Warburg 효과(Warburg effect, 호기성 해당과정 증가)부터 시작해,
최근 연구에서 밝혀진 oncometabolites,
지질 대사,
아미노산 대사 등이
어떻게 T 세포, NK 세포,
수지상세포(DC), MDSC(myeloid-derived suppressor cells),
Treg(regulatory T cells) 등에 영향을 미치는지 설명합니다.


https://pmc.ncbi.nlm.nih.gov/articles/PMC7238960/
| 1. 면역항암제(면역 체크포인트 억제제)의 기본 원리 우리 몸의 T세포(특히 암을 공격하는 세포독성 T세포, CD8+ T세포)는 암세포를 인식하고 죽이는 역할을 합니다. 하지만 T세포가 과도하게 활성화되면 자가면역 질환이 생길 수 있어서, 몸에는 '브레이크(억제 신호)' 역할을 하는 면역 체크포인트 단백질이 있습니다.  암세포는 이 브레이크를 교묘하게 이용해 면역 회피(immune evasion)를 합니다. 면역항암제는 이 브레이크를 풀어주는 약물로, T세포가 다시 암세포를 강하게 공격할 수 있게 만듭니다. 대표적인 체크포인트:
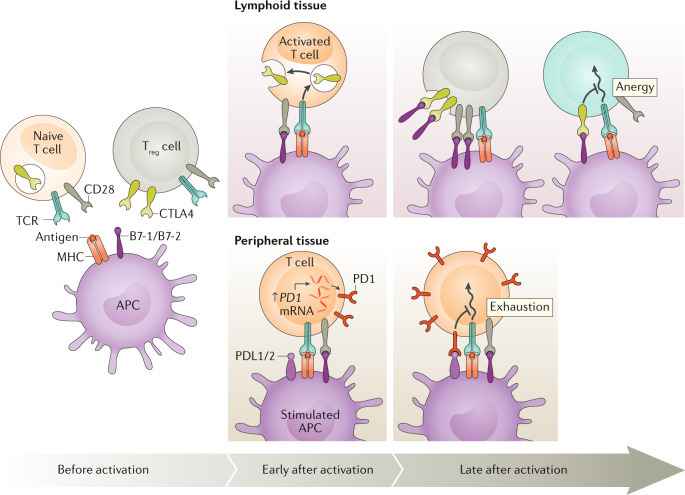 1. Before activation (활성화 전)
2. Early after activation (활성화 직후) – 주로 Lymphoid tissue (림프조직)
(1) Lymphoid tissue (림프조직)에서 – CTLA-4 중심 (Anergy: 무기력 상태)
(2) Peripheral tissue (말초 조직, 종양 부위)에서 – PD-1 중심 (Exhaustion: 소진 상태)
2. CTLA-4 억제제의 기전 (예: Ipilimumab)
3. PD-1/PD-L1 억제제의 기전 (예: Pembrolizumab, Nivolumab - PD-1 억제제 / Atezolizumab - PD-L1 억제제)
4. CTLA-4 vs PD-1/PD-L1 비교 (논문 Fig. 2 참조)
|

식이 요인(특히 식이섬유 발효)이
면역항암제(주로 immune checkpoint inhibitors, ICI like anti-PD-1/PD-L1)의 효과를
어떻게 영향을 미치는지 종합적으로 검토한 리뷰.
발효가 면역항암제 효과를 증진하는 주요 기전 (논문 중심 요약)
발효의 핵심은
장내 미생물(gut microbiota)에 의한 식이섬유(dietary fiber)의 발효로,
단쇄지방산(Short-Chain Fatty Acids, SCFAs)
특히 부티레이트(butyrate) 같은 대사물이 생성되는 과정입니다.
이 SCFAs가
면역 체계를 조절해 종양 미세환경(Tumor Microenvironment, TME)을 개선하고,
IC(면역항암제)I의 반응성을 높입니다.
주요 기전:
- SCFAs 생산 증가: 섬유질이 장내 세균에 의해 발효되면 butyrate, acetate, propionate 등이 생성됨. 이들은 항염증 효과를 발휘하며, Treg (조절 T세포) 수를 늘리고, IFN-γ 같은 프로염증 사이토카인을 줄임.
- 면역 세포 조절: SCFAs가 T세포 분화와 기능을 자극 → 효과기 T세포(effector T cells) 활성화 ↑, 항종양 면역 강화.
- HDAC 억제: Butyrate는 histone deacetylase(HDAC)를 억제해 유전자 발현을 변화시켜 면역 활성화를 돕습니다.
- GPR 수용체 활성화: SCFAs가 G-protein-coupled receptor (예: GPR43)를 통해 신호를 전달해 Treg 기능과 면역 균형을 조절.
- TME 개선: PD-L1 발현 억제 가능성, 면역억제 신호 감소 → ICI(anti-PD-1 등)가 T세포를 더 잘 "풀어주는" 환경 조성.
- 장내 미생물 변화: 섬유질 풍부 식이가 SCFA 생산균을 증가시켜 전체적인 면역 기능을 강화 → ICI 반응성 향상 (동물 모델에서 고섬유 식이 + anti-PD-1 병용 시 종양 성장 억제 효과 ↑).
결과적으로,
고섬유 식이 → 장 발효 → SCFAs ↑ → 면역 강화 → ICI 효과 증진의 축이
핵심입니다.
논문 그림 위주 설명
Fig. 5A가 발효 기전을 가장 잘 보여주는 핵심 그림입니다. 나머지 그림들은 배경 설명 역할을 합니다.
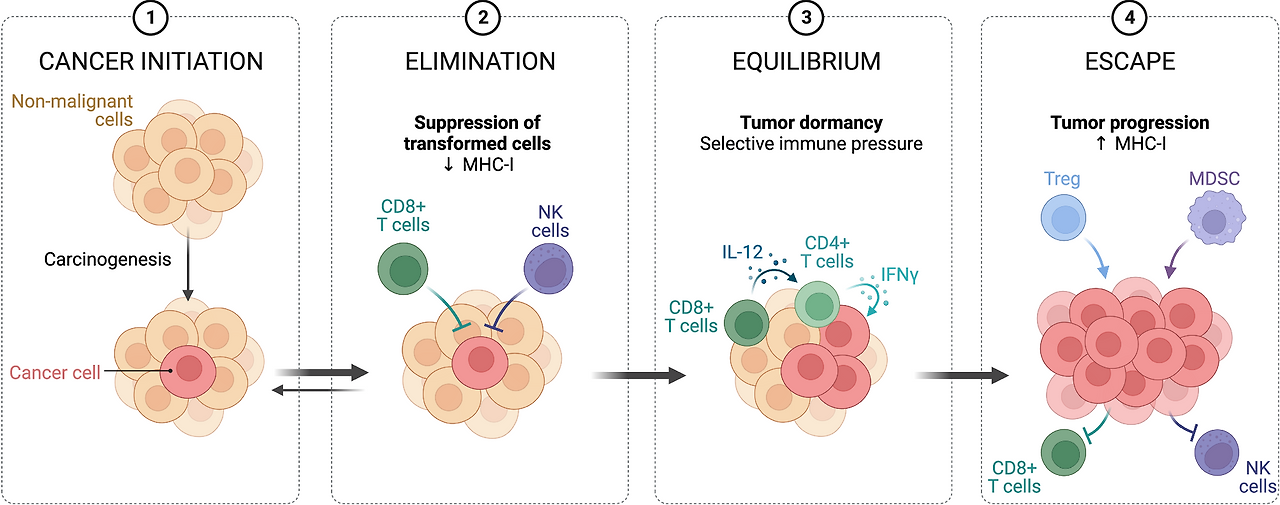
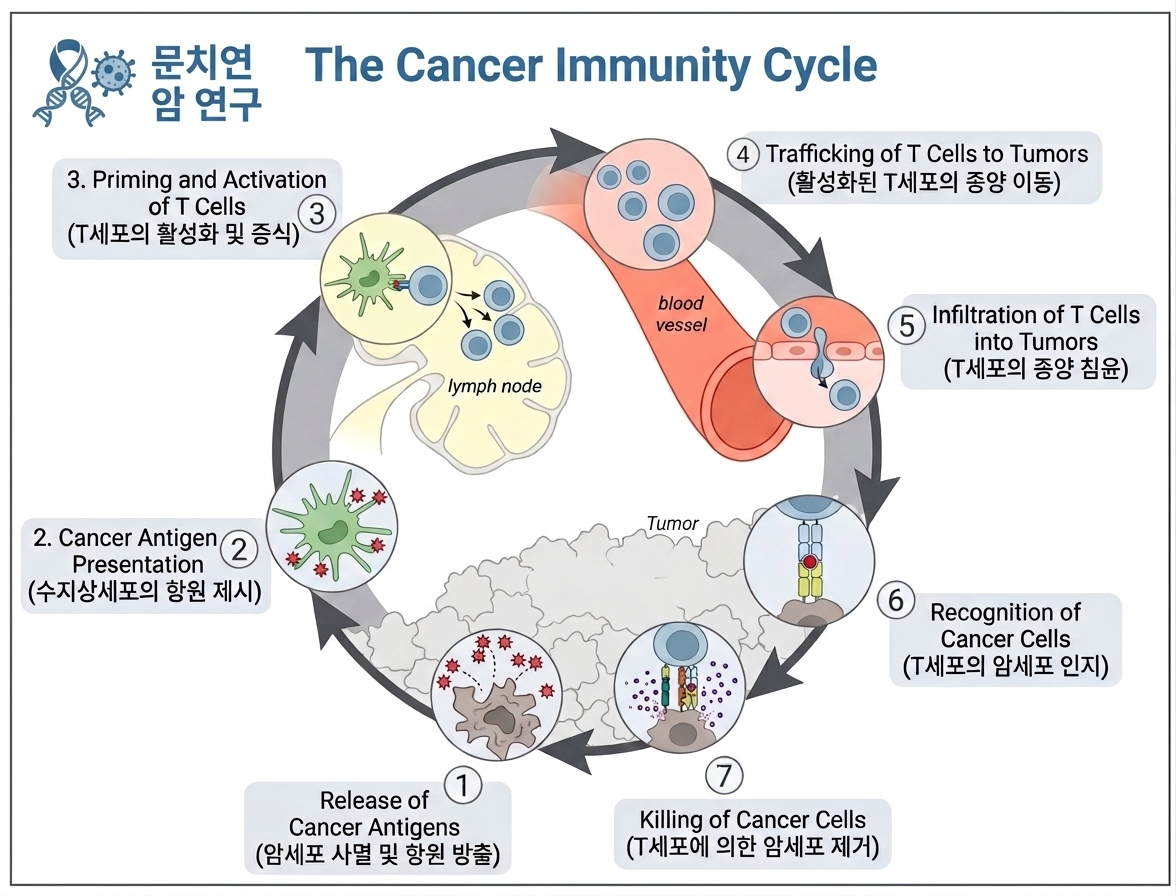
- Figure 1: Immune surveillance in controlling malignant cells 암 면역 주기(immune surveillance)를 3단계로 보여줍니다:
- Elimination (제거): 면역세포가 암세포를 인식·제거.
- Equilibrium (평형): 암세포가 면역을 피하며 균형.
- Escape (탈출): 면역억제 TME 형성으로 암 진행·전이. 발효/SCFAs는 주로 Escape 단계에서 개입해 TME를 덜 억제적으로 만들어 ICI가 더 효과적으로 작동하도록 돕습니다.
| 면역감시(immune surveillance)가 암을 어떻게 제어하다가 실패하는지를 4단계로 보여주는 모식도. 간단 요약:
|
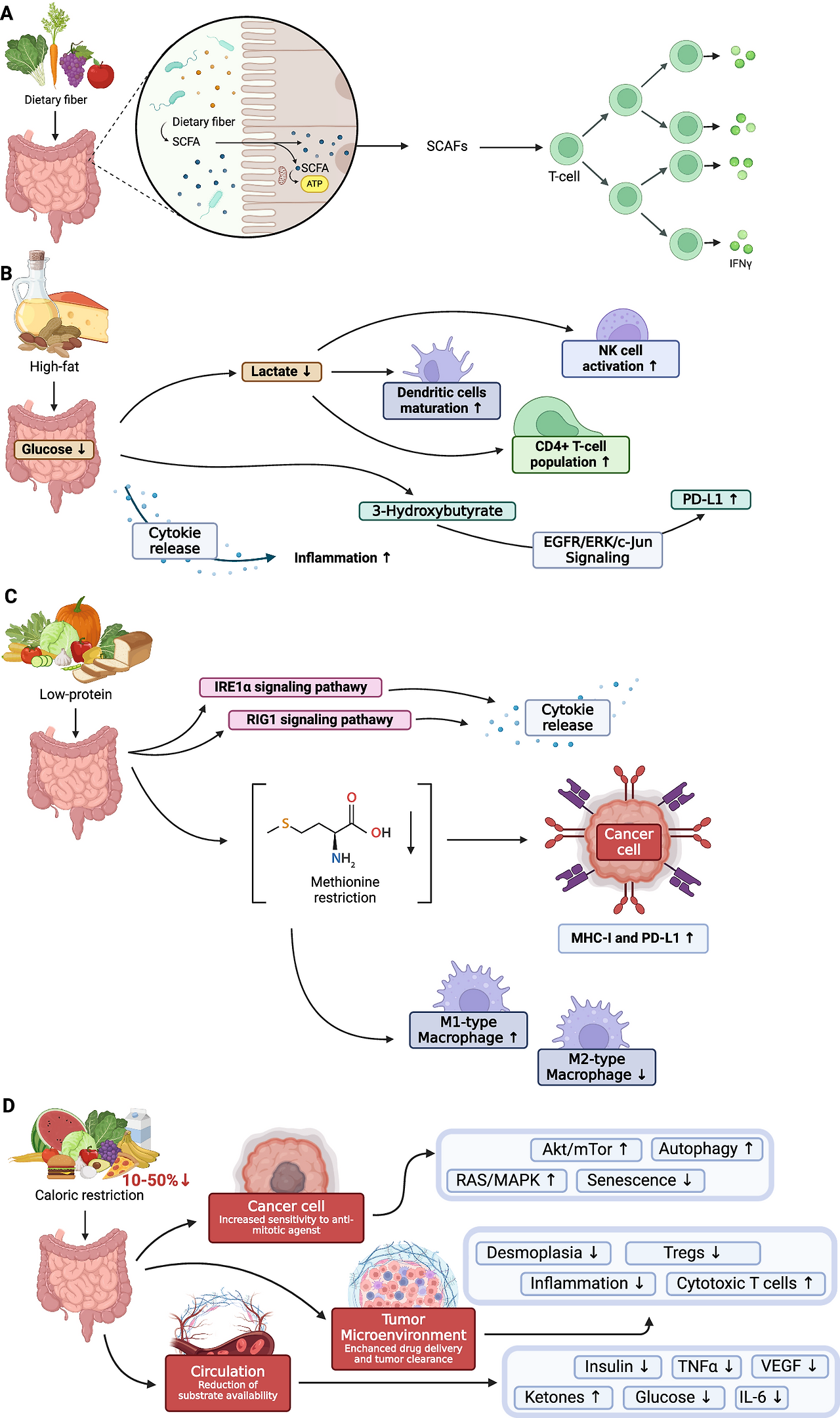
- Figure 5: Overview of diet impact on the immune system (가장 중요!) 여러 식이의 영향을 패널로 나눠 설명합니다.
- Fig. 5A (식이섬유 풍부 식이의 영향): 섬유질 음식이 prebiotics로 작용 → 장 미생물이 발효 → SCFAs (butyrate 등) 생산. 결과:
- 항염증 사이토카인 ↑
- 프로염증 사이토카인 ↓
- Treg 수 증가
- IFN-γ 수준 감소
- 면역세포(T세포, 대식세포 등) 분화·기능 자극 → ICI 효능 향상 (고섬유 식이로 anti-PD-1 반응성 ↑, 동물 모델에서 확인). 이 그림이 발효 기전의 핵심 시각 자료예요. (고지방 식이, 저단백 식이, 칼로리 제한과 비교됨)
- Fig. 5A (식이섬유 풍부 식이의 영향): 섬유질 음식이 prebiotics로 작용 → 장 미생물이 발효 → SCFAs (butyrate 등) 생산. 결과:
A: Dietary fiber (식이섬유 풍부 식이) — 발효와 가장 직접 관련된 부분
|
- Figure 3 & 4: Cancer immunotherapy strategies Fig. 3은 면역치료 분류(수동/능동, 조합 등). Fig. 4A는 ICI 메커니즘 (PD-1/PD-L1 차단으로 T세포 활성화)을 자세히 보여줍니다. 발효는 이 ICI를 "보조"하는 역할을 하며, SCFAs가 T세포 기능을 강화해 PD-1 차단 효과를 증폭시킵니다.
- Figure 6: Modulation of PD-L1/PD-1 by phytochemicals 다양한 식물성 화합물이 PD-L1을 억제하는 경로를 보여주지만, 발효와 직접 연결은 약합니다. (SCFAs도 간접적으로 PD-L1 조절에 기여할 수 있음)
- Figure 2: 영양 + 면역치료 관련 임상시험 추이 (2013~2023). 발효/식이섬유 연구도 이 트렌드에 포함됩니다.

https://pmc.ncbi.nlm.nih.gov/articles/PMC11365797/

초록 요약
암세포의 대사 재배선(metabolic rewiring)은
단순히 암세포의 증식과 미세환경 적응을 돕는 데 그치지 않고,
항종양 면역(antitumour immunity)을 회피하는 데 핵심적인 역할을 합니다.
이 리뷰는
암세포 내재적(intrinsic) 및 외재적(extrinsic) 메커니즘을 통해
metabolic rewiring된 암세포 대사가
선천성(innate) 및 적응성(adaptive) 면역 기능을
어떻게 방해하는지 체계적으로 정리합니다.
특히
종양 미세환경(TME)에서의
영양 경쟁, 면역억제성 대사물질 생산, 면역세포 기능 억제를 중점적으로 다루며,
이러한 대사 변화를 표적으로 한 치료 전략의 잠재력을 논의합니다.
