman’s disease (a rare B-cell lymphoproliferative disorder) [72]. Studies on siltuximab have shown its efficacy in the treatment of various cancers in both mono- or combination-therapy with other anti-cancer drugs [[73], [74], [75], [76], [77], [78], [79], [80], [81]]. Based on results derived from the preclinical studies, siltuximab has prevented the IL-6-mediated progression of ovarian cancer [74], increased survival in advanced prostate cancer [82], and attenuated lung cancer development [80]. Administration of siltuximab into patients with relapsed refractory multiple myeloma was also associated with the complete remission[83]. In contrast, lack of efficacy has also been reported in clinical trials on the patients with advanced multiple myeloma, ovarian cancer, hormone-refractory prostate cancer, renal cancer, and non-Hodgkin’s lymphoma [78]. Although there is no preclinical or clinical data investigating the efficacy of this mAb in patients with breast cancer, it is reported that siltuximab could suppress the expansion of tumor cells implying its possible efficacy in vivowhich requires further investigations [84].
6.2. Sirukumab (CNTO 136)
Sirukumab, another anti–IL-6 mAb, binds with high specificity and affinity to IL-6 and blocks its function. Majority of studies have evaluated the efficacy of sirukumab on patients with autoimmune disorders including systemic lupus erythematosus (SLE), rheumatoid arthritis (RA), and cutaneous lupus erythematosus [85] which was associated with desirable pharmacokinetic, and low immunogenicity [86]. Regarding these features, it seems that it would be a good candidate for breast cancer therapy.
6.3. Olokizumab (CP6038)
Although the therapeutic potential of olokizumab, a humanized anti–IL-6 mAb, has been evaluated in patients with RA [85], there is no data about its potential as a therapeutic drug in patients with breast cancer. It binds to the gp130 binding site on IL-6 and blocks the functional activities of IL-6.
6.4. mAb 1339 (OP-R003)
The mAb 1339, a fully human anti-IL-6 mAb, is derived from the previously developed murine anti-human IL-6 mAb B-E8 (Elsilimomab) [87]. Anti-cancer activity of mAb 1339 has been demonstrated in the multiple myeloma, in vitro[88]. However, it is not investigated in the treatment of other cancers, including breast cancer.
6.5. Clazakizumab (BMS945429, ALD518)
Clazakizumab, a humanized anti–IL-6 mAb, is used in the phase II clinical trial for the treatment of RA patients [89]. It has also been evaluated for the treatment of non-small cell lung cancer (NSCLC) and patients with head and neck cancer [90]. However, it has not been evaluated against breast cancer.
PF-04236921 (a humanized anti-IL-6 mAb) [85], MEDI 5117 (a humanized anti-IL-6 mAb) [85], and C326 (AMG-220, anti-IL-6/anti-IgG avimer protein) [91] are the other agents against IL-6, but none of them have been evaluated for treatment of breast cancer.
7. IL-6R direct inhibitors
Blockage of IL-6R is considered as another effective anti-cancer therapeutic approach which is discussed below (Table 2).
Table 2. Studies related to blockage of IL-6R for cancer therapy.
