Abstract
Diabetes is associated with an increased risk of developing and dying from cancer. This increased risk may be due to hyperglycemia, hyperinsulinemia, and insulin resistance or other factors. Metformin has recently gained much attention as it appears to reduce cancer incidence and improve prognosis of patients with diabetes. In vitro data and animal studies support these findings from human epidemiological studies. Metformin has multiple potential mechanisms by which it inhibits cancer development and growth. For example, metaformin inhibits hepatic gluconeogenesis, thus decreasing circulating glucose levels, and it increases insulin sensitivity, thus reducing circulating insulin levels. Intracellularly, metformin activates AMPK, which decreases protein synthesis and cell proliferation. Metaformin also reduces aromatase activity in the stromal cells of the mammary gland. Finally, metformin may diminish the recurrence and aggressiveness of tumors by reducing the stem cell population and inhibiting epithelial to mesenchymal transition. Here, we discuss the metabolic abnormalities that occur in tumor development and some of the mechanisms through which metformin may alter these pathways and reduce tumor growth.
Introduction
For over a century, an association between diabetes and cancer has been recognized.1Initially, many were skeptical that the association was real, including the statistician Karl Pearson, who wrote: “I must frankly admit that at first I viewed Dr. Maynard's conclusion as in some‐way based on disregarded spurious correlation, and due to non‐allowance for population, age or general unhealthiness factors. But I have been gradually forced by the pressure of these statistical results to consider it something very real.”2 Decades of epidemiological evidence have now accumulated, supporting the link between diabetes and an increased incidence of certain cancers in different populations, after adjusting for age and other confounding factors such as obesity. In addition, epidemiological studies report that those with diabetes who develop cancer have a worse prognosis after treatment with chemotherapy or surgery and have a greater mortality than those without diabetes.3-5
Meta‐analyses have pooled data from case‐control and cohort studies to examine the association between diabetes and cancer at specific sites. These studies have demonstrated that having diabetes increases the risk of pancreatic, hepatocellular, and endometrial cancer to approximately twice that of the nondiabetic population.6-9 Some studies have reported a similar increase in the risk of bladder cancer, although others have reported a more modest increase in risk of approximately 20%.10, 11 The risk of kidney cancer is reported to be approximately 40% higher, and colorectal cancer approximately 30% higher, in diabetic individuals compared to those without diabetes.12, 13 Diabetes is associated with a 20% increased risk of breast cancer;14 those with diabetes are more likely to present with advanced stage breast cancer and are more likely than those without diabetes to die from breast cancer.4, 15 Notably, epidemiological studies have reported that the risk of developing prostate cancer is lower in those with diabetes, although in men who develop prostate cancer, diabetes is associated with a greater risk of mortality, recurrence, and treatment failure.16, 17 Across all cancers, it remains to be determined whether diabetes is truly associated with greater cancer mortality. Having diabetes almost doubles the age‐adjusted mortality in the general population without cancer; therefore, the data reporting that diabetes is associated with increased cancer mortality could simply be a reflection of the increased mortality related to having diabetes, rather than a specific increase related to having diabetes and cancer.18 Furthermore, although the body of epidemiological evidence supporting the association between diabetes and certain cancers is substantial, the data from these studies merely demonstrate that diabetes and certain cancers are associated, not that there is a causal link between the two conditions. To further understand the connection between the two conditions and to determine whether a causal relationship between diabetes and cancer development is plausible, some of the biological factors that are common to the two conditions have been identified and studied in humans, as well as in animals and in vitro systems.
