
확실한 것은 췌장 소화효소를 먹으면
췌장암 환자가 소화, 흡수 기능이 며칠내로 좋아진다는 것이다.

https://www.youtube.com/watch?v=7wV1sdOIY20
또 한가지
고용량 비타민 c를 정맥주사하면
췌장 암세포는 사멸하고
정상세포는 H202를 H2O+O2로 전환해버린다는 사실이다.
https://www.youtube.com/watch?v=4m9DFttpREI
Gastroenterology. Author manuscript; available in PMC 2023 Aug 1.
Published in final edited form as:
Gastroenterology. 2022 Aug; 163(2): 386–402.e1.
Published online 2022 Apr 7. doi: 10.1053/j.gastro.2022.03.056
PMCID: PMC9516440
NIHMSID: NIHMS1833771
PMID: 35398344
Pancreatic Cancer: Pathogenesis, Screening, Diagnosis, and Treatment
Laura D. Wood,1 Marcia Irene Canto,2 Elizabeth M. Jaffee,3 and Diane M. Simeone4
Author information Copyright and License information PMC Disclaimer
The publisher's final edited version of this article is available at Gastroenterology
Associated DataSupplementary Materials
Abstract
Pancreatic ductal adenocarcinoma (PDAC) is a clinically challenging cancer, due to both its late stage at diagnosis and its resistance to chemotherapy. However, recent advances in our understanding of the biology of PDAC have revealed new opportunities for early detection and targeted therapy of PDAC. In this review, we discuss the pathogenesis of PDAC, including molecular alterations in tumor cells, cellular alterations in the tumor microenvironment, and population-level risk factors. We review the current status of surveillance and early detection of PDAC, including populations at high risk and screening approaches. We outline the diagnostic approach to PDAC and highlight key treatment considerations, including how therapeutic approaches change with disease stage and targetable subtypes of PDAC. Recent years have seen significant improvements in our approaches to detect and treat PDAC, but large-scale, coordinated efforts will be needed to maximize the clinical impact for patients and improve overall survival.
췌관 선암종(PDAC)은
진단 시기가 늦고 화학 요법에 대한 내성이 있기 때문에
임상적으로 어려운 암입니다.

그러나
최근 PDAC의 생물학에 대한 이해가 발전하면서
PDAC의 조기 발견과 표적 치료를 위한 새로운 기회가 열렸습니다.
이 리뷰에서는
종양 세포의 분자적 변화,
종양 미세 환경의 세포적 변화,
인구 수준 위험 인자를 포함한 PDAC의 발병 기전에 대해 논의합니다.
고위험군 및 스크리닝 접근법을 포함하여
PDAC의 감시 및 조기 발견 현황을 검토합니다.
PDAC에 대한 진단 접근법을 개괄적으로 설명하고
질병 단계와 표적 가능한 PDAC 하위 유형에 따라 치료 접근법이
어떻게 달라지는지 등 주요 치료 고려 사항을 강조합니다.
최근 몇 년 동안
PDAC를 발견하고 치료하는 접근법이 크게 개선되었지만, 환
자의 임상적 효과를 극대화하고 전반적인 생존율을 개선하기 위해서는
대규모의 조율된 노력이 필요합니다.
Keywords: Pancreatic Cancer, Pancreatic Ductal Adenocarcinoma, Cancer Screening
Pancreatic ductal adenocarcinoma (PDAC) is a deadly disease that is predicted to be the second leading cause of cancer death in the United States by 2030.1 One reason for the dismal prognosis of pancreatic cancer is that 90% of tumors are diagnosed at a late stage after they have spread beyond the pancreas, with systemic metastases in >50%.2,3 This underscores the need for tools to detect PDAC earlier, before it has spread beyond the pancreas, and for therapies that more effectively kill PDAC cells after they have metastasized. In the past decade, basic and translational investigations have significantly improved our understanding of the biological processes that drive pancreatic tumorigenesis, and this understanding is beginning to be leveraged to improve the care of patients with pancreatic cancer. In this review, we discuss the pathogenesis, screening, diagnosis, and treatment of PDAC, focusing on both fundamental concepts and recent advances.
췌관 선암종(PDAC)은
2030년까지 미국에서 암 사망 원인 2위가 될 것으로 예상되는 치명적인 질환입니다.1
췌장암의 예후가 좋지 않은 이유 중 하나는
종양의 90%가 췌장을 넘어
전이된 후 말기에 진단되고
이는 PDAC가 췌장 밖으로 퍼지기 전에 조기에 발견할 수 있는 도구와 전이된 후 PDAC 세포를 더 효과적으로 죽이는 치료법이 필요하다는 것을 강조합니다. 지난 10년 동안 기초 및 중개 연구를 통해 췌장 종양 형성을 주도하는 생물학적 과정에 대한 이해가 크게 향상되었으며, 이러한 이해는 췌장암 환자의 치료를 개선하는 데 활용되기 시작했습니다. 이 리뷰에서는 기본적인 개념과 최근의 발전에 초점을 맞추어 PDAC의 발병 기전, 선별 검사, 진단 및 치료에 대해 논의합니
Pathogenesis of Pancreatic Cancer
Pathology of Pancreatic Neoplasia
PDAC arises from noninvasive precancerous lesions that are curable if detected and treated early enough (Figure 1). These precancerous lesions are classified by their size and involvement with the pancreatic ductal system. Most PDACs arise from microscopic pancreatic intraepithelial neoplasia (PanIN), a neoplasm involving pancreatic ducts, which is by definition <5 mm.4 A much smaller proportion of PDACs (<10%) arise from intraductal papillary mucinous neoplasms (IPMNs), macrocystic lesions that involve the pancreatic ductal system.4–6 The least common precancerous neoplasm, mucinous cystic neoplasm, is clinically and pathologically distinct. Mucinous cystic neoplasms do not involve the ductal system and have a characteristic ovarian-type stroma. They are much more common in women and involve the pancreatic body/tail.7 The unique pathological features of each lesion lead to distinct clinical challenges. For example, because of their larger size, IPMNs are commonly detected incidentally on abdominal imaging studies, requiring data-driven approaches to surveillance and intervention for these patients.8 In contrast, PanINs are rarely detected incidentally. Although the pathological features of these precancers are well characterized, the cell of origin of pancreatic cancer remains controversial. The anatomic location of precancers in the ductal system could support a ductal cell of origin, but numerous studies in murine models have shown that acinar cells can give rise to PanINs following pancreatic injury and metaplasia. However, it is challenging to confirm such findings in human samples. This controversy was recently reviewed in detail elsewhere.9
PDAC는
조기에 발견하여 치료하면
완치가 가능한 비침습적 전암성 병변에서 발생합니다(그림 1).
이러한 전암성 병변은 크기와 췌관 시스템과의 관련성에 따라 분류됩니다. 대부분의 PDAC는 췌관과 관련된 종양인 미세 췌장 상피내 종양(PanIN)에서 발생하며, 이는 정의상 5mm 미만입니다.4 훨씬 적은 비율(<10%)의 PDAC는 췌관 시스템을 포함하는 거대 낭성 병변인 췌관 내 유두상 점액성 종양(IPMN)에서 발생합니다.4-6 가장 흔한 전암성 종양인 점액성 낭성 종양은 임상적으로나 병리학적으로 뚜렷하게 구분됩니다. 점액성 낭성 신 생물은 관 시스템을 포함하지 않으며 특징적인 난소형 기질을 가지고 있습니다. 여성에게 훨씬 더 흔하며 췌장 몸체/꼬리를 침범합니다.7 각 병변의 고유한 병리학적 특징은 뚜렷한 임상적 어려움을 초래합니다. 예를 들어, IPMN은 크기가 크기 때문에 복부 영상 검사에서 우연히 발견되는 경우가 많기 때문에 이러한 환자를 감시하고 중재하기 위해서는 데이터에 기반한 접근법이 필요합니다.8 반면, PanIN은 우연히 발견되는 경우가 거의 없습니다. 이러한 전암의 병리학적인 특징은 잘 알려져 있지만 췌장암의 기원 세포는 여전히 논란의 여지가 있습니다. 관 시스템에서 전암의 해부학적 위치는 관 세포의 기원 세포를 뒷받침할 수 있지만, 쥐 모델을 대상으로 한 수많은 연구에 따르면 췌장 손상 및 전이 후 선세포가 PanIN을 생성할 수 있는 것으로 나타났습니다. 그러나 인간 샘플에서 이러한 결과를 확인하기는 어렵습니다. 이 논란은 최근 다른 곳에서 자세히 검토한 바 있습니다.9
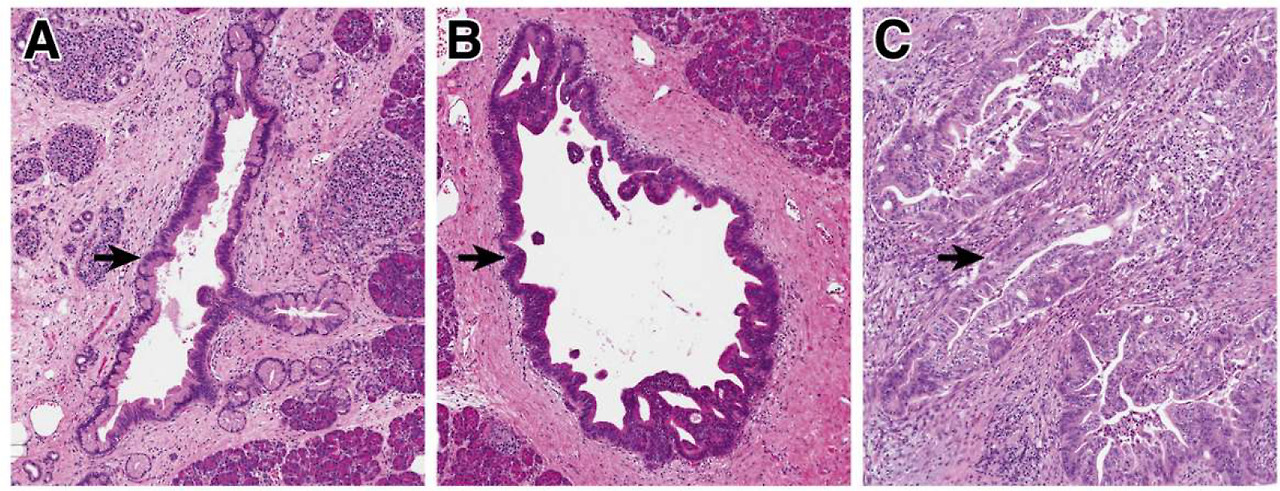
Precancerous neoplasia in the pancreas.
(A) Low-grade PanINs involve the pancreatic ducts. They have apical mucin and mild cytologic atypia, with the nuclei still retaining their polarity. (B) High-grade PanIN shows increase architectural and cytologic atypia, which architectural complexity, nuclear pleomorphism, and loss of nuclear polarity. (C) Invasive PDAC shows severe architectural and cytologic atypia with a striking desmoplastic response in the stroma.
췌장의 전암성 신생물.
(A) 저등급 PanIN은 췌관을 침범합니다. 이들은 정점 뮤신과 경미한 세포학적 이형성을 가지며, 핵은 여전히 극성을 유지합니다.
(B) 높은 등급의 PanIN은 구조적 복잡성, 핵 다형성, 핵 극성 상실과 같은 구조적 및 세포학적 이형성이 증가합니다.
(C) 침습성 PDAC는 기질에서 현저한 탈형성 반응과 함께 심각한 구조적 및 세포학적 이형성을 보입니다.