| Target | Agent name | structure | Study type | Models/cell lines | Cancer type | Result(s) | Ref. |
|---|---|---|---|---|---|---|---|
| IL-6R | Tocilizumab (Actemra, RoActemra) | Humanized mAb | In vivo | HT-29 and COLM-5 cell lines/ mouse xenograft model | Colon cancer | Inhibits tumor angiogenesis | [67] |
| In vitro, in vivo | 786-O cell line/ mouse xenograft model | Renal cell carcinoma | Suppression of tumor growth | [92] | |||
| In vivo | Mouse xenograft model | Lung cancer | Enhanced the survival of mice | [93] | |||
| In vivo | Mouse xenograft model | Breast cancer | Decreased tumor growth, metastasis, and inflammation | [60] | |||
| NRI | Anti-IL-6R scFv of tocilizumab fused to IgG1 Fc | In vivo | S6B45 cell line | Multiple myeloma | Inhibited cell growth | [100] | |
| SANT-7 | super-antagonist of IL-6 | invitro, invivo | INA-6 and XG-1 cell lines | Multiple myeloma | Arrested cell-cycle and induced apoptosis | [102] | |
| ERBF | IL-6R–antagonist | In vitro | CTLL-2, Baf3, L929, U-937, PC-12 | Various | Suppressed cancer cell growth | [103] | |
| ERBA | IL-6R–antagonist | In vitro, in vivo | – | Colon cancer | Inhibited tumor growth | [104] | |
| sgp130Fc | Soluble gp130-Fc fusion protein | In vivo | Colo357 cell line/ mouse xenograft model | Pancreatic cancer | Decreased microvessel density and significant reduction of tumor weight and metastases | [99] |
7.1. Tocilizumab (RoActemra or Actemra)
Tocilizumab, which is a humanized anti–IL-6R mAb, inhibits both sIL-6R and mIL-6R and is licensed by the FDA for treatment of patients with RA [85]. The anti-cancer effects of tocilizumab have been demonstrated in various cancer types including a colon cancer xenograft model [67], renal cell carcinoma [92], lung cancer [93] and breast cancer [42,60,94]. A rodent analog of tocilizumab, MR16-1, had also promising effect on IL-6-induced cachexia in lung cancer [95] and also inhibited the growth of the fibrosarcoma, in vivo [96]. However, there is no clinical study on the efficacy of tocilizumab in the treatment of breast cancer.
7.2. Sarilumab (SAR153191 or REGN88)
Sarilumab (KEVZARA®), a human anti-IL-6R, received the FDA license for treatment of RA on 22 May 2017 [85].
7.3. Soluble gp130-Fc fusion protein (sgp130Fc or FE 999301)
As mentioned earlier, sgp130 is an inhibitor of IL-6 trans-signaling and only binds to the complex of IL-6/sIL-6R (not IL-6 or IL-6R alone) [97]. Similar to sgp130, soluble gp130 linked to IgG-Fc (sgp130Fc) is competitive inhibitor of IL-6/sIL-6R complex and trans-signaling [33]. Regarding its specific function for targeting pro-inflammatory trans-signaling [98] and its suppressive effect on xenograft models of pancreatic cancer [99], sgp130Fc may be considered as an effective therapeutic strategy for treatment breast cancer.
There are some other agents with therapeutic potential for targeting the IL-6R/sIL-6R including ALX-0061 (a bi-specific nanobody) [98], NRI (an antiIL-6R scFv of tocilizumab fused to IgG1 Fc) [100], SANT-7 (a super-antagonist of IL-6) [101,102], ERBF (an IL-6R–antagonist) [103] and ERBA (an IL-6R–antagonist) [104], however none of them are considered for treatment of breast cancer.
8. Gp130 direct inhibitors
Regarding the tumorigenic potential of gp130 signaling, blockage of gp130 is considered as an effective therapeutic approach for cancer therapy (Table 3). Anti-gp130 mAbs, gp130 antagonists, and some chemical small molecules have been developed, however, little is known regarding their effectiveness in cancer.
Table 3. Studies related to blockage of gp130 for cancer therapy.