Warburg 효과(Warburg effect, 호기성 해당과정 증가)부터 시작해,
최근 연구에서 밝혀진 oncometabolites,
지질 대사,
아미노산 대사 등이
어떻게 T 세포, NK 세포,
수지상세포(DC), MDSC(myeloid-derived suppressor cells),
Treg(regulatory T cells) 등에 영향을 미치는지 설명합니다.

https://cafe.daum.net/panicbird/RnFZ/488

주요 배경 및 개념
암세포는
유전자 변이(예: KRAS, IDH, p53 관련)로 인해 대사가 재프로그래밍되어
ATP 생산, 생합성 전구체(biosynthetic precursors), redox 균형을 유지합니다.
이는
TME의 저산소(hypoxia), 영양 부족 조건에서도 생존을 돕지만,
동시에 면역세포와의 대사 경쟁(metabolic competition)을 유발합니다.
- 암세포 내재적 메커니즘: 암세포가 직접 면역억제성 대사물을 분비하거나, 자신의 대사 경로를 통해 MHC-I 발현을 줄이거나, autophagy를 이용해 면역 회피.
- 암세포 외재적 메커니즘: TME에서 glucose, glutamine, fatty acids 등을 독점적으로 소비하여 effector T 세포(특히 CD8⁺ T cell)의 기능을 억제하고, suppressor cells(MDSC, Treg, M2 macrophage)를 활성화.
https://pmc.ncbi.nlm.nih.gov/articles/PMC10023409/
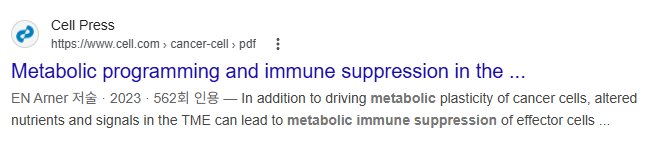
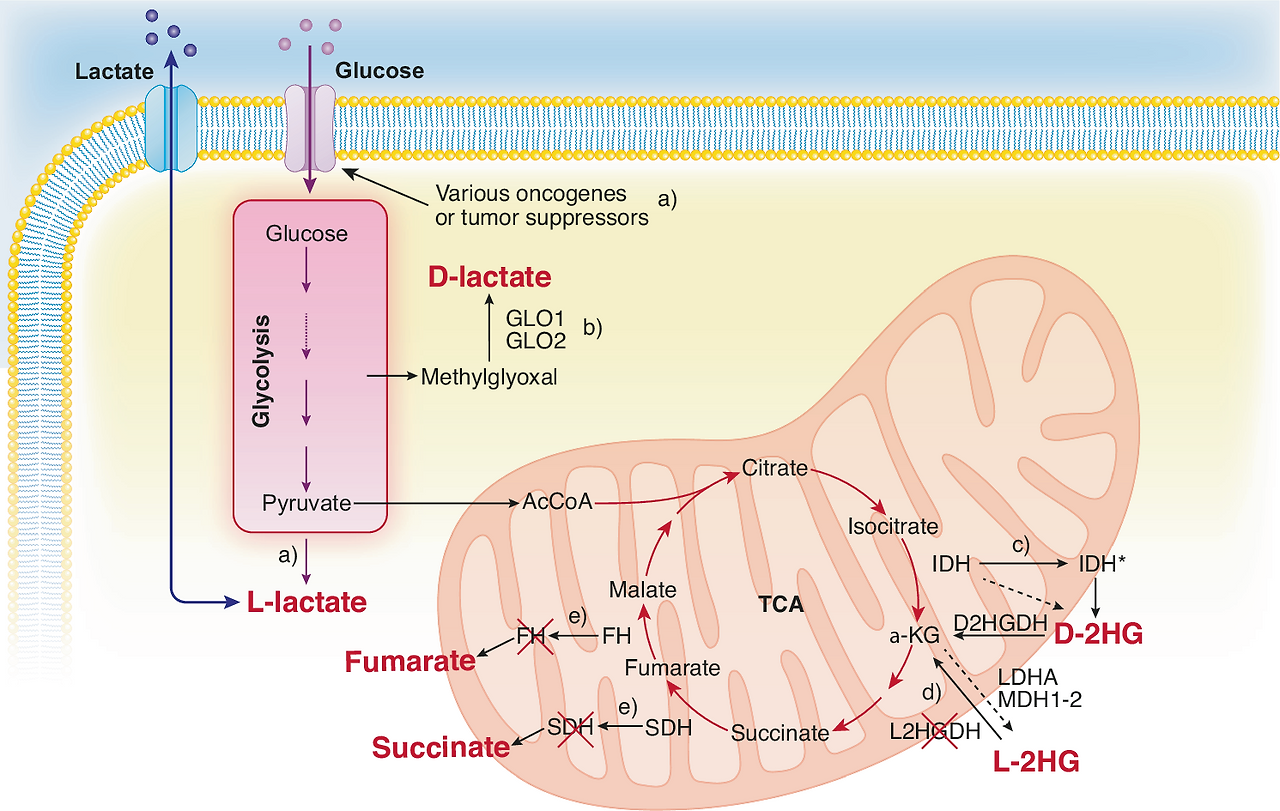
| 종양은 위치(저산소 지역 vs. 혈관 근처), 영양소 가용성, 세포 구성(암세포, 면역세포, 섬유아세포 등)에 따라 대사적으로 매우 이질적(heterogeneous). 이러한 대사 이질성이 면역세포의 기능을 억제하여 항종양 면역을 무력화시킴.  종양의 4가지 주요 지역과 특징
주요 대사 물질과 면역 억제 기전
주요 기전 (Metabolic Immune Suppression)
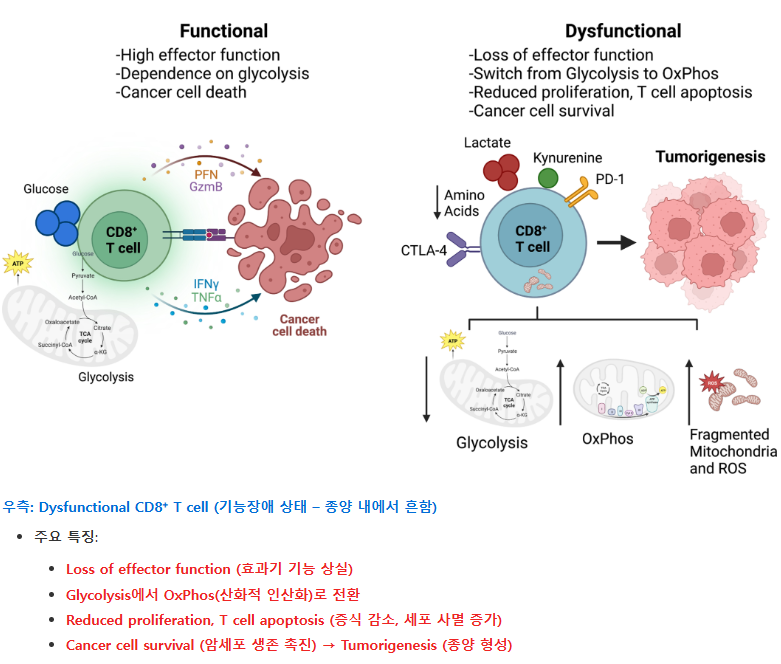
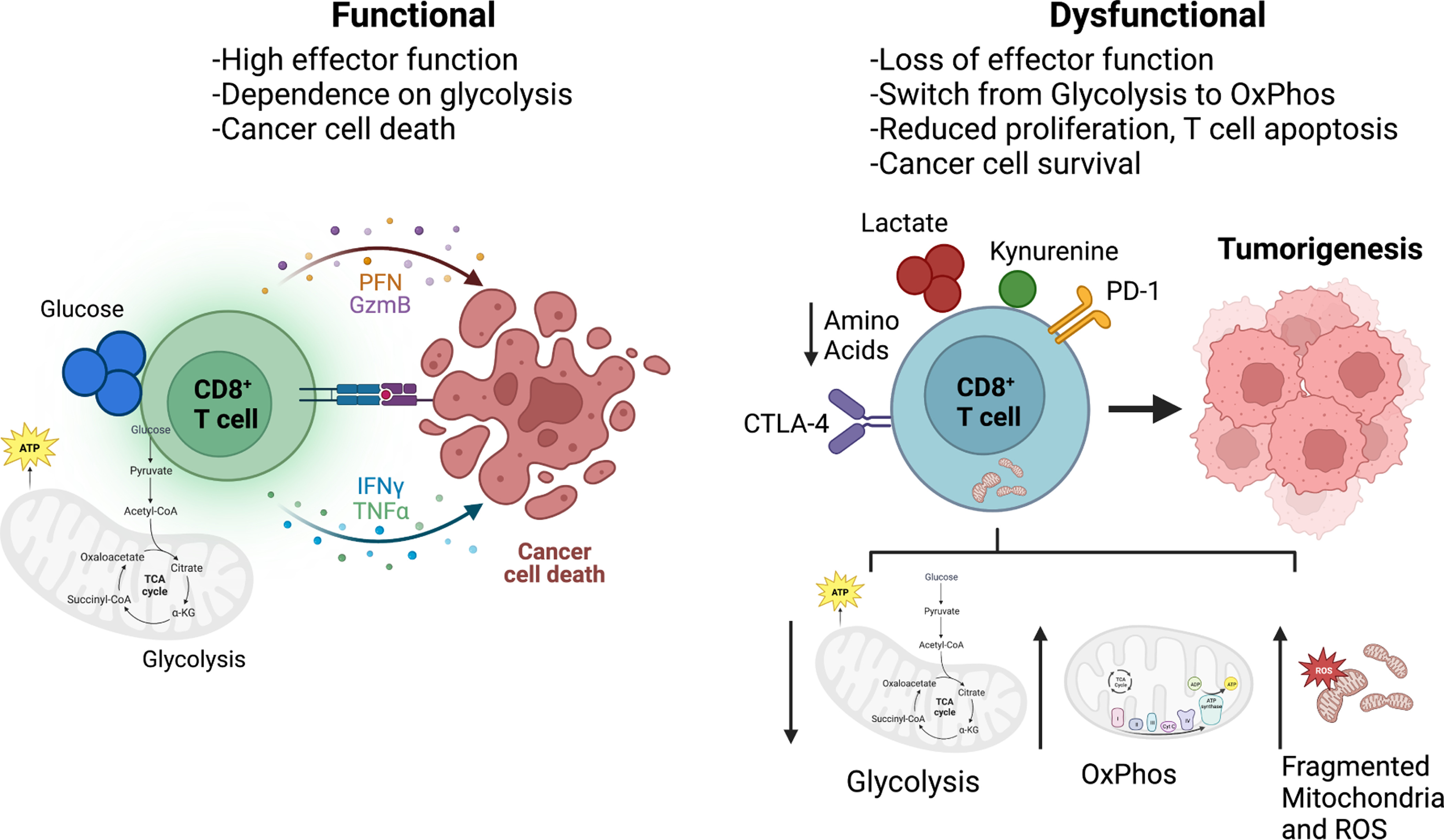 우측: Dysfunctional CD8⁺ T cell (기능장애 상태 – 종양 내에서 흔함)
좌측: Functional CD8⁺ T cell (정상적으로 작동하는 상태)
우측: Dysfunctional CD8⁺ T cell (기능장애 상태 – 종양 내에서 흔함)
정상 상태에서는 CD8⁺ T 세포가 granzyme, perforin 등을 분비하며 암세포를 직접 죽이고, glycolysis에 의존해 강한 염증 반응을 일으킵니다. 종양 미세환경(TME)에서는:
T 세포는 사이토카인을 제대로 분비하지 못하고, 암세포를 공격하지 못하게 됩니다 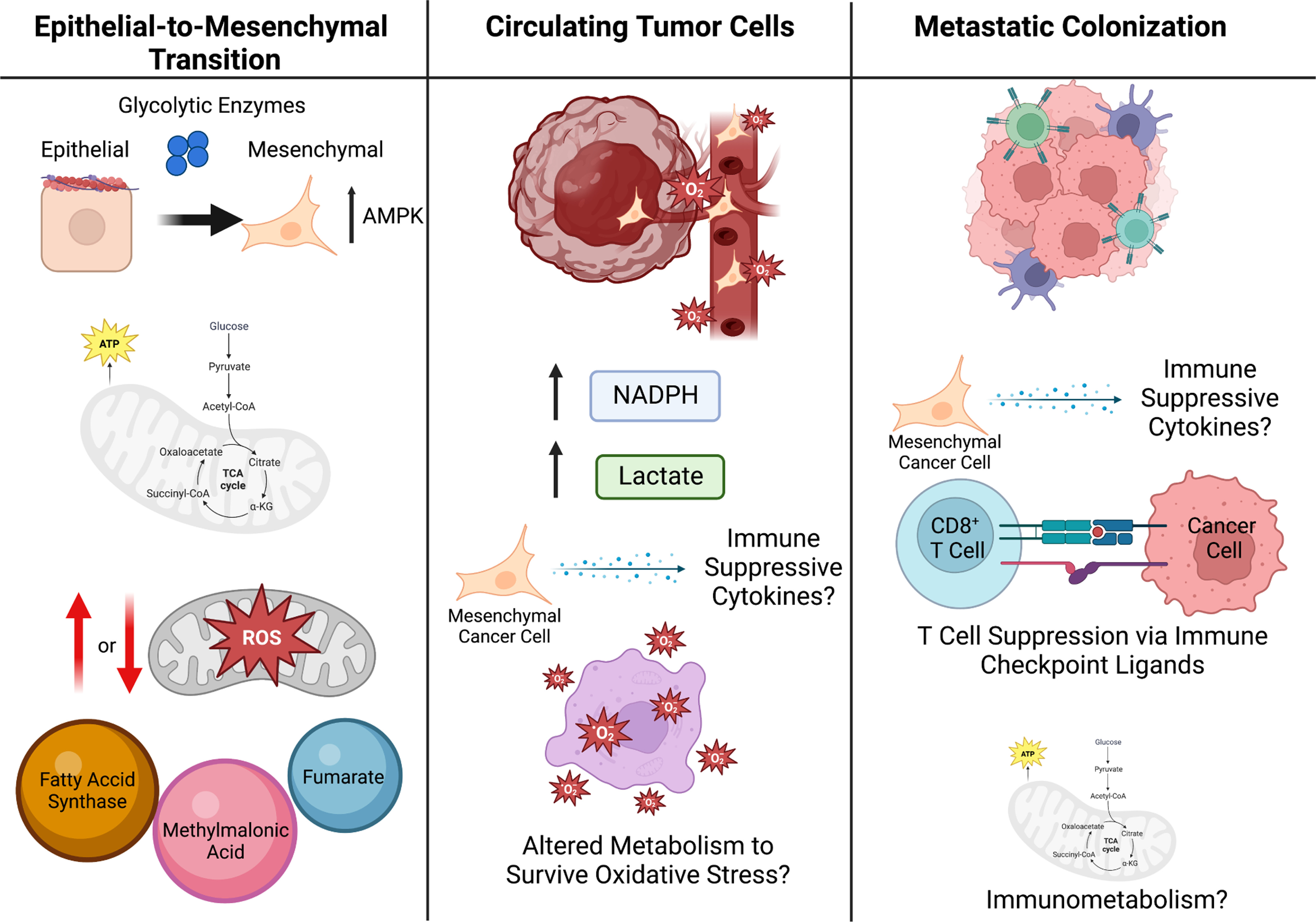 종양 미세환경의 대사 이질성 → T 세포 기능 장애 → CAR-T 대사 리프로그래밍에 이어, 암세포 스스로가 전이를 위해 대사를 재설계하는 모습을 보여주는 중요한 부분. 1. Epithelial-to-Mesenchymal Transition (EMT: 상피-간엽 전이)
한 줄 요약 종양 미세환경의 대사 경쟁과 폐기물(락트산 등)이 T 세포 등 면역세포를 억제하여 종양이 면역을 회피하게 만들며, 이를 대사적으로 타겟팅하면 면역치료 효과를 크게 높일 수 있다. |
핵심 대사 경로와 면역 영향 (주요 메커니즘)
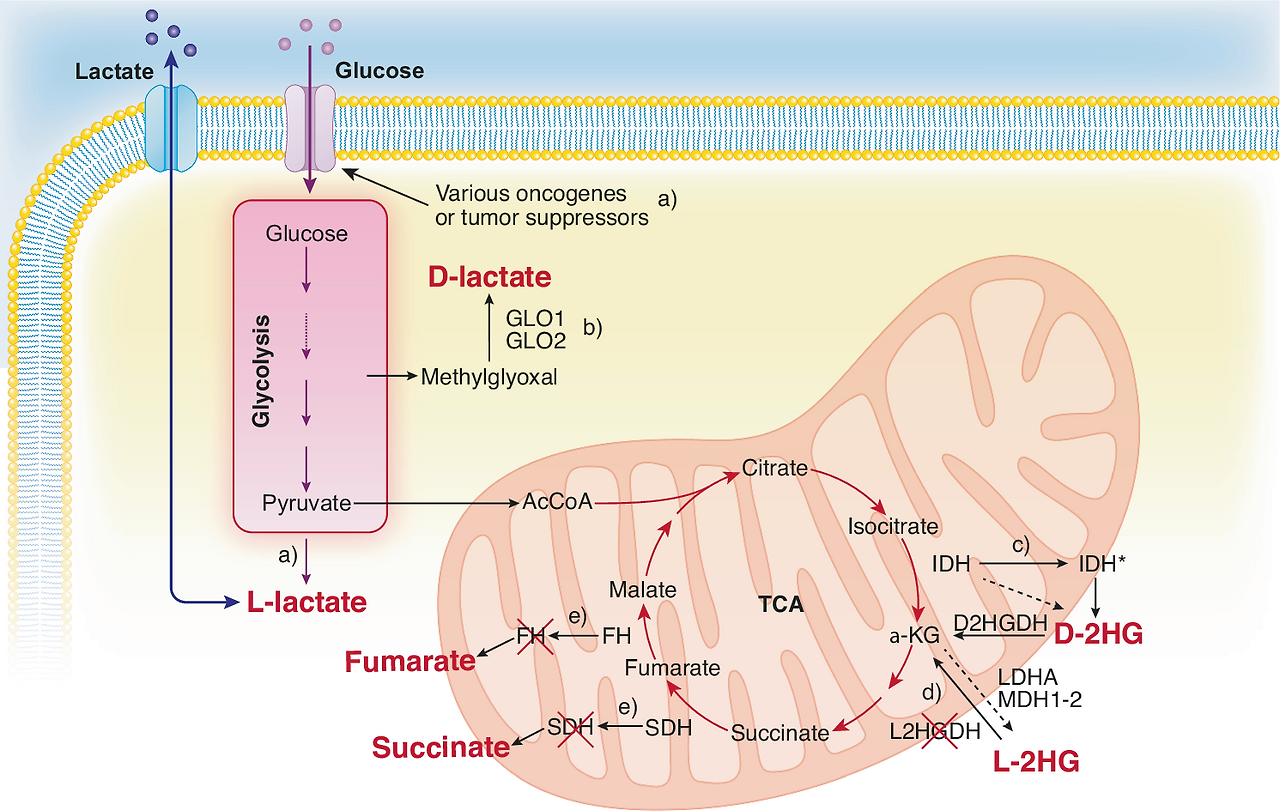
Glucose 및 Lactate 대사 (Glycolysis 중심, Fig. 1)
- 암세포의 고갈소 소비 → TME 내 glucose 고갈 → effector T 세포의 glycolysis 억제 → T 세포 증식/효능 기능 저하.