Precancerous pancreatic neoplasms are incredibly common and increase in prevalence with age; for example, one autopsy study in older adults identified PanINs in >75% of completely sampled pancreata.10,11 Although these lesions have a risk for progression to PDAC, most will not progress to cancer, and by themselves they have no risk of spread beyond the pancreas.12,13 Morphologically, these lesions are categorized as low-grade or high-grade based on architectural and cytological atypia (Figure 1).4 This 2-tiered grading system (low-grade vs high-grade) is a relatively recent revision of a previously used 3-tiered grading system for pancreatic precancers; the change to a 2-tiered system was driven by increased interobserver reproducibility as well as clinical impact of the revised system.4 Low-grade precancers have basally oriented nuclei and mild to moderate cytologic atypia (Figure 1A), and high-grade precancers have marked architectural alterations (cribriforming, micropapillae, budding), loss of nuclear polarity, and severe cytologic atypia (Figure 1B).4 In IPMNs, the grade of dysplasia is also correlated with the direction of differentiation of the neoplastic cells, with gastric-type IPMNs enriched for low-grade dysplasia and intestinal-type and pancreatobiliary-type more likely to be high-grade.14 When stratified by grade of dysplasia, low-grade precancerous lesions are far more common, whereas high-grade lesions are mostly found in pancreata associated with PDAC, suggesting that high-grade precancers have a higher risk for progression to invasive carcinoma.10,11 Moreover, associated carcinomas are much more frequently associated with high-grade rather than low-grade IPMNs, and recent evolutionary analysis defined high-grade IPMNs as the direct precursor of PDAC.6,15 This evidence highlights that the morphological grade of dysplasia correlates with risk of transformation to invasive carcinoma.
Pathologically, PDAC consists of malignant glands with haphazard architecture embedded in a dense desmoplastic stroma (Figure 1C). This pauci-cellularity significantly complicates the molecular analysis of primary PDAC samples, as most cells in a tissue fragment are likely non-neoplastic; without enrichment for neoplastic cells, often fewer than 10% of cells may be malignant. There are also several morphological variants, including adenosquamous carcinoma and undifferentiated carcinoma with osteoclast-like giant cells, some of which have unique clinical and/or molecular features.16 Most PDACs are not localized at diagnosis, and >50% of patients present with distant metastases.2 The most common site of metastasis is the liver, and increasing evidence suggests that distinct metastatic sites carry different prognostic features, with lung-only metastases having an improved prognosis compared with other sites.17,18 Moreover, autopsy studies suggest that distinct molecular alterations may increase the likelihood of local vs systemic disease progression, with distant metastases more common in patients with somatic mutations in the gene SMAD4.19
전암성 췌장 신 생물은
매우 흔하며 나이가 들수록 유병률이 증가합니다.
예를 들어, 고령자를 대상으로 한 부검 연구에서는 완전히 샘플링된 췌장의 75% 이상에서 PanIN이 확인되었습니다.10,11 이러한 병변은 PDAC로 진행될 위험이 있지만, 대부분은 암으로 진행되지 않으며 그 자체로는 췌장 밖으로 퍼질 위험이 없습니다.12,13 형태학적으로 이러한 병변은 구조적 및 세포학적 이형성에 따라 저등급 또는 고등급으로 분류됩니다(그림 1).4 이 2단계 등급 시스템(저등급 대 고등급)은 이전에 췌장 전암에 사용되었던 3단계 등급 시스템을 비교적 최근에 수정한 것으로, 2단계 시스템으로의 변경은 관찰자 간 재현성 증가와 개정된 시스템의 임상 영향에 의해 주도되었습니다.4 저등급 전암은 기본적으로 핵이 배향되어 있고 경증에서 중등도의 세포학적 이형성이 있으며(그림 1A), 고등급 전암은 현저한 구조적 변화(융모 형성, 미세 유두, 발아), 핵 극성 상실, 심각한 세포학적 이형성이 있습니다(그림 1B). 4 IPMN에서 이형성증의 등급은 종양 세포의 분화 방향과도 상관관계가 있으며, 위형 IPMN은 저등급 이형성증, 장형 및 췌담도형은 고등급일 가능성이 더 높습니다.14 이형성증의 등급별로 계층화하면 저등급 전암 병변이 훨씬 더 흔한 반면, 고등급 병변은 대부분 PDAC와 관련된 췌장에서 발견되어 고등급 전암이 침습성 암종으로 진행될 위험이 더 높다는 것을 시사합니다.10,11 또한, 관련 암종은 저등급보다 고등급 IPMN과 훨씬 더 자주 연관되며, 최근 진화 분석에서는 고등급 IPMN을 PDAC의 직접적인 전구체로 정의했습니다.6,15 이러한 증거는 이형성의 형태학적 등급이 침습성 암종으로의 전환 위험과 관련이 있음을 강조합니다.
병리학적으로 PDAC는 치밀한 탈형성 기질에 우연한 구조를 가진 악성 샘으로 구성됩니다(그림 1C). 조직 조각에 있는 대부분의 세포는 비종양일 가능성이 높고, 종양 세포에 대한 농축이 없으면 10% 미만의 세포가 악성일 수 있기 때문에 이러한 유세포성은 일차 PDAC 샘플의 분자 분석을 상당히 복잡하게 만듭니다. 또한 선편평암종과 파골세포와 유사한 거대 세포를 가진 미분화암종 등 여러 가지 형태학적 변이가 있으며, 이 중 일부는 독특한 임상 및/또는 분자적 특징을 가지고 있습니다.16 대부분의 PDAC는 진단 시 국소화되지 않으며, 환자의 50% 이상이 원격 전이를 동반합니다.2 가장 흔한 전이 부위는 간이며, 폐에만 전이된 경우 다른 부위에 비해 예후가 개선되는 등 전이 부위에 따라 예후가 다르다는 증거가 증가하고 있습니다.17,18 또한 부검 연구에 따르면 뚜렷한 분자적 변화가 국소 대 전신 질환 진행 가능성을 높일 수 있으며, SMAD4 유전자에 체세포 변이가 있는 환자에서 원거리 전이가 더 흔하다는 사실이 밝혀졌습니다.19
Molecular Alterations in Neoplastic Cells
PDAC is caused, at least in part, by somatic mutations in oncogenes and tumor suppressor genes (Figure 2). The most commonly mutated genes in PDAC, the oncogene KRAS and the tumor suppressor genes CDKN2A, TP53, and SMAD4, were originally discovered in the 1980s and 1990s through targeted molecular biology and sequencing approaches.20–23 The first comprehensive look at the PDAC exome came in 2008 with the first whole exome sequencing study of 24 PDACs.24 This study revealed that the PDAC genome landscape is composed of these 4 previously described “mountains” (KRAS, CDKN2A, TP53, SMAD4) as well as a larger number of less frequently mutated “hills.”24 Subsequent large-scale PDAC sequencing studies have refined this landscape in great detail, including efforts by The Cancer Genome Atlas and The International Cancer Genome Consortium.25–28 Several important groups of “hills” have been characterized in these studies, including genes involved in DNA repair, chromatin remodeling, and axon guidance, some of which delineate clinically important groups that respond to specific therapies. Recent efforts have also identified kataegis, a process leading to clustered nucleotide substitutions, in PDAC; this process is likely associated with the activity of APOBEC enzymes.29
PDAC는 적어도 부분적으로는
발암 유전자와 종양 억제 유전자의 체세포 돌연변이에 의해 발생합니다(그림 2).
PDAC에서 가장 흔하게 돌연변이가 발생하는 유전자인
발암 유전자 KRAS와 종양 억제 유전자 CDKN2A, TP53, SMAD4는
1980년대와 1990년대에 표적 분자생물학 및 시퀀싱 접근법을 통해 처음 발견되었습니다.20-23
PDAC 엑솜에 대한 포괄적인 첫 연구는 2008년에 24개의 PDAC에 대한 최초의 전체 엑솜 시퀀싱 연구를 통해 이루어졌습니다.24 이 연구는 PDAC 게놈 지형이 이전에 설명한 4개의 "산"(KRAS, CDKN2A, TP53, SMAD4)과 덜 자주 변이되는 더 많은 "언덕"으로 구성되어 있음을 밝혀냈습니다.24 이후 대규모 PDAC 시퀀싱 연구는 암 게놈 아틀라스와 국제 암 게놈 컨소시엄의 노력을 포함하여 이 지형을 매우 상세하게 개선했습니다.25 -28 이러한 연구에서 DNA 복구, 염색질 리모델링 및 축삭 유도에 관여하는 유전자를 포함하여 몇 가지 중요한 "언덕" 그룹이 특징 지어졌으며, 그중 일부는 특정 치료법에 반응하는 임상적으로 중요한 그룹을 묘사합니다. 최근의 노력으로 PDAC에서 클러스터 뉴클레오타이드 치환으로 이어지는 과정인 카타지스도 확인되었는데, 이 과정은 APOBEC 효소의 활성과 관련이 있을 가능성이 높습니다.29
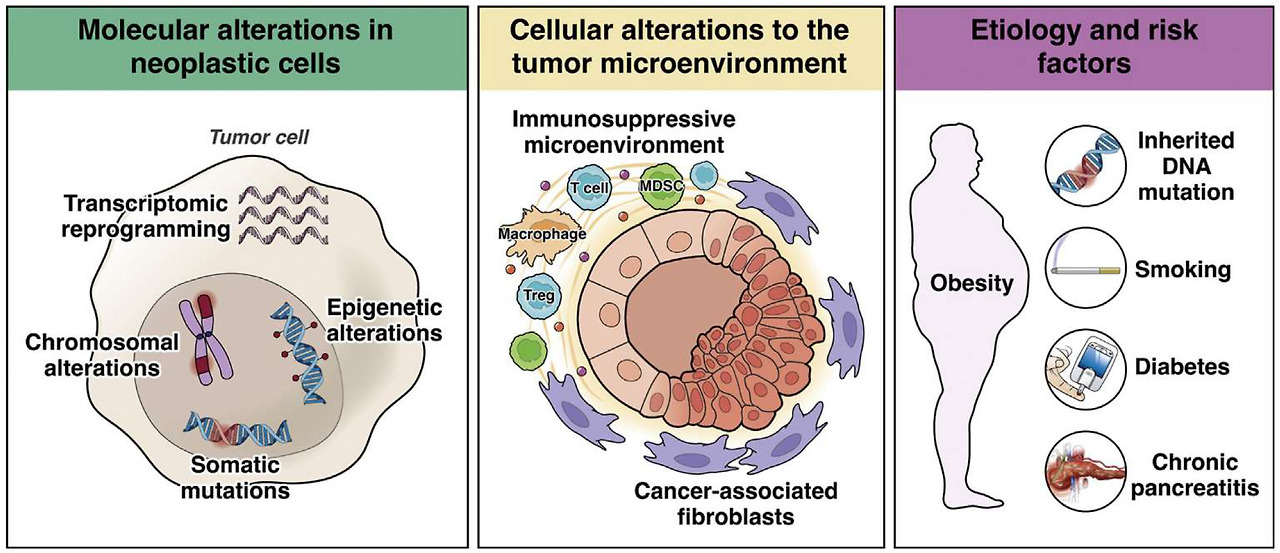
Mechanisms of pancreatic cancer pathogenesis. Molecular contributions to pathogenesis include somatic mutations in driver genes, chromosomal alterations, epigenetic alterations, and transcriptional reprogramming, all occurring in tumor cells. Cellular contributions from the non-neoplastic tumor microenvironment include alterations in cancer-associated fibroblasts and the immune microenvironment. Population-level contributions include inherited DNA mutations, diabetes, obesity, smoking, and chronic pancreatitis.
췌장암 발병 기전.
발병 기전에 대한 분자적 기여에는
종양 세포에서 발생하는
드라이버 유전자의 체세포 돌연변이,
염색체 변화,
후성유전학적 변화 및 전사 재프로그래밍이 포함됩니다.
비종양성 종양 미세 환경의 세포적 기여에는
암 관련 섬유아세포 및
면역 미세 환경의 변화가 포함됩니다.
인구 수준에서의 기여에는
유전적 DNA 돌연변이,
당뇨병,
비만,
흡연,
만성 췌장염이 포함됩니다.