| Target | Agent name | Structure | Study type | Models/cell lines | Cancer type | Result(s) | Ref. |
|---|---|---|---|---|---|---|---|
| Gp130 | B-R3 | mouse mAb | In vitro | – | – | Blocked the responses to all the IL-6 cytokine family | [105] |
| B-P4 | mouse mAb | In vitro | – | – | Inhibition of gp130-induced STAT3 phosphorylation in hepatic adenomas | [106] | |
| Madindoline A | Gp130 antagonist | In vitro, in vivo | – | – | IL-6 enhances amyloid A generation and bone resorption in an experimental model of postmenopausal osteoporosis | [107] | |
| SC144 | Synthetic molecule | In vitro, in vivo | OVCAR-8, OVCAR-5, and OVCAR-3 cell lines/ mouse xenograft model | Ovarian cancer | Inhibition of gp130 suppresses STAT3 signaling and tumor growth in mouse xenograft model | [109] | |
| Raloxifene | Synthetic molecule | In vitro/ clinical trial- Phase II, III | SUM159 cell line | Breast cancer | Inhibition of STAT3 phosphorylation and IL-6-signaling prevent breast cancer | [110,111] | |
| Bazedoxifene | synthetic molecule | In vitro/ clinical trial- Phase IV, Cohort | SUM159 and MCF-7 cell lines | Breast cancer | Inhibition of constitutive STAT3 phosphorylation and IL-6-induced breast cancer cells proliferation | [111,112] | |
| LMT-28 | Synthetic molecule | In vitro, in vivo | TF-1 cells | Erythroleukemia | Suppression of IL-6–mediated cell proliferation | [113] |
8.1. Anti-gp130 mAb
B-R3, a mouse anti-gp130 mAb, has been used in the preclinical studies [105]. B-P4, which is an anti-gp130 mAb, blocks gp130-mediated activation of STAT3 and is upregulated in hepatic adenomas [106]. There is no data regarding the evaluation of this therapeutic in clinical trials.
8.2. Chemical small molecules
Madindoline A (MDL-A), a gp130 antagonist, attaches to the extracellular domain of gp130, which leads to blockage of the IL-6/IL-6R signaling pathway. It can suppress IL-6-induced osteoclastogenesis, in vitro [107,108].
SC144, another gp130 inhibitor, prevents STAT3 activation which leads to tumor growth arrest in ovarian cancer xenograft models [109].
Bazedoxifene and Raloxifene, the selective ER-modulators (SERMs), inhibit the IL-6-gp130 interaction and are evaluated clinically to treat or suppress ER-positive invasive breast cancer [110,111]. Bazedoxifene with conjugated estrogens has an inhibitory effect on breast cancer cell line [112].
LMT-28, a novel IL-6 blocker synthetic compound, can also bind to gp130 and blocks the sIL-6R/IL-6 complex signaling [113].
Although the above-mentioned agents potentially can suppress IL-6/JAK/STAT3 signaling, most of them require more additional preclinical and clinical studies for breast cancer treatment.
9. JAK inhibitors
The JAK inhibitors have been evaluated in the treatment of various cancer types (Table 4). TG101209, a JAK2 inhibitor, had synergistic effects with radiation in lung cancer [114]. Similarly, other JAK2 inhibitors including WP1066 and CEP 3379 could inhibit the tumor growth in gastric and colorectal cancer via suppressing IL-6/JAK2/STAT3 pathway [115,116]. A multi-kinase inhibitor, sorafenib, is another therapeutic that dephosphorylates STAT3 and suppresses AKT signaling in glioblastoma and prostate cancer [117]. AG490 is a potent JAK inhibitor which blocks STAT3 signaling, attenuates the invasive potential of human pancreatic cancer cells, induces apoptosis in gastric cancer cells [118,119], and enhances the secretion of anti-tumor cytokines [120]. Ruxolitinib, another JAK inhibitor, is now evaluating clinically for the treatment of leukemia [121]. Little is known about the efficacy of JAK inhibitors for the treatment of breast cancer, and this issue needs further investigations.
Table 4. Studies related to blockage of JAK signaling for cancer therapy.