- Lactate (LDHA에 의해 생산): 산성 환경(pH 저하) 유발 → T/NK 세포 기능 억제, NAD⁺/NADH redox 불균형.
- Treg 안정화(promotes Foxp3 expression, PD-1 upregulation).
- MDSC 축적 촉진(CEBPB 경로 등).
- Hexokinase 2 (HK2) 등 효소가 IκBα 인산화 → NF-κB 활성화 → 면역억제 사이토카인 생산.
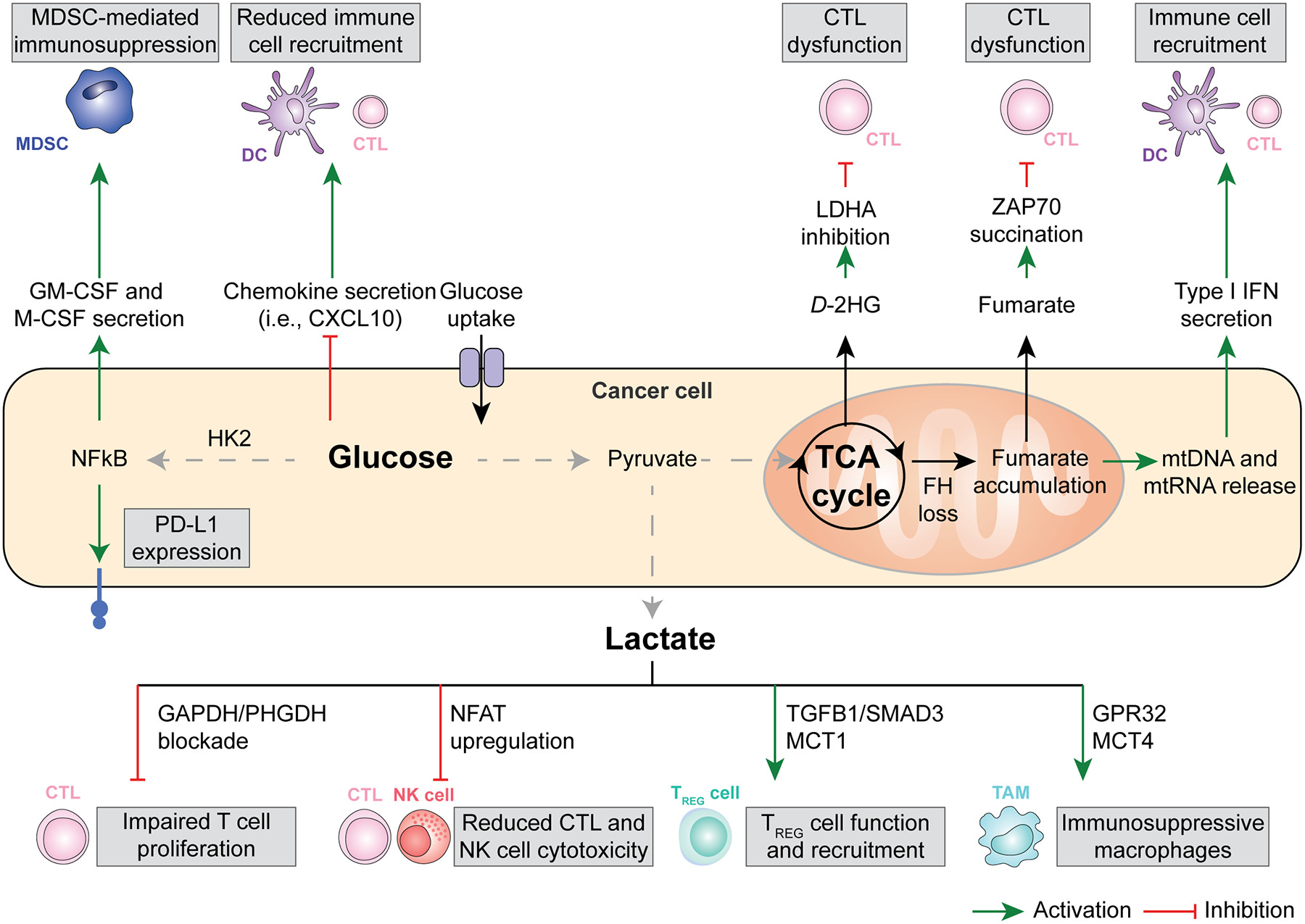
1. 암세포 내부 대사 흐름 (중앙 상단)
 3. 하단: Lactate가 면역세포에 미치는 직접적인 영향 (가장 중요) Lactate가 아래로 내려가면서 여러 면역세포를 공격합니다:
|
TCA cycle 및 Oncometabolites
- Fumarate, Succinate: CD8⁺ T 세포의 항종양 능력 억제. Fumarate는 mtDNA release → cGAS-STING 활성화 (역설적으로 면역 자극 가능하지만, 맥락에 따라 억제).
- D-2-hydroxyglutarate (D-2HG, IDH mutation 관련 oncometabolite): CD8⁺ T 세포 대사 재프로그래밍 → effector 기능 저하, T 세포 피로(exhaustion) 유발.
- Succinate dehydrogenase (SDH) 억제 등으로 mitochondrial dysfunction.
- 글루코스 과섭취 → HK2 활성화 → NF-κB 활성화 →
- PD-L1 발현 증가 (T 세포 억제)
- GM-CSF, M-CSF 분비 → MDSC (골수유래 억제세포) 모집 및 활성화 → 면역억제
- Chemokine (CXCL10 등) 분비 감소 → DC와 CTL (세포독성 T 세포) 모집 감소
- TCA cycle 이상 (FH loss 등) →
- Fumarate 축적 → ZAP70 succination → CTL 기능 장애
- D-2HG (oncometabolite) 생산 → LDHA 억제 관련 → CTL 기능 장애
- mtDNA/mtRNA 방출 → Type I IFN 분비 → 면역세포 모집 (긍정적 효과도 있지만, 전체적으로는 억제 우세)
- CTL (세포독성 T 세포): GAPDH/PHGDH 차단 → T 세포 증식 저하
- CTL / NK 세포: NFAT upregulation → 세포독성(cytotoxicity) 감소
- Treg 세포: TGFβ1/SMAD3 + MCT1 경로 → Treg 기능 강화 및 모집 증가 (면역억제 강화)
- TAM (종양연관 대식세포): GPR32 + MCT4 경로 → 면역억제성 대식세포로 변화
- 효과적 면역세포(CTL, NK, DC)의 모집·활성화·증식이 줄어듦
- 억제성 세포(MDSC, Treg, TAM)가 늘어나서 항종양 면역 전체가 약화됨
- 글루코스 과섭취 → HK2 활성화 → NF-κB 활성화 →
지방산(Fatty acid) 및 Eicosanoid 대사 (Fig. 2)
- Fatty acid oxidation (FAO, CPT1A 등): 암세포 radioresistance 증가, CD47 upregulation → phagocytosis 회피.
- PGE₂ (COX-2 경로): cDC1 (conventional DC type 1)와 NK 세포 모집 억제 → cross-presentation 저하.
- CD36-mediated lipid uptake: Treg 생존 촉진, T 세포 ferroptosis 유발 → exhaustion.
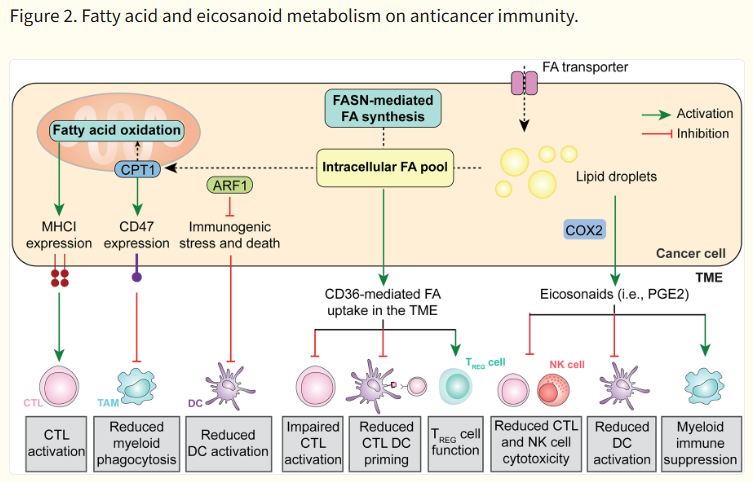
1. 암세포 내부 지방산 대사
|

| 자가포식(Autophagy)이란 세포가 손상된 미토콘드리아, 단백질 등을 스스로 분해하여 재활용하는 lysosome 의존적 과정입니다. 모든 세포에 필수적이지만, 암세포에서는 면역 반응을 크게 좌우합니다. 1. 긍정적 효과 (항암 면역 촉진)
암세포의 자가포식은 항암 면역의 중심 조절자(central regulator)입니다. 상황에 따라 면역을 돕기도 하지만, 대부분의 경우 면역억제 쪽으로 작용하여 종양이 살아남는 데 유리하게 작동 |
Nucleotide 및 Amino acid 대사 (Fig. 3)
- Adenosine (CD39/CD73 경로): A2AR signaling → T 세포 억제 (많은 임상 시험 대상).
- Glutamine: Glutaminase 억제 시 MDSC 기능 저하 + T 세포 활성화 향상.
- Methionine: T 세포 exhaustion 관련.
- Tryptophan (IDO1 경로): Kynurenine 생산 → T 세포 apoptosis, Treg 분화.
- Nucleotide release: 초기에는 "find-me" signal로 phagocytosis 촉진하지만, adenosine로 전환되면 억제.
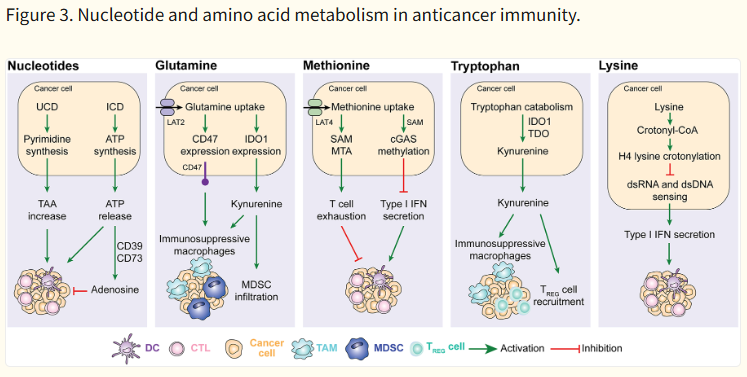
| 암세포는 핵산(누클레오티드)과 아미노산(글루타민, 메티오닌, 트립토판, 라이신) 대사를 조작하여 T 세포, NK 세포, DC 등을 억제하고 면역억제 환경을 만듭니다. 각 부분별 간단 설명
암세포는 글루타민·트립토판·메티오닌 등 아미노산을 독점하고 분해하면서 kynurenine, adenosine 등을 만들어 T 세포를 소진시키고, Type I IFN을 억제하여 항암 면역 전체를 약화시킵니다. |
Autophagy 및 기타
- 암세포 autophagy: MHC-I degradation → CD8⁺ T 세포 인식 회피.
- Hypoxia 하 NK 세포 lysis 저항성 증가.
이러한 메커니즘은 종양 유형(예: triple-negative breast cancer, lung adenocarcinoma, glioblastoma)과 heterogeneity에 따라 다르게 나타납니다.
Nat Rev Immunol
. Author manuscript; available in PMC: 2024 Sep 1.
Published in final edited form as: Nat Rev Immunol. 2024 Apr 22;24(9):654–669. doi: 10.1038/s41577-024-01026-4
Cancer cell metabolism in tumor-targeting immunity
Mara De Martino 1, Jeffrey C Rathmell 2, Lorenzo Galluzzi 1,3,4,*, Claire Vanpouille-Box 1,3,*
- Author information
- Article notes
- Copyright and License information
PMCID: PMC11365797 NIHMSID: NIHMS1989213 PMID: 38649722
The publisher's version of this article is available at Nat Rev Immunol
This article has been corrected. See Nat Rev Immunol. 2024 May 28.
Abstract
Accumulating evidence suggests that metabolic rewiring in malignant cells supports tumor progression not only by providing them with a superior proliferative potential and an increased adaptability to adverse microenvironmental conditions, but also by favoring the evasion of natural and therapy driven anticancer immunosurveillance. Here, we review cancer cell-intrinsic and extrinsic mechanisms through which alterations of metabolism in malignant cells interfere with innate and adaptive immune functions in support of accelerated disease progression, and we discuss the potential of targeting such alterations to enhance anticancer immunity for therapeutic purposes.
Abstract (초록)
암세포의 대사 재배선(metabolic rewiring)에 대한 증거가 점점 쌓이면서,
이는 종양 진행을 단순히 더 빠른 증식 능력과
불리한 미세환경 조건에 대한 적응력을 높이는 데 그치지 않고,
자연적·치료 유발 항암 면역감시(anticancer immunosurveillance)를 회피하는 데에도 기여한다는 것이
밝혀지고 있다.
본 리뷰에서는
악성 세포의 대사 변화가 어떻게 세포 내(intrinsic) 및 세포 외(extrinsic) 기전을 통해
선천성 및 적응성 면역 기능을 방해하여
질병 진행을 가속화하는지 검토하고,
이러한 변화를 표적화하여 항암 면역을 강화할 수 있는 치료적 잠재력을 논의한다.
Keywords: autophagy, fatty acid synthesis, glycolysis, immunometabolism, T cells, tumor-associated macrophages
Introduction
Malignant transformation and tumor progression are accompanied by numerous alterations in metabolic pathways that emerge in the context of at least three conceptually different (but not mutually exclusives) scenarios1,2. First, metabolites that accumulate because of mutations in enzyme-coding genes are the primary drivers for oncogenesis. As an example, this occurs downstream of gain-of-function isocitrate dehydrogenase (NADP(+)) 1 (IDH1) or IDH2 mutations (which are common in patients with glioblastoma and leukemia), resulting in the accumulation of 2-hydroxyglutarate [2HG], which has bona fide tumor-promoting activity3. Second, genetic or epigenetic alterations in well-established oncoproteins or oncosuppressors drive oncogenesis along with a direct influence on metabolism. Indeed, multiple cancer-initiating events such as activating mutations in KRAS proto-oncogene, GTPase (KRAS) as well as the genetic or epigenetic inactivation of the tumor protein p53 (TP53, best known as TP53) have been shown to directly impact catabolism or anabolism4,5. Third, cancer cells acquire metabolic alterations as tumors evolve in response to spatiotemporally changing microenvironmental conditions, one of the major driver of intra- and inter-tumor heterogeneity6. As an example, malignant cells not located in the close proximity of blood vessels respond to hypoxia with a global metabolic reconfiguration orchestrated by hypoxia inducible factor 1 subunit alpha (HIF1A)7. Of note, though conceptually distinct, all these scenarios generate metabolic vulnerabilities that (at least theoretically) may be targeted for therapeutic purposes8,9.
Initiated a century ago by the German physiologist Otto H. Warburg with the observation that malignant cells take up an increased amount of glucose as compared to their normal counterparts10, the field of cancer metabolism has by now revealed that malignant transformation and tumor progression involve a cell-wide metabolic rewiring that goes way beyond the so-called Warburg effect2. Indeed, cancer cells often exhibit complex metabolic alterations that also affect oxidative phosphorylation (OXPHOS), the tricarboxylic acid (TCA) cycle, multiple biosynthetic cascades, as well as global catabolic pathways such as autophagy2,11. Such changes (which often influence the tumor stroma, Box 1) provide malignant cells with the metabolic substrates that are required to enable accelerated proliferation, including (but not limited to) nucleotides, lipids and amino acids as needed for cellular growth and division2,11. Moreover, the metabolic rewiring that characterize most (if not all) cancer cells endow them with a superior adaptability to changing microenvironmental conditions, de facto fostering tumor evolution and diversification6,12. Accumulating data indicate that malignant cells also benefit from metabolic changes that counteract a major selective pressure in the host-tumor co-evolution, namely, anticancer immunosurveillance13. Indeed, it is now widely accepted that oncogenesis is not a merely cancer cell-intrinsic phenomenon driven by genetic and/or epigenetic alterations that support malignancy, but it also involves a prominent cancer cell-extrinsic component, i.e., the acquisition of phenotypic, secretory and behavioral features that enable cancer cells to evade recognition and killing by the host immune system13.
Introduction (서론)
악성 변형(malignant transformation)과 종양 진행은
대사 경로의 수많은 변화와 동반되며,
이는 개념적으로 서로 다르지만 배타적이지 않은
최소 세 가지 시나리오에서 나타난다.
첫째,
효소 코딩 유전자의 돌연변이로 인해 축적되는 대사 산물이
종양 발생의 주요 원동력이 되는 경우이다.
