BEYOND REASON
Ferroptosis의 개념
Iron and cancer: recent insights
Abstract
Iron is an essential dietary element. However, the ability of iron to cycle between oxidized and reduced forms also renders it capable of contributing to free radical formation, which can have deleterious effects, including promutagenic effects that can potentiate tumor formation. Dysregulation of iron metabolism can increase cancer risk and promote tumor growth. Cancer cells exhibit an enhanced dependence on iron relative to their normal counterparts, a phenomenon we have termed iron addiction. Work conducted in the past few years has revealed new cellular processes and mechanisms that deepen our understanding of the link between iron and cancer. Control of iron efflux through the combined action of ferroportin, an iron efflux pump, and its regulator hepcidin appears to play an important role in tumorigenesis. Ferroptosis is a form of iron-dependent cell death involving the production of reactive oxygen species. Specific mechanisms involved in ferroptosis, including depletion of glutathione and inhibition of glutathione peroxidase 4, have been uncovered. Ferritinophagy is a newly identified mechanism for degradation of the iron storage protein ferritin. Perturbations of mechanisms that control transcripts encoding proteins that regulate iron have been observed in cancer cells, including differences in miRNA, methylation, and acetylation. These new insights may ultimately provide new therapeutic opportunities for treating cancer.
Introduction
Iron is an essential nutrient. However, the ability of iron to cycle between oxidized and reduced forms enables it to contribute to the formation of potentially damaging free radical species, including reactive oxygen species (ROS). Iron is therefore very carefully regulated at both a systemic and cellular level (see recent reviews and Fig. 1).1,2 Multiple experimental and epidemiological studies have associated disruption of iron regulatory pathways and attendant accumulation of excess iron with mutagenesis and enhanced tumor growth (see recent reviews3–6). Cancer cells exhibit an enhanced dependence on iron for growth and are dramatically more susceptible to iron depletion than noncancer cells, a phenomenon we have termed iron addiction. Nevertheless, despite extensive study, specific links between iron and cancer remain incompletely defined. This review briefly highlights recent insights into novel processes and mechanisms through which iron may become dysregulated and potentially contribute to tumor growth. Although not yet fully articulated, the concepts described below provide new perspectives on mechanistic links between iron and cancer that may ultimately lead to new approaches to cancer prognosis and therapy.

Overview of iron absorption and metabolism. Enterocytes absorb dietary iron through the combined action of ferric reductases, such as Dcytb and divalent metal transporter DMT1. Ferrous iron taken up by enterocytes is exported into the circulation by ferroportin (FPN-1). Concurrently, the Fe2+ is oxidized to Fe3+ by hephaestin (HEPH), which is functionally associated with FPN-1. Fe3+ is loaded on to circulating apo-TF in plasma. Cells take up diferric TF through the cell surface transferrin receptor (TFR1). The TFR1–TF–(Fe+3)2 complex is endocytosed, and iron is released from TF. Fe3+ is reduced by the ferric reductase STEAP3, and Fe2+ is then transported to the cytosol by DMT1, where it enters the cytosolic labile iron pool (LIP). The LIP is utilized by cells for various metabolic needs. Excess iron is stored in ferritin or effluxed to the circulation through FPN-1. Hepatocytes synthesize hepcidin (HAMP) in response to systemic iron levels; the binding of hepcidin to FPN-1 triggers FPN-1 degradation, thus inhibiting iron efflux.