In 2010, 100 years after the first publications on the subject of diabetes and cancer by Maynard and Pearson, a consensus report was published by the American Diabetes Association (ADA) and the American Cancer Society (ACS) to (1) review the scientific knowledge regarding the association between diabetes and cancer; (2) explore the risk factors common to both conditions; (3) examine their possible biological links; and (4) determine whether treatments for diabetes influence cancer risk or prognosis. The report acknowledged that for more common cancers, diabetes is associated with an increased risk, but there is less evidence for less common cancers; therefore, it calls for more research to determine whether there is an association between diabetes and less common tumors. The risk factors cited as common to both conditions and the proposed biological links are outlined in Table 1; they include nonmodifiable (age, sex, and race/ethnicity) and modifiable (overweight/obesity, smoking, alcohol intake, physical activity level, and diet) risk factors. The prevalence of most cancers increases with age, with 78% of newly diagnosed cancers occurring in those over 55 years of age. Similarly, the prevalence of diabetes increases with age, from 10.8% in those aged 40–59 years up to 23.8% in those aged 60 years or more. Apart from tumors that are sex‐specific or almost completely sex‐specific (prostate, testicular, cervical, endometrial, and breast cancers), men are more likely than women to develop cancer and men also have a slightly higher risk of developing diabetes. Cancer and diabetes risks are also different in different ethnic groups. African Americans are more likely to develop and die from cancer and are also more likely to develop diabetes than other ethnic groups. Highlighting these risk factors can identify individuals at greater risk of developing diabetes and cancer and direct efforts toward reducing their modifiable risk factors; however, it does not answer the question of whether diabetes and cancer are more likely to occur in an individual because of risk factors common to both conditions, or if having the metabolic disturbances that occur in diabetes increase the risk of cancer.19
| Factors linking diabetes and cancer from the American Diabetes Association/American Cancer Society consensus report 2010 | ||
|---|---|---|
| Nonmodifiable risk factors | Modifiable risk factors | Biological links |
| Age | Overweight/obesity | Hyperglycemia |
| Sex | Physical activity | Insulin |
| Race/ethnicity | Diet | IGF‐1 |
| Estrogen and androgen bioavailability | ||
| Cytokines | ||
Type 2 diabetes (T2DM) is characterized by insulin resistance and endogenous hyperinsulinemia, before the eventual development of the frank hyperglycemia by which we define the condition.20, 21 Both hyperglycemia and hyperinsulinemia have been cited as possible mechanisms through which diabetes may stimulate tumor growth.19 Obesity has been known for many years to increase the risk of T2DM and is itself associated with an increased risk of cancer.22, 23 Obesity potentially contributes to tumor progression through the effects of hyperinsulinemia, increased circulating estrogen, and chronic inflammation with altered regulation of cytokines and adipokines such as tumor necrosis factor alpha (TNF‐α), interleukin‐6 (IL‐6), fatty acid synthase, resistin, leptin, and adiponectin.24, 25Therefore, in many individuals with diabetes and obesity there are multiple physiological elements that may contribute to tumor development. Diabetes treatments may also influence the risk of developing cancer. Recent population‐based cohort studies have reported that insulin secretagogues and insulin analogs may increase the risk of developing cancer, whereas metformin treatment may decrease the risk.26-30 None of these studies are randomized controlled trials and there are multiple factors that may be confounding the data, such as comorbidities, duration of diabetes, glycemic control, and preexisting undiagnosed tumors. In view of these potential confounding factors, the ADA/ACS have concluded that the data on these medications is inconclusive and further studies need to be conducted to investigate these potential links.19
In this review, we will examine the clinical and scientific studies that explore the roles of hyperglycemia and hyperinsulinemia in cancer development as well as those that are investigating whether metformin reduces cancer development and progression. There are many factors apart from hyperinsulinemia and hyperglycemia that are important in the relationship between diabetes and cancer metabolism, including oncogenes and tumor suppressor genes, glutamine metabolism, inflammation, and obesity; these have recently been reviewed elsewhere.25, 31-34
Hyperinsulinemia, hyperglycemia, and cancer
Insulin
Insulin resistance and hyperinsulinemia are important factors in the development of type 2 diabetes. Insulin is known to stimulate cell proliferation and injection of insulin in rats promoted carcinogen‐induced colon cancer.23, 35, 36 Some large prospective studies in humans have enrolled cohorts of men and women, gathered anthropometric data and serum at baseline, and followed them periodically for many years to identify factors that may influence future cancer development. One such study, the Women's Health Initiative Observational Study (WHI‐OS), was conducted in 40 clinical centers across the United States. The study reported that women with endogenous insulin levels in the highest quartile of the normal range and those with insulin resistance had a higher risk of developing postmenopausal breast cancer compared to those with lower insulin levels and without insulin resistance.37 Another smaller study examined insulin levels in women with early stage breast cancer and reported that those with insulin levels in the highest quartile had a risk of recurrence and death that was two to three times that of women with insulin levels in the lowest quartile.38 The Physicians Health Study and Nurses Health Study both measured C‐peptide (as a reflection of insulin secretion) and found that increased levels were strongly associated with an increased risk of colorectal cancer in men and weakly associated in women.39, 40 In Finnish men, who were a subset of the Alpha‐Tocopherol, Beta‐Carotene Cancer Prevention Study cohort, fasting insulin concentrations were found to be higher, as was insulin resistance in men who subsequently developed prostate cancer.41 Although most studies have reported a positive association between insulin levels and breast cancer, the Nurse's Health Study II, which included predominantly premenopausal women, did not find an association between insulin and breast cancer. The discrepant associations between insulin and breast cancer in pre‐ and postmenopausal women may be due to differences in tumor hormone receptor status and the significance of insulin's indirect contribution to circulating estrogen levels in pre‐ and postmenopausal women. Additionally, premenopausal women may have a shorter duration of exposure to hyperinsulinemia and have other risk factors for breast cancer.42 Overall, the majority of human epidemiological studies suggest that elevated insulin levels promote the development of certain cancers. Human tissue and animal in vitro, studies provide evidence that insulin may have both direct and indirect actions that promote cancer progression.