예를 들어,
교모세포종과 백혈병에서 흔한 이소시트레이트 탈수소효소(IDH1 또는 IDH2)의 기능 획득 돌연변이는
2-하이드록시글루타레이트(2HG)의 축적을 초래하며,
이는 진정한 종양 촉진 활성을 갖는다.
둘째,
잘 알려진 종양유전자(oncoprotein)나 종양억제유전자(oncosuppressor)의
유전적·후성유전적 변화가 종양 발생과 동시에
대사에 직접적인 영향을 미치는 경우이다.
KRAS 활성화 돌연변이나 TP53의 불활성화가
대사(이화작용 또는 합성작용)에 직접 영향을 미치는 것이 대표적이다.
셋째,
종양이 시공간적으로 변화하는 미세환경 조건에 적응하면서
대사 변화가 획득되는 경우로,
이는 종양 내·종양 간 이질성의 주요 원동력 중 하나이다.
예를 들어,
혈관에서 멀리 떨어진 악성 세포는
저산소증(hypoxia)에 반응하여
HIF1A에 의해 조절되는 전반적인 대사 재구성을 일으킨다.
이 세 시나리오는 개념적으로 구분되지만,
모두 치료적으로 표적화할 수 있는
대사 취약점(metabolic vulnerabilities)을 생성한다.
약 100년 전
독일 생리학자 오토 H. 바르부르크(Otto H. Warburg)가
악성 세포가 정상 세포에 비해 포도당을 훨씬 많이 섭취한다는 관찰을 시작으로,
암 대사 분야는 이제 ‘바르부르크 효과’를 훨씬 넘어선
세포 전반의 대사 재배선(metabolic rewiring)을 밝혀냈다.
암세포는
산화적 인산화(OXPHOS), TCA 회로, 다양한 생합성 경로,
그리고 자가포식(autophagy) 같은 전반적인 이화 경로에도 복잡한 대사 변화를 보인다.
이러한 변화(종양 기질에도 영향을 미침, Box 1 참조)는
악성 세포에게 핵산, 지질, 아미노산 등 빠른 증식에 필요한 대사 기질을 공급한다.
또한
변화하는 미세환경 조건에 대한 우수한 적응력을 부여하여
종양 진화와 다양화를 촉진한다.
더 나아가, 축적되는 데이터에 따르면
악성 세포는 숙주-종양 공진화 과정에서
주요 선택 압력인 항암 면역감시(anticancer immunosurveillance)를
회피하는 대사 변화로부터도 이득을 얻는다.
이제 종양 발생은
단순히 암세포 내부의 유전적·후성유전적 변화에 의한 것이 아니라,
숙주 면역계로부터의 인식과
살상을 회피하는 표현형, 분비물, 행동적 특징을 획득하는 암세포 외적 요소가
크게 관여한다는 것이 널리 받아들여지고 있다.
Box 1. Influence of cancer cell metabolism on the tumor stroma.
Malignant lesions developed in the context of an intimate crosstalk with stromal cells including cancer-associated fibroblasts (CAFs) that exhibits a considerable metabolic component. For instance, ovarian cancer cells have been shown to supply lactate and glutamate to CAFs, hence fostering the synthesis and release of glutamine by CAFs in support of their own proliferation173. A similar mechanism also appears to be operational in prostate cancer models174. Along similar lines, pancreatic cancer cells have been reported to promote autophagic responses in stromal pancreatic stellate cells, resulting in a local release of alanine that relieves the malignant cell dependency on glucose and serum-derived nutrients for proliferation175. Moreover, pancreatic cancer cells can reportedly harness proline derived from the breakdown of CAF-produced collagen to survive under nutrient-limited conditions176, highlighting yet another metabolic circuitry connecting malignant cells and their stroma. To which extent proline secreted by CAFs to accommodate redox stress as induced by transforming growth factor beta 1 (TGFB1) signaling177 contributes to cancer cell survival, however, remains to be demonstrated. Importantly, other stromal cells have also been shown to support cancer cell proliferation via metabolic circuitries. For instance, fatty acid binding protein 4 (FABP4) expression in ovarian cancer cells appears to underlie a mechanism that promote lipolysis in cancer-associated adipocytes. This results in the secretion of fatty acids that are avidly taken up by malignant cells and used for bioenergetic purposes via fatty acid oxidation178. Collectively, these observations nicely exemplify the existence of multiple metabolic exchanges between neoplastic cells and non-transformed components of the tumor microenvironment, notably stromal cells.
Here, we discuss mechanisms through which alterations of core metabolism in neoplastic cells influence natural and therapy-driven immunosurveillance as we analyze the potential of targeting such changes to enhance anticancer immune responses for therapeutic purposes. Importantly, macromolecular metabolic pathways including DNA replication, DNA-to-RNA transcription and protein synthesis, despite their potential impact on tumor-targeting immunity,14,15 go beyond the scope of this review. Along similar lines, the influence of immune cell metabolism on tumor-targeting immunity has been extensively reviewed elsewhere16–18, and hence will not be discussed here.
Box 1. 암세포 대사가 종양 기질에 미치는 영향
악성 병변은
암 관련 섬유아세포(CAFs)를 비롯한 기질 세포와 밀접한 상호작용 속에서 발생하며,
여기에는 상당한 대사적 요소가 포함된다.
예를 들어
난소암 세포는 CAFs에 락트산과 글루타메이트를 공급하여
CAFs가 글루타민을 합성·분비하게 만들고,
이를 통해 자신의 증식을 지원한다.
유사한 기전이 전립선암 모델에서도 관찰된다.
췌장암 세포는
기질 췌장성상세포의 자가포식을 촉진하여 알라닌을 방출하게 만들고,
이는 악성 세포의 포도당과 혈청 유래 영양소 의존성을 줄여준다.
또한
췌장암 세포는
CAFs가 생산한 콜라겐 분해로부터 유래한 프롤린을 활용하여 영양 부족 조건에서 생존한다.
중요하게도,
다른 기질 세포도 대사 회로를 통해 암세포 증식을 지원한다.
난소암 세포의 FABP4 발현은
암 관련 지방세포의 지방분해를 촉진하여 지방산을 분비하게 만들고,
암세포는 이를 지방산 산화(fatty acid oxidation)를 통해 에너지원으로 이용한다.
이러한 관찰들은
종양 미세환경의 비변형 세포(특히 기질 세포)와
악성 세포 사이에 다양한 대사 교환이 존재한다는 것을 잘 보여준다.
Glucose, lactate, and the TCA cycle
To meet their increased energy demand, cancer cells generally exhibit an accelerated and diversified bioenergetic metabolism, involving an enhanced glucose flux through glycolysis as well as alterations of the TCA cycle. All these metabolic changes influence tumor-targeting immunity (Fig. 1).
Glucose, lactate, and the TCA cycle (포도당, 락트산, 그리고 TCA 회로)
증가된 에너지 수요를 충족하기 위해 암세포는
일반적으로 해당과정(glycolysis)을 통한 가속화된 포도당 흐름과
TCA 회로의 변화를 보인다.
이러한 모든 대사 변화는
종양 표적 면역(tumor-targeting immunity)에 영향을 미친다(Fig. 1 참조).
Figure 1. Glucose, lactate, and intermediate metabolism in anticancer immunity.
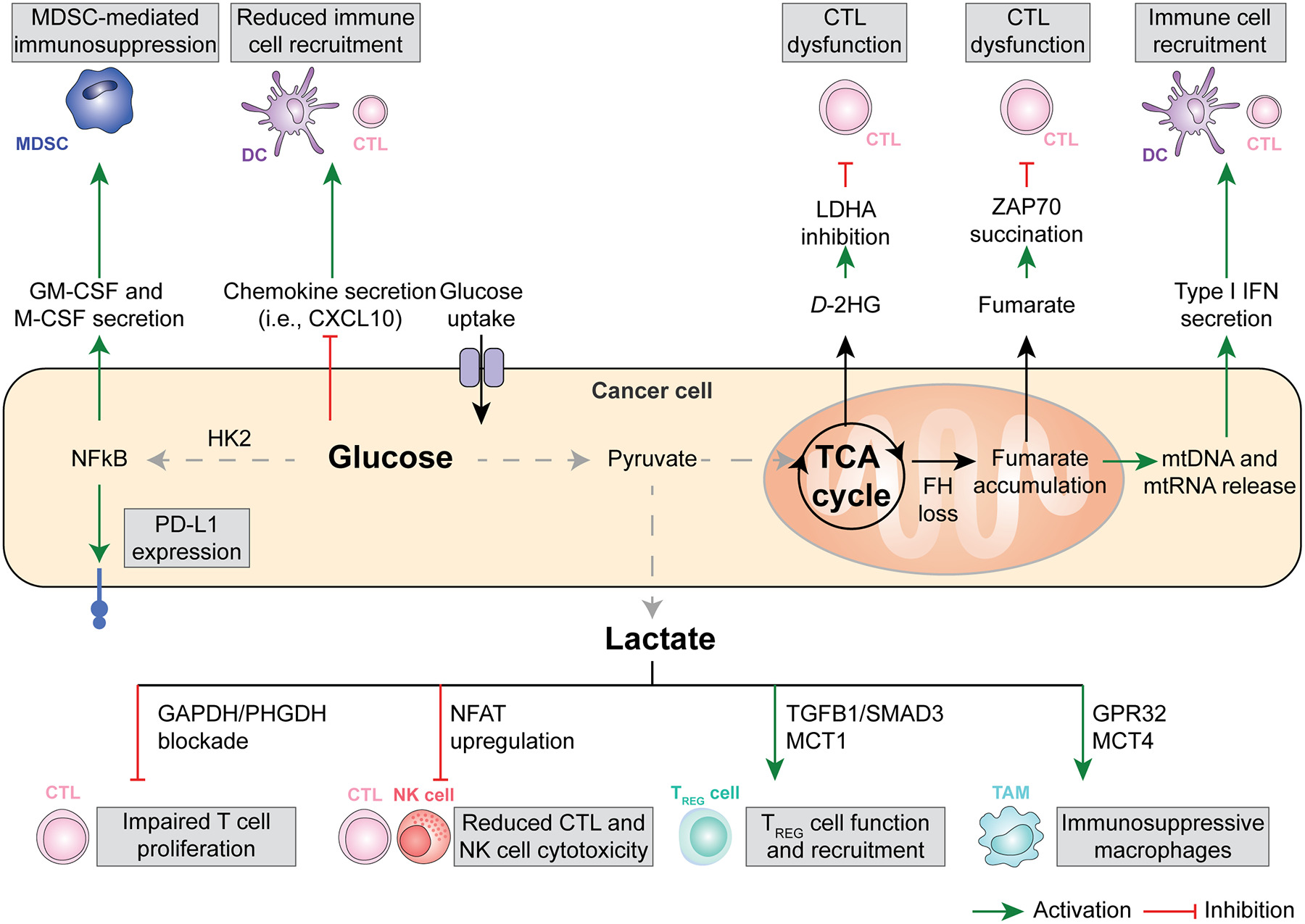
The bioenergetic metabolism of cancer cells, characterized by increased glucose uptake coupled with abundant lactate secretion as well as alterations in the tricarboxylic acid (TCA) cycle has a major impact on the immunological tumor microenvironment (TME). For instance, an increased glycolytic flux in cancer cells has been associated with the NF-κB-dependent upregulation of PD-L1 and the secretion of myeloid-derived suppressor cell (MDSC)-recruiting cytokines like GM-CSF and M-CSF, as well as with the reduced release of the cytotoxic T lymphocyte (CTLs)-recruiting and pro-inflammatory chemokine CXCL10. Along similar lines, microenvironmental lactate has been shown to limit the proliferation and activation of CTLs and natural killer (NK) cells while promoting the recruitment and immunosuppressive function of regulatory T (TREG) cells and tumor-associated macrophages (TAMs). Finally, mitochondrial alterations emerging from TCA cycle defects have been linked with the secretion of metabolic intermediates with direct CTL-suppressive effects, including fumarate and D-2-hydroxyglutarate (D-2HG), as well as with the cytosolic accumulation of cytosolic mitochondrial DNA (mtDNA) and mitochondrial RNA (mtRNA), instead culminating with the secretion of immunostimulatory type I interferon (IFN). DC, dendritic cell.
Glucose.
Malignant cells use glucose for bioenergetic purposes upon conversion to pyruvate, mitochondrial uptake and entry in the TCA cycle, as well as for anabolic purposes via the pentose phosphate pathway (PPP) and the serine synthesis pathway (SSP)19. Thus, cancer cells may be in competition with immune cells, notably CD8+ cytotoxic T lymphocytes (CTLs), for glucose uptake in the tumor microenvironment (TME) of some malignancies, as demonstrated in immunocompetent mouse models of sarcoma20. However, it appears that immune cells generally consume more glucose than cancer cells themselves, and that glutamine (rather than glucose itself) is the limiting nutrient that determines differential glucose uptake by malignant vs immune compartments of the TME21. That said, an increased glycolytic flux in melanoma cells has been associated with limited expression of chemotactic factors involved in CTL recruitment, such as C-X-C motif chemokine ligand 10 (CXCL10)22. In line with this observation, genetic signatures of glycolysis have been shown to inversely correlate with immune cell infiltration in patients with melanoma and non-small cell lung carcinoma (NSCLC), in the former setting especially amongst patients refractory to adoptive T cell transfer22. Moreover, elevated glucose intake has been linked with the hexokinase 2 (HK2)-dependent activation of an NF-κB transcriptional response culminating with the expression of the co-inhibitory ligand CD274 (best known as PD-L1) in models of glioblastoma23. Finally, increase glycolytic flux has been linked with the overexpression of colony stimulating factor 1 (CSF1, best known as M-CSF) and CSF2 (best known as GM-CSF) by triple negative breast cancer (TNBC) cells, resulting in the repolarization of the TME towards an immunosuppressive state dominated by myeloid-derived suppressor cells (MDSCs)24. Interestingly, patients with TNBC from the METABRIC public dataset with an elevated expression of lactate dehydrogenase A (LDHA, encoding for the final enzyme of anerobic glycolysis, which diverts pyruvate from mitochondrial uptake to conversion into lactate and secretion) were found to exhibit an enrichment in gene signatures associated with MDSCs coupled with an underrepresentation of gene signatures representative of T cell infiltration, and exhibited poor disease outcome24. Most likely, however, these observations do not stem only from the immunosuppressive effects of lactate (see below), but also reflects the elevated LDHA levels found in myeloid cells including MDSCs themselves.
In line with an immunosuppressive role for glycolysis in cancer cells, several pharmacological or genetic strategies for glycolysis inhibition have been shown to mediate immunostimulatory effects and restore (at least partially) immunosurveillance in preclinical tumor models. For instance, genetic inhibition of glycolysis in mouse lung carcinoma LLC cells and mouse pancreatic cancer Panc02 cells as imposed by the deletion of solute carrier family 2 (facilitated glucose transporter), member 1 (Slc2a1, best known as Glut1), which encodes a plasma membrane glucose channel, or glucose-6-phosphate isomerase 1 (Gpi1), which encodes an isomerase catalyzing the interconversion of glucose-6-phosphate (G6P) and fructose-6-phosphate (F6P), in has been shown to increase the sensitivity of malignant cells to CTLs25. Mechanistically, such an immune sensitization originated from accelerated OXPHOS coupled with reactive oxygen species (ROS) overproduction and increased sensitivity to tumor necrosis factor (TNF)-driven cell death25. Whether Glut1 or Gpi1 deletion also alters antigen presentation by cancer cells remains to be investigated. Irrespective of this incognita, both GLUT1 levels and genetic signatures of glycolysis were associated with limited T cell infiltration in patients with various tumors25. Moreover, in patients with lung or pancreatic adenocarcinoma, transcriptional markers of elevated glycolysis and reduced TNF signaling were linked with poor overall survival25. These findings exemplify the clinical relevance of suppressed anticancer immunity as driven by glycolysis in cancer cells.
In summary, glucose metabolism in cancer cells may have immunosuppressive effects on the TME. Of note, glucose-dependent immunosuppression at least partially originates from lactate secretion, potentially offering an improved target for therapeutic interventions (as discussed here below).
Lactate.
Lactate is abundant in most solid tumors, mediating not only trophic functions26, but also eliciting multiple mechanisms of immunosuppression27. For instance, lactate has been shown to inhibit CTL cytotoxicity by limiting the replenishment of TCA cycle intermediates via pyruvate carboxylase (PC), resulting in the accumulation of pyruvate dehydrogenase (PDH), limited secretion of succinate and hence poor pro-inflammatory autocrine/paracrine signaling via succinate receptor 1 (SUCNR1), at least in transplantable mouse models of melanoma and colorectal carcinoma (CRC)28. Lactate also represses effector T cell proliferation by blocking glyceraldehyde 3-phosphate dehydrogenase (GAPDH) and phosphoglycerate dehydrogenase (PHGDH), which results in the deprivation of key post-GAPDH glycolytic intermediates and serine29, as well as by promoting lysosomal acidification, which interferes with diacylglycerol-dependent protein kinase C theta (PRKCQ) signaling30. In line with this notion, LDHA-deficient mouse B16 melanomas exhibit a decreased growth rate than their wild-type counterparts when established subcutaneously in immunocompetent (but not immunodeficient Rag2−/−Il2Rg−/−) syngeneic hosts, an altered immunological control associated with increased tumor infiltration by interferon gamma (IFNG)-producing CTLs and natural killer (NK) cells31. That said, systemic inhibition of LDHA with the small molecule NCI-006 reportedly mediates anticancer effects in athymic mice bearing human pancreatic cancer MIA PaCa-2 xenografts32. Whether such an anticancer activity emerges from a direct effect on malignant cells or instead involves immune TME compartments other than T cells, however, remains to be formally established. Of note, high-dose daily i.p. lactate administration has been shown to control the growth of mouse I3TC mammary and MC38 colorectal cancers established subcutaneously in immunocompetent syngeneic mice33. Supporting a role for CD8+ CTLs in these observations, the subcutaneous administration of lactate (but not glucose) reportedly elicits the CTL-dependent control of MC38 tumors evolving in immunocompetent syngeneic mice as a consequence of improved CD8+ T cell stemness34. These latter findings suggest that the detrimental effects of lactate on anticancer immunity may at least partially stem by local acidification.