In addition to small somatic mutations involving coding regions, the expansion of PDAC genomic analysis to include whole genome sequencing has also catalogued large chromosomal alterations, including copy number alterations, chromosomal rearrangements, and chromothripsis. Chromothripsis, a term derived from the Greek for “shattering,” refers to a phenomenon in which 1 or a few chromosomes contain hundreds of clustered genomic rearrangements.29 For the latter, some investigators propose that these alterations are acquired through catastrophic DNA damage events, resulting in punctuated rather than gradual evolution.30 An ongoing challenge in the interpretation of these alterations is the identification of potential driver genes in large altered regions and the distinction of driver alterations from passengers that occur due to PDAC genomic instability. Copy number alterations and chromothripsis have also been reported in precancerous lesions, with a higher prevalence in high-grade lesions, suggesting these chromosomal alterations are a late event in pancreatic tumorigenesis.31
In addition to the identification of genetic drivers, DNA sequencing studies have also revealed other important features of pancreatic tumorigenesis. For example, multiregion sequencing of precancerous lesions, primary tumors, and metastases have allowed evolutionary modeling of various steps in pancreatic tumorigenesis. Studies in IPMNs suggest a period of almost 4 years between the development of high-grade dysplasia and invasive carcinoma.15 Similarly, in PanIN lesions, modeling from sequencing data suggests an interval of approximately 4 years between the common ancestral cell and the founder cell of the associated PDAC.32 Relatedly, modeling from multiregion sequencing of metastases and primary PDACs suggests that many years elapse between tumor initiation and metastasis.33 Intriguingly, these studies show no differences in driver gene mutations between primary and metastatic tumors, suggesting that alterations other than DNA mutations may promote metastasis.33,34 Recent studies in precancerous lesions have revealed surprising genetic heterogeneity, even with respect to well-characterized driver gene mutations.15,35–37 These studies suggest a polyclonal origin for at least a subset of precancerous lesions, convergent evolution in later driver gene mutations, and distinct selective forces at different time points in tumorigenesis (Figure 3). In addition, multifocal neoplasia is also quite common in patients with precancerous pancreatic lesions; co-occurring PanINs and IPMNs/carcinomas are frequently genetically independent (ie, sharing no somatic mutations, which suggests initiation from separate cells), as are IPMNs and their recurrences.6,38–40
코딩 영역과 관련된 작은 체세포 돌연변이 외에도 전체 게놈 시퀀싱을 포함하도록 PDAC 게놈 분석이 확장되면서 복제 수 변경, 염색체 재배열, 크로모트립시스 등 큰 염색체 변경도 목록화되었습니다. '산산조각'을 뜻하는 그리스어에서 유래한 용어인 크로모트립시스는 1개 또는 몇 개의 염색체에 수백 개의 군집된 게놈 재배열이 나타나는 현상을 말합니다.29 후자의 경우, 일부 연구자들은 이러한 변경이 치명적인 DNA 손상 사건을 통해 획득되어 점진적인 진화가 아니라 점점이 찍힌 결과라고 제안합니다.30 이러한 변경을 해석하는 데 있어 지속적인 과제는 대규모 변경 영역에서 잠재적인 드라이버 유전자를 식별하고 PDAC 게놈 불안정으로 인해 발생하는 드라이버 변경과 패신저를 구별하는 것입니다. 전암성 병변에서도 복제수 변이와 염색체 변이가 보고되었는데, 높은 등급의 병변에서 유병률이 더 높았으며, 이는 이러한 염색체 변이가 췌장 종양 발생의 후기 사건임을 시사합니다.31
유전적 동인의 확인 외에도 DNA 시퀀싱 연구는 췌장 종양 형성의 다른 중요한 특징도 밝혀냈습니다. 예를 들어 전암성 병변, 원발성 종양 및 전이에 대한 다중 영역 시퀀싱을 통해 췌장 종양 형성의 다양한 단계에 대한 진화적 모델링이 가능해졌습니다. IPMN에 대한 연구는 고등급 이형성증과 침습성 암종 발생 사이에 약 4년의 기간이 있음을 시사합니다.15 마찬가지로, PanIN 병변에서 시퀀싱 데이터를 통한 모델링은 공통 조상 세포와 관련 PDAC의 창립자 세포 사이에 약 4년의 간격이 있음을 시사합니다.32 이와 관련하여 전이 및 원발 PDAC의 다중 지역 시퀀싱을 통한 모델은 종양 시작과 전이 사이에 수년이 경과한다는 것을 시사합니다.33 흥미롭게도 이러한 연구는 원발성 종양과 전이성 종양 간에 드라이버 유전자 돌연변이에 차이가 없음을 보여주며, DNA 돌연변이 이외의 변화가 전이를 촉진할 수 있음을 시사합니다.33,34 전암성 병변에 대한 최근 연구는 잘 특성화된 드라이버 유전자 돌연변이에서도 놀라운 유전적 이질성을 밝혀냈습니다.15,35-37 이러한 연구는 적어도 전암성 병변의 하위 집합에 대한 다클론 기원, 후기 드라이버 유전자 돌연변이의 수렴적 진화, 종양 발생의 다른 시점에서의 뚜렷한 선택적 힘을 시사합니다(그림 3 참조). 또한 다초점 신생물도 전암성 췌장 병변이 있는 환자에서 매우 흔하며, 동시 발생하는 PanIN과 IPMN/암종은 종종 유전적으로 독립적이며(즉, 별도의 세포에서 시작되었음을 시사하는 체세포 변이를 공유하지 않음), IPMN과 그 재발도 마찬가지입니다.6,38-40
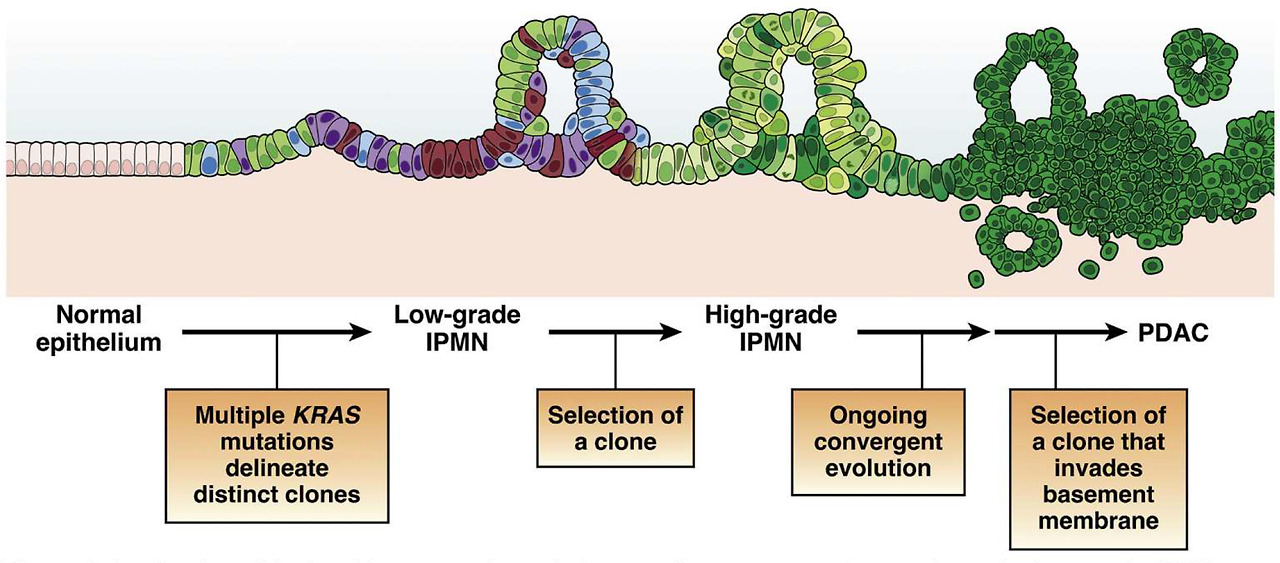
A refined model of multistep tumorigenesis in premalignant pancreatic tumorigenesis. Low-grade IPMNs are characterized by multiple mutations in the initiating oncogene KRAS, suggesting polyclonal evolution of multiple genetically independent precancers. Selection of a clone leads to the development of a high-grade IPMN, which then undergoes ongoing evolution, at times with distinct alterations in the same gene arising in distinct subclones. Selection of an advanced clone that invades the basement membrane leads to the development of invasive PDAC. While this model has been validated in human IPMN samples, its applicability to microscopic human PanINs requires further investigation.
전악성 췌장 종양 발생에서 다단계 종양 발생의 정교한 모델.
저등급 IPMN은 초기 발암 유전자 KRAS의 다중 돌연변이가 특징이며, 이는 유전적으로 독립적인 여러 전암의 다클론 진화를 시사합니다. 클론을 선택하면 고급 IPMN이 개발되고, 그 후 지속적인 진화를 거치며 때로는 동일한 유전자에 뚜렷한 변이가 다른 하위 클론에서 발생하기도 합니다. 기저막을 침범하는 고급 클론을 선택하면 침습성 PDAC로 발전하게 됩니다. 이 모델은 인간 IPMN 샘플에서 검증되었지만, 미세한 인간 PanIN에 적용할 수 있는지는 추가 조사가 필요합니다.
Although alterations in DNA sequence are the most extensively characterized, other types of molecular alterations also contribute to PDAC pathogenesis. Epigenetic modifications to DNA (such as methylation) and histones (such as acetylation and methylation) can heritably modulate chromatin structure and gene expression. Epigenetic inactivation via DNA methylation has been frequently identified for the tumor suppressor gene CDKN2A, but methylation is not a common inactivation mechanism for other PDAC drivers, such as TP53 and SMAD4.41,42 Comprehensive methylation profiling in The Cancer Genome Atlas study identified 2 clusters of PDACs based on the extent of their DNA hypermethylation, and integrated analysis of DNA methylation and messenger RNA expression data identified almost 100 genes that were recurrently silenced by DNA methylation, including ZPF82, PAPR6, and DNAJC15.28 In addition, global epigenomic reprogramming modulates the PDAC genome. Studies in human autopsy samples have shown global alterations in histone states in metastases compared with primary tumors, suggesting that epigenetic alterations may play a role in driving metastasis.43 Such epigenetic alterations also have been reported in precancerous PanIN and IPMN lesions.44–47
Multiple studies have categorized the transcriptomes of human PDACs, with several unique subtyping schemes proposed.26,28,48–51 Integrated analyses of these data suggest that there are likely 2 distinct transcriptional subtypes of PDAC, termed classical and basal, with the basal phenotype enriched in metastases and correlated with a worse prognosis.28,52 Classical tumors are enriched for epithelial and pancreatic lineage transcriptomic programs, whereas basal tumors are enriched for epithelial to mesenchymal transition, cell cycle progression, and transforming growth factor-β signaling.53 Some studies suggest heterogeneity in transcriptional subtype within PDAC tumors, but it is not clear whether basal tumors are inherently more metastatic or the switch to a basal phenotype drives metastasis.52,54 In addition, some studies have identified PDACs with intermediate transcriptomic subtypes or subtypes discordant with different classifiers, but the clinical implications of these intermediate tumors remain to be comprehensively investigated.53,55 Moreover, the timing of acquisition of these transcriptional programs in premalignant pancreatic tumorigenesis is not yet known, highlighting an important area for future research.
DNA 염기 서열의 변화가 가장 광범위하게 특징지어지지만, 다른 유형의 분자 변형도 PDAC 발병에 기여합니다. DNA(예: 메틸화)와 히스톤(예: 아세틸화 및 메틸화)에 대한 후성유전학적 변형은 염색질 구조와 유전자 발현을 유전적으로 조절할 수 있습니다. DNA 메틸화를 통한 후성유전학적 비활성화는 종양 억제 유전자 CDKN2A에서 자주 확인되었지만 메틸화는 TP53 및 SMAD4와 같은 다른 PDAC 드라이버에 대한 일반적인 비활성화 메커니즘은 아닙니다. 41,42 암 게놈 아틀라스 연구에서 포괄적인 메틸화 프로파일링을 통해 DNA 과메틸화 정도에 따라 2개의 PDAC 클러스터를 확인했으며, DNA 메틸화 및 메신저 RNA 발현 데이터를 통합 분석한 결과 ZPF82, PAPR6, DNAJC15 등 DNA 메틸화에 의해 반복적으로 침묵하는 거의 100개의 유전자를 확인했습니다.28 또한 글로벌 에피게놈 재프로그램이 PDAC 게놈을 조절하는 것으로 나타났습니다. 인간 부검 샘플을 대상으로 한 연구에 따르면 원발성 종양에 비해 전이성 종양에서 히스톤 상태가 전반적으로 변화하는 것으로 나타나 후성유전학적 변화가 전이를 촉진하는 역할을 할 수 있음을 시사합니다.43 이러한 후성유전학적 변화는 전암성 PanIN 및 IPMN 병변에서도 보고되었습니다.44-47
여러 연구에서 인간 PDAC의 전사체를 분류하고 몇 가지 독특한 하위 유형 체계를 제안했습니다.26,28,48-51 이러한 데이터의 통합 분석에 따르면 PDAC에는 고전적 및 기저라고 하는 두 가지 다른 전사적 하위 유형이 있을 수 있으며 기저 표현형은 전이가 풍부하고 예후 악화와 상관관계가 있습니다.28,52 고전적 종양은 상피 및 췌장 계통 전사체 프로그램이 풍부한 반면, 기저 종양은 상피에서 중간엽 전이, 세포 주기 진행 및 변형 성장 인자-β 신호 전달이 풍부합니다.53 일부 연구에서는 PDAC 종양 내 전사 아형의 이질성을 시사하지만 기저 종양이 본질적으로 더 전이성이 높은지 아니면 기저 표현형으로의 전환이 전이를 유발하는지는 명확하지 않습니다.52,54 또한 일부 연구에서는 중간 전사체 아형 또는 다른 분류자와 불일치하는 아형을 가진 PDAC를 확인했지만 이러한 중간 종양의 임상적 의미는 종합적으로 조사되어야 합니다.53,55 또한 전악성 췌장 종양 형성에서 이러한 전사 프로그램의 획득 시기는 아직 알려지지 않아 향후 연구의 중요한 영역으로 부각되고 있습니다.