Brief overview of iron metabolism
Systemic iron balance is maintained by careful control of iron intake and recycling. Both heme and non-heme dietary iron are largely present in the oxidized state (Fe3+). For non-heme sources of dietary iron, Fe3+ is reduced to Fe2+ in the intestinal lumen by ferric reductases, such as duodenal cytochrome b reductase (Dcytb), and transported into enterocytes by divalent metal transporter 1 (DMT1)2,7,8 (Fig. 1). Heme iron is also taken up by enterocytes through an as-yet unknown mechanism and metabolized by heme oxygenase 1 (HO-1) to liberate Fe2+. The iron absorbed through enterocytes is exported across the basolateral membrane into the circulation by ferroportin (FPN-1), a highly conserved multitransmembrane protein and the only known iron efflux protein in mammals.9 Fe2+ exported through FPN-1 is re-oxidized at the basolateral surface of the enterocyte by the membrane-bound multicopper oxidase hephaestin (HEPH) before loading onto transferrin (TF), a protein that circulates in the plasma and delivers iron to peripheral tissues.10 Holotransferrin (transferrin bound to two atoms of iron) delivers iron to cells by binding to transferrin receptor (TFR1), which is expressed on the surface of most cells, and the TFR1/TF-(Fe3+)2 complex subsequently undergoes endocytosis. In the endosome, Fe3+ is reduced by the ferric reductase STEAP3 to Fe2+ and transported across the endosomal membrane by DMT1. Imported iron enters a metabolically available, transient cytosolic labile iron pool (LIP). Cells use this iron pool for (1) incorporation into prosthetic groups of iron-dependent enzymes and proteins, (2) incorporation into heme (after transport across the mitochondrial membrane) and iron–sulfur cluster biogenesis, and (3) storage in ferritin. Excess iron is exported back to the circulation by FPN-1.4 Iron taken up through these processes is efficiently recycled in macrophages, which engulf senescent red blood cells, extract iron from hemoglobin, and deliver iron back into the circulation by FPN-1–mediated efflux.11 Both intestinal iron uptake and iron recycling in the reticuloendothelial system are negatively regulated by the systemic peptide hormone hepcidin, which binds to FPN-1, promoting its phosphorylation and subsequent lysosomal degradation.12
Like systemic iron, cellular iron homeostasis is also tightly regulated. In this case, regulation is achieved by a network of iron-dependent proteins. Iron-regulatory proteins (IRP1 and IRP2) are the principal components of this iron-regulatory network 13. IRPs are cytosolic proteins that bind to iron-responsive elements (IREs), stem-loop structures found in either the 5′ or 3′ untranslated regions of mRNAs for proteins involved in iron import (TFR1, DMT1), storage (ferritin (both FTH1 and FTL subunits)) and export (FPN-1).14 Under conditions of iron deficiency, IRPs bind to 5′ IREs present in both ferritin and FPN-1 mRNAs to repress their translation, and to 3′ IREs in TfR1 mRNA to prevent its degradation. Excess cytosolic iron destabilizes IRP1 and IRP2, preventing them from binding to IREs, resulting in increased synthesis of ferritin and FPN-1 and enhanced degradation of TfR1 mRNA. By controlling the import, storage, and efflux of iron, IRPs ensure that metabolic needs for iron are met while minimizing the toxic effects of excess iron.
Recently discovered intracellular processes involving iron and their relation to cancer
The reader is referred to reviews that provide a more global overview of the literature on dysregulation of iron in cancer cells and tissues.4–6 In this review, we restrict our focus to a limited number of recent findings that have begun to suggest novel processes and mechanisms through which iron may be dysregulated in cancer (Fig. 2).

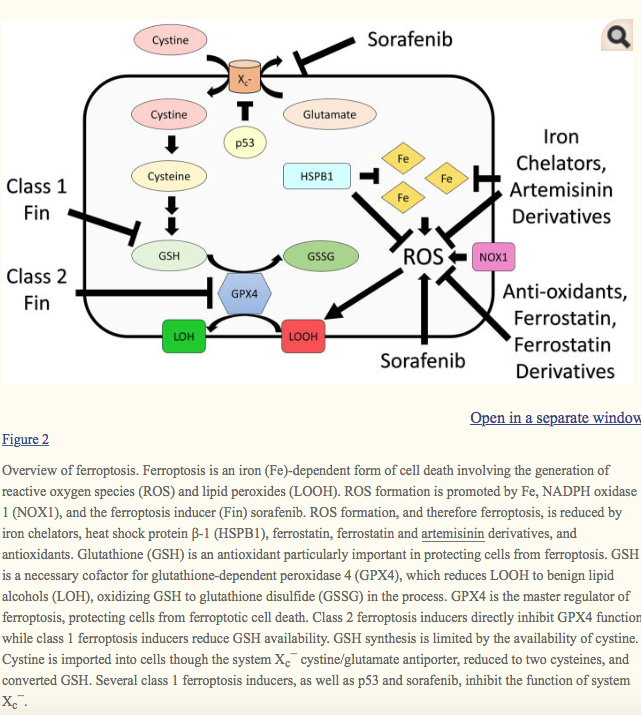
Overview of ferroptosis. Ferroptosis is an iron (Fe)-dependent form of cell death involving the generation of reactive oxygen species (ROS) and lipid peroxides (LOOH). ROS formation is promoted by Fe, NADPH oxidase 1 (NOX1), and the ferroptosis inducer (Fin) sorafenib. ROS formation, and therefore ferroptosis, is reduced by iron chelators, heat shock protein β-1 (HSPB1), ferrostatin, ferrostatin and artemisinin derivatives, and antioxidants. Glutathione (GSH) is an antioxidant particularly important in protecting cells from ferroptosis. GSH is a necessary cofactor for glutathione-dependent peroxidase 4 (GPX4), which reduces LOOH to benign lipid alcohols (LOH), oxidizing GSH to glutathione disulfide (GSSG) in the process. GPX4 is the master regulator of ferroptosis, protecting cells from ferroptotic cell death. Class 2 ferroptosis inducers directly inhibit GPX4 function while class 1 ferroptosis inducers reduce GSH availability. GSH synthesis is limited by the availability of cystine. Cystine is imported into cells though the system Xc− cystine/glutamate antiporter, reduced to two cysteines, and converted GSH. Several class 1 ferroptosis inducers, as well as p53 and sorafenib, inhibit the function of system Xc−.