As noted previously, insulin resistance and hyperinsulinemia occur prior to the onset of hyperglycemia in the setting of type 2 diabetes. Hyperinsulinemia may have direct effects on tumor growth through its action on the insulin receptor (IR) and/or IGF‐1 receptor (IGF‐1R).43 Additionally, hyperinsulinemia leads to increased hepatic IGF‐1 production indirectly by increasing hepatic growth hormone receptor (GHR) levels, through which GH stimulates IGF‐1 expression.44 Higher circulating IGF‐1 levels, even when within the normal range, were associated with an increased risk of cancer mortality in men over the age of 50 years in the Rancho Bernardo Study, and meta‐analyses of other epidemiological studies have reported an increased risk of colorectal cancer, premenopausal breast cancer, and prostate cancer with increased IGF‐1 levels.45-49 Animal studies have demonstrated that low circulating IGF‐1 levels, induced by either caloric restriction or genetic manipulation that resulted in liver IGF‐1 deficiency, protected against cancer development, while administration of IGF‐1 reversed this protective effect.50, 51 Therefore, in the setting of type 2 diabetes, insulin and IGF‐1 may be involved in tumor development.
Insulin directly signals in cells by binding to the IR and/or IGF‐IR, resulting in autophosphorylation of the beta subunit of the receptor and activation of downstream signaling pathways. Many tumors are known to express the IR, and studies of human breast cancer tissue demonstrate an increase in IR content in the tumors compared to normal breast tissue.52-54 Similarly, human prostate and hepatocellular cancers have been shown to have greater IR content than benign prostate and normal hepatic tissue, respectively.55, 56 A subset of chronic lymphocytic leukemia (CLL) lymphocytes was recently shown to have significantly increased IR expression, compared to normal B lymphocytes. The increased IR expression occurred in the CLL subset that carried a mutation in the long arm of chromosome 11 (11q) and is associated with a more aggressive course.57 In the same study, having higher IR expression was associated with shorter time to first therapy and overall survival than in those patients with CLL lymphocytes with lower IR content.57 In breast cancer tissue from patients, the presence of detectable IR by immunohistochemistry was associated with a better prognosis than having no detectable IR. But there is contradictory data regarding how higher IR content affects prognosis, with one study reporting that higher IR content was associated with greater disease‐free survival and another reporting that very high IR content is linked to decreased disease‐free survival.53, 54 Differences in methodologies and arbitrary cutoffs for high versus low IR content most likely account for the conflicting results, and thus it remains to be determined how IR content is linked to prognosis in human breast cancer. In vitro studies demonstrate that the IR has a significant role in cell transformation and tumor progression. One such study reported that knocking out the IGF‐IR in mouse embryonal fibroblasts (MEFs) increased the sensitivity of the IR to insulin and promoted downstream signaling by concentrations of insulin that were close to physiological levels.58 Other studies have demonstrated that signaling through the IR can compensate for a nonfunctioning IGF‐IR and enhance tumor progression.58-61 In T2DM, metabolic tissues demonstrate insulin resistance; in contrast, in one study of women with insulin resistance and breast cancer, the IR content of the tumors was not decreased compared to patients without insulin resistance.54 In a nonobese mouse model, insulin‐resistant, hyperinsulinemic mice developed greater breast cancer growth than control mice. The tumors of the hyperinsulinemic mice were found to have increased activation of the IR/IGF‐IR signaling pathway and the mammary epithelial cells had increased expression of the IR isoform A, the more mitogenic isoform of the IR.43 Therefore, the IR may play a role in tumor growth and proliferation, and in the setting of hyperinsulinemia, tumor cells do not develop insulin resistance. Therefore, in individuals with hyperinsulinemia, signaling downstream of the IR/IGF‐IR may be increased.