| Target | Agent name | Structure | Study type | Models/cell lines | Cancer type | Result(s) | Ref. |
|---|---|---|---|---|---|---|---|
| JAK | TG101209 | Synthetic molecule | In vitro, in vivo | H460 and HCC2429 cell lines/ mouse xenograft model | Lung cancer | Inhibition of STAT3 signaling enhanced apoptosis and reduced proliferation and vascular density | [114] |
| WP1066 | Synthetic molecule | In vitro, in vivo | AGS cells/ Mouse Model | Gastric Cancer | Inhibited tumor growth | [116] | |
| CEP 3379 | Synthetic molecule | In vivo | Mouse Model of Colitis-Induced Colorectal Cancer | Colorectal cancer | Inhibited tumor growth | [115] | |
| Sorafenib | Synthetic molecule | In vitro | U87, U251, PBT015 and PBT022 cell lines | Glioblastoma | Inhibited tumor growth through inhibition of upstream kinases JAK1 and JAK2 | [117] | |
| AG490 | Synthetic molecule | In vitro | SW1990 and Capan-2 cell lines | Pancreatic cancer | Inhibition of STAT3 activation suppressed the cancer cell growth | [118] | |
| In vitro, in vivo | OV2944-HM-1 cell line/ Mouse Model | Ovarian cancer | Suppressed tumor growth and improved survival | [120] | |||
| Ruxolitinib | Synthetic molecule | Clinical trial- Phase I, II | – | Myeloid leukemia | Without satisfactory clinical benefit | [121] |
10. STAT3 inhibitors
The STAT3 inhibitors are other therapeutic tools for blocking IL-6/JAK/STAT3 signaling through inhibition of STAT3 phosphorylation (Table 5). For instance, JSI-124 prevents STAT3 phosphorylation at serine 727 leading to induction of apoptosis in leukemic B cells [122]. The similar function has reported about the effect of Stattic and Eriocalyxin B (a diterpenoid) in MDA-MB-231 and HepG2 cells through blocking STAT3 phosphorylation [123,124].The 6a compound, a selective inhibitor of STAT3 phosphorylation, is a pyrrolidinesulphonylaryl synthetic molecule with potent inhibitory effect on the IL-6/STAT3 signaling in IL-6 stimulated HeLa and MDA-MB-231 breast cancer cell lines [125]. Further studies and investigation are needed in breast cancer cell lines and patients to introduce 6a as an anticancer therapeutic agent. G-quartet oligodeoxynucleotides target the DNA-binding site in STAT3, which leads to attenuation of cancer cell expansion in breast cancer [126]. Similarly, S3I-201 (a.k.a. NSC74859) prevents STAT3 DNA-binding domain in vitro, which blocks the expansion and survival of human breast carcinoma cells [127]. Similar results were also reported regarding the effect of STA-21 on the growth inhibition and apoptosis induction in human breast carcinoma and rhabdomyosarcoma model [128,129].
Table 5. Studies related to blockage of STAT3 signaling for cancer therapy.
| Target | Agent name | Structure | Study type | Models/cell lines | Cancer type | Result(s) | Ref. |
|---|---|---|---|---|---|---|---|
| STAT3 | JSI-124 | Synthetic molecule | In vitro | BJAB, I-83, and NALM-6 cell lines | B-leukemia | Inhibition of STAT3 phosphorylation (serine 727) leads to apoptosis and cell-cycle arrest | [122] |
| Stattic | Synthetic molecule | In vitro | HepG2 cell line(liver)/ MDA-MB-231, MDA-MB-468, and MDA-MB-453 cell lines(breast) | Breast and liver cancer | Apoptosis induction | [123] | |
| Eriocalyxin B | Synthetic molecule | In vitro | HepG2 cell line(liver)/ MDA-MB-231, MDA-MB-468, and MDA-MB-453 cell lines(breast) | Breast and liver cancer | Apoptosis induction | [124] | |
| 6a | Pyrrolidinesulphonylaryl synthetic molecule | In vitro | MDA-MB-231 and HeLa cell lines | Breast cancer, cervical cancer | Selectively inhibits phosphorylation of STAT3 | [125] | |
| G-quartet | Oligodeoxynucleotides | In vitro, in vivo | MDA-MD-468(breast) and PC-3(prostate) cell lines/ mouse xenograft model | Breast and prostate cancer | Tumor growth inhibition | [126] | |