Iron uptake and storage
Transferrin receptor 1 (TFRC1), a transmembrane glycoprotein homodimer, binds diferric transferrin and is essential for iron uptake and the regulation of cell growth. Many studies have shown that cancer cells have increased expression of transferrin receptor 1 when compared to their normal counterparts.15,16 This observation has led to the exploration of TFRC1-targeted therapeutic approaches that target TRFC1 either directly, using antibodies, or indirectly, using transferrin conjugates that bind to TFRC1 to achieve tumor-specific drug delivery.15,16 High levels of TFRC1 expression in cancers such as breast,17 renal cell,18 and nonsmall cell lung cancer19 are correlated with poorer patient prognosis. The expression of TFRC1 is regulated at the posttranscriptional level by IRPs in response to intracellular iron levels.20 TFRC1 is also a direct transcriptional target of the oncoprotein c-Myc.21 Recently it was reported that cancer stem-like cells (CSCs) in glioblastoma express increased levels of TFRC1 and its ligand transferrin compared to non-CSCs.22 Iron-tracing experiments on these CSCs showed increased iron uptake in CSCs compared with non-CSCs, indicating that enhanced iron uptake may also be a property of cancer stem-like cells.
In addition to increasing iron uptake, cancer cells can increase intracellular iron by modulating levels of the iron-storage protein ferritin. Ferritin sequesters excess iron for storage, allowing iron to accumulate intracellularly while simultaneously preventing iron-mediated free radical–generating reactions. Ferritin is overexpressed in many cancer tissues, including breast,23–25 pancreatic,25 and hepatocellular cancers,26Hodgkin’s lymphoma,27 and glioblastoma multiforme.22 However, the specific cell types within the tumor that overexpress ferritin have not always been examined. In tumors of the breast, there is strong ferritin expression in stromal cells within tumor tissue.28 Further analysis revealed that the ferritin-rich stromal cells are CD68+ macrophages that infiltrate into tumors and can secrete ferritin.29 These tumor-associated macrophages may release ferritin locally. Interestingly, locally secreted ferritin appeared to contribute to tumor cell proliferation through an iron-independent mechanism.29 An iron-independent function of ferritin in the induction of inflammatory pathways has been previously suggested.30 In addition to stromal cells, ferritin is increased in glioblastoma CSCs compared to non-CSCs and the non-neoplastic brain, similar to the results for TFRC1 described above.22 Targeting both the H and L subunits of ferritin with small hairpin RNA (shRNA) resulted in dramatic reduction in CSC growth both in vitro and in vivo, suggesting that storage of iron is crucial for tumor growth.22
Iron efflux
Iron efflux mediated by FPN-1 and controlled by the peptide hormone hepcidin plays an important role in tumor growth and metastasis. Many cancer types exhibit an altered regulation of FPN-1. FPN-1 is significantly diminished in breast, prostate, and hepatocellular cancer cells as compared to the normal counterparts.31–34. In breast cancer, diminished FPN-1 is associated with reduced metastasis-free and disease-specific survival and is predictive of patient outcome. Low levels of FPN-1 are significantly correlated with poor prognostic markers, such as absence of estrogen receptors, high histological grade, and lymph node metastasis.32 Studies from several groups have shown that forced expression of FPN-1 in breast cancer cells inhibits growth of cancer xenografts in mice.32,35 Further, FPN-1 overexpression in breast cancer cells impaired metastasis to lung and liver in mice and significantly decreased the levels of epithelial mesenchymal transition (EMT) markers, such as Snail1, Twist1, ZEB2, and Vimentin.35