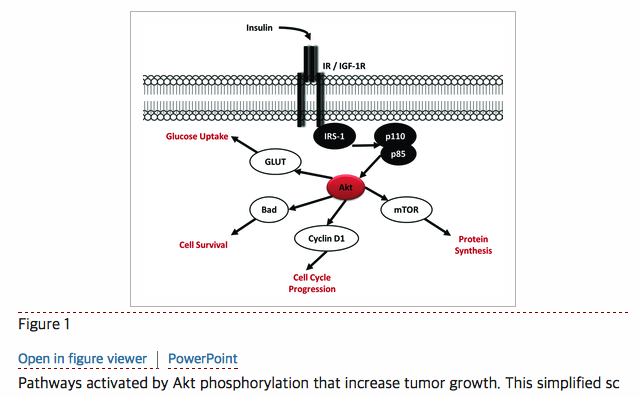
Insulin binding to the IR leads to autophosphorylation of the IR beta subunit tyrosine kinase, which results in recruitment and tyrosine phosphorylation of the insulin receptor substrates (IRS) and then activation of downstream signaling pathways, including the phosphatidylinositol 3‐kinase (PI3K)/Akt/mammalian target of rapamycin (mTOR) pathway and the mitogen‐activated protein kinase (MAPK) pathway. Hyperinsulinemia has been shown to cause increased Akt phosphorylation in breast cancer xenografts in animal models43 and is associated with increased tumor growth and, ex vivo, with CLL lymphocytes from humans that overexpress the IR.57 Akt phosphorylation leads to the phosphorylation of mTOR. mTOR complex 1 (mTORC1), formed by the complex of mTOR, GbL, Raptor, and PRAS40, functions as a nutrient/energy sensor. mTOR is inhibited by the complex formed by tuberous sclerosis protein 1 (TSC1) and tuberous sclerosis protein 2 (TSC2) through the inhibition of the small GTPase Rheb. Activated Akt phosphorylates TSC2, which releases the inhibition of the TSC1/TSC2 complex on Rheb, allowing for the activation of mTORC1.62 Akt also suppresses PRAS40, which leads to binding of 14‐3‐3 regulatory proteins and activation of mTORC1.63 mTORC1 stimulates protein synthesis by activating S6 kinase and 4E‐BP1/eukaryotic translation factor 4E (eIF4E).
Akt also activates many other signaling pathways (Fig. 1). It inhibits cell apoptosis by phosphorylating and thereby inhibiting the pro‐apoptotic protein Bad, and by phosphorylating the forkhead transcription factors (FOXO), sequestering Bad and FOXO in the cytoplasm, rendering them unable to initiate the expression of apoptotic genes, such as FasL, TRAIL, and Bcl‐XL. FOXO1 is phosphorylated by Akt in cancer cells lacking phosphatase and tensin homolog (PTEN), while FOXO3 and FOXO4 currently appear to be phosphorylated by Akt‐independent pathways; however a detailed understanding of the FOXO proteins and their regulators remains to be determined.64 In addition, Akt phosphorylation promotes the progression of cells from G1 to S phase of the cell cycle, when DNA replication occurs,65 by phosphorylating p21 and increasing cyclin D translation, which drives the cell through the G1/S transition.66 Activation of Akt also promotes glucose uptake into cells by promoting translocation of the glucose transporters (GLUT) to the cell membrane. Therefore, insulin signaling and activation of Akt potentially activates multiple pathways involved in tumor growth.

Pathways activated by Akt phosphorylation that increase tumor growth. This simplified schematic shows insulin binding to the insulin receptor (IR) and IGF‐1R (insulin‐like growth factor receptor), leading to phosphorylation of insulin‐receptor substrate‐1 (IRS‐1), the phosphatidylinositol 3‐kinase (p110 and p85), and activation of Akt. Akt then activates multiple pathways, as shown.
Hyperglycemia
Glucose is a crucial nutrient for proliferating cells.67 In the 1920s, Otto Warburg proposed that cancer cells develop an increased ability to produce energy from aerobic glycolysis and fermentation of pyruvate to lactate, even in the presence of abundant oxygen.68 This observation, known as the “Warburg hypothesis,” has become recognized as a common feature of many tumors. Producing energy from glucose in this manner contrasts the metabolism of glucose in normal, differentiated adult cells that have low rates of cell proliferation. Normal, nondividing adult cells derive most of their energy in the form of adenosine triphosphate (ATP) in two phases: first by the glycolysis of glucose to pyruvate, and second, by the oxidation of pyruvic acid to carbon dioxide and water. Although this method of glucose metabolism is the most efficient for producing ATP in nondividing cells, cancer cells are proliferating, and so are thought to rely on aerobic glycolysis as a mechanism of generating glycolytic intermediates that can then be used for protein synthesis and cell division.31 By using this means of energy production, tumor cells have a greater uptake of glucose than do normal cells, a phenomenon that can be clinically exploited to diagnose cancers by visualization of fluorodeoxyglucose (FDG) uptake with positron emission tomography (PET).68, 69 Because tumor cells display such avid uptake of glucose, it is worth examining whether hyperglycemia in the setting of T2DM can feed tumor growth.