Cellular Alterations to the Tumor Microenvironment
In addition to the extensive molecular alterations described previously in neoplastic cells, interactions with non-neoplastic cells in the tumor microenvironment also play a crucial role in pancreatic tumorigenesis (Figure 2). This topic was recently reviewed in depth by others.56 PDACs exhibit multiple alterations to immune cell subtypes, resulting in an immunosuppressive or “cold” microenvironment. These alterations include increased immunosuppressive regulatory T cells and myeloid-derived suppressor cells, preventing immune-mediated targeting of cancer cells. In addition, tumor-associated macrophages can promote progression of precancerous lesions by secretion of interleukin-6.57 However, these features of the immune microenvironment are not universal across human PDACs, and some investigators have proposed subtyping schemes for the immune microenvironment that include distinct “escape” and “exhausted” phenotypes.58 Immune alterations in PDAC likely influence disease progression and contribute to the lack of response to immunotherapy approaches, such as checkpoint inhibition. Greater mechanistic insights into the signals driving the immunosuppressive microenvironment may reveal novel strategies to enhance therapeutic efficacy, as recently reviewed in depth.59,60
Interactions with nonimmune cells also contribute significantly to the development of PDAC. PDACs have a dense desmoplastic stroma that is produced by cancer-associated fibroblasts (CAFs). CAFs have been studied extensively in vitro, in murine models, and in human tissue samples. Perhaps counterintuitively, CAFs can have both tumor-promoting and tumor-suppressive effects. CAFs can provide metabolic support to PDAC cells, including amino acids and lipids, as well as paracrine signaling to promote growth and survival.61
In addition to direct interactions with tumor cells, CAFs also modulate the immune microenvironment. CAFs may mediate immunosuppression by promoting T cell exclusion, and recent studies suggest that CAFs can present antigens to immune cells, highlighting an additional possible mechanism for immune modulation by CAFs.62,63 Netrin G1 was recently identified as a regulator of both metabolic and immunosuppressive functions of CAFs, suggesting that shared molecular mechanisms underlie distinct CAF functions.64 Despite these seemingly tumor-promoting effects, targeting of CAFs in mice has led to increased tumor growth, and clinical trials of Hedgehog pathway inhibitors that target stromal cells have shown no benefit.65–68 These dual tumor-promoting and tumor-suppressive effects of CAFs may be explained in part by functional heterogeneity of different CAF populations. Recent studies have identified spatially and molecularly distinct subtypes of CAFs, including myofibroblastic and inflammatory CAFs, that likely play distinct roles in pancreatic tumorigenesis.69,70 One recent study showed that inhibition of Hedgehog signaling decreased the proportion of myofibroblastic CAFs and altered the immune cell composition, suggesting intricate crosstalk between different non-neoplastic cell types in the PDAC microenvironment.71 A newly described CAF subtype, antigen-presenting CAFs, express major histocompatibility complex class II and present antigens to T cells, further demonstrating this CAF-immune crosstalk.62
The provision of metabolic support to PDAC cells is not limited to CAFs. Peripheral axons innervating pancreatic tumors release amino acids to support PDAC growth in a nutrient-poor microenvironment.72 Ablation of nerves can slow PDAC development in model systems, highlighting the role of this component of the microenvironment in pancreatic tumorigenesis.73,74
앞서 설명한 종양 세포의 광범위한 분자적 변화 외에도 종양 미세환경에서 비종양 세포와의 상호작용도 췌장 종양 발생에 중요한 역할을 합니다(그림 2). 이 주제는 최근 다른 연구자들에 의해 심도 있게 검토되었습니다.56 PDAC는 면역 세포 아형에 여러 가지 변화를 일으켜 면역 억제 또는 "차가운" 미세 환경을 초래합니다. 이러한 변화에는 면역 억제 조절 T 세포와 골수 유래 억제 세포가 증가하여 암세포의 면역 매개 표적화를 방해하는 것이 포함됩니다. 또한 종양 관련 대식세포는 인터루킨-6을 분비하여 전암성 병변의 진행을 촉진할 수 있습니다.57 그러나 이러한 면역 미세환경의 특징은 인간 PDAC에서 보편적으로 나타나는 것은 아니며 일부 연구자들은 면역 미세환경의 하위 유형화 체계를 제안하여 뚜렷한 "탈출" 및 "소진" 표현형을 포함합니다.58 PDAC의 면역 변화가 질병 진행에 영향을 주고 체크포인트 억제 같은 면역 치료 접근법에 대한 반응 부족에 기여할 가능성이 높습니다. 최근 심도 있게 검토된 바와 같이 면역 억제성 미세 환경을 유발하는 신호에 대한 메커니즘적 통찰력이 높아지면 치료 효능을 향상시킬 수 있는 새로운 전략을 발견할 수 있습니다.59,60
비면역 세포와의 상호작용도 PDAC의 발달에 크게 기여합니다. PDAC는 암 관련 섬유아세포(CAF)에 의해 생성되는 치밀한 탈형성 기질을 가지고 있습니다. CAF는 시험관 내, 쥐 모델 및 인간 조직 샘플에서 광범위하게 연구되었습니다. 직관적이지 않을 수도 있지만, CAF는 종양 촉진 효과와 종양 억제 효과를 모두 가질 수 있습니다. CAF는 성장과 생존을 촉진하는 파라크린 신호뿐만 아니라 아미노산과 지질을 포함한 신진대사 지원을 PDAC 세포에 제공할 수 있습니다.61
CAF는 종양 세포와의 직접적인 상호 작용 외에도 면역 미세 환경을 조절합니다. CAF는 T세포 배제를 촉진하여 면역 억제를 매개할 수 있으며, 최근 연구에 따르면 CAF가 면역 세포에 항원을 제시할 수 있어 CAF에 의한 면역 조절의 또 다른 가능한 메커니즘을 강조합니다.62,63 Netrin G1은 최근 CAF의 대사 및 면역 억제 기능을 모두 조절하는 것으로 확인되었으며, 이는 공유 분자 메커니즘이 다른 CAF 기능의 근간을 이루고 있음을 시사합니다.64 이러한 종양 촉진 효과에도 불구하고 생쥐에서 CAF를 표적으로 삼으면 종양 성장이 증가했으며, 기질 세포를 표적으로 하는 헤지혹 경로 억제제의 임상 시험에서는 이점이 없는 것으로 나타났습니다.65-68 CAF의 이러한 이중 종양 촉진 및 종양 억제 효과는 부분적으로는 서로 다른 CAF 집단의 기능 이질성으로 설명할 수 있습니다. 최근 연구에 따르면 췌장 종양 형성에서 뚜렷한 역할을 하는 근섬유아세포 및 염증성 CAF를 포함하여 공간적, 분자적으로 뚜렷한 하위 유형의 CAF가 확인되었습니다.69,70 최근 한 연구에 따르면 헤지혹 신호의 억제가 근섬유아세포 CAF의 비율을 감소시키고 면역 세포 구성을 변화시켜 PDAC 미세환경에서 다양한 비종양 세포 유형 간의 복잡한 교류를 시사합니다.71 새로 기술된 CAF 하위 유형인 항원 제시 CAF는 주요 조직 적합성 복합체 클래스 II를 발현하고 T 세포에 항원을 제시하여 이러한 CAF 면역 교류를 더욱 입증합니다.62
PDAC 세포에 대한 대사 지원 제공은 CAF에만 국한되지 않습니다. 췌장 종양을 자극하는 말초 축삭은 영양이 부족한 미세 환경에서 PDAC 성장을 지원하기 위해 아미노산을 방출합니다.72 신경을 제거하면 모델 시스템에서 PDAC 발달이 느려져 췌장 종양 발생에서 미세 환경의 이 구성 요소의 역할을 강조할 수 있습니다.73,74
Etiology and Risk Factors
Studies in large patient cohorts have revealed several environmental and chronic disease-related factors that increase the risk of development of PDAC (Figure 2). Cigarette smoking is an established risk factor for pancreatic cancer, with an odds ratio of 1.74 for current smokers compared with never smokers and a population attribute risk of 11% to 32%.75 The mechanisms by which smoking increases the PDAC risk are likely multifactorial and include DNA damage as well as inflammation and fibrosis.76 Similarly, heavy alcohol consumption (>6 drinks per day) increases PDAC risk by 1.6-fold compared with nondrinking controls.
Patients with diabetes mellitus also have an increased risk of PDAC; this increased risk persists among patients with new-onset diabetes as well as those with long-standing diabetes.77 Long-standing diabetes of >3 years increases risk by 1.5- to 2.4-fold.75 Although long-standing diabetes may play a causal role in pancreatic tumorigenesis, it appears that new-onset diabetes (NOD) is an effect rather than a cause of PDAC, and recent studies have focused on this symptom to define a high-risk group for screening.78,79 Approximately 1% or less of patients with NOD develop PDAC within 3 years.80,81 The incidence of PDAC in patients with NOD may increase to as high as 3% in white men after age 50 or range between 1.5% and 6% when a predictive model that considers age at onset, change in weight, and change in blood glucose (Enriching New-Onset Diabetes for Pancreatic Cancer or ENDPAC score82) is used. A large NOD prospective cohort study consisting of subjects 50 years or older is under way to determine the 3-year incidence of PDAC and collect biospecimens as early detection initiative (CPDPC16-01).79
Obesity correlates with increased PDAC risk, although it is challenging to separate cancer risk due solely to obesity from that due to concomitant diabetes mellitus, which is increased in obese patients.77,83 Still, the increased risk associated with obesity persisted even after adjustment for history of diabetes in several studies, suggesting the obesity-associated risk is not solely driven by confounding diabetes.84 Thus, obesity is currently considered an independent risk factor for PDAC (odds ratio 1.72 for body mass index >30 kg/m2).85
Chronic pancreatitis is also associated with increased risk of pancreatic cancer. Like diabetes, a challenge in interpreting co-occurrence of chronic pancreatitis and PDAC is the ability of PDAC to cause chronic pancreatitis via obstruction of the ductal system. Although the risk declines over time after diagnosis of chronic pancreatitis, the elevated risk persists even after long-term follow-up, suggesting that this association is not solely a result of chronic pancreatitis caused by PDAC.86 In addition, smoking and alcohol consumption are risk factors for both chronic pancreatitis and PDAC, further complicating the assessment of specific risk associated with chronic pancreatitis.87
Inherited DNA variants play an important role in PDAC risk, and approximately 10% of PDACs occur in patients with a family history of pancreatic cancer. Increased risk of PDAC is a component of several inherited syndromes. These include familial cancer predisposition syndromes, such as familial atypical mole melanoma syndrome (caused by constitutional CDKN2A mutations) and Peutz-Jeghers syndrome (caused by constitutional STK11 mutations), both of which have strikingly elevated risks of PDAC.88,89 Patients with these syndromes are at increased risk of cancer in the pancreas as well as other organs. Patients with hereditary pancreatitis have an increased cancer risk limited to the pancreas, likely due to the repeated bouts of inflammation and repair. Patients with constitutional mutations in PRSS1, which encodes cationic trypsinogen and is the best characterized hereditary pancreatitis gene, have an astounding cumulative risk of PDAC of approximately 50% by age 75.90 Mutations in PRSS1 result in inappropriate intrapancreatic activation of trypsin, either by enhanced autoactivation or impaired inactivation, leading to pancreatic tissue destruction.91 Outside of these well-characterized inherited syndromes, patients with a family history of pancreatic cancer are at increased risk for PDAC, with the greatest risk among first-degree relatives of patients with PDAC.92 “Familial pancreatic cancer” is defined as families with a pair of affected first-degree relatives.93 Although some cases of familial pancreatic cancer are associated with constitutional alterations in well-characterized cancer predisposition genes (such as BRCA2, ATM), the genetic basis for most familial pancreatic cancer remains unknown.94–96 Pathogenic germline variants in pancreatic cancer susceptibility genes are identified in patients with seemingly sporadic PDAC, in almost 4% of unselected patients with PDAC in one study.97 These results highlight that inherited DNA alterations can increase PDAC risk even in patients without a family history of pancreatic cancer. Taken together, inherited mutations likely play a role in 10% to 15% of pancreatic cancers.