The expression of FPN-1 is regulated at transcriptional, posttranscriptional, and posttranslational levels.11In macrophages, FPN-1 is transcriptionally regulated by heme, which increases FPN-1 transcription by modulating Bach1.36 Bach1 binds to the antioxidant response element (ARE) present in the promoter sequence of FPN-1 and represses its transcription.36 Heme causes the degradation of Bach1, leading to the derepression of transcription. Another factor that affects FPN-1 transcription is hypoxia; activation of hypoxia-inducible factor (HIF) in enterocytes upregulates FPN-1 expression.37 In breast cancer cells, transcription of FPN-1 is activated by the transcription factors Nrf2 (Nuclear factor erythroid 2–like 2)38and MZF1 (myeloid zinc finger-1).31 Posttranscriptionally, FPN-1 is regulated by IRPs. Most cell types predominantly express FPN-1 isoform 1a mRNA, which contains a 5′ IRE. Under conditions of iron deficiency, IRPs bind to a 5′ IRE, repressing FPN-1 translation and decreasing expression of FPN-1 on the cell membrane.39 However, IRP regulation of FPN-1 is finely tuned to meet cellular and organismal needs. In enterocytes, depletion of FPN-1 by iron deficiency would reduce uptake from the diet and be detrimental to an organism that was iron deficient. Hence, enterocytes express FPN-1 isoform 1b mRNA, which lacks the 5′ IRE. FPN-1 is also regulated posttranslationally by the peptide hormone hepcidin, which binds to FPN-1 resulting in FPN-1 internalization and proteolysis in lysosomes.40 Mechanisms underlying the reduction in FPN-1 expression in cancer cells are incompletely understood. However, aberrant DNA methylation of the FPN-1 gene has been reported to contribute to the downregulation of FPN-1 in breast tumors.31
Hepcidin–ferroportin axis in cancer
Hepcidin, the negative regulator of FPN-1, is also deregulated in neoplastic disease.32,33,40,41 Serum hepcidin is elevated in patients with many cancers, including prostate cancer,42 breast cancer,43 multiple myeloma,44 non-Hodgkin’s lymphoma,45 hepatocellular carcinoma (HCC),46 ovarian cancer, and upper gastrointestinal tract tumor.47 In addition to systemic hepcidin, cancer cells also synthesize hepcidin. For example, breast, colorectal, and prostate cancer cells synthesize higher amounts of hepcidin than their non-malignant counterparts.32,33,41
Hepcidin is regulated by a number of signaling pathways, with major roles played by the bone morphogenetic protein (BMP) and interleukin-6 (IL-6) inflammatory pathways (see Refs. 12 and 48–50 for recent reviews). Dysregulation of these pathways in malignant cells33,51–53 likely contributes to the dysregulation of hepcidin in cancer.33,34,54 In particular, BMP, IL-6, and Wnt signaling all contribute to enhanced hepcidin production in prostate cancer.33 BMP signaling through SMADs (vertebrate homologs of Sma and mothers against decapentaplegic) and IL-6 signaling through STAT3 (signal transducers and activators of transcription 3) have been further implicated in breast cancer.34,54
Dysregulation of hepcidin in cancer results in altered iron homeostasis. High plasma hepcidin blocks the mobilization of iron into the circulation from enterocytes and macrophages (contributing to the anemia seen in cancer) and can also cause the accumulation of iron in tumor cells by degrading FPN-1, resulting in the activation of signaling pathways such as Wnt55 and NF-κB,56 which contribute to tumor progression.57The tumor-promoting role of circulating hepcidin was demonstrated using hepcidin (Hamp−/−) knockout mice. Hamp−/− mice implanted with Lewis lung cancer cells (LL/2 cells) displayed improved survival and fewer metastases than wild-type mice.35 In addition to systemic hepcidin, cancer cells also synthesize hepcidin. For example, breast, colorectal, and prostate cancer cells synthesize higher amounts of hepcidin than their nonmalignant counterparts.32,33,41. Hepcidin generated by cancer cells initiates a local autocrine signaling to degrade membrane FPN-1.32,33 High hepcidin expression in tumors has been associated with metastatic progression and poor prognosis.32,58 Accumulating evidence suggests that the pro-oncogenic nature of hepcidin may be due to its ability to increase intracellular iron content in tumor cells by initiating internalization and degradation of FPN-1.