In epidemiological studies of individuals with diabetes, hyperglycemia and elevated hemoglobin A1c (A1c) have not been consistently associated with greater cancer incidence and mortality. A recent meta‐analysis of prospective diabetes studies (VADT, ACCORD, UKPDS33, and UKPDS34) compared complications in patients with intensive glucose controls (A1c 6.9%, 6.4%, 7%, and 7.9%, respectively) compared to standard diabetes controls (A1c 8.4%, 7.5%, 7.9%, and 8.5%, respectively) and determined there was no difference in cancer incidence between standard and intensive glucose control groups.70 Notably, cancer incidence was not the primary outcome of these studies, and the results may be confounded by higher rates of sulfonylurea and insulin usage, in addition to higher doses of insulin in the intensively treated group with lower A1c levels, which some studies have suggested may increase tumor incidence.26, 28 In contrast, a prospective study on over 60,000 individuals in the Vasterbotten Intervention Project in Sweden, and the Hong Kong diabetes registry study both reported an association between hyperglycemia and increased cancer risk. However, in these two studies, insulin resistance and other factors that could lead to increased tumor growth in the setting of hyperglycemia, such as chronic inflammation, were not analyzed.71, 72 Thus, human studies assessing the effect of hyperglycemia on tumor growth are inconclusive due to confounding effects of comorbidities and medications.
In vitro studies and animal models have been used to study the effects of hyperglycemia in the absence of these other factors. In cell lines, for example, including MCF‐7 breast cancer and BxPC‐3 and MIA PACA‐2 pancreatic cell lines, increasing concentrations of glucose stimulate proliferation.73, 74 In a hyperglycemic mouse model, after tumor induction by carcinogens, the fatless diabetic (A‐ZIP/F‐1) mouse developed more aggressive skin and mammary tumors than the nondiabetic mouse.75 The tumor tissues from the A‐ZIP/F‐1 mouse were found to have increased activation of PI3K/Akt/mTOR and MAPK signaling pathways. These mice, however, also have elevated circulating insulin levels and inflammation cytokines, so it is not possible to conclude that the hyperglycemia alone is driving the accelerated tumor growth.75 In rats, inducing insulin‐deficient hyperglycemia using alloxan did not increase carcinogen‐induced mammary tumor growth.76 Therefore, despite the increased uptake of glucose in tumor cells, if glucose supplies are adequate to keep up with the demand of the tumor, studies do not conclusively demonstrate that hyperglycemia will further stimulate the growth of tumor cells.
The avid uptake of glucose and aerobic glycolysis seen in tumors can occur due to loss of tumor suppressor genes, oncogenic mutations, and the increased activation of the IR signaling pathway (Fig. 2).68, 77, 78 GLUT1, GLUT3, and GLUT4, are known to be upregulated in certain tumors. GLUT1 is expressed in many cancers (gynecological, thyroid, gastrointestinal, lung, renal, brain, and skin79-84), whereas GLUT 4 is expressed less commonly but is found in some breast, gastric, and lung cancers, osteosarcomas, and rhabdomyosarcomas.85-89 The expression of these transporters and their translocation to the cell membrane are regulated in different cells by the tumor suppressor gene p53, the oncogene c‐Myc, and by insulin signaling.31, 32, 86, 89-91 The tumor suppressor genes p53 and PTEN and the oncogenes Myc and RAS, as well as Akt signaling, also regulate many points in the glycolytic pathway (Fig. 2). Normally, p53 induces the transcription of TP53‐induced glycolysis and apoptosis regulator (TIGAR), which lowers cellular levels of fructose 2,6 bisphosphate (a stimulator of glycolysis) and inhibits glucose‐6‐phosphate dehydrogenase, the rate‐limiting enzyme in the pentose phosphate pathway.92 In addition, p53 enhances the transcription of the gene for cytochrome c oxidase 1 (SCO2), which is a component of the mitochondrial oxidative phosphorylation complexes (Fig. 2A).93 Loss of p53 therefore results in decreased SCO2, which prevents oxidative phosphorylation, promotes glycolysis by increasing fructose 2,6 bisphosphate, and activates the anabolic pentose phosphate pathway by activating glucose‐6‐phosphate dehydrogenase (Fig. 2B).31, 94 p53 also regulates the transcription of PTEN, TSC2, and the beta subunit of the AMP‐activated protein kinase (AMPK), which are negative regulators of Akt and mTOR.31 The PTEN gene encodes for the protein phosphatidylinositol‐3,4,5‐triphosphate 3‐phosphatase, which dephosphorylates phosphatidylinositol 3,4,5‐triphosphate (PIP3) and results in the inhibition of Akt signaling. Loss of PTEN commonly occurs in many cancers and leads to phosphorylation and activation of Akt (Fig. 2B). Phosphorylation of Akt activates many pathways, as described earlier. In addition, the oncogene Myc and hypoxia‐inducible factor‐1 (HIF1) independently increase GLUT1 expression increasing glucose uptake by tumors. They activate hexokinase and pyruvate dehydrogenase kinase1 (PDK1), and increase lactate dehydrogenase A (LDH‐A), which leads to conversion of pyruvate to lactate.31, 95-98 PDK phosphorylates and inactivates pyruvate dehydrogenase, which prevents pyruvate from entering Kreb's cycle and instead allows its conversion to lactate by LDH‐A.31 Higher tumor lactate levels have been demonstrated in cervical tumors with metastatic spread and are associated with decreased survival.96 In the estrogen receptor (ER) positive MCF‐7 breast cancer cell line, lactate increased in the expression of transcription factors, known to be associated with stem cells, which may contribute to tumor metastases.99 The mechanisms through which lactate promote tumor spread are incompletely understood. Proposed mechanisms include the conversion of lactate to acetyl‐CoA, which can then be used to acetylate proteins, including histones, and thereby increase gene expression that leads to tumor metastasis. Alternatively, lactate may be converted to acetyl‐CoA which then undergoes oxidative mitochondrial metabolism to yield ATP that can increase tumor growth and spreading.99 In summary, insulin signaling, the loss of tumor suppressor genes, and overexpression of oncogenes lead to increased uptake of glucose into tumor cells where it undergoes aerobic glycolysis. This is an inefficient method for producing ATP, but results in the production of metabolic intermediates that can be used for cell growth and synthesis, and lactate production which creates an environment that promotes metastatic tumor spread. Aerobic glycolysis increases the tumor's demand for glucose and is enabled by the activation of transcription factors, which may occur due to mutations of the tumor suppressor genes or activation of components of the insulin signaling pathway, such as mTOR.