At odds with their effector counterparts, immunosuppressive CD4+CD25+FOXP3+ regulatory T (TREG) cells are considerably resistant to the antiproliferative effects of lactate, at least in part as a direct consequence of the metabolic reprogramming imposed by forkhead box P3 (FOXP3), which involves active glycolysis suppression in favor of NADH oxidation and OXPHOS35. TREG cells actually appear to abundantly import extracellular lactate via solute carrier family 16 member 1 (SLC16A1, best known as MCT1) resulting in increased PD-1 expression via a nuclear factor of activated T cells 1 (NFATC1)-dependent mechanism, as documented in immunocompetent models melanoma and CRC in mice36. In the same models as well as in transplantable models of head and neck squamous cell carcinoma (HNSCC), MCT1 expression appears indeed to be required for intratumoral (but not circulating) TREG cells to preserve their immunosuppressive and tumor-promoting functions37, at least in part reflecting the ability of intracellular lactate accumulation to promote the lactylation of moesin (MSN), hence favoring immunosuppressive transforming growth factor beta 1 (TGFB1) signaling via SMAD family member 3 (SMAD3)38. Of note, histone lactylation has also been proposed to mediate immunosuppressive and hence tumor-promoting effects in the myeloid compartment of transplantable (B16) mouse melanomas as well transplantable (MC38) and carcinogen-induced mouse CRCs39. The translational relevance of these observations, however, remains to be defined.
Interestingly, in mouse melanomas, hepatocellular carcinomas (HCCs) and CRCs that are naturally glycolytic or are genetically engineered to exhibit an elevated glycolysis (but not in their poorly glycolytic counterparts), programmed cell death 1 (PDCD1, best known as PD-1) blockage actively promotes the immunosuppressive activity of TREG cells and hence has no therapeutic effects, a resistance phenotype that can be successfully reverted by pharmacological or genetic LDHA inhibition36,38. On the contrary, mouse TNBCs with reduced glycolytic activity have been shown to respond to immune checkpoint inhibitors (ICIs) specific for cytotoxic T lymphocyte-associated protein 4 (CTLA4) along with the destabilization of tumor-infiltrating TREG cells and their shift toward an effector-like state characterized by the secretion of TNF and IFNG40. The relative contribution of extracellular lactate (vs. glucose) availability to these latter observations, however, remains to be clearly defined.
Of note, lactate also influences intratumoral myeloid cells. For instance, lactate appears to signal to tumor-associated macrophages (TAMs) via G protein-coupled receptor 132 (GPR132), resulting in their repolarization towards an immunosuppressive “M2-like” phenotype associated with increased metastatic dissemination in mouse models of TNBC41. Supporting the clinical relevant of preclinical these observations, GPR132 levels have been shown to positively correlated with genetic signatures of M2-like TAMs as well as increased metastatic dissemination and poor disease outcome in a cohort of patients with TNBC41. Similar results have been obtained in preclinical models of lung carcinoma as driven by the loss of serine/threonine kinase 11 (Stk11), although in this latter case extracellular lactate accumulated downstream of solute carrier family 16 member 4 (SLC16A4, best known as MCT4) overexpression and appeared to signal to TAMs (and CTLs) via hydroxycarboxylic acid receptor 1 (HCAR1, also known as GPR81)42. While the reasons underlying such an apparent discrepancy have not yet been clarified, it is plausible that TAMs infiltrating different neoplasms like TNBC and lung carcinoma might express different lactate-sensitive receptors or signal transducers thereof, reflecting the well-established heterogeneity of TAMs at large43.
Taken together, these observations suggest that at least part of the immunosuppressive effects of deregulated glucose metabolism originate from the intratumoral accumulation of lactate.
The TCA cycle.
The TCA is critical not only to provide reducing equivalent for OXPHOS but also to regulate the pool of numerous metabolites that have both metabolic and signaling functions, such as acetyl-CoA, citrate, fumarate, α-ketoglutarate (α-KG), and succinate44,45. Not surprisingly, many of these metabolic intermediates have also direct or indirect immunomodulatory effects. For instance, mouse B16 melanomas depleted of the TCA cycle enzyme fumarate hydratase (FH) exhibit high intratumoral levels of fumarate irrespective of potential alterations in glycolysis, resulting in acute T cell dysfunction as a consequence of the non-enzymatic succination of zeta chain of T cell receptor associated protein kinase 70 (ZAP70)46. In line with this notion, engineered CD19-specific CAR T cells for FH overexpression has been shown to result in superior therapeutic efficacy against human CD19-expressing leukemia cells expanding in immunocompromised mice46. The loss of FH also destabilizes the mitochondrial network to promote the release of small vesicles containing mitochondrial nucleic acids via a sorting nexin 9 (SNX9)-dependent mechanism, at least in preclinical models of hereditary leiomyomatosis and renal cell carcinoma (HLRCC)47. In this setting, the release of mitochondrial DNA (mtDNA) and mtRNA into the cytosol was shown to drive type I interferon (IFN) secretion upon activation of cyclic GMP-AMP synthase (cGAS) and RNA sensor RIG-I (RIGI)47, a chronic, indolent inflammatory response potentially supporting oncogenesis in the context of compromised immunosurveillance48,49.
Interestingly, a normal flow of electrons through the mitochondrial respiratory complexes that mediate OXPHOS has been recently shown to limit the recognition of melanoma cells by CTLs50. Such an immunosuppressive effect originates from the ability of respiratory complex II to efficiently convert succinate into fumarate, hence preventing the activation of a succinate-dependent epigenetic mechanism resulting in the upregulation of MHC class I molecules and other components of the antigen-presenting machinery50. These findings suggest that defects in mitochondrial electron flow that may emerge in some cancer cells during tumor evolution may be the target of negative selective pressure by the host immune system. That said, succinate accumulation in mouse TNBC cells as driven by TAM-derived TGFB1 has been shown to promote glycolysis and hence an overall immunosuppressive phenotype (see above)51. While the reasons for such an apparent discrepancy remain to be defined, it is plausible that tumor-specific mechanisms beyond glycolysis may underlie the immunosuppressive effects of succinate accumulation in TNBC but not melanoma cells, potentially including the succinate-dependent accumulation of hypoxia inducible factor 1 subunit alpha (HIF1A) (Box 3).
Box 3. Microenvironmental hypoxia and tumor-targeting immunity.
Solid tumors are often characterized by at least some areas where oxygen tension falls below physiological values184. Malignant cells adapt to these abnormal metabolic conditions by a variety of mechanisms that are often orchestrated by the transcriptional regulator hypoxia inducible factor 1 subunit alpha (HIF1A)184. The major metabolic shift imposed by HIF1A involves the redirection of glucose flux from oxidative phosphorylation (OXPHOS) to anaerobic glycolysis coupled with abundant lactate secretion184, which has major immunosuppressive effects (see main text). Moreover, HIF1A promotes the upregulation of vascular endothelial growth factor A (VEGFA), which besides promoting neoangiogenesis supports the accumulation and immunosuppressive activity of CD4+CD25+FOXP3+ regulatory T (TREG) cells185. Moreover, HIF1A promotes immunosuppressive nucleotide metabolism (see main text) by upregulating not only ectonucleoside triphosphate diphosphohydrolase 1 (ENTPD1, best known as CD39) and ectonucleotidase 5’-nucleotidase ecto (5NTE, best known as CD73), hence resulting in accelerated extracellular ATP degradation, but also adenosine A2a receptor (ADORA2A) and ADORA2B, further fostering adenosinergic signaling186. Of note, hypoxia also mediate immunosuppressive effects that do not directly involve cancer cell metabolism, including the activation of a CD39-dependent exhaustion program in tumor-infiltrating T lymphocytes187, the repolarization of tumor-associated macrophages (TAMs) towards an M2-like state188, and the elimination of tumor-targeting γδ T cells189. Thus, hypoxia represent a major driver of immunosuppression in the tumor microenvironment,
2HG also mediates bona fide oncogenic functions via epigenetic mechanisms, especially (but not exclusively) upon the inhibition of multiple α-KG-dependent dioxygenases, prolyl-hydroxylases and histone demethylases3. Moreover, the D enantiomer of 2HG (but not its L counterpart) can be actively taken up by tumor-infiltrating T lymphocytes, resulting in a dose-dependent and fully reversible inhibition of proliferation and effector functions, including IFNγ secretion, in immunocompetent mouse models of melanoma and CRC52. At least in part, such an immunosuppressive effect appears to originate from the ability of D-2HG to (1) inhibit LDHA, resulting in a metabolic shift towards OXPHOS as driven by an increased mitochondrial uptake of pyruvate and lowered NAD+/NADPH ratio52, and (2) to disrupt nuclear factor of activated T cells 1 (NFCAT1) activity and polyamine synthesis53.
Interestingly, another metabolic intermediate that inhibits α-KG-dependent dioxygenases, notably glutarate, has been shown to have a positive, rather than negative, influence on T cell functions54. Specifically, the administration of the cell permeant glutarate precursor diethyl-glutarate has been associated with improved T cell cytotoxicity against mouse B16 melanoma and SKOV3 ovarian carcinoma cells downstream of the PDH glutarylation and consequent inhibition of OXPHOS in favor of anaerobic glycolysis54. These findings point to glutarate metabolism as a potential target to improve T cell-dependent anticancer immune responses. Along similar lines, it has recently been shown that blocking acyl-CoA synthetase short chain family member 2 (ACSS2) – which converts acetate into acetyl-CoA – in breast cancer cells converts them from consumers to producers of acetate, resulting in abundant acetate accumulation in the TME55. Such microenvironmental acetate can be avidly taken up by tumor-infiltrating T lymphocytes supporting a therapeutically actionable improvement in T-cell effector functions and proliferation55. As these effects are observed under pharmacological ACSS2 inhibition, they are unlikely to emerge from epigenetic alterations in T cells as driven by the ACSS2-dependnent conversion of acetate into acetyl-CoA, but may instead relate to acetate signaling via free fatty acid receptor 2 (FFAR2, best known as GPR43)56. Of note, the TCA intermediate itaconate also appears to mediate multipronged immunosuppressive effects in a variety of cell types, including immune cell themselves57,58. In line with this notion, the itaconate-dependent activation of NFE2 like bZIP transcription factor 2 (NFE2L2, best known as NRF2) by a cell-permeant precursor of itaconate has been shown to suppress type I IFN responses downstream of stimulator of interferon response cGAMP interactor 1 (STING1) activation in cultured human NSCLC A549 cells59. To the best of our knowledge, however, the precise impact of cancer cell-derived itaconate on anticancer immune responses as emerging in immunocompetent mice bearing syngeneic tumors remain to be formally investigated.
In summary, multiple cancer-associated alterations of metabolism have been shown to elicit microenvironmental perturbations negatively affecting immunosurveillance.
Lipid metabolism
Accelerated proliferation as exhibited by transformed cells heavily relies on enhanced lipid metabolism, not only as an extra energy source downstream of fatty acid oxidation (FAO)-driven OXPHOS, but also as a source of cellular membranes and other lipid constituents generated by fatty acid synthesis. Cancer-associated alterations of FAO and fatty acid synthesis have been shown to impact tumor-targeting immune responses (Fig. 2)
Figure 2. Fatty acid and eicosanoid metabolism on anticancer immunity.
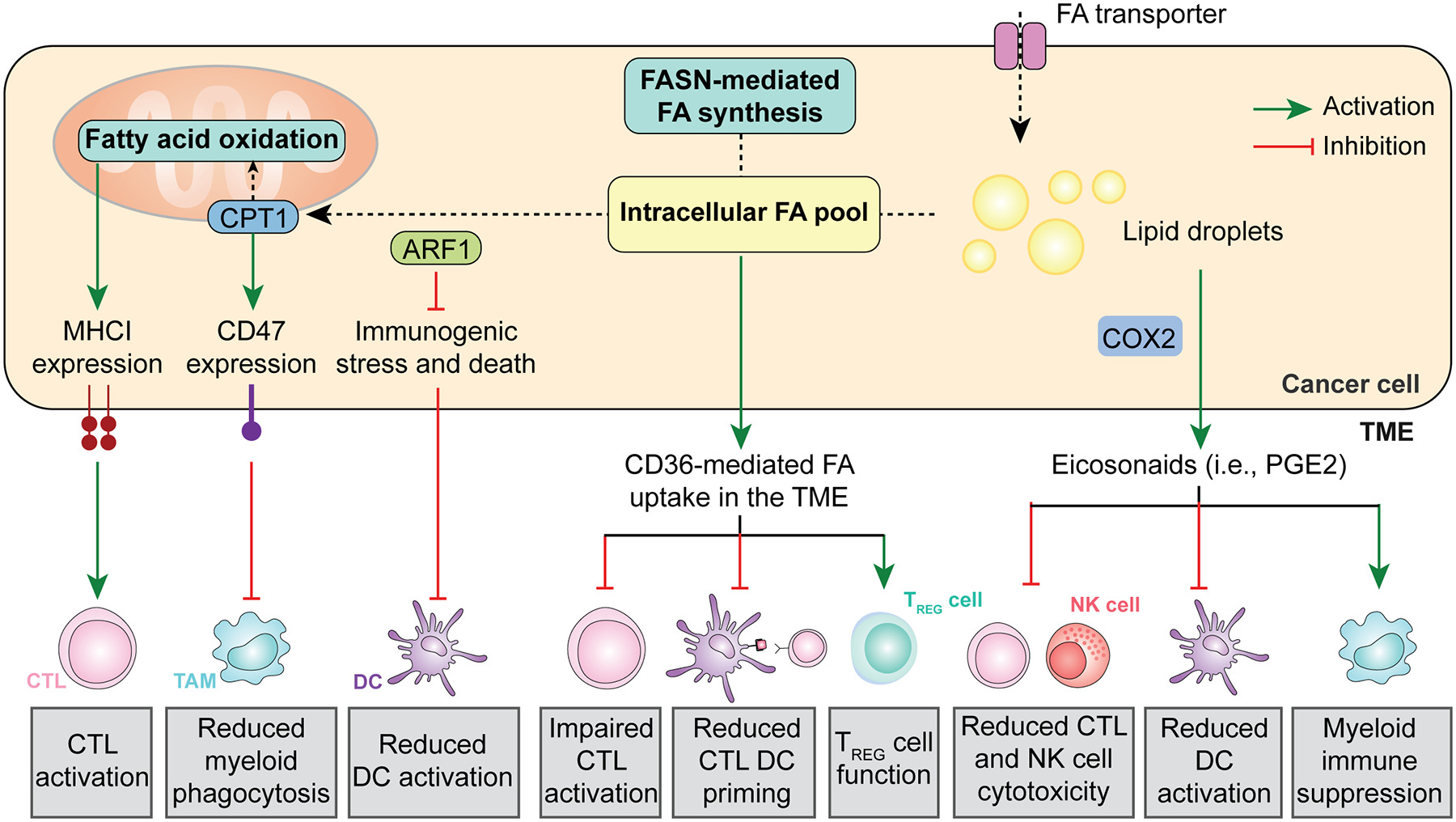
Cancer cells generally exhibit an increase in both fatty acid (FA) intake from the tumor microenvironment (TME) and endogenous fatty acid synthesis. This results in immunomodulatory effects emerging from (1) increased MHC Class I exposure on the cancer cell surface, resulting in improve recognition by cytotoxic T lymphocytes (CTLs), (2) elevated CD47 expression, limiting phagocytic uptake by myeloid cells; and (3) inhibited immunogenic cell death (ICD), preventing pronounced dendritic cell (DC) activation. Moreover, high levels of FAs in the TME have a direct immunosuppressive effect on CTLs and DCs, coupled with an increased in regulatory T (TREG)-mediated immunosuppression. Finally, cancer cells can convert FAs stored as lipid droplets into immunosuppressive eicosanoids such as prostaglandin E2 (PGE2). NK, natural killer.
Fatty acid oxidation.
FAO consists in the catabolism of long chain fatty acids into acetyl-CoA as a substrate for the TCA cycle to fuel OXPHOS60. Multiple cancer types such as glioblastoma (GBM) exhibit elevated rates of FAO in support of aggressive disease progression61. In this setting, the co-upregulation of multiple FAO-related enzymes such as carnitine palmitoyltransferase 1A (CPT1A), CPT2 and acyl-CoA dehydrogenase family member 9 (ACAD9) has appear to occur along with an increased exposure of CD47 on the GBM cell membrane, resulting in a potent inhibition of phagocytosis by myeloid cells and radioresistance, mechanistically reflecting the ability of FAO-derived acetyl-CoA to drive NF-κB signaling62. Supporting the therapeutic relevance of these observations, combining radiotherapy with a CPT1 inhibitor and a CD47 blocker was shown to result in superior tumor control in preclinical models of GBM62. Of note, acetyl-CoA is also a potent inhibitor of autophagy63, which also has potent immunomodulatory activities (Box 2), potentially implicating autophagy modulation in the immunological effects of FAO. Of note, CPT1A expression also appears to promote cancer cell resistance to CTL-derived IFNG by promoting antiapoptotic signaling (irrespective of changes in antigen presentation), at least in mouse melanoma B16 and mouse prostate cancer RM1 cells64.
Box 2. Autophagic responses in cancer cells and tumor-targeting immunity.
Autophagy is a lysosome-dependent catabolic mechanisms that dispose of potentially cytotoxic and/or dysfunctional cytoplasmic entities (e.g., permeabilized mitochondria)179. Autophagy serves major homeostatic and metabolic functions in all nucleated cells, de facto supporting the differentiation and functions of numerous immune cell types180. Moreover, both natural and therapy-driven autophagic responses in malignant cells have been shown to influence tumor-targeting immune responses via multiple, context-dependent mechanisms. On the one hand, autophagic responses as driven by immunogenic chemotherapy have been shown to be required for the optimal release of ATP by dying cells140, hence orchestrating the recruitment and activation of dendritic cells (DCs) and ultimately promoting CD8+ cytotoxic T lymphocyte (CTL)-dependent anticancer immunity. On the other hand, autophagy has been reported to mediate robust immunosuppressive effects including: (1) the inhibition of type I interferon (IFN) responses as elicited in malignant cells by radiotherapy or as driven spontaneously in neoplasms with elevated mutational burden, resulting in limited cell death adjuvanticity and hence preferential tumor infiltration by regulatory T (TREG) cells coupled with CTL exhaustion170,172, (2) the inhibition of MHC Class I molecule exposure on the cancer cell surface, resulting in reduced antigenicity171, and (3) the degradation of natural killer (NK)-produced granzyme B (GZMB), resulting in limited susceptibility to lysis by immune effector cells181. Thus, autophagic responses in malignant cells stand out as central regulators of all aspects of anticancer immunity. That said, the development of pharmacological modulators of autophagy present multiple challenges that have not been successfully addressed yet182,183.