Ferroptosis
Ferroptosis, named for its dependence on iron,59,60 is a recently characterized form of non-apoptotic cell death (Fig. 2). Ferroptosis was first identified in a high-throughput screen of small molecules that selectively kill cells overexpressing oncogenic HRAS,61 although subsequent work has shown that ferroptosis is not limited to cells with activated HRAS.60,62 In this study, the small molecule erastin induced cell death with distinct morphological and mechanistic characteristics,61 differentiating ferroptosis from other forms of cell death.25 Ferroptosis is distinguished from apoptosis by the lack of chromatin margination, nuclear fragmentation, plasma membrane blebbing, and caspase activation.60,61,63Ferroptosis is differentiated from autophagy owing to a lack of autophagosome formation and changes in microtubule-associated protein light chain 3 (LC3).61,64,65 Ferroptosis is also distinct from classical necrosis as there is no rapid depletion of ATP.60 Furthermore, ferroptosis was not repressed by known inhibitors of apoptosis, autophagy, or necrosis.60 Instead, ferroptosis is morphologically characterized by reduced mitochondrial size with increased membrane density.60,63 Mechanistically, ferroptosis causes cell death through iron-mediated accumulation of lipid ROS.59,60,66 Accumulation of lipid ROS interferes with cellular integrity and membrane fluidity and permeability, effects which can be lethal.67
Iron, which plays a role in several types of cell death,66 has long been known to participate in the generation of ROS through the Fenton reaction.4 It is likely that iron-catalyzed ROS production is primarily responsible for ferroptotic death, although iron may also function as a cofactor for enzymes involved in ROS production.66 Indeed, the sensitivity of cells to ferroptosis can be modulated by cellular iron status (Fig. 2). Increasing intracellular iron by iron treatment or through transferrin and transferrin receptor promotes ferroptosis.60,68 Heme oxygenase-1, which promotes iron availability69 but also functions as an antioxidant,70 has an unclear effect on ferroptosis. Conversely, reducing intracellular iron through iron chelation reduces ferroptosis.59 Heat shock protein β-1 (HSPB1) protects cancer cells from ferroptosis through reduction of intracellular iron and lipid ROS.71. In vivo, HSPB1 knockdown enhanced the antitumor activity of the ferroptosis inducer erastin.71 Iron loading of cancer cells, such as through aminoferrocence-based therapies,72,73 may prove useful therapeutically by enhancing ferroptosis in cancer cells.
In addition to iron status, other sources of ROS seem to play roles in ferroptosis. In some cells, ROS are derived from nicotinamide adenine dinucleotide phosphate (NADPH) generated by NADPH oxidase 1 (NOX1).60 NAPDH is produced by several cellular reactions, but the pentose phosphate pathway is particularly important for ferroptosis in RAS-mutated cancer cells.60,74 However, ROS generated from the mitochondrial electron transport chain, which was originally found to not contribute to ferroptosis,60,65may contribute under certain circumstances.75
The importance of ROS in ferroptosis led to the study of antioxidant pathways as potential regulators of ferroptosis. Glutathione (GSH) is a potent antioxidant76 whose production is limited by the availability of cystine/cysteine.77 Erastin reduced GSH production through inhibiting cystine uptake by system Xc−.60Recently, p53, a well-characterized tumor suppressor,78 was also shown to sensitize cells to ferroptosis. Similar to erastin, p53 reduces cystine uptake and GSH by repression of the SLC7A11 component of system Xc−.79,80 In contrast, increases in cysteine and GSH following knockdown of cysteinyl-tRNA synthetase (CARS) protected cancer cells from erastin-induced ferroptosis.81 Furthermore, GSH is a cofactor essential for glutathione-dependent peroxidase (GPX) function.82 GPX4, which normally reduces lipid peroxides formed by ROS,82 is particularly important in ferroptosis.83 GPX4 was identified in a screening for target proteins of a known ferroptosis inducer, RSL3.83 GPX4 overexpression rescued ferroptotic cell death induced by a panel of 12 structurally diverse ferroptosis inducers, indicating a central role for GPX4 in regulation of ferroptosis83 (Fig. 2). In addition to its role in cancer, modulation of ferroptosis by GPX4 seems to play an important role in ischemia/reperfusion-induced tissue damage62, 84as well as motor neuron survival.31