Aerobic glycolysis and its regulating enzymes and genes. This simplified schematic highlights important elements of the glycolytic pathway and oxidative phosphorylation that are regulated by tumor suppressor genes and the insulin‐signaling pathway (Akt). Arrows indicate activation of one component by another, and circles indicate a component inhibiting another. Panel (A) indicates the elements that are regulating glycolysis and oxidative phosphorylation in normal cells. Panel (B) shows elements that are upregulated in tumor cells with loss of the tumor suppressor genes p53 and PTEN, the oncogene Myc, HIF, and increased Akt phosphorylation. Grayed out lines are pathways that are lost in tumor cells. Red arrows indicate components that are increased (↑) or decreased (↓).
The interaction of glucose and insulin with estrogens and androgens
Hyperinsulinemia and hyperglycemia may indirectly affect estrogen and androgen signaling in tumor cells by increasing the circulating bioavailability of these hormones, particularly in the setting of obesity. By expressing aromatase, adipose tissue is capable of forming estrogens from androgenic precursors. In fact, adipose tissue is the main source of estrogens in men and postmenopausal women. Obese postmenopausal women are known to have circulating levels of estrogens 50–100% higher than their lean counterparts.100 In addition, hyperglycemia and hyperinsulinemia reduce the hepatic production of sex hormone binding globulin (SHBG), which normally binds to circulating estrogens and androgens.101 The increased production of estrogens and decreased SHBG leads to increased bioavailable estrogen and may contribute to the increased risk of hormone responsive cancers in women with T2DM and obesity. Additionally, in vitro studies of cell lines including breast cancer cell lines, have demonstrated that there is significant cross talk between ER, insulin receptor, and IGF‐IR signaling, with IR and IGF‐IR signaling enhancing ER signaling pathways.102, 103 Therefore, insulin resistance, hyperglycemia, hormonal alterations, and obesity all potentially interact in individuals with T2DM to increase the risk of hormone responsive cancers.
Considering the potential biological links between T2DM and cancer, is it possible that metformin inhibits tumor growth? Although human epidemiological data are limited, the ADA/ACS consensus statement reported that there is some support for the protective role of metformin in certain cancers. While further studies are needed to determine whether metformin truly improves cancer outcomes in humans, there are numerous proposed mechanisms through which metformin may work to reduce tumor growth and metastases.19 Our present knowledge of the actions of metformin is incomplete and many of its effects are only now being elucidated. Therefore, we will discuss the current understanding of how metformin affects cancer growth.