대규모 환자 코호트를 대상으로 한 연구에서 PDAC 발병 위험을 높이는 몇 가지 환경 및 만성 질환 관련 요인이 밝혀졌습니다(그림 2). 흡연은 췌장암의 확고한 위험 요인으로, 현재 흡연자의 경우 비흡연자에 비해 발병 확률이 1.74이고 인구 속성 위험은 11%에서 32%입니다.75 흡연이 PDAC 위험을 증가시키는 메커니즘은 다인성이며 염증 및 섬유증뿐만 아니라 DNA 손상도 포함할 수 있습니다.76 마찬가지로, 과도한 음주(하루 6잔 이상)는 비음주에 비해 PDAC 위험을 1.6배 높입니다.
당뇨병 환자도 PDAC 위험이 증가하며, 이러한 위험 증가는 새로 발병한 당뇨병 환자뿐만 아니라 오래 앓고 있는 당뇨병 환자에서도 지속됩니다.77 3년 이상의 장기 당뇨병은 위험을 1.5~2.4배 증가시킵니다.75 장기 당뇨병이 췌장 종양 발생에 인과적 역할을 할 수 있지만, 신규 발병 당뇨병(NOD)은 PDAC의 원인보다는 결과로 보이며, 최근 연구에서는 이 증상에 중점을 두고 선별 검사 고위험군을 정의하고 있습니다.78,79 NOD 환자의 약 1% 이하에서 3년 이내에 PDAC가 발생합니다.80,81 NOD 환자의 PDAC 발생률은 50세 이후 백인 남성에서 3%까지 증가하거나 발병 연령, 체중 변화, 혈당 변화를 고려한 예측 모델(췌장암 신규 발병 당뇨병 강화 또는 ENDPAC 점수82)을 사용할 경우 1.5%에서 6% 사이로 증가할 수 있습니다. 50세 이상의 피험자로 구성된 대규모 NOD 전향적 코호트 연구가 진행 중이며, PDAC의 3년 발병률을 파악하고 조기 발견 이니셔티브(CPDPC16-01)로 생체 검체를 수집하고 있습니다.79
비만은 PDAC 위험 증가와 관련이 있지만, 비만 환자에서 증가하는 당뇨병으로 인한 암 위험과 비만만으로 인한 암 위험을 분리하기는 어렵습니다.77,83 그럼에도 불구하고 여러 연구에서 당뇨병 병력을 조정한 후에도 비만과 관련된 위험 증가가 지속되어 비만 관련 위험이 당뇨병으로 인한 혼동만으로 인한 것이 아님을 시사합니다.84 따라서 비만은 현재 PDAC의 독립적 위험 요인으로 간주됩니다(체질량 지수 >30 kg/m2에 대한 오즈비 1.72).85
만성 췌장염은 또한 췌장암의 위험 증가와 관련이 있습니다. 당뇨병과 마찬가지로 만성 췌장염과 PDAC의 동시 발생을 해석하는 데 있어 어려운 점은 PDAC가 관 시스템을 방해하여 만성 췌장염을 유발할 수 있다는 점입니다. 만성 췌장염 진단 후 시간이 지남에 따라 위험은 감소하지만, 장기 추적 관찰 후에도 높은 위험은 지속되며, 이는 이러한 연관성이 PDAC로 인한 만성 췌장염의 결과만은 아님을 시사합니다.86 또한 흡연과 음주는 만성 췌장염과 PDAC 모두의 위험 요인으로 만성 췌장염과 관련된 특정 위험의 평가를 더욱 복잡하게 합니다.87
유전성 DNA 변이는 PDAC 위험에 중요한 역할을 하며, 췌장암 가족력이 있는 환자에서 약 10%의 PDAC가 발생합니다. PDAC의 위험 증가는 여러 유전 증후군의 구성 요소입니다. 여기에는 가족성 비정형 점 흑색종 증후군(체질성 CDKN2A 돌연변이로 인한)과 푸츠-제거스 증후군(체질성 STK11 돌연변이로 인한)과 같은 가족성 암 소인 증후군이 포함되며, 두 증후군 모두 PDAC 위험이 현저히 높습니다.88,89 이러한 증후군 환자는 췌장뿐만 아니라 다른 장기에서 암 위험이 높습니다. 유전성 췌장염 환자는 췌장에 국한된 암 위험이 증가하는데, 이는 염증과 회복이 반복되기 때문일 가능성이 높습니다. 양이온성 트립시노겐을 암호화하고 가장 잘 특징지어지는 유전성 췌장염 유전자인 PRSS1에 체질적 변이가 있는 환자는 75세까지 약 50%의 놀라운 PDAC 누적 위험이 있습니다.90 PRSS1의 변이는 자가 활성화 강화 또는 비활성화 장애로 인해 트립신의 부적절한 췌장 내 활성화를 초래하여 췌장 조직 파괴로 이어집니다.91 이러한 특징적인 유전 증후군 외에도 췌장암 가족력이 있는 환자는 PDAC의 위험이 높으며, PDAC 환자의 1촌 친척이 가장 큰 위험에 노출됩니다.92 "가족성 췌장암"은 영향을 받은 한 쌍의 일차 친척이 있는 가족으로 정의됩니다.93 가족성 췌장암의 일부 사례는 잘 특성화 된 암 소인 유전자 (예 : BRCA2, ATM)의 체질 변화와 관련이 있지만 대부분의 가족성 췌장암의 유전 적 기초는 아직 알려지지 않았습니다.94 -96 췌장암 감수성 유전자의 병원성 생식세포 변이는 한 연구에서 선택되지 않은 PDAC 환자의 거의 4%에서 산발적으로 나타나는 PDAC 환자에서 확인되었습니다.97 이러한 결과는 췌장암 가족력이 없는 환자에서도 유전된 DNA 변이가 PDAC 위험을 높일 수 있음을 강조합니다. 종합하면, 유전성 돌연변이는 췌장암의 10~15%에서 중요한 역할을 하는 것으로 보입니다.
Screening and Early Detection of Pancreatic Cancer
Rationale for Pancreatic Cancer Screening
Although PDAC comprises only 3% of new cancer cases in the United States,98 the prediction that by 2030 it will become the second leading cause of cancer death reflects its high mortality.1 The primary reason for this high mortality is that only a minority (11%) of patients are diagnosed with localized disease, and 52% of PDACs are already metastatic at presentation, with little hope of cure.2,98 This stage distribution implies that any successful strategy in improving PDAC mortality should include screening and early detection of potentially curable cancers. The long-term survival of patients with PDAC is greatly dependent on small tumor size and early disease stage.99
Population-based screening for PDAC is not currently justified due to its relatively low incidence in the United States (3% of new cancers),98 compared with breast (30%), colon (8%), or lung cancer (12%).2 The lifetime risk (age 85) of PDAC for Americans is only 1.7% 98 Enriching the population by targeting subgroups at greater risk for PDAC might improve the diagnostic yield of screening.
Surveillance of High-Risk Individuals
Currently, PDAC surveillance focuses on genetically predisposed individuals. Approximately 10% to 15% of PDAC cases are due to inherited mutations. High-risk individuals (HRIs) with a significantly increased risk for PDAC can be identified and offered surveillance (Table 1). Thresholds for risk over the general population can be set to design screening studies. Currently, an accepted threshold to consider PDAC surveillance is a lifetime risk of 5%. Table 1 summarizes the estimated lifetime risk, relative risk, and recommended age to start surveillance for HRIs supported by the multidisciplinary International Cancer of the Pancreas Consortium in 2019.100 New onset diabetes in an HRI should initiate surveillance regardless of age. The goal of surveillance is to detect and treat stage I PDAC or its high-grade precursor lesions (PanIN or IPMN) to prevent malignant progression.
Table 1.
Candidates for Pancreatic Cancer Screening, Estimated Risk for PDAC, and Recommended Age to Initiate Screening
Deleterious mutationGenetic syndromeLifetime risk of PDAC (age 70 y)Relative riskPDAC in affected blood relativeWhen to start
| Group 1 Low to Moderate Risk | |||||
| BRCA1 | Breast-Ovarian Cancer | 1.2% | 2.6 (1.6–4.7) | ≥1 FDR or any 2 | 50–55 a |
| BRCA2 | Breast-Ovarian Cancer | 2%–5% | 3.5 (2.2–12.8) | ≥1 FDR or any 2 | 50–55 ya |
| PALB2 | ? | ? | ≥1 FDR or any 2 | 50–55 ya | |
| ATM | ? | 5.7 | ≥1 FDR or any 2 | 50–55 ya | |
| MLH1, MSH2, MSH6 (DNA mismatch repair) | Lynch | 3.7% | 6.6–8.6 | ≥1 FDR or any 2 | 50–55 ya |
| Unknown | Familial PC | 8%–12% | 6.4 | 2 FDR | 50–55 ya |
| Group 2 High Risk | |||||
| Unknown | Familial PC | 40% | 32 | 3 FDR | 50–55 ya |
| CDKN2A | FAMMM | 10%–17% | 12–34 | None required | 40 y |
| P16 Leiden variant | FAMMM | 17%–58% | 48–80 | None required | 40 y |
| PRSS1 | Hereditary pancreatitis | 25%–40% | 50–80 | None required | 40 yb |
| STK11/LKB1 | Peutz-Jeghers | 30%–60% | 132 | None required | 40 y |
ATM, ataxia telangiectasia mutated; BRCA, breast-related cancer; CDKN2A, cyclin-dependent kinase inhibitor 2A; FAMMM, familial atypical multiple mole melanoma; PALB2, partner and localizer breast cancer 2; PRSS1, cationic trypsinogen gene; STK11, serine threonine kinase 11 gene.
aOr start 10 years younger than the youngest affected blood relative. Early age of onset of PDAC <50 years increases risk 9.3-fold.
bOr 20 years after first attack of acute pancreatitis.
Nineteen cohort studies (ranging in size from 15–262 HRIs) conducted around the world in the past 2 decades performing endoscopic ultrasound (EUS), magnetic resonance imaging (MRI), and/or computed tomography (CT)-based screening of HRIs report frequent detection of pancreatic lesions. Cystic lesions are commonly identified during screening in HRIs (10.5%–87.5%).101 A solid component within a cyst and main pancreatic duct dilation are detected in 5.8% (95% confidence interval 3%–9%) and 20.2% (95% confidence interval 14%–28%), respectively.102 In addition to conventional imaging, annual glucose testing or assessment of HbA1C is recommended to diagnose NOD in HRIs.100
The risk of PDAC is higher in germline mutation carriers. Hence, genetic testing is recommended for individuals with a PD AC-associated genetic syndrome (Table 1) and at-risk relatives of patients with PDAC. Two large, well-characterized high-risk cohorts with long-term follow-up of more than 350 patients showed significantly greater risk of neoplastic progression to high-grade dysplasia (HGD) or PDAC in mutation carriers compared with familial pancreatic cancer relatives (15% vs 9%, 9% vs 0%).