Several modulators of ferroptosis have been identified through the study of ferroptotic mechanisms (Fig. 2). Most ferroptosis inducers can be classified based on their mechanism of action83 (Table 1). Class I ferroptosis inducers indirectly reduce GPX4 function by depleting GSH. Class I ferroptosis inducers include erastin, erastin derivatives (aldehyde erastin, morpholine erastin II, and piperazine erastin), buthionine sulfoximine, DPI2, glutamate, lanperisone, sulfasalazine, SRS13-45, SRS13-60, and sorafenib.60,70,83,85,86 Class 2 ferroptosis inducers directly inhibit GPX4 function and include DPI7, DPI10, DPI12, DPI13, DPI17, DPI18, DPI19, ML162, and RSL3.59,83,87 Ferroptosis inducers that function by mechanisms not involving direct depletion of GSH or inhibition of GPX have also been identified. Sorafenib, a small molecule kinase inhibitor that is approved by the U.S. Food and Drug Administration for treatment of renal cell, hepatocellular, and differentiated thyroid carcinomas was also found to induce ferroptotic cell death by inducing ROS.75,88 Similarly, artemisinin derivatives, including artesunate, induce ferroptosis through iron-mediated ROS generation.89,90
Table 1
Ferroptosis modulators
| Type | Class | Name | References |
|---|---|---|---|
| Ferroptosis inducers | Class 1: Reduce glutathione | Erastin | 60, 70 |
| Aldehyde erastin | 83 | ||
| Morpholine erastin II | |||
| Piperazine erastin | |||
| DPI2 | |||
| Lanperisone | 85 | ||
| Sulfasalazine | 60 | ||
| Buthionine sulfoximine | 70, 83 | ||
| SRS13-45 | 86 | ||
| SRS13-60 | 86 | ||
| Inhibits system Xc− and induces reactive oxygen species (ROS) | Sorafenib | 70, 75, 86, 88 | |
| Iron-induced ROS | Artemisinin derivatives | 89, 90 | |
| Class 2: Inhibit glutathione-dependent peroxidase 4 (GPX4) | DPI7 | 83 | |
| DPI10 | 83, 87 | ||
| DPI12 | 83 | ||
| DPI13 | |||
| DPI17 | |||
| DPI18 | |||
| DPI19 | |||
| ML162 | 87 | ||
| RSL3 | 59 | ||
| Ferroptosis inhibitors | Antioxidant | Vitamin E | 83 |
| Trolox | 60 | ||
| U0126 | 60, 83 | ||
| Inhibit ROS formation | Ferrostatin-1 | 60 | |
| SRS8-72 | |||
| SRS11-92 | 91 | ||
| SRS12-45 | |||
| SRS13-35 | |||
| SRS13-37 | |||
| SRS16-86 | 62 | ||
| Iron chelators | Desferoxamine, | 60 | |
| 2,2-bipyridyl | |||
| Ciclopirox olamine | |||
| Protein synthesis inhibitor | Cycloheximide | 60 | |
| Transaminase inhibitor | Aminooxyacetic acid | 60 | |
| GPX mimetic | Ebselen | 60, 92 | |
| Promotes cystine uptake | β-mercaptoethanol | 60, 86 |
Ferroptosis inhibitors have also been identified (Table 1). While ferroptosis inducers are envisioned as cancer therapeutics, inhibitors of ferroptosis may prove useful in minimizing ischemia/reperfusion-induced tissue damage,84 or perhaps in protecting normal cells from being targeted by ferroptosis inducers. Ferroptosis inhibitors exhibit varied mechanisms of action, all primarily involving a reduction of ROS. Antioxidants (vitamin E, Trolox, U0126)60, 83 and inhibitors of ROS production, such as ferrostatin-1 and its more potent derivatives (SRS8-72, SRS11-92, SRS12-45, SRS13-35, SRS13-37, and SRS16-86),60,62,91 also show antiferroptotic function. β-Mercaptoethanol inhibits ferroptosis by increasing cystine uptake.60,86 The iron chelators desferoxamine, 2,2-bipyridyl, and ciclopiroxolamine are able to inhibit ferroptosis by reducing iron and ROS.60 Cycloheximide, aminooxyacetic acid, and ebselen––a protein synthesis inhibitor, a transaminase inhibitor, and a GPX mimetic, respectively––also inhibit ferroptosis.60,92 These agents may provide leads to clinically relevant therapeutics.