Apparently at odds with these observations, deletion of the FAO-relevant gene acetyl-CoA acetyltransferase 1 (Acat1) from mouse melanoma cells has been reported to compromise immune recognition and elimination, at least in part reflecting reduced expression of MHC Class I molecules on the cancer cell surface coupled with reduced T cell activation in the TME65. At least theoretically, such an apparent discrepancy may reflect the role of ACAT1 in cholesterol esterification66, potentially leading to disruptions in plasma membrane lipid rafts associated with MHC molecules67,68. Despite this unknown, profiling the proteome of clinical samples from advanced stage melanoma patients undergoing either tumor infiltrating lymphocyte (TIL)-based or receiving an ICI specific for PD-1 revealed that proteomic signatures of lipid metabolism and OXPHOS to be enriched amongst responders65. It remains to be demonstrated whether these observations can be generalized to other tumor types.
Lipid mobilization upstream of FAO, as mediated by ADP ribosylation factor 1 (ARF1) has been implicated in the robust immunoevasive properties of cancer stem cells (CSCs). Specifically, Arf1 deletion in a genetically engineered model of CRC has been shown to considerably slow down disease onset and progression as a consequence of the activation of immunogenic stress and death15 in the CSC compartment, resulting in the DC-dependent activation of a tumor-targeting immune response69.
Altogether, these observations exemplify the impact of FAO in cancer cells on the immunological contexture of the TME and cancer sensitivity to immunotherapy.
Lipid synthesis.
Fatty acid synthase (FASN) is the rate-limiting enzyme of de novo lipid biosynthesis and its expression levels generally correlate with advanced cancer stage and metastatic dissemination70. In patients with ovarian carcinoma, high FASN levels are also associated with decreased T cell infiltration, owing not only T cell inhibition as directly mediate by the CD36-dependent uptake of fatty acids accumulating in the TME upon FASN overexpression71,72, but also (1) to a lipid-driven defect of T cell cross-priming by DCs73, as well as (2) to the ability of CD36 to promote TREG cell functions74. In line with this notion, abundant tumor infiltration by CD36-expressing CD8+ T cells has been associated with poor disease outcome in patients with NSCLC receiving immunogenic chemotherapy75.
Eicosanoid synthesis.
Increased FASN activity also promotes the accumulation of intracellular lipid droplets that are associated with prostaglandin-endoperoxide synthase 2 (PTGS2, best known as COX2), a key enzyme in the synthesis of eicosanoids including prostaglandin E2 (PGE2)76. PGE2 mediates direct mitogenic functions on malignant cells77 as well as multipronged immunosuppressive effects that involve DC dysfunction as a consequence of prostaglandin E receptor 2 (PTGER2) and PTGER4 signaling coupled with intracellular cyclic AMP elevations78,79, inhibition of NK cytotoxic and secretory activity (which also affect the recruitment of conventional type 1 DCs)80, as well as T cell suppression upon the NF-κB-mediated upregulation of PD-1 (Ref. 81), an effect that is aggravated by the ability of PGE2 to elicit the upregulation of PD-L1 on myeloid cells82,83. Of note, PGE2 resembles hypoxia (which also has multipronged immunosuppressive effects, Box 3) in its ability to promote the upregulation of the ectonucleotidase 5’-nucleotidase ecto (5NTE, best known as CD73) on myeloid cells of the TME84, which contributes to the immunosuppressive effects of nucleotide metabolism (see below). Interestingly, MFSD2 lysolipid transporter A, lysophospholipid (MFSD2A) has been shown to operate as an endogenous COX2 inhibitor in human and mouse gastric cancer cells, resulting in suppressed release of PGE2 and the immunosuppressive cytokine transforming growth factor beta 1 (TGFB1)85. In this setting, MFSD2A overexpression by malignant cells has been shown to circumvent resistance to PD-1 inhibition along with signs of improved CD8+ CTL reactivity in the TME85.
Collectively, these observations highlight the complex interplay between fatty acid metabolism and immune cell function in the TME. An improved understanding of these mechanisms will provide valuable insights for the development of novel therapeutic strategies to enhance the efficacy of immunotherapy.
Other metabolic pathways
Additional metabolic circuitries that are altered along with malignant transformation have been shown to influence anticancer immunosurveillance. These include (but are not limited to) the biochemical cascades involved in the metabolism of nucleotides and various amino acids (Fig. 3).
Figure 3. Nucleotide and amino acid metabolism in anticancer immunity.
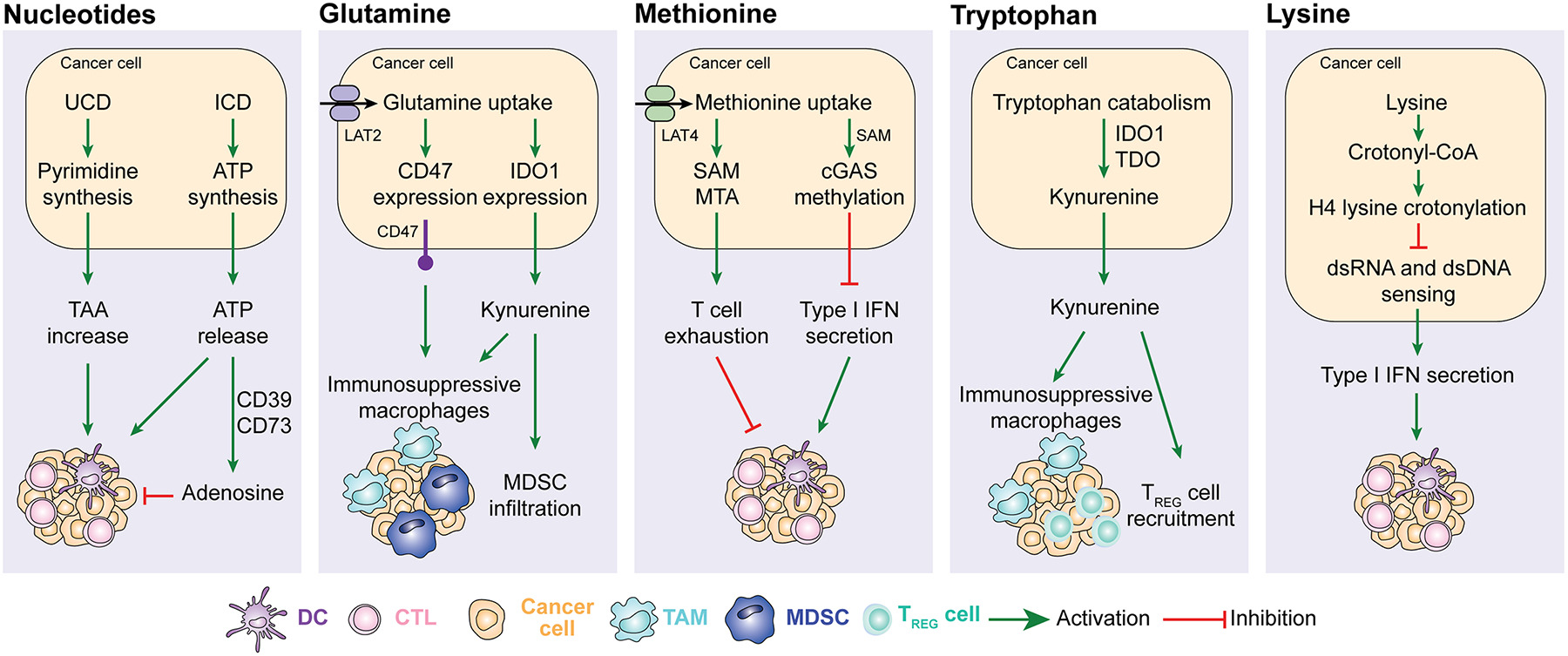
Alterations in the urea cycle promote cancer cell immunogenicity by favoring the expression of tumor-associated antigens (TAA), while ATP released in the context of immunogenic cell death (ICD) mediates potent chemotactic and immunostimulatory effects on myeloid cells that are actively counteracted when extracellular ATP is converted into immunosuppressive adenosine by CD39 and CD73. Increased glutamine metabolism favors the accumulation of immunosuppressive tumor-associated macrophages (TAMs) and myeloid-derived suppressor cells (MDSCs) by promoting CD47 and IDO1 upregulation. Methionine uptake by cancer cells promotes cytotoxic T lymphocyte (CTL) exhaustion via 5-methylthioadenosine (MTA) and S-adenosylmethionine (SAM) as it inhibits type I interferon (IFN) secretion by methylating CGAS. Tryptophan degradation as mediated in cancer cells and myeloid cells by IDO1 results in the accumulation of immunosuppressive metabolites including kynurenine. Finally, lysine has been shown to suppress type I IFN production by malignant cells upon histone H4 lysine crotonylation. DC, dendritic cell; TREG, regulatory T; UCD, urea cycle dysregulation.
Nucleotides.
Cancer cells exhibit increased nucleotide synthesis as compared to their normal counterparts, and this has a considerable impact on anticancer immunity86. For instance, alterations of urea cycle enzymes reportedly lead to so-called “urea cycle dysregulation” (UCD), which redirects nitrogen flux toward carbamoyl-phosphate synthetase 2, aspartate transcarbamylase, and dihydrooratase (CAD)87. This results in excess pyrimidine synthesis promoting a distinctive genomic signature that is characterized by R→Y transversions and is associated with an increase in hydrophobic tumor antigens linked to improved sensitivity to ICIs in patients with melanoma87. Similar findings were obtained in preclinical models of CRC driven into the UCD by the depletion of argininosuccinate synthase 1 (ASS1)87. Moreover, the release of nucleotides by stressed and dying malignant cells has a major impact on the immunological contexture of the TME and the immunological reaction to cancer cell death15. For instance, extracellular ATP can be detected by myeloid cells including DCs and their precursors via purinergic receptor P2Y2 (P2RY2), which promotes chemotaxis88,89, hence attracting them to the proximity of dying cells90, or purinergic receptor P2X 7 (P2RX7), which promotes DC activation via inflammasome signaling and interleukin 1 beta (IL1B, best known as IL-1β) secretion91. Such an immunostimulatory effect is actively counteracted by extracellular ATP degradation through the sequential activity of ectonucleoside triphosphate diphosphohydrolase 1 (ENTPD1, best known as CD39) and CD73, which collectively promote the generation of adenosine, which has a potent and multipronged immunosuppressive activity92,93. Thus, intratumoral levels of CD39 and CD73 (which exhibit considerable variations not only across different neoplasms, but also in distinct cellular compartments of a the same tumor) are critical determinants of the immunostimulatory (low CD39 and CD73 expression) vs immunosuppressive (high CD39 and CD73 expression) effects of extracellular ATP93.
Collectively, these observations point to the possibility to target nucleotide metabolism in cancer cells to achieve immunotherapeutic effects, an approach that is being investigated in clinical trials with promising results (see below).
Glutamine.
Microenvironmental glutamine represents an important source of energy and intermediate metabolites for rapidly proliferating cancer cells and immune cells94. In line with this notion, most cancer cells consume glutamine at a high rate relative to glucose and exhibit at least some degree of non-oncogene addiction to glutamine availability21. Suggesting an immunosuppressive function for this metabolic adaptation, human basal-like breast cancers exhibiting high transcriptional signatures of glutamine metabolism appear to be characterized by a scarce immune infiltrate, which correlates with poor disease outcome95. Accordingly, deletion of glutaminase (Gls) – which encodes the first enzyme of glutamine catabolism – in mouse TNBC cells has been shown to promote in vivo tumor control by a T cell-dependent mechanism95. Of note, pharmacological inhibition of GLS by a pro-drug that is preferentially activated in the TME (i.e., JHU083) appears to drive potent anticancer responses in mice bearing MC38 CRCs by suppressing oxidative and glycolytic metabolism in cancer cells, but at the same time promoting OXPHOS and hence eliciting a long-lasting activated phenotype in CTLs96. These findings point to the existence of therapeutic strategies that efficiently target glutamine metabolism in cancer cells while sparing intratumoral CTLs. Glutamine metabolism in cancer cells also influences myeloid cells recruitment and activation. For instance, inhibiting GLS with a pharmacological agent has been reported to limit tumor infiltration by MDSCs and to favor to repolarization of TAMs toward an immunostimulatory “M1-like” profile in preclinical models of TNBC97. At least partially, this originated from the downregulation of indoleamine 2,3-dioxygenase 1 (IDO1) in malignant (and immune) cells, leading to a marked decrease in the abundance of immunosuppressive kynurenine97. Moreover, malignant cells appear to compete with type I conventional DCs (cDC1s) – which are key for antigen cross-presentation to T cells – for intratumoral glutamine availability via solute carrier family 38 member 2 (SLC38A2), at least in mouse models of melanoma and CRC98. These findings point to SLC38A2 on neoplastic cells as a potential target for the development of novel (immuno)therapeutic agents against cancer. Finally, increased glutamine uptake by cancer cells via solute carrier family 7 member 8 (SLC7A8, best known as LAT2) has been associated with CD47 upregulation in preclinical osteosarcoma models, resulting in inhibited phagocytosis and accelerated tumor progression99. Taken together, these findings exemplify the multipronged immunomodulatory functions of glutamine metabolism in cancer and immune cells.
Methionine.
Methionine is an essential amino acid that – besides contributing to protein synthesis – in involved in enzymatic methylation reactions100. Elevated levels of methionine-recycling enzymes and their products including 5-methylthioadenosine (MTA) and S-adenosylmethionine (SAM) have been linked to T cell exhaustion in mouse and human models of HCC101. In this setting, deletion of methionine adenosyltransferase 2A (Mat2a), which encodes a key enzyme in SAM synthesis, resulted in restored T cell activation and in vivo HCC control101. Along similar lines, methionine has been shown to impair CGAS activity in a methylation-dependent manner102. Of note, cancer cells generally express high levels of the methionine transporter solute carrier family 43 member 2 (SLC43A2, best known as LAT4), hence competing with T cells for this essential amino acid103. This results in the loss of demethylation of histone H3 at lysine 79 (H3K79me2), reduced signal transducer and activator of transcription 5A (STAT5) signaling and suppressed T cell functions103. Both LAT4 inhibition and methionine supplementation have been shown to circumvent this defect and restore anticancer immunity in mice bearing CRCs103. Tumor-specific LAT4 inhibition coupled with STING1 activation as achieved by a bimetallic nanoplatform bearing a SLC43A2-targeting CRISP/Cas9 construct plus Zn2+ ions has also been shown to mediate promising immunotherapeutic effects in preclinical TNBC models104. These examples point to the LAT4 as a potential target for the development of novel immunostimulatory agents with clinical applications. In this context, targeted approaches must be envisioned to enable the selective depletion of methionine from cancer cells102,105, but not immune cells, which also heavily rely on methionine uptake for their anticancer function103.
Tryptophan.
Tryptophan catabolism initiated by the enzymes indoleamine 2,3-dioxygenase 1 (IDO1) and tryptophan 2,3-dioxygenase (TDO) has attracted considerable attention as a potential target for the development of novel immunotherapeutic strategies106. IDO1 hyperactivation as occurring in some malignant cells (as well as in tolerogenic DCs) mediates indeed multipronged immunosuppressive effects that largely originate from the accumulation of kynurenine, which (amongst other activities) potently inhibits T cells107 and promotes TREG cell differentiation108. Indeed, while tryptophan is an essential amino acid, its concentration does not fall below a limiting threshold in the TME109, implying that tryptophan shortage does not contribute to IDO1-driven immunosuppression as initially proposed110. Additional immunosuppressive products of the IDO1 pathway include quinolinic acid, which has been shown to promote M2-like TAM polarization downstream of forkhead box O1 (FOXO1) and peroxisome proliferator activated receptor gamma (PPARG) signaling in the GBM setting111.
Lysine.
GBM stem cells have been shown to reprogram lysine catabolism, resulting in an intracellular accumulation of crotonyl-CoA and consequent histone H4 lysine crotonylation112. In this context, inhibition of lysine crotonylation enhances type I IFN signaling elicited by double stranded RNA (dsRNA) and dsDNA, culminating with restored CD8+ T cell infiltration, and impaired disease progression112. These data point to lysine metabolism as a potential target for the development of novel immunotherapies. Whether this mechanism is operational in cancer types other than GBM, though, remains unclear.
Targeting metabolic cancer vulnerabilities to restore immunosurveillance
The development of small molecules and monoclonal antibodies targeting cancer metabolism has been initiated decades ago, including various agents that are currently under clinical evaluation113. Accumulating data indicate that at least some of these agents represent promising tools to restore cancer immunosurveillance and increase tumor sensitivity to approved cancer therapeutics that engage anticancer immunity, including not only immunotherapy, but also immunogenic chemotherapy114, some targeted anticancer agents115, and radiotherapy (at least when used focally and according to specific dose and fractionation protocols)116. Importantly, a number of dietary interventions are also being investigated for their ability to alter cancer cell metabolism in support of enhanced anticancer immunity, but owing to space limitations they are not further discussed here (Supplemental Information).
Glucose and lactate.