Five studies have concluded that imaging-based PC screening is cost-effective in appropriately identified HRIs.103–106 A meta-analysis of all prospective studies on surveillance of 7085 HRI estimated that the number needed to screen (NNS) to identify one patient with a high-risk pancreatic lesion (with HGD or PDAC) was 135. Specifically, the NNS is 71 for patients with Peutz-Jegher syndrome and 51 patients with constitutional CDKN2A mutations.101 These low numbers might suggest a high priority for screening in HRIs; however, important data are lacking in determining the NNS to prevent one pancreatic cancer death.
A 2019 systematic review of studies from January 2002 to March 2019 in patients with and without risk factors for PDAC published by the U.S. Preventive Services Task Force concluded that imaging-based screening in groups with a high familial risk can detect PDAC with minimal harm (18 cases of PDAC detected in 1156 familial HRIs). However, the U.S. Preventive Services Task Force recommended against screening for PDAC in asymptomatic adults107 because of the limited evidence to assess the benefits vs harms of surgical intervention, and effect of screening on mortality of screen-detected cancers.
Although the diagnostic yield of cross-sectional imaging is high, few studies report on the effect of screening on the morbidity, mortality, effectiveness of treatment, and quality of life of screen-detected PDAC.108 However, data supporting PDAC surveillance continue to accumulate and provide continued hope for positive outcomes from surveillance of HRIs. One meta-analysis of all prospective studies confirmed a stage shift of PDAC diagnosed within formal surveillance programs of HRI, with only 20% of PDAC at stage IV.101 Although limited by the small number of PDACs, there is also evidence that screen-detected PDACs in HRIs are more likely to be resectable (60%–90%)109–112 than sporadic PDAC in the general population (15%). Furthermore, surgical treatment of detected PDAC is associated with a high R0 resection rate and zero mortality.110,111,113,114 PDAC screening also has a positive effect on quality of life, psychological distress, and cancer worry.115,116
Currently, despite advances in surgical and oncologic treatment approaches, the 5-year survival of sporadic PDAC is only 10.8%.98 In contrast, PDAC surveillance programs in Europe and the United States report 5-year survival rates with asymptomatic screen-detected PDAC that are increased compared with the average PDAC (25%109 and 60%,113 respectively). These extended long-term survival rates are not attributable to lead-time bias, but appear to reflect the combined benefits of surgery for earlier stage disease, with adjuvant therapy in patients with better performance status. Overall, we maintain guarded optimism that there is a benefit for surveillance on PDAC survival when performed within formal multidisciplinary programs with state-of-the-art imaging and expertise in genetics, endoscopy, radiology, pancreatic surgery, and pathology.110,111,113,117
Challenges and Future Approaches for HRIs
Current challenges in conventional imaging-based pancreatic cancer screening/surveillance include high cost, invasiveness (at least for EUS), and suboptimal sensitivity and specificity. False positives may lead to unnecessary surgery for findings that do not represent HGD or PDAC114 (Figure 4). False negatives (nonspecific abnormalities such as a mildly dilated pancreatic duct, low-risk cystic lesions) may in turn lead to a delayed recommendation for treatment (Figure 5) or advanced incurable disease. Although PDAC resectability improves in HRI screening, only a handful of potentially curable stage I PDACs have been detected thus far in screening programs. Although screen-detected PDAC resectability is higher, there are still patients in screening who have identification of disease at a later stage than the desired target lesions (high-grade dysplasia or stage I PDAC) (Figure 5). We use well-known conventional cyst imaging features for IPMN progression to PDAC (such as solid mass, marked duct dilation), similar to those used for management of branch-duct IPMN, to recommend operative treatment, but these do not address microscopic PanIN progression.118 The ideal surveillance interval is yet to be known and interval missed cancers still develop,112 particularly in CDKN2A mutation carriers.117
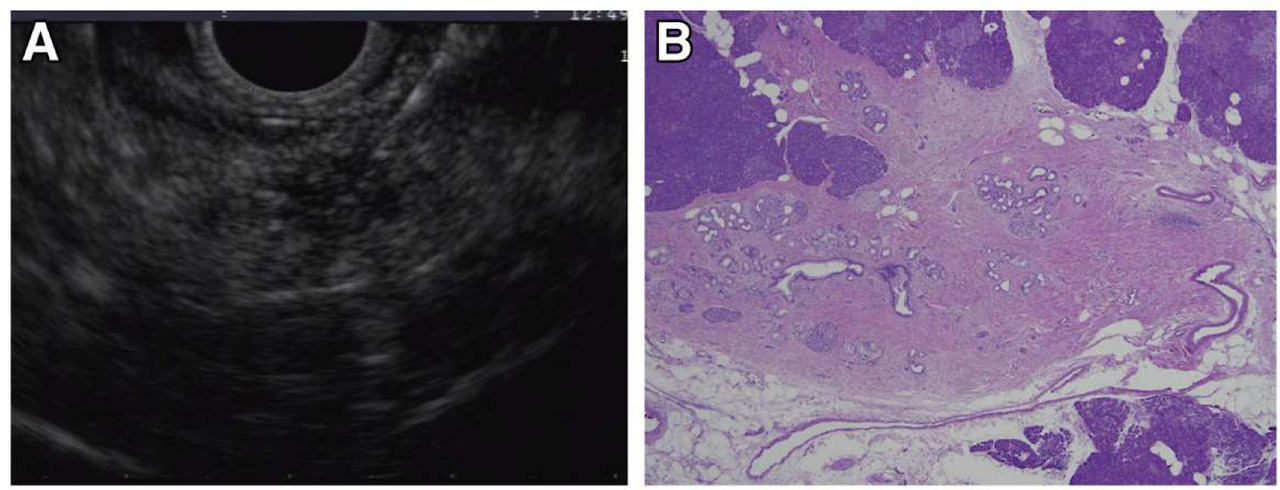
Incident indeterminate solid nodule: low-grade PanIN lesion. EUS and microscopic images from a familial pancreatic cancer relative who developed a 5-mm hypoechoic nodule in the tail (A). EUS-fine needle aspiration performed at 2 separate visits showed atypical cells. EUS-guided tattooing was performed to localize the lesion for surgical resection and histologic evaluation. Distal pancreatectomy with pathology examination of the specimen revealed a focal cluster of multiple low-grade PanIN with adjacent fibrosis (B), corresponding to the location of the preoperative tattoo.
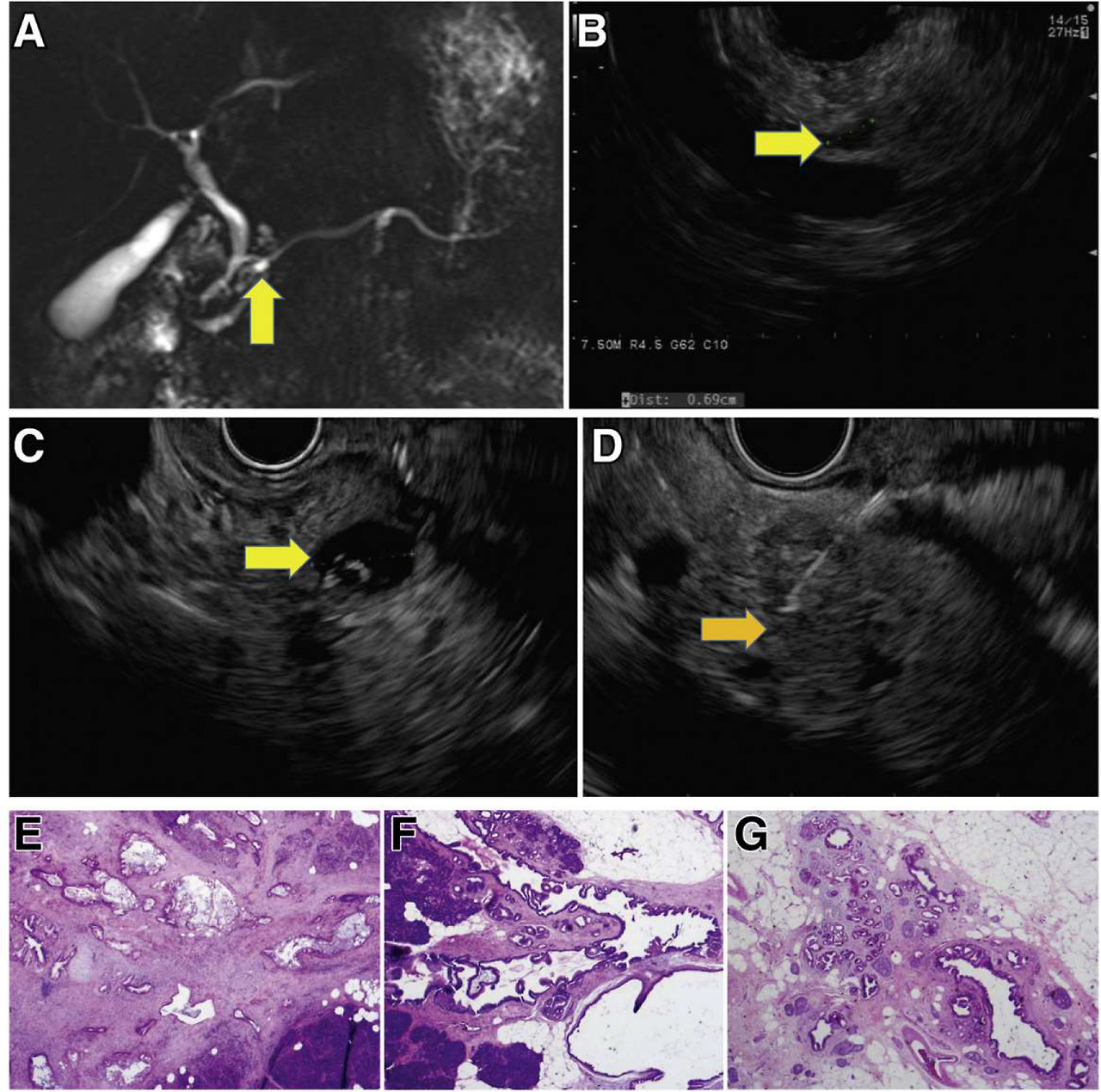
Incident screen-detected PDAC arising in a low-risk BD-IPMN. A familial pancreatic cancer patient with 3 affected FDR, who at baseline screening had subcentimeter cysts in the head (7 mm, yellow arrow) by MRI (A) and EUS (B). Surveillance continued with annual MRI alternating with EUS. Seven years from baseline, EUS showed a mildly dilated main pancreatic duct (4 mm) in the neck of the gland without a mass and 5 low-risk subcentimeter cystic lesions in the head, body, and tail. These changes did not meet international consensus guideline criteria for resection of IPMN. Repeat EUS at the next scheduled visit 1 year later showed the IPMN in the head (C, yellow arrow) associated with an asymptomatic PDAC (orange arrow, D), not visible on CT. Pancreaticoduodenectomy revealed a moderately differentiated T2N1 PDAC (hematoxylin-eosin stained pathology, image E) arising in an IPMN (F), low-grade IPMN, and extensive low-grade PanIN and associated lobulocentric atrophy (G). The patient is alive and well 4 years later.
Early Detection Biomarkers
Biomarkers may help detect microscopic high-grade PanIN precursors or early stage PDAC (Figure 1), clarify ambiguous screen-detected lesions, or prevent interval cancers in patients with a normal pancreas or trivial abnormalities on imaging. We look forward to continued progress with research on the discovery, optimization, and clinical validation of biomarkers in pancreatic juice (such as mutant TP53/SMAD4 DNA119) and blood (such as CA19–9, HbA1C plus other tumor markers,120–122 circulating tumor cells,123 exosomes,123 circulating-tumor DNA [ctDNA],124 or cell-free DNA [cfDNA]124). For the latter, the biggest challenge is the very low levels of peripheral DNA, which affects sensitivity,123 particularly for early-stage disease. A PDAC-specific antibody-based microarray test (IMMray PanCan-d, Immunovia, Marlborough, MA) measuring 8 serum biomarkers plus CA19–9, is being tested to determine the presence of a high-risk signature for PDAC. Although the details of the biomarkers are not publically available, they include proteins in multiple functional classes, including tumor-associated, hormone transport, bone metabolism, complement, coagulation, and protease inhibitor targets. Designed for use in HRIs, it serves to aid risk stratification and identify HRIs needing further clinical evaluation. Although initial results are promising,125 results from the PanFAM-1 longitudinal study in HRIs are pending (NCT03693378). Targeted methylation analysis of circulating cfDNA from pancreatic cancer (as well as multiple other tumor types, for example Galleri, Grail) is another promising approach still undergoing evaluation.126 Cancer-SEEK (Exact Sciences) assays ctDNA for 16 genes and 8 proteins shows promise in detecting cancers across 10 organs, including 7 that have no recommended standard-of-care screening tests, such as PDAC.127 The test by Blue Star Genomics, which received a Breakthrough Device designation from the Food and Drug Administration (FDA) and is undergoing clinical validation studies, involves analysis of 5-hydroxymethylcytosine signatures in cfDNA in various cancer types.128 In addition to these blood-based biomarkers, strategies deploying artificial intelligence129,130 are being developed in parallel to improve risk stratification for neoplastic progression and patient selection for surgery, high priorities for the next decade.
