beyond reason
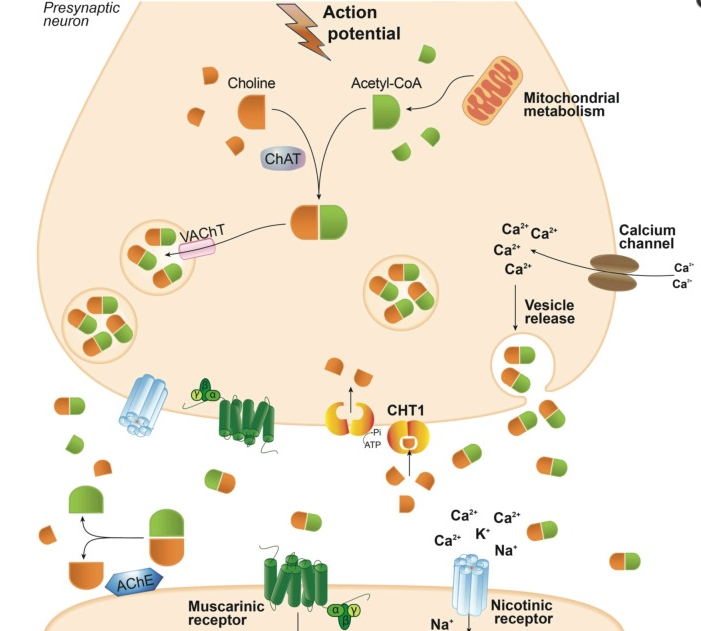