Pharmacological GLUT1 inhibition with BAY-876 has been harnessed for increasing therapeutic responses to an ICI targeting PD-1 in preclinical models of pancreatic and lung cancer25. In this setting though, solute carrier family 2 member 3 (SLC2A3, best known as GLUT3) overexpression appeared to compensate (at least in part) for GLUT1 inhibition25, pointing to dual GLUT1/GLUT3 inhibition as a potentially superior strategy. PD-1 blockage has been shown to synergize with PKF-015, a pharmacological inhibitor of the glycolytic enzyme 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3 (PFKFB3), in mouse models of melanoma and colorectal carcinoma (CRC), an effect that could be attributed to the ability of PFKFB3 inhibition to elicit PD-L1 expression117. Preliminary results from a dose-escalation Phase I clinical trial testing a PKF-015 analog in patients with solid tumors (NCT02044861) confirmed the feasibility of this approach118. That said, targeting glucose uptake or consumption for cancer therapy remains challenging as multiple healthy cells including neurons abundantly rely on glucose metabolism for their normal functions, calling for the development of targeted delivery strategies. At least theoretically, inhibiting lactate secretion or uptake in the TME may present less challenges than blocking glycolytic metabolism as a whole. However, past drug development efforts focused on lactate, including the development of the MCT1 inhibitor AZD3965 (which demonstrated a good tolerability in patients with advanced solid tumors)119 have been discontinued. Whether recent preclinical findings demonstrating a positive interaction of multiple lactate-targeting strategies including MCT4 inhibitors with various forms of immunotherapy120,121 will reinvigorate these efforts remains unclear.
Glutamine.
Telaglenastat (a GLS-targeting agent also known as CB-839) has been reported to synergize with radiotherapy (which can mediate robust immunostimulatory effects)122,123 against human HNSCC and NSCLC xenografts124,125. Whether such a radiosensitizing effect involved any degree of innate immune activation, however, remains unclear. That said, telaglenastat has also been shown to synergize with CTLA4 and PD-1 blockers in immunocompetent models of melanoma126, suggesting that this agent mediates indeed therapeutically relevant immunostimulatory effects. Irrespective of this unresolved possibility, clinical data from a few independent studies in patients with metastatic renal cell carcinoma suggest that telaglenastat can be safely combined with standard-of-care chemotherapy in this patient population, although with minimal therapeutic benefits127–129. These clinical findings considered reduced the interest in the development of telaglenastat as a novel anticancer agent, with only one study in patients with NSCLC remaining open for recruitment as of Feb 2024 (NCT03831932, source www.clinicaltrials.gov).
Pharmacological inhibition of glutamine uptake via LAT2 with BCH 2-aminobicyclo-(2,2,1)-heptane-2-carboxylic acid has been shown to enhance the therapeutic effect of the immunogenic chemotherapeutic doxorubicin against osteosarcoma cells, an effect that at least partially reflected CD47 downregulation and restored cancer cell phagocytosis99. Along similar lines, V-9302, a pharmacological inhibitor of glutamine uptake, has been shown to mediate T cell-dependent tumor control in preclinical models of TNBC95. That said, V-9302 administration to mouse lung cancer and CRC cells has also been associated with PD-L1 upregulation via NF-κB, de facto suppressing tumor-targeting immune responses130. Such an immunosuppressive response, however, was accompanied by the upregulation of Fas cell surface death receptor (FAS)131 on malignant cells, rendering them more sensitive to T cell responses as pharmacologically reactivated with a PD-L1 blocker130. Whether these apparently discrepant observations reflect specificities of glutamine metabolism in different cancer cell types remains to be clarified.
Tryptophan.
Preclinical data generated in a large panel of immunocompetent tumor models demonstrate that pharmacologically or genetically blocking IDO1 and/or TDO activity promotes robust immunotherapeutic effects that can generally be amplified with ICIs132,133 These findings spurred considerable interest in the development of clinically testable IDO1 inhibitors such as epacadostat134. Preliminary findings from non-randomized early phase clinical trials suggested that epacadostat can be safely and effectively combined with PD-1 blockers in patients with advanced solid tumors135. However, despite considerable expectations, a randomized Phase III clinical study enrolling subject with advanced melanoma demonstrated no therapeutic advantages for epacadostat plus the PD-blocker pembrolizumab over pembrolizumab alone136. Whether this reflects the existence of alternative tryptophan degradation pathways that have been shown to mediate immunosuppressive effects in non-oncological settings (notably autoimmune disorders)137 and may be upregulated in the context of IDO1 inhibition remains to be demonstrated. Obviously, these negative results considerably decreased the interest of pharma companies to develop IDO1 inhibitors138, with only few clinical trials still open to recruitment as of Feb 2021 (source www.clinicaltrials.gov). Whether novel approaches targeting IDO1 such as the inhibition of ubiquitin specific peptidase 14 (USP14), which effectively restores T cell-dependent disease control in preclinical models of CRC139, will reinvigorate such interest remains unclear.
Nucleotides.
Standalone pharmacological inhibition of CD39, CD73 and/or adenosine receptors have all been associated with restored anticancer immunity and improved disease control in a variety of preclinical cancer models140–142. Moreover, the small CD73-targeting molecule AB680 reportedly sensitize mouse pancreatic carcinoma to ICIs specific for PD-1, a therapeutic effect reflecting decreased tumor infiltration by TREG cells143. Along similar lines, a monoclonal antibody targeting CD73 has been reported to improve the therapeutic effects of focal radiotherapy combined with a CTLA4 blocker in preclinical models of breast cancer, at least partially reflecting superior DC recruitment to the TME and activation144. Similar results have been obtained by combining pegylated adenosine deaminase (ADA), which converts extracellular adenosine into inosine, with a PD-1 blocker in preclinical models of TNBC and pancreatic carcinoma84. Finally, adenosine A2a receptor (ADORA2A) antagonists have been shown to positively cooperate with several immunotherapeutic strategies in preclinical tumor models, including (but not limited to) CAR T cell therapies in leukemia models145 as well as PD-1 blockers in models of breast cancer and melanoma146,147. In line with these preclinical findings, various phase I clinical trials have evaluated ADORA2A or ADORA2B antagonists in patients with CRC, NSCLC and castration-resistant prostate cancer with encouraging results148,149. Moreover, two parallel Phase II studies reported promising activity for a monoclonal antibody neutralizing CD73 (i.e., oleclumab) in combination with the PD-L1 blocker durvalumab delivered as neoadjuvant interventions to patients with operable NSCLC150 or as part of the management of unresectable NSCLC151. Conversely, while numerous studies have evaluated and are evaluating CD39 blockers in patients with cancer (source www.clinicaltrials.gov), the clinical applicability of this approach remains uncertain.
Fatty acids.
Inhibiting FAO via the carnitine palmitotransferase 1 (CPT1) blocker etomoxir reportedly synergizes with CD47-targeting antibodies and radiotherapy against otherwise radioresistant mouse GBMs established intracranially, along with the restoration of macrophage-dependent cancer cell phagocytosis62. Whether these findings can be translated to human GBM, however, remains unclear. Pharmacological inhibition of FASN with cerulenin has been shown to restore DC activation and tumor infiltration by effector T lymphocytes coupled with at least partial tumor control in preclinical models of ovarian cancer73. In line with these findings, the FASN inhibitor denifanstat (also known as TVB-2640) has been shown to be well tolerated in patients with advanced tumors152 and high-grade astrocytoma153, prompting the initiation of clinical trials in patients with various neoplastic conditions (NCT02980029; NCT03179904; NCT03808558; NCT05743621). CD36 blockers have also demonstrated promising activity in combination with immunogenic immunotherapy in preclinical models of pancreatic cancer154 as well as in combination with PD-1 blockers in preclinical models of melanoma74, but their development into clinically available drugs is still in its infancy, with only one agent (i.e., VT1021) being under evaluation for the treatment of GBM (NCT03970447).
Eicosanoids.
Corroborating the ability of PGE2 to potently suppress anticancer immune responses, pharmacological strategies for the inhibition of COX2, PTGER2 or PTGER4 have been associated with improved tumor control along with restored immune effector functions and positive cooperativity with ICIs in multiple preclinical tumor models, including models of CRC, melanoma, breast cancer and NSCLC78,79,155–157. Since agonists of the so-called liver X receptors (LXRs) have been demonstrated to promote MFSD2A expression158,159, these agents might provide an appealing tool to limit COX2-dependent immunosuppression. However, recent evidence indicates that LXR activation also results in the expression of sphingomyelin phosphodiesterase acid like 3A (SMPDL3A), which actively degrades the CGAS product 2′3′-cyclic GMP-AMP (cGAMP) to suppress STING1 activation, at least in myeloid cells160. Along similar lines, conventional COX2 inhibitors including celecoxib and aspirin mediate multipronged immunosuppressive effects encompassing direct CGAS inhibition161. Thus, the restoration of optimal anticancer immunity by PGE2-directed strategies may benefit from agents that antagonize PGE2 receptors such as PTGER2 and PTGER4. Along these lines, TPST-1495 (a novel dual antagonist of PTGER2 and PTGER4) is currently being investigated as standalone therapeutic agent of in combination with pembrolizumab in patients with advanced solid tumors (NCT04344795). Additional trials testing celecoxib plus pembrolizumab in patients with colorectal or rectal cancer are also underway (NCT03638297, NCT03926338, NCT05731726), largely based on the proven oncopreventive effects of COX2 inhibitors in this oncological indication162. Based on the aforementioned considerations, it will be interesting to see these studies will document any degree of cooperativity between COX2 inhibition and PD-1 blockers.
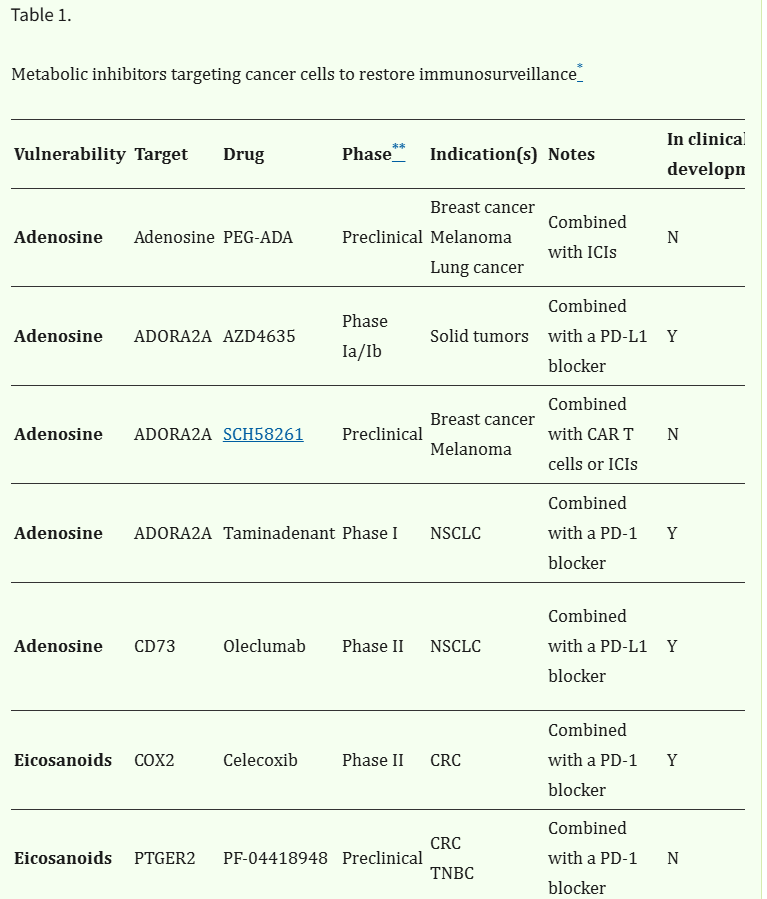
These findings collectively emphasize the potential of metabolic inhibitors not only as cancer-targeted drugs, but also as immunostimulants that may synergize with other therapeutic strategies that restore immunosurveillance (Table 1).
당뇨병 vs. 암대사 치료의 차이점
- 당뇨병처럼: 혈당 조절 약(인슐린, 메트포르민 등)을 평생 복용하면서 혈당을 안정적으로 관리하고, 합병증을 예방하며 만성질환으로 잘 살아가는 모델입니다. → 암대사 프로그램을 바꾸는 약(예: Table 1의 MCT1 억제제, IDO1 억제제, FASN 억제제, GLS 억제제 등)이 성공한다면, 암을 완전히 없애는 대신 “조절 가능한 만성질환”으로 바꿀 가능성이 커집니다.
- 하지만 차이점:
- 암은 유전적으로 불안정하고, 대사 경로가 매우 가소성(plasticiity)이 높아서 약 하나만으로는 쉽게 저항성이 생깁니다.
- 당뇨는 “혈당 조절”이 주목표라면, 암대사 치료는 면역을 다시 깨우는 것(immunosurveillance 회복)이 핵심입니다. 단순히 암세포를 굶기는 게 아니라, 락트산·아데노신·키누레닌 등을 줄여 T세포·NK세포 기능을 되살리는 방향입니다.
- 대부분의 약물이 아직 Preclinical ~ Phase II 단계(Table 1 참조)이며, 단독으로 완치 효과를 내는 경우는 거의 없습니다. 주로 PD-1/PD-L1 억제제(ICI), CAR-T, 화학요법과 병용으로 개발되고 있습니다.
현재 현실 (2026년 기준)
- 아데노신 경로(ADOAR2A, CD73), 락트산 수송(MCT1), 글루타민 분해(GLS), 트립토판 분해(IDO1) 등을 타겟으로 하는 약들이 임상에서 테스트 중입니다.
- 일부는 이미 병용 요법으로 효과를 보이고 있지만(예: Oleclumab + PD-L1 blocker, Telaglenastat + 표준치료), 아직 당뇨약처럼 표준 치료로 자리 잡은 것은 없습니다.
- 성공하면 암을 완치보다는 오래 조절하면서 생존 기간을 크게 연장하고, 삶의 질을 높이는 방향으로 갈 가능성이 큽니다. 일부 고형암(특히 면역 저항성 암)에서 “만성 관리형”으로 바뀔 수 있다는 기대가 있습니다.
비유를 더 정확히 하자면
“당뇨처럼 평생 먹는 약으로 암을 안정적으로 관리한다”는 말은 희망적인 미래 방향으로는 맞지만, 현재 단계에서는 “면역치료를 더 잘 듣게 만드는 보조 치료”에 가깝습니다.
결국 목표는 암을 완전 관해(complete response)로 유도하거나, 재발을 오래 지연시키면서 만성질환처럼 관리하는 것입니다. Table 1에 나오는 약들이 성공하면, 암 환자도 “특정 대사 경로를 평생 또는 장기간 조절하는 약”을 먹는 시대가 올 수 있어요.
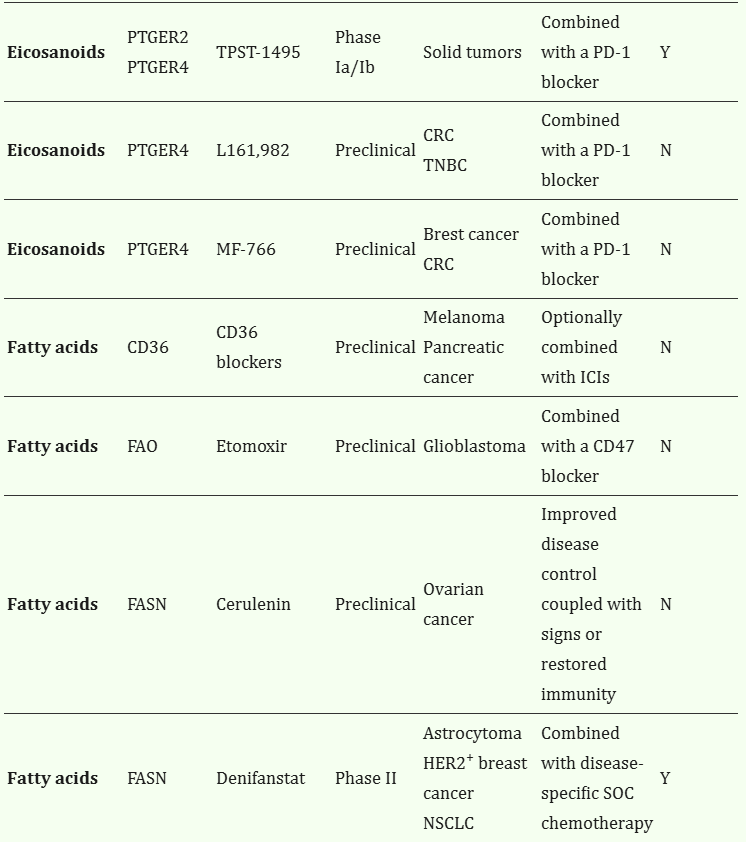
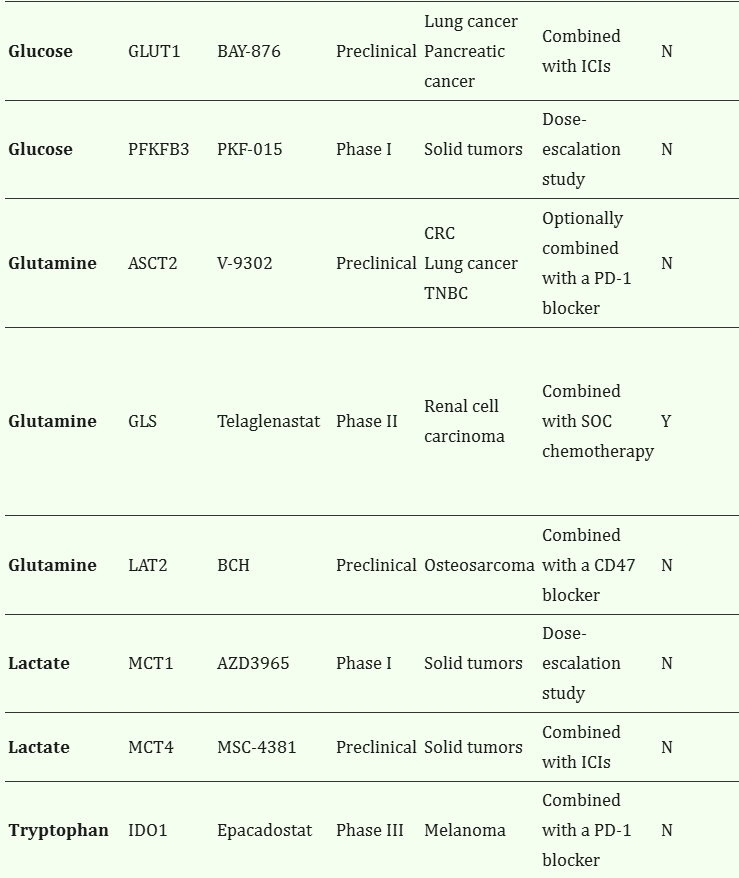
Table 1.