Diagnosis of Pancreatic Cancer
Clinical Presentation
Most patients with pancreatic cancer do not develop symptoms until the advanced stage of the disease. When patients develop symptoms, they are often vague and nonspecific, leading to a delay in diagnosis that may extend several months.131 The most commonly reported symptoms are fatigue, weight loss, anorexia, and abdominal or back pain. Tumors that arise in the pancreatic head and neck may be associated with pruritis and jaundice from biliary obstruction, whereas tumors in the body and tail of the gland more commonly present with pain. The development of NOD in the setting of any of the previously listed symptoms should prompt the patient’s physician to consider pancreatic cancer in the differential diagnosis. A thorough family history of cancer should be obtained to assess whether the patient may be a member of a familial pancreatic cancer kindred, or may carry a pathogenic germline mutation, especially if there are close family members with breast, colon, or skin (melanoma) cancers. Less common presenting symptoms include steatorrhea due to malignant obstruction of the main pancreatic duct, acute pancreatitis, gastric outlet obstruction, and venous thromboembolism.
Diagnostic Evaluation
After a complete history and physical examination, patients should undergo lab work to include a complete blood count, electrolyte panel, and liver function tests, the latter of which can help assess if an obstructive biliary process is present. A serum amylase is obtained for patients who present with epigastric and/or back pain to evaluate for the presence of acute pancreatitis. The tumor marker sialylated Lewisa blood group antigen CA 19–9 is a commonly used serum biomarker for PDAC, with a sensitivity and specificity of 80% to 90% in patients presenting with symptomatic disease.132 CA 19–9 levels are related to tumor size, presence of metastasis, and patient prognosis.132 Serial monitoring of CA 19–9 is useful to track response to systemic therapy in patients who present with elevated levels. Ten percent of the population do not express Lewis antigens,133 and in this situation the CA 19–9 level is deemed uninformative. CA 19–9 also can be elevated in the setting of benign pancreaticobiliary disease and biliary obstruction. Thus, in patients with PDAC with biliary obstruction who undergo biliary stent placement, CA 19–9 levels should be re-checked to more accurately assess true tumor burden before the initiation of therapy. In patients with localized, resectable disease and high CA 19–9 levels,134 consideration should be given to a neoadjuvant first approach, although National Comprehensive Cancer Network (NCCN) guidelines do not recommend a specific CA 19–9 cutoff level. Although CA 19–9 alone lacks adequate sensitivity for the early detection of pancreatic cancer, its use in combination with other biomarkers may provide added benefit in a blood-based early detection blood test in the future.122,135
Imaging
The imaging test of choice for the evaluation of a patient with a suspected pancreatic adenocarcinoma is a thin-cut, pancreatic protocol CT scan of the abdomen, providing a sensitivity in the detection of pancreatic cancer ranging from 76% to 96%.136 CT, compared with MRI, provides better anatomic detail of the relationship of the tumor to the surrounding vasculature, important in tumor staging. Pancreatic cancer typically appears as hypodense compared with the surrounding pancreatic parenchyma, reflecting poor penetration of contrast into these tumors, which possess high interstitial pressures due to extensive desmoplasia.137 Standard guidelines regarding the technical aspects of the performance of the pancreatic cancer CT protocol are included in NCCN guidelines, and include submillimeter slice thickness, proper timing of arterial and venous imaging, and 3-dimentional reconstruction for full vascular assessment.138 MRI is reserved for use in patients with contraindications to CT scanning, significant renal insufficiency, or a severe contrast allergy. MRI may also be a useful adjunct in patients to determine the etiology of indeterminate liver lesions. Positron emission tomography scans are not routinely used in the diagnostic evaluation of patients with PDAC, but may be helpful to detect occult metastatic disease, particularly in the setting of a high CA 19–9 disproportionately elevated based on tumor staging. Endoscopic ultrasound is used primarily for tissue acquisition to confirm the diagnosis of pancreatic cancer, but may be useful for diagnosis of small, isodense lesions.
Staging
PDAC is formally staged using a tumor nodes metastasis (TNM) system based on the eighth edition of the American Joint Committee on Cancer Staging Manual, with TNM staging used to provide prognostic information regarding patient outcomes. In practicality, clinicians use a 4-category staging system to classify tumors and guide treatment decisions: resectable, borderline resectable, locally advanced, and metastatic disease. NCCN guidelines (shown in Supplementary Table 1) define resectable disease as lacking arterial and venous involvement, with no distant spread.139 Tumors with venous contact >180 degrees, venous contour irregularity, or thrombosis are considered borderline resectable as long as the venous segment is reconstructible. Unreconstructable involvement of the superior mesenteric vein or portal vein is categorized as locally advanced.138 Arterial contact <180 degrees is considered borderline resectable, whereas contact >180 degrees is considered locally advanced. Resectability status should be determined in the context of a multidisciplinary team evaluation.
Treatment of Pancreatic Cancer
Pancreatic cancer has a very poor prognosis with a median survival of approximately 10 to 12 months with treatment and 5 to 6 months without treatment due to the advanced stage of diagnosis for many patients and the low percentage of patients eligible for surgical resection. The current standard of care for first-line therapy is FOLFIRONOX (a combination of 5-fluorouracil [5-FU], leucovorin, irinotecan, and oxaliplatin) or gemcitabine plus albumin-bound (nab) paclitaxel. Other combinations to date have not shown significant survival benefits over these treatments and/or result in treatment-limiting toxicities. However, current research on the molecular and tumor microenvironmental changes driving pancreatic cancer development, progression, and metastasis is uncovering distinct targetable pathways in certain patient subpopulations. By identifying these populations early in diagnosis, it may be possible to personalize therapy for some patients and improve their treatment outcome. As we learn more about these PDAC driving pathways, it should be possible to develop new drugs and combinations that further improve treatment outcomes for most patients.
Targetable Subtypes of Pancreatic Cancer
Many of the subtypes of PDAC susceptible to targeted therapies are associated with specific genetic alterations. Approximately 10% to 15% of PDACs are thought to be associated with inherited mutations, and all PDACs have somatic mutations in distinct combinations of driver genes. These mutations affect a range of cellular processes, including DNA repair, cellular proliferation pathways, and transcriptional activation of genes involved in cancer progression.
BRCA1/BRCA2 mutations.
BRCA1 (breast cancer type 1 susceptibility protein) and BRCA2 genes, which are involved in repairing double-strand DNA breaks via homologous DNA repair (HDR), are the most extensively characterized mutations. Mutations in these 2 genes were initially implicated as risk factors in breast and ovarian cancer and were later linked to pancreatic cancer. Mutations of other genes involved in the homologous DNA repair pathway, such as PALB2 and ATM, have also been implicated in PDAC, as well as what is referred to as a “BRCAness” HDR impaired phenotype. Mutations in BRCA1/2 are found in 5% to 10% of PDACs and occur in both familial and nonfamilial cases. In the Ashkenazi Jewish population, up to 20% patients may harbor these mutations.140
Breast and ovarian cancers with BRCA1, BRCA2, and/or related HDR mutations are susceptible to drugs that cause double-stranded DNA breaks, such as platinum salts and topoisomerase inhibitors. Recent retrospective141,142 and prospective143 studies examining the use of cisplatin in patients with PDAC with altered BRCA1/2 have shown clinical benefit, although superiority over standard of care treatments and optimal dosing and combination therapies are still being investigated.
PARP inhibitors such as niraparib and olaparib have also shown promise as treatment for tumors with BRCA mutations. These inhibitors act by preventing single-strand break repair, resulting in double-stranded breaks that are unable to be repaired by HDR deficient tumors, causing cell cycle arrest and apoptosis. Olaparib was shown to increase progression-free survival when used as a maintenance therapy for patients with pancreatic cancer who responded to first-line platinum-based therapy, leading to its FDA approval in 2019.144 Although these therapies show promise, they are also plagued by high cost and development of drug resistance. The development of optimal combination therapies for PDAC with the “BRCAness” phenotype should be pursued and may prove to be of great benefit to this subset of patients.140,145
Microsatellite instability.
Another distinct pancreatic cancer subtype is characterized by microsatellite instability (MSI). These tumors are defective in DNA mismatch repair, and can occur in inherited disorders such as Lynch Syndrome. These patients with PDAC (1% of cases) have been found to differ from patients with microsatellite stable tumors by being less responsive to 5-FU and gemcitabine and more responsive to FOLFIRINOX.146,147
In many cancers, these MSI tumors are more responsive to immune-based therapies and have a favorable prognosis.148 Since 2017, pembrolizumab, an inhibitor of the immune checkpoint protein PD-1 (programmed cell death protein-1) has been approved for mismatch-repair deficient cancers at all tumor sites. It acts as an immune checkpoint blockade preventing the binding of PD-1 to PD-L1 (programmed death ligand-1), which is selectively expressed on the surface of cancer cells but not normal cells. PD-1/PD-L1 binding suppresses the proliferation of antigen-specific T cells and causes apoptosis of regulatory T cells. Therefore, blocking of this interaction increases the patient’s innate immune response to the tumor.
PDAC, however, has proven to be more difficult to treat with immune-based therapies due to large quantities of immune-suppressor cells and characteristics of the stromal tissue. Pembrolizumab has shown some success in patients with MSI-high PDAC but not MSI-low tumors. Numerous phase I/II clinical trials have sought to increase the efficacy of PD-1/PD-L1 inhibitors in MSI-low PDACs by combining them with chemotherapy, radiotherapy, other immunotherapies, and vaccines to increase their efficacy. The COMBAT trial (NCT02826486) recently demonstrated that immunomodulation with a CXCR4 antagonist and pembrolizumab may improve clinical response to chemotherapy.149 There is also evidence suggesting that the CD40 agonist selicrelumab may modulate the tumor microenvironment in pancreatic cancer making it more immunologically active.150 Ghidini et al151 recently published a review of the current immune-based treatment approaches for patients with PDAC with regard to MSI. Although significant increases in overall survival have yet to be observed from these combinations, measurable changes in tumor immunogenicity indicate that further research in this area may lead to significantly improved immune therapies for pancreatic cancer.151
KRAS mutations.
In contrast to the preceding mutations and phenotypes that only affect a small subset of PDACs, mutations in KRAS (Kirsten rat sarcoma viral oncogene homolog) are found in 90% to 95% of PDACs and may be a driving force in pancreatic tumorigenesis. KRAS is a membrane-bound GTPase that signals cellular growth through the MAPK and PI3K pathways in its GTP form. The most prevalent KRAS oncogenic mutations have been shown to disrupt the balance of GTP- and GDP-bound KRAS.
Although mutant KRAS has been pursued as a therapeutic target for many years, its high picomolar binding affinity for GTP/GDP have made small molecule inhibition efforts of the nucleotide binding pocket difficult. However, in 2013, Ostrem et al152 published crystallographic evidence of an unknown allosteric binding pocket of KRASG12C, a common KRAS mutation in many cancers. In 2018, Janes et al153 published the discovery of a small molecular compound, ARS-1620, that is specific to this binding pocket and acts to sequester KRAS in its inactive GDP bound form. This proof-of-concept study spurred renewed interest in selective inhibition of mutated KRAS. Just 4 years later, 2 next-generation KRASG12C inhibitors have advanced to clinical use. In 2021 Sotorasib (AMG510) was FDA approved154 and Adagrasib (MRTX849) received FDA breakthrough therapy designation.155 A timely review of the clinical progress of KRASG12C inhibitors was recently published in January 2022.156
Although the advancement of KRASG12C inhibitors is impactful for treatment of certain cancers, such as lung cancer, this mutation is relatively rare in pancreatic cancer, which primarily has G12D and G12V mutations. Other innovative KRAS inhibition approaches, including Pan KRAS inhibitors that target the guanine exchange factor SOS1 (BI 1701963),157 a number of SHP2 inhibitors, and combination therapies including KRAS-specific vaccines158 may benefit patients with pancreatic cancer in the future. A promising G12D-specific inhibitor, MRTX1133, is also poised to undergo clinical testing in 2022.159 Active work in this area is focusing on the development of inhibitors to mutations relevant to PDAC and combination therapies to combat compensatory signaling pathways.