Ferritinophagy
Autophagy is a lysosomal-mediated cellular self-digestion process that is upregulated in response to stress stimuli. Autophagy involves formation of double-membrane autophagosomes, which engulf cellular components and deliver them to lysosomes for degradation.93 Autophagy helps in maintaining normal cellular homeostasis by acting as a recycling mechanism: cells degrade damaged organelles and protein aggregates by autophagy and use the products to refuel supplies of amino acids, lipids, nutrients, and metals.94 Recent evidence suggests that selective autophagy is critical for iron homeostasis.95,96 The iron-storage protein ferritin plays a key role in this process. Ferritin is a 24-subunit protein comprised of ferritin light and heavy chains. Ferritin is able to store up to 4500 iron atoms.97 Under conditions of low intracellular iron, ferritin is degraded to release iron for use by the cell. This degradation of ferritin occurs in lysosomes through a targeted autophagic process referred as ferritinophagy.95,96 Nuclear receptor coactivator 4 (NCOA4) was recently identified as an important protein in ferritinophagy. NCOA4 binds to ferritin and delivers it to lysosomes for degradation. Ncoa4−/− mice exhibited significant accumulation of iron in spleen, indicating the disruption of iron storage and recycling.95 The role of this process in cancer has not been studied; however, we speculate that activation of this pathway may represent an additional source of iron in malignant cells.
New mechanisms for control of iron
In addition to the well-established mechanisms that regulate changes in iron metabolism in cancer,4,98novel regulatory mechanisms that may contribute cancer-specific alterations in iron metabolism have recently been identified.
MicroRNAs
MicroRNAs (miRNAs) are a class of small, noncoding RNAs that function to regulate gene expression posttranscriptionally by sequence-specific binding to target mRNAs, resulting in their degradation.99 Since deregulation of miRNAs is implicated in cancer, alterations of miRNAs that control iron metabolism may represent a novel mechanism through which cancer cells alter their iron phenotype.100
One study designed specifically to identify miRNAs that posttranscriptionally regulate FPN-1 identified 44 miRNAs that were differentially expressed after exogenous addition of iron by ferric ammonium citrate (FAC) or chelation of iron by deferoxamine (DFO).101 After analyzing the predicted targets of these miRNAs, two miRNAs, miR-485-3p and miR-194, were predicted to target the 3′ UTR of FPN-1. Of these two, miR-485-3p was the most induced during cellular iron deficiency across cell types. The functional significance of FPN-1 targeting by miR-485-3p was demonstrated by an increase in ferritin, a protein that is translationally induced by intracellular iron and is frequently used as an indirect indicator of high intracellular iron. Presumably, the miR-485-3p–mediated decrease in FPN-1–dependent iron efflux was the cause of increased ferritin. Thus, miR-485-3p may act as a mechanism to restore iron homeostasis in cells challenged by iron deficiency. Reduced FPN-1 is also observed in cancer cells, as tumor cells require increased iron to support their proliferative demands.32,33 It is plausible that this may in part be due to increased levels of miR-485-3p. It will be interesting to determine if miR-485-3p is upregulated in cancer cells, and if that contributes to the basal reduction of FPN-1 present in tumors.
In addition to controlling iron export, miRNAs also regulate proteins that control iron import. TFR1 was shown to be a direct target of miRNA-210: overexpression of miRNA-210 inhibited TFR1 expression and decreased uptake of transferrin-bound iron and proliferation.102 In addition, TFR1 was identified as a target of miRNA-152 in a computational analysis of HCC.103 Using the TCGA database, an inverse correlation was identified between TFR1 and miRNA-152, where miRNA-152 levels were reduced and TFR1 was overexpressed in human HCC tissue compared to non-tumor liver tissue.103 This inverse relationship was confirmed experimentally, as transfection of HCC with miRNA-152 resulted in suppression of TFR1, identifying a novel means to target TFR1 and restore normal iron levels in cancer cells.103 MicroRNAs also control expression of iron-storage proteins, as miR-200b was shown to target the iron-storage protein ferritin heavy chain (FTH).104 This study revealed that FTH is upregulated in the aggressive, triple-negative breast cancer cell line MDA-MB-231.104 The increase of FTH corresponds to decreased levels of miR-200b, and reintroduction of miR-200b experimentally resulted in decreased FTH expression, suggesting that FTH is a direct target of miR-200b.104 Interestingly, the reduction in FTH by miR-200b was associated with increased sensitivity to doxorubicin chemotherapy treatment, suggesting that reducing iron storage may sensitize cancer cells to apoptosis.104 Other miRNAs that may prove useful in targeting the disrupted iron metabolism observed in cancer cells are displayed in Table 2.