Metabolic inhibitors targeting cancer cells to restore immunosurveillance*
VulnerabilityTargetDrugPhase**Indication(s)NotesIn clinical development***Ref.
| Adenosine | Adenosine | PEG-ADA | Preclinical | Breast cancer Melanoma Lung cancer | Combined with ICIs | N | 84 |
| Adenosine | ADORA2A | AZD4635 | Phase Ia/Ib | Solid tumors | Combined with a PD-L1 blocker | Y | NCT02740985 149 |
| Adenosine | ADORA2A | SCH58261 | Preclinical | Breast cancer Melanoma | Combined with CAR T cells or ICIs | N | 145 147 |
| Adenosine | ADORA2A | Taminadenant | Phase I | NSCLC | Combined with a PD-1 blocker | Y | NCT02403193 148 |
| Adenosine | CD73 | Oleclumab | Phase II | NSCLC | Combined with a PD-L1 blocker | Y | NCT03794544 NCT03822351 150 151 |
| Eicosanoids | COX2 | Celecoxib | Phase II | CRC | Combined with a PD-1 blocker | Y | NCT03638297 NCT03926338 NCT05731726 |
| Eicosanoids | PTGER2 | PF-04418948 | Preclinical | CRC TNBC | Combined with a PD-1 blocker | N | 79 |
| Eicosanoids | PTGER2 PTGER4 | TPST-1495 | Phase Ia/Ib | Solid tumors | Combined with a PD-1 blocker | Y | NCT04344795 |
| Eicosanoids | PTGER4 | L161,982 | Preclinical | CRC TNBC | Combined with a PD-1 blocker | N | 79 |
| Eicosanoids | PTGER4 | MF-766 | Preclinical | Brest cancer CRC | Combined with a PD-1 blocker | N | 156 |
| Fatty acids | CD36 | CD36 blockers | Preclinical | Melanoma Pancreatic cancer | Optionally combined with ICIs | N | 74 154 |
| Fatty acids | FAO | Etomoxir | Preclinical | Glioblastoma | Combined with a CD47 blocker | N | 62 |
| Fatty acids | FASN | Cerulenin | Preclinical | Ovarian cancer | Improved disease control coupled with signs or restored immunity | N | 73 |
| Fatty acids | FASN | Denifanstat | Phase II | Astrocytoma HER2+ breast cancer NSCLC | Combined with disease-specific SOC chemotherapy | Y | NCT03032484 NCT03179904 NCT03808558 153 |
| Fatty acids | THBS1 | VT1021 | Phase II/III | Glioblastoma | Compared to SOC chemotherapy | Y | NCT03970447 |
| Glucose | GLUT1 | BAY-876 | Preclinical | Lung cancer Pancreatic cancer | Combined with ICIs | N | 25 |
| Glucose | PFKFB3 | PKF-015 | Phase I | Solid tumors | Dose-escalation study | N | NCT02044861 118 |
| Glutamine | ASCT2 | V-9302 | Preclinical | CRC Lung cancer TNBC | Optionally combined with a PD-1 blocker | N | 95 130 131 |
| Glutamine | GLS | Telaglenastat | Phase II | Renal cell carcinoma | Combined with SOC chemotherapy | Y | NCT03163667 NCT03428217 NCT03831932 127 128 129 |
| Glutamine | LAT2 | BCH | Preclinical | Osteosarcoma | Combined with a CD47 blocker | N | 99 |
| Lactate | MCT1 | AZD3965 | Phase I | Solid tumors | Dose-escalation study | N | NCT01791595 119 |
| Lactate | MCT4 | MSC-4381 | Preclinical | Solid tumors | Combined with ICIs | N | 120 |
| Tryptophan | IDO1 | Epacadostat | Phase III | Melanoma | Combined with a PD-1 blocker | N | NCT02752074 136 |
| Tryptophan | IDO1 | IU1 | Preclinical | CRC | Combined with a PD-1 blocker | N | 139 |
Abbreviations: BCH, 2-aminobicyclo-(2,2,1)-heptane-2-carboxylic acid; CRC, colorectal carcinoma; ICI, immune checkpoint inhibitor; NSCLC, non-small cell lung carcinoma; PEG-ADA, PEGylated adenosine deaminase; SOC, standard of care; TNBC, triple negative breast cancer.
*
Main examples;
**
most advanced;
***
as of February 24, according to www.clinicaltrials.gov.
Concluding remarks
The term “immunometabolism” has recently been coined to refer to the metabolic configuration of immune cells, which – perhaps not surprisingly – is very dynamic, critical for immune effector functions, and extremely sensitive to microenvironmental cues16–18. The abundance and function of tumor-infiltrating immune cells is also influenced by the metabolic alterations that accompany malignant transformation and tumor progression, as discussed herein. Importantly, while multiple clinically relevant agents have been shown to mediate immunostimulatory effects114–116, the potential contribution of altered cancer cell metabolism to immunostimulation has generally been overlooked.
An expanding preclinical literature points indeed to the possibility of targeting cancer cell metabolism to achieve immunostimulatory effects that can be maximized with various forms of immunotherapy, notably ICIs. The clinical translation of this paradigm, however, presents multiple challenges. First, most metabolic modulators developed so far exhibit limited (if any) specificity for cancer cells, implying that they may be toxic for healthy tissues and/or directly impair immune functions9. This calls for the development of strategies for the targeted delivery of metabolic inhibitors to malignant cells, such as drug-containing liposomes expressing one or more ligands for cancer cell receptors, or drug-associated nanoparticles with physicochemical features that promote their selective uptake by cancer cells163. Second, while modern omics technologies may be harnessed to investigate potentially actionable metabolic liabilities in diagnostic biopsies, several therapeutics commonly employed in clinical cancer management have major metabolic consequences, either by directly promoting a metabolic rewiring in malignant cells164, or indirectly by favoring the selection of neoplastic cells with specific metabolic traits165, often in the context of extensive intratumoral heterogeneity6. In this respect, it will be important to acquire as much information as possible on the metabolic changes imposed by conventional therapies and their immunomodulatory correlates from pre- and post-treatment biopsies (for instance by longitudinal monitoring of intratumoral metabolites in the setting of window-of-opportunity clinical trials). Third, the vast majority of current preclinical tumor models fail to recapitulate the metabolic and immunological heterogeneity of human neoplasms166. With all limitations that apply167, we surmise that humanized mice hosting non-dissociated patient-derived material and colonized with patient-derived hemopoietic precursors may at least partially circumvent such issue. Finally, a number of host-related factors have been shown to influence cancer cell metabolism and/or immune functions, including not only fairly obvious systemic conditions such as obesity and diabetes168, but also less recognizable variables such as the abundance and composition of the intratumoral and intestinal microbiome169. Additional work is needed to mechanistically decipher the intricate links between these factors, cancer cell metabolism and tumor-targeting immunity.
Despite these and other challenges, cancer cell metabolism stands out as a promising target to restore immunosurveillance and hence convert immunologically cold tumors into hot lesions that respond to immunotherapy.
Supplementary Material
1
NIHMS1989213-supplement-1.pdf (330.4KB, pdf)
Acknowledgements.
We apologize to the authors of several high-quality articles dealing with cancer cell metabolism its implications in anti-tumor immune responses that we were not able to discuss and cite owing to space limitations. MDM is supported by the Future Leaders 2023 Postdoctoral Fellowship from the Brain Tumor Charity (#BTC224874-01). JCR receives support related to this work from one R01 grant from the NIH/NCI (#CA217987). LG is/has been supported (as a PI unless otherwise indicated) by one R01 grant from the NIH/NCI (#CA271915), by two Breakthrough Level 2 grants from the US DoD BCRP (#BC180476P1, #BC210945), by a grant from the STARR Cancer Consortium (#I16-0064), by a Transformative Breast Cancer Consortium Grant from the US DoD BCRP (#W81XWH2120034, PI: Formenti), by a U54 grant from NIH/NCI (#CA274291, PI: Deasy, Formenti, Weichselbaum), by the 2019 Laura Ziskin Prize in Translational Research (#ZP-6177, PI: Formenti) from the Stand Up to Cancer (SU2C), by a Mantle Cell Lymphoma Research Initiative (MCL-RI, PI: Chen-Kiang) grant from the Leukemia and Lymphoma Society (LLS), by a Rapid Response Grant from the Functional Genomics Initiative (New York, US), by a pre-SPORE grant (PI: Demaria, Formenti) and a Clinical Trials Innovation Grant from the Sandra and Edward Meyer Cancer Center (New York, US); by startup funds from the Dept. of Radiation Oncology at Weill Cornell Medicine (New York, US), by industrial collaborations with Lytix Biopharma (Oslo, Norway), Promontory (New York, US) and Onxeo (Paris, France), as well as by donations from Promontory (New York, US), the Luke Heller TECPR2 Foundation (Boston, US), Sotio a.s. (Prague, Czech Republic), Lytix Biopharma (Oslo, Norway), Onxeo (Paris, France), Ricerchiamo (Brescia, Italy), and Noxopharm (Chatswood, Australia). CVB is supported from one R01 grant from the NIH/NINDS (#NS131945-01), one R21 grant from the NIH/NCI (#CA280787-01), one grant from the St. Baldrick’s Foundation Pray for Dominic Funds (#SBF222633-01) and one Uncle Kory Foundation seed grant.
GlossaryCatabolism
Set of metabolic pathways that breakdown large molecules into smaller units for recycling or ATP production purposes
Anabolism
Set of metabolic pathways that build large molecules from smaller units in support of cell growth and proliferation
OXPHOS
Mitochondrial pathway that generates ATP from a series of oxidation reactions that culminate with the generation of H2O
TCA cycle
Mitochondrial circuitry that ensure adequate levels of key metabolites involved in several catabolic and anabolic reactions
PPP
Metabolic shunt that diverts glycolytic intermediates towards the synthesis of nucleotides, some amino acids and antioxidants
Lactylation
Post-translational modification of lysine residues by lactate
De novo lipid biosynthesis
Metabolic cascade converting acetyl-CoA into long-chain lipids for cellular anabolism
Urea cycle
Metabolic pathway to convert excess ammonia into urea for excretion
Crotonylation
Post-translational modification of lysine residues by crotonyl-CoA
Footnotes
Competing Interests. JCR is a founder and scientific advisory board member of Sitryx Therapeutics. LG is/has been holding research contracts with Lytix Biopharma, Promontory and Onxeo, has received consulting/advisory honoraria from Boehringer Ingelheim, AstraZeneca, OmniSEQ, Onxeo, The Longevity Labs, Inzen, Imvax, Sotio, Promontory, Noxopharm, EduCom, and the Luke Heller TECPR2 Foundation, and holds Promontory stock options. The other authors have no conflicts of interest to declare.
References
- 1.Hanahan D Hallmarks of Cancer: New Dimensions. Cancer Discov 12, 31–46 (2022). [DOI] [PubMed] [Google Scholar]
- 2.Izzo LT, Affronti HC & Wellen KE The Bidirectional Relationship Between Cancer Epigenetics and Metabolism. Annu Rev Cancer Biol 5, 235–257 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pirozzi CJ & Yan H The implications of IDH mutations for cancer development and therapy. Nat Rev Clin Oncol 18, 645–661 (2021). [DOI] [PubMed] [Google Scholar]
- 4.Kerk SA, Papagiannakopoulos T, Shah YM & Lyssiotis CA Metabolic networks in mutant KRAS-driven tumours: tissue specificities and the microenvironment. Nat Rev Cancer 21, 510–525 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kruiswijk F, Labuschagne CF & Vousden KH p53 in survival, death and metabolic health: a lifeguard with a licence to kill. Nat Rev Mol Cell Biol 16, 393–405 (2015). [DOI] [PubMed] [Google Scholar]
- 6.Vitale I, Shema E, Loi S & Galluzzi L Intratumoral heterogeneity in cancer progression and response to immunotherapy. Nat Med 27, 212–224 (2021). [DOI] [PubMed] [Google Scholar]
- 7.Singleton DC, Macann A & Wilson WR Therapeutic targeting of the hypoxic tumour microenvironment. Nat Rev Clin Oncol 18, 751–772 (2021). [DOI] [PubMed] [Google Scholar]
- 8.Petroni G, Buqué A, Coussens LM & Galluzzi L Targeting oncogene and non-oncogene addiction to inflame the tumour microenvironment. Nat Rev Drug Discov 21, 440–462 (2022). [DOI] [PubMed] [Google Scholar]
- 9.Stine ZE, Schug ZT, Salvino JM & Dang CV Targeting cancer metabolism in the era of precision oncology. Nat Rev Drug Discov 21, 141–162 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Warburg O, Posener K & Negelein E Über den stoffwechsel der carcinomzelle. Naturwissenschaften 12, 1131–1137 (1924). [Google Scholar]
- 11.Debnath J, Gammoh N & Ryan KM Autophagy and autophagy-related pathways in cancer. Nat Rev Mol Cell Biol 24, 560–575 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim J & DeBerardinis RJ Mechanisms and Implications of Metabolic Heterogeneity in Cancer. Cell Metab 30, 434–446 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kroemer G, Chan TA, Eggermont AMM & Galluzzi L Immunosurveillance in clinical cancer management. CA Cancer J Clin (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Klapp V et al. The DNA Damage Response and Inflammation in Cancer. Cancer Discov 13, 1521–1545 (2023). [DOI] [PubMed] [Google Scholar]
- 15.Kroemer G, Galassi C, Zitvogel L & Galluzzi L Immunogenic cell stress and death. Nat Immunol 23, 487–500 (2022). [DOI] [PubMed] [Google Scholar]
- 16.Voss K et al. A guide to interrogating immunometabolism. Nat Rev Immunol 21, 637–652 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bantug GR & Hess C The immunometabolic ecosystem in cancer. Nat Immunol 24, 2008–2020 (2023). [DOI] [PubMed] [Google Scholar]
- 18.Leone RD & Powell JD Metabolism of immune cells in cancer. Nat Rev Cancer 20, 516–531 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lunt SY & Vander Heiden MG Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu Rev Cell Dev Biol 27, 441–464 (2011). [DOI] [PubMed] [Google Scholar]
- 20.Chang CH et al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell 162, 1229–1241 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Reinfeld BI et al. Cell-programmed nutrient partitioning in the tumour microenvironment. Nature 593, 282–288 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]; This article elegantly demonstrates that intratumoral myeloid cells exhibit superior glucose (but inferior glutamine) uptake as compared to malignant cells.
- 22.Cascone T et al. Increased Tumor Glycolysis Characterizes Immune Resistance to Adoptive T Cell Therapy. Cell Metab 27, 977–987.e974 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guo D et al. Aerobic glycolysis promotes tumor immune evasion by hexokinase2-mediated phosphorylation of IκBα. Cell Metab 34, 1312–1324.e1316 (2022). [DOI] [PubMed] [Google Scholar]
- 24.Li W et al. Aerobic Glycolysis Controls Myeloid-Derived Suppressor Cells and Tumor Immunity via a Specific CEBPB Isoform in Triple-Negative Breast Cancer. Cell Metab 28, 87–103.e106 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wu L et al. Tumor aerobic glycolysis confers immune evasion through modulating sensitivity to T cell-mediated bystander killing via TNF-α. Cell Metab 35, 1580–1596.e1589 (2023). [DOI] [PubMed] [Google Scholar]
- 26.Galluzzi L, Kepp O, Vander Heiden MG & Kroemer G Metabolic targets for cancer therapy. Nat Rev Drug Discov 12, 829–846 (2013). [DOI] [PubMed] [Google Scholar]
- 27.Claps G et al. The multiple roles of LDH in cancer. Nat Rev Clin Oncol 19, 749–762 (2022). [DOI] [PubMed] [Google Scholar]
- 28.Elia I et al. Tumor cells dictate anti-tumor immune responses by altering pyruvate utilization and succinate signaling in CD8(+) T cells. Cell Metab 34, 1137–1150.e1136 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Quinn WJ 3rd et al. Lactate Limits T Cell Proliferation via the NAD(H) Redox State. Cell Rep 33, 108500 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ma J et al. Lithium carbonate revitalizes tumor-reactive CD8(+) T cells by shunting lactic acid into mitochondria. Nat Immunol (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brand A et al. LDHA-Associated Lactic Acid Production Blunts Tumor Immunosurveillance by T and NK Cells. Cell Metab 24, 657–671 (2016). [DOI] [PubMed] [Google Scholar]
- 32.Oshima N et al. Dynamic Imaging of LDH Inhibition in Tumors Reveals Rapid In Vivo Metabolic Rewiring and Vulnerability to Combination Therapy. Cell Rep 30, 1798–1810 e1794 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rundqvist H et al. Cytotoxic T-cells mediate exercise-induced reductions in tumor growth. Elife 9 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Feng Q et al. Lactate increases stemness of CD8 + T cells to augment anti-tumor immunity. Nat Commun 13, 4981 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]; This report shows that lactate may also mediate immunostimulatory effects by promoting CD8+ T cell stemness.
- 35.Angelin A et al. Foxp3 Reprograms T Cell Metabolism to Function in Low-Glucose, High-Lactate Environments. Cell Metab 25, 1282–1293.e1287 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kumagai S et al. Lactic acid promotes PD-1 expression in regulatory T cells in highly glycolytic tumor microenvironments. Cancer Cell 40, 201–218.e209 (2022). [DOI] [PubMed] [Google Scholar]
- 37.Watson MJ et al. Metabolic support of tumour-infiltrating regulatory T cells by lactic acid. Nature 591, 645–651 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gu J et al. Tumor metabolite lactate promotes tumorigenesis by modulating MOESIN lactylation and enhancing TGF-β signaling in regulatory T cells. Cell Rep 39, 110986 (2022). [DOI] [PubMed] [Google Scholar]
- 39.Xiong J et al. Lactylation-driven METTL3-mediated RNA m(6)A modification promotes immunosuppression of tumor-infiltrating myeloid cells. Mol Cell 82, 1660–1677 e1610 (2022). [DOI] [PubMed] [Google Scholar]