ARID1A mutations.
Another commonly mutated gene across a range of cancers is AR1D1A (AT-rich interaction domain 1A), a subunit of the chromatin remodeling complex SWI/SNF. In PDAC, ARID 1A deficiency correlates with poorly differentiated tumors and is a poor prognostic indicator. ARID1A is functionally complex, having both tumor suppressor and oncogenic properties across a number of cellular processes, including telomere maintenance, DNA damage repair, and transcriptional control.160 Recent studies have found that loss of ARID1A function correlates with the development of MSI and is associated with high mutational burden, high PD-L1 expression, and an increase in tumor infiltrating lymphocytes, which are all predictive of sensitivity to immune checkpoint blockade therapy.161 Indeed, increased progression-free and overall survival has been observed retrospectively in patients with ARID1A-deficient tumors that have undergone immunotherapy.162,163 Clinical and mouse model data also indicate increased sensitivity to PARP inhibition.164 Preclinical studies also have implicated that EZH2, HDAC6, and PI3K/AKT/mTOR inhibitors may also have increased efficacy against ARIDlA-deficient tumors.161 The exact role of ARID 1A specifically in PDAC and the use of its mutational status as an indicator for targeted therapy are still actively being investigated.
Treatment Based on Disease Stage
Other promising targeted PDAC therapies are based on disease stage. Localized tumors are categorized as resectable, borderline resectable, or locally advanced. The standard of care for resectable tumors, which lack of distant metastases and have limited vascular invasion, is partial surgical removal of the affected region of the pancreas followed by adjuvant chemotherapy. Partial removal includes pancreaticoduodenectomy surgery, also known as Whipple procedure, to remove the head of the pancreas or distal pancreatectomy to remove the tail of the organ. A total pancreatectomy is performed when the entire pancreas is affected. In addition, neoadjuvant treatment is sometimes used in this patient group. Randomized data comparing outcomes in up-front surgery vs neoadjuvant treatment followed by surgery in resectable patients is not yet available.
Neoadjuvant chemotherapy and/or radiotherapy before surgery is used in locally advanced and in most borderline resectable tumors to improve margin-negative resection rates and to potentially induce tumor downstaging. Improvements in surgical techniques have led to increased use of venous and arterial reconstruction to render some locally advanced tumors resectable. Tumor tissue acquisition pretreatment (via biopsy) and at surgery offers a unique opportunity to observe the intratumoral changes in the tumor and TME after various treatment regimens.165,160 A recent analysis by Farren et al167 that examined gene expression and spatial distribution of numerous immunologically relevant cells and proteins showed distinct differences in resected tumors from untreated pancreatic tumors vs tumors treated with neoadjuvant therapy. As a retrospective study, the relevance of these changes to patient outcome and response to further adjuvant therapy are not addressed; however, the results indicate further research in this area is needed.167
Adjuvant therapy typically occurs within 4 to 8 weeks of resection once the patient has recovered from surgery. Selection of the proper treatment regimen for each patient may vary based on the patient’s health status and cancer stage. Recent progress in the identification of genetic biomarkers, the use of ctDNA as a disease state indicator, and improved imaging methods are beginning to allow for improved ability to optimize adjuvant therapy selection. In a recent review, Turpin et al168 gives a comprehensive overview of the current state of adjuvant therapy selection. This is an active area of research that may eventually allow for the personalization of adjuvant therapy for every patient.
The treatment of nonresectable and metastatic disease varies greatly depending on treatment response, the existence of any of the aforementioned genetic mutations or phenotypes, and the overall health of the patient. In most cases, the standard first-line treatment, as with localized disease, is FOLFIRINOX or gemcitabine plus albumin-bound (nab) paclitaxel. The select subgroups noted previously may benefit from alternative targeted treatments at this stage. Second-line treatment for patients capable of more combination therapy would be the alternate treatment, that is, fluoropyrimidine-based treated patients would receive gemcitabine plus albumin-bound (nab) paclitaxel and gemcitabine-treated patients would get FOLFIRONOX or a modified 5-FU-based regimen such as FOLFIRI (5-FU, folic acid, and irinotecan) or FOLFOX (leucovorin, 5-FU, oxaliplatin). Single-agent treatments may be given to patients with a lower performance status.
Recent research is also showing that maintenance therapy may be beneficial to select patient groups. Olaparib was approved as a maintenance therapy in 2019 for patients with BRCA1 or BRCA2 mutations and metastatic disease.144 Clinical trials examining a combination of 5-FU and leucovorin or gemcitabine as PDAC maintenance therapy have also shown encouraging results.169,170 Many clinical trials are now incorporating maintenance therapy for pancreatic cancer minimal residual disease detected by ctDNA to improve long-term prognosis. As more targetable subtypes of pancreatic cancer are discovered, the treatment of both localized and metastatic disease should improve.
Future Treatment Considerations
To date, the few FDA-approved chemotherapy and targeted therapies have only increased the 5-year overall survival of pancreatic cancer patients from about 2% a decade ago to 11% in 2022. However, an increase in our understanding of the biology of subtypes of PDAC have led to more targeted approaches. New clinical trial designs that incorporate drug lead-in, neoadjuvant testing of investigational agents, and platform studies that allow rapid testing of combinations are facilitating progress. Over the next decade, we should expect to see improved clinical outcomes for a greater number of patients who will be treated with patient-specific drug combinations.
Conclusions
An improved understanding of the biologic processes driving pancreatic tumorigenesis is taking place. With the advent of increased use of genetic testing, individuals at elevated risk of pancreatic cancer are being identified and entered into screening programs, and targeted therapies for subtypes of patients with pancreatic cancer are coming into clinical use. Although these efforts show promise, large-scale, coordinated efforts will be needed to maximize the clinical impact for patients and improve overall survival.
Supplementary Material
1
Click here to view.(230K, pdf)
Abbreviations:
| CAF | cancer-associated fibroblast |
| cfDNA | cell-free DNA |
| CT | computed tomography |
| ctDNA | circulating-tumor DNA |
| EUS | endoscopic ultrasound |
| FDA | Food and Drug Administration |
| HDR | homologous DNA repair |
| HGD | high-grade dysplasia |
| HRI | high-risk individual |
| IPMN | intraductal papillary mucinous neoplasm |
| MRI | magnetic resonance imaging |
| MSI | microsatellite instability |
| NCCN | National Comprehensive Cancer Network |
| NNS | number needed to screen |
| NOD | new-onset diabetes |
| PanIN | pancreatic intraepithelial neoplasia |
| PD-1 | programmed cell death protein-1 |
| PDAC | pancreatic ductal adenocarcinoma |
| PD-L1 | programmed death ligand-1 |
Footnotes
Conflict of interest
These authors disclose the following: Elizabeth M. Jaffee is the Dana and Albert “Cubby” Professor of Oncology. She is a paid consultant for Adaptive Biotech, CSTONE, Achilles, DragonFly, Candel Therapeutics, NextCure, STIMIT, and Genocea. She receives funding from Lustgarten Foundation, AstraZeneca, Genentech, and Bristol Myer Squibb. She is the Chief Medical Advisor for Lustgarten and SAB advisor to the Parker Institute for Cancer Immunotherapy (PICI) and for the Break Through Cancer Institute. She is a founding member of Abmeta. Diane M. Simeone is the Laura and Isaac Perlmutter Professor of Surgery. She serves in an advisory capacity for Merck, Bluestar Genomics, and Interpace, and receives funding from the Pancreatic Cancer Action Network, Cyteir Therapeutics, Tempus, Micronoma and Novartis. Marcia Irene Canto is a Professor of Medicine and Oncology. She is a consultant for Castle Biosciences, BlueStar Genomics, and Pentax Medical Corporation. She receives royalties from UpToDate. She receives funding for research from Pentax Medical Corporation and Endogastric Solutions. The remaining author discloses no conflicts.
Supplementary Material
Note: To access the supplementary material accompanying this article, visit the online version of Gastroenterology at www.gastrojournal.org, and at https://doi.org/10.1053/j.gastro.2022.03.056.
References
1. Rahib L, Smith B, Aizenberg R, et al. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res 2014;74:2913–2921. [PubMed] [Google Scholar]
2. Siegel RL, Miller KD, Fuchs HE, et al. Cancer Statistics, 2021. CA Cancer J Clin 2021;71:7–33. [PubMed] [Google Scholar]
3. Kommalapati A, Telia SH, Goyal G, et al. Contemporary management of localized resectable pancreatic cancer. Cancers (Basel) 2018;10:24. [PMC free article] [PubMed] [Google Scholar]
4. Basturk O, Hong SM, Wood LD, et al. A revised classification system and recommendations from the baltimore consensus meeting for neoplastic precursor lesions in the pancreas. Am J Surg Pathol 2015;39:1730–1741. [PMC free article] [PubMed] [Google Scholar]
5. Winter JM, Cameron JL, Campbell KA, et al. 1423 pancreaticoduodenectomies for pancreatic cancer: a single-institution experience. J Gastrointest Surg 2006;10:1199–1210; discussion 1210–1191. [PubMed] [Google Scholar]
6. Felsenstein M, Noe M, Masica DL, et al. IPMNs with cooccurring invasive cancers: neighbours but not always relatives. Gut 2018;67:1652–1662. [PMC free article] [PubMed] [Google Scholar]
7. Zamboni G, Scarpa A, Bogina G, et al. Mucinous cystic tumors of the pancreas: clinicopathological features, prognosis, and relationship to other mucinous cystic tumors. Am J Surg Pathol 1999;23:410–422. [PubMed] [Google Scholar]
8. Laffan TA, Horton KM, Klein AP, et al. Prevalence of unsuspected pancreatic cysts on MDCT. AJR Am J Roentgenol 2008;191:802–807. [PMC free article] [PubMed] [Google Scholar]
9. Grimont A, Leach SD, Chandwani R. Uncertain beginnings: acinar and ductal cell plasticity in the development of pancreatic cancer. Cell Mol Gastroenterol Hepatol 2021;13:369–382. [PMC free article] [PubMed] [Google Scholar]
10. Matsuda Y, Furukawa T, Yachina S, et al. The prevalence and clinicopathological characteristics of high-grade pancreatic intraepithelial neoplasia: autopsy study evaluating the entire pancreatic parenchyma. Pancreas 2017;46:658–664. [PubMed] [Google Scholar]
11. Andea A, Sarkar F, Adsay VN. Clinicopathological correlates of pancreatic intraepithelial neoplasia: a comparative analysis of 82 cases with and 152 cases without pancreatic ductal adenocarcinoma. Mod Pathol 2003;16:996–1006. [PubMed] [Google Scholar]
12. Rezaee N, Barbon C, Zaki A, et al. Intraductal papillary mucinous neoplasm (IPMN) with high-grade dysplasia is a risk factor for the subsequent development of pancreatic ductal adenocarcinoma. HPB (Oxford) 2016;18:236–246. [PMC free article] [PubMed] [Google Scholar]
13. Oyama H, Tada M, Takagi K, et al. Long-term risk of malignancy in branch-duct intraductal papillary mucinous neoplasms. Gastroenterology 2020;158:226–237.e225. [PubMed] [Google Scholar]
14. Furukawa T, Kloppel G, Adsay NV, et al. Classification of types of intraductal papillary-mucinous neoplasm of the pancreas: a consensus study. Virchows Archiv 2005;447:794–799. [PubMed] [Google Scholar]
15. Noë M, Niknafs N, Fischer CG, et al. Genomic characterization of malignant progression in neoplastic pancreatic cysts. Nat Commun 2020;11:4085. [PMC free article] [PubMed] [Google Scholar]