Calorie restriction and cancer
In cancer cells, metformin mimics many of the effects of calorie restriction. Although diabetes, obesity, and insulin resistance are associated with increased cancer risk,5, 23, 104, 105 caloric restriction has been demonstrated for many years to reduce tumor growth.51, 106 The effect was first demonstrated in rodent models and later in the rhesus monkey.107Caloric restriction can potentially reduce tumor growth by multiple mechanisms. Insulin sensitivity increases with calorie restriction, therefore a decrease in circulating insulin and IGF‐1 levels occurs. Calorie restriction also reduces PI3K/Akt/mTOR signaling, which decreases protein synthesis and cell proliferation.108 Decreased caloric intake is associated with lower glucose levels and a decrease in ATP levels. As intracellular ATP levels decrease, the intracellular ratio of AMP:ATP increases, leading to activation of AMPK. AMPK phosphorylates TSC2 and raptor, leading to inhibition of mTORC1. Calorie restriction has many other effects on adipokines and inflammatory cytokines that may protect against cancer and are reviewed elsewhere.109
Metformin
In the past few years, some epidemiological studies have suggested that metformin is associated with a reduced cancer risk.29, 30, 110-114 These studies report that both new and long‐term users of metformin appear to be at lower risk of developing cancer.29, 30, 110 Individuals who take metformin alone or in addition to sulfonylureas are also at lower risk compared to those taking either sulfonylureas or insulin alone, according to one cohort study from the Netherlands.115 In one case‐control study of insulin‐treated patients with T2DM, taking metformin in addition to insulin appeared to protect against cancer development.116 Furthermore, diabetic patients with breast cancer who take metformin along with neoadjuvant chemotherapy have been reported to have higher rates of pathological complete response than those not taking metformin.117 Finally, cancer mortality appears to be decreased in diabetic metformin users.115, 118, 119 Most of these epidemiological data are retrospective, and there are as yet no published randomized controlled trials designed to ascertain if metformin truly lowers cancer incidence and mortality, or improves the response to chemotherapy in those with cancer. There are ongoing metformin studies that are examining the clinical outcomes; those that are currently registered as “active” or “recruiting” (at http://clinicaltrials.gov/) are outlined in Table 2. There are in vitro and in vivo data from cell lines and animal models that support the hypothesis that metformin is protective against cancer. Some human studies are also beginning to examine the effects of metformin treatment on the molecular pathways in cancer cells.120
| Trial number | Type of trial | Phase | Tumor type | Stage | Primary outcome |
|---|---|---|---|---|---|
| NCT01266486 | Single arm, open label | Phase II | Breast cancer | Early stage | pS6K, p4E‐BP‐1, pAMPK |
| NCT01302002 | NonRandomized, open label | Phase 0 | Breast cancer | Operable stage I and II | Proliferation and apoptosis |
| NCT00897884 | Single arm, nonrandomized | – | Breast cancer | Operable T1–4 (T1 ≥ 1cm), Nx | Proliferation |
| NCT00984490 | Single arm, open label | – | Breast cancer | Stage I and II | Proliferation |
| NCT01310231 | Randomized, double‐blind, placebo controlled | Phase II | Breast cancer | Metastatic | Progression‐free survival |
| NCT01101438 | Randomized, double blind, placebo controlled | Phase III | Breast cancer | Early stage | Invasive disease‐free survival |
| NCT00930579 | Nonrandomized, open label | Phase II | Breast cancer | DCIS or operable invasive breast cancer | AMPK/TOR signaling |
| NCT01205672 | Single arm, open label | – | Endometrial cancer | All candidates for surgical staging | Insulin/glucose metabolism and mTOR signaling |
| NCT01333852 | Randomized, double blind, placebo controlled | – | Head & neck cancer | Metastatic or recurrent | Disease control at 12 weeks |
| NCT01210911 | Randomized, placebo controlled | Phase II | Pancreatic cancer | Locally advanced or metastatic | Six‐month survival |
| NCT01167738 | Randomized, open label | Phase II | Pancreatic cancer | Metastatic | Progression‐free survival at six months |
| NCT01215032 | Single arm, open label | – | Prostate cancer | Castration resistant | PSA response |
| NCT01243385 | Single arm, open label | Phase II | Prostate cancer | Locally advanced or metastatic | Progression‐free survival at 12 weeks |
- Active and recruiting clinical trials examining tumor outcomes of cancer tissue signaling from the http://ClinicalTrials.gov registry (last accessed July 18, 2011).
A recent randomized, unblinded study of women with stage I or II primary breast cancer without diabetes examined the effect of two weeks of metformin treatment on tumor proliferation and gene expression. Subjects had core biopsies of tumors taken before starting metformin and two weeks later. In the metformin treated group they found a decrease in cell proliferation and alterations in the gene expression for pathways involved in cell proliferation and phosphodiesterase 3B, a regulator of cyclic AMP and activator of AMPK,120 which inhibits protein synthesis and cell cycle progression. Metformin does not appear to activate AMPK directly, but inhibits mitochondrial complex I of the respiratory chain, leading to an increase in the AMP:ATP ratio and thus activation of AMPK.121 In MCF7 breast cancer cells, metformin was found to inhibit cell cycle progression by downregulating cyclin D1 and inhibiting the cell cycle G1/S phase transition of cells. This effect was lost by inhibiting AMPK.122, 123 A similar effect has also been described in ovarian cancer cells.124However, it has been reported that metformin in prostate cancer cells can inhibit cell cycle progression in an AMPK‐independent manner, with one study suggesting that this occurs through the activation of the HIF target gene REDD1 (RTP801/Dig2/DDIT4), which leads to mTOR inhibition and cell cycle arrest.125, 126 Further evidence that metformin has AMPK‐independent effects comes not from tumors but from AMPK‐deficient hepatocytes and mice with a liver deficiency of AMPK. These mice had no difference in hepatic glucose output, or gluconeogenic gene transcription.127 Metformin suppresses glucose‐6‐phosphatase expression in the liver by inhibiting mitochondrial complex I, an affect that also appears to be independent of AMPK.128 Although the effects of metformin in normal metabolic tissue and tumor cells may differ, these data suggest metformin may potentially affect AMPK‐dependent and ‐independent pathways in cancers.