| S3I-201 | Synthetic molecule | In vitro, in vivo | MDA-MB-231 cell line/ mouse xenograft model | Breast cancer | Apoptosis induction and inhibited proliferation | [127] | |
| STA-21 | Synthetic molecule | In vitro | Saos-2, U2OS, and SJSA(Osteosarcoma), RH30, RH3 and RD2(rhabdomyosarcoma), and SK-LMS-1(leiomyosarcoma) Cell lines | Sarcoma | Apoptosis induction and inhibited proliferation | [128] | |
| In vitro | MDA-MB-231, MDA-MB-435 s, MDA-MB-453, MDA-MB-468, and MCF7 Cell lines | Breast cancer | Apoptosis induction | [129] |
11. Conclusion
Cancer cells use several mechanisms for evading anti-tumor immune responses. They recruit and induce various suppressor cells such as regulatory T cells [130], MDSCs [131], TAMs [132], and type II NKT cells [133]. Similarly, they may upregulate expression of suppressive mediators such as adenosine, negative checkpoint molecules or tumor promoting cytokines [134,135]. Among the cytokines, IL-6 is known as a key immune escape mechanism by which cancer cells can freely expand. IL-6 can promote several aspects of tumor growth and induce therapeutic resistance [69,136]. Regarding its importance, it seems that targeting IL-6/IL-6R and/or their signaling pathway (IL-6/JAK/STAT3) can be considered as a novel promising therapeutic approach for cancer therapy. Several studies have used the blockage of IL-6 or IL-6R or suppression of JAK/STAT3 to inhibit tumor growth. Use of mAbs against IL-6 or IL-6R was associated with hopeful outcomes in preclinical and clinical studies. Moreover, phase I and II clinical trials have demonstrated the efficiency of mAbs in both mono- or combination-therapy with other therapeutics such as chemo- or radiotherapy in various cancers. Despite the promising results in various cancers, little is known regarding the therapeutic potential of this approach in the treatment of breast cancer. Evaluation of various targeting approaches in the breast cancer is highly required, and future studies should focus on the specific targeting of IL-6/IL-6R or its signaling pathway (JAK/STAT) in the tumor microenvironment to elude unfavorable adverse effects.
Conflict of interest statement
None of the authors has any conflict of interest to declare. This study was supported in part by grant no. 972850 from National Institute for Medical Research Development (NIMAD).
Acknowledgments
None.
References
- [1]
- N. Harbeck, M. GnantBreast cancerLancet, 389 (10074) (2017), pp. 1134-1150
- [2]
- W.D. Foulkes, I.E. Smith, J.S. Reis-FilhoTriple-negative breast cancerN. Engl. J. Med., 363 (20) (2010), pp. 1938-1948
- [3]
- F. Jadidi-Niaragh, F. Atyabi, A. Rastegari, N. Kheshtchin, S. Arab, H. Hassannia, M. Ajami, Z. Mirsanei, S. Habibi, F. MasoumiCD73 specific siRNA loaded chitosan lactate nanoparticles potentiate the antitumor effect of a dendritic cell vaccine in 4T1 breast cancer bearing miceJ. Control. Release, 246 (2017), pp. 46-59
- [4]
- F. Jadidi-Niaragh, F. Atyabi, A. Rastegari, E. Mollarazi, M. Kiani, A. Razavi, M.Yousefi, N. Kheshtchin, H. Hassannia, J. HadjatiDownregulation of CD73 in 4T1 breast cancer cells through siRNA-loaded chitosan-lactate nanoparticlesTumor Biol., 37 (6) (2016), pp. 8403-8412
- [5]
- N. Kheshtchin, S. Arab, M. Ajami, R. Mirzaei, M. Ashourpour, N. Mousavi, N.Khosravianfar, F. Jadidi-Niaragh, A. Namdar, F. NoorbakhshInhibition of HIF-1α enhances anti-tumor effects of dendritic cell-based vaccination in a mouse model of breast cancer, Cancer immunologyImmunotherapy (2016), pp. 1-9
- [6]
- F. Hosseini, H. Hassannia, A. Mahdian‐Shakib, F. Jadidi‐Niaragh, S.E. Enderami, M. Fattahi, A. Anissian, A. Mirshafiey, P. KokhaeiTargeting of crosstalk between tumor and tumor microenvironment by β‐D mannuronic acid (M2000) in murine breast cancer modelCancer Med., 6 (3) (2017), pp. 640-650
- [7]