Table 2
Micro RNAs suggested to play a role in regulation of iron homeostasis.
| miRNA | Iron-related target mRNA | Evidence | Reference |
|---|---|---|---|
| miR-Let-7d | DMT1-IRE, BACH1 | Cell culture | 105, 106 |
| miR-31 | TFRC | Correlative | 107 |
| miR-122 | HFE, HJV | Animal models | 108 |
| miR-152 | TFRC | Cell culture | 103 |
| miR-194 | FPN1 | Correlative | 107 |
| miR-196 | BACH1 | Cell culture | 109 |
| miR-200b | FTH | Cell culture | 104 |
| miR-210 | ISCU/TFR/FECH | Cell culture | 102, 110, 111 |
| miR-214 | Lactoferrin | Cell culture | 112 |
| miR-320 | TFRC | Cell culture | 113 |
| miR-485-3p | FPN1 | Cell culture | 114 |
| miR-584 | Lactoferrin receptor | Cell culture | 115 |
Epigenetic control
Epigenetic mechanisms alter the accessibility of chromatin to transcriptional regulation through modifications of DNA or modification or rearrangement of nucleosomes.116 Alterations in epigenetic profiles are an important mechanism through which cancer cells modify gene expression.116 Specific epigenetic alterations to iron-related genes have been reported in cancer cells.
DNA hypermethylation observed at CpG islands in proximal promoters is an epigenetic mechanism of gene silencing.105 One study observed CpG island methylation at the FPN-1 promoter, which resulted in reduced basal FPN expression in breast cancer cells.31 Hepcidin, the negative regulator of FPN-1, is also controlled by promoter hypermethylation, although in this case the regulation is exerted indirectly, through methylation of HAMP regulators.33 One such regulator, SOSTDC1, a negative regulator of hepcidin, was found to be epigenetically silenced through promoter hypermethylation in prostate cancer.33 SOSTDC1 silencing increased expression of hepcidin, reduced FPN-1, and increased intracellular iron in prostate tumor cells. Methylation of certain histone sites, such as histone H3 at lysine 4 (H3K4me), are epigenetic markings for gene activation and have also been observed in the HAMP promoter.117 Specifically, exogenous addition of TGF-β or BMP-4, positive regulators of hepcidin, resulted in increased H3K4 methylation at the hepcidin promoter, consistent with increased hepcidin expression associated with this activating mark.118
Acetylation of histones is another epigenetic mechanism for controlling gene expression.116 Generally, histone acetylation corresponds to transcriptional activation by providing an open chromatin structure for active transcription of genes.119 Malignant cells often exhibit upregulation of histone deacetylases (HDAC), enzymes responsible for removing acetyl groups from histones, promoting closed chromatin state and thus repressing transcription of tumor suppressor genes.119 For this reason, HDAC inhibitors have emerged as promising anticancer therapeutics. HDAC inhibitors function by inhibiting HDAC enzyme activity, thus maintaining histone acetylation and subsequent gene expression of critical tumor suppressor genes.119 Interestingly, the H subunit of the iron-storage protein ferritin was found to be induced by HDAC inhibitors, through recruitment of NF-Y transcription factor to the ferritin H promoter.120 In this case, restoring histone acetylation results in increased ferritin H. Increased ferritin H was hypothesized to promote apoptosis through its ability to sequester and bind iron, limiting metabolically active iron cancer cells require to proliferate. It is expected that other genes encoding proteins involved in iron metabolism may be modified through epigenetic mechanisms in cancer to allow for increased iron availability.
Conclusions
The past few years have deepened and expanded our insights into the regulation of iron in cancer. Novel processes involving iron, such as ferritinophagy and ferroptosis, have been identified, and efforts to harness these insights for therapeutic advantage have begun. The FPN/HP axis, responsible for control of iron efflux, appears to be a major node through which iron is controlled in cancer, and understanding mechanisms through which this occurs is an important area of future investigation. The epigenetic and miRNA-mediated pathways through which iron is controlled are just beginning to be understood, but may ultimately offer additional potential therapeutic windows.
Acknowledgments
This work was supported in part by Grants R01 CA188025 (S.V.T), R01 CA171101 (F.M.T), and T90 DE021989 (D.H.M.) from the U.S. National Institutes of Health.