Metformin treats diabetes by inhibiting hepatic glucose output, and therefore reduces glucose levels.129 It also improves insulin sensitivity by lowering circulating insulin levels.130, 131 Metformin decreases Akt phosphorylation, in contrast to other inhibitors of mTOR, such as rapamycin, which actually increases Akt phosphorylation. In MCF7 cells, AMPK phosphorylates IRS‐1 at serine 789 and has been reported by some to dampen signal transduction through IRS‐1.132 Metformin was also seen to reduce IGF‐IR and IRS‐1 levels in MCF7 cells.132 As insulin signals through the IR and IGF‐IR, it is possible that metformin may reduce insulin signaling by decreasing the level of the IGF‐IR. In an animal model of colon cancer metformin reduced the accelerated growth of tumors induced by a high fat diet. Tumors from these animals treated with metformin were found to have decreased Akt phosphorylation and decreased expression of fatty acid synthase, along with activated AMPK. Akt stimulates GLUT1‐ and GLUT4‐mediated glucose uptake, and therefore inhibition of Akt will reduce glucose uptake in cells. Along with the decreased circulating level of glucose due to decreased hepatic gluconeogenesis, metformin will reduce the supply of glucose to the tumor cells by preventing its uptake (Fig. 3). As mentioned, in tumors p53 mutations result in activation of the pentose phosphate pathway and production of precursors for the synthesis of fatty acids, amino acids, and nucleic acids.94 Therefore, a decreased supply of glucose will result in decreased ATP production by tumor cells and less metabolic intermediates, such as glucose 6‐phosphate to enter the pentose phosphate pathway. Metformin also appears to inhibit cross talk between IR/IGF‐IR and G protein‐coupled receptor (GPCR) signaling pathways in pancreatic cancer by activating AMPK and thus inhibiting mTOR‐mediated stimulation of GPCR signaling.133 Additionally, AMPK activation by metformin has been shown to inhibit aromatase expression by inhibiting the activity of its promoter in human breast adipose tissue.134 The inhibition of aromatase could also explain the reduced tumor incidence in diabetic patients taking metformin.

Effect of metformin on glucose and insulin signaling. This schematic represents some of the potential AMPK‐dependent and ‐independent effects of metformin in tumors. Arrows indicate activation of one component by another; circles show one component that inhibits another.
In summary, metformin may (1) decrease insulin signaling in tumor cells by decreasing circulating insulin levels; (2) inhibit the insulin signaling pathway in tumor cells by activating AMPK, which reduces glucose uptake; (3) prevent cell cycle progression through AMPK‐dependent and ‐independent mechanisms; and (4) inhibit cross talk between receptors and decrease local estrogen production by breast adipose tissue by inhibiting aromatase expression (Fig. 3).
Metformin has also recently been shown to suppress breast cancer stem cells, which are thought to exist within tumors. These stem cells have the ability to self‐renew and give rise to differentiated cells and to form new tumors. After treatment with traditional chemotherapy, tumor size will decrease but the proportion of cancer stem cells will increase. Metformin has been shown to decrease the number of breast cancer stem cells and impair their ability to self‐renew and proliferate.135-138 Many of the pathways through which metformin is active in tumor cells and metabolic tissues are still being determined, therefore many studies are being performed to determine the clinical efficacy of metformin in human cancers and to uncover the details of its mechanism of action.
Conclusions
In conclusion, diabetes is associated with an increased risk of developing cancers at a variety of sites and is associated with worse outcomes, whereas calorie restriction in animal studies reduces tumor incidence. Elevated circulating insulin levels in animals and humans are associated with increased tumor growth, while hyperglycemia alone has proven difficult to assess due to confounding factors. Intracellular insulin signaling and the aerobic glycolysis of glucose promote the production of metabolic intermediates that can be used to form amino acids and nucleic acids needed for cell proliferation. Metformin reduces tumor proliferation by multiple mechanisms that involve the circulating levels of insulin and glucose, the expression of aromatase in surrounding tissues, and the intracellular regulation of insulin signaling and glucose metabolism. Randomized trials are anticipated to determine whether the potential of metformin to prevent tumor development, metastasis, and recurrence is real. Further studies are ongoing to expand our understanding of the pathways linking diabetes and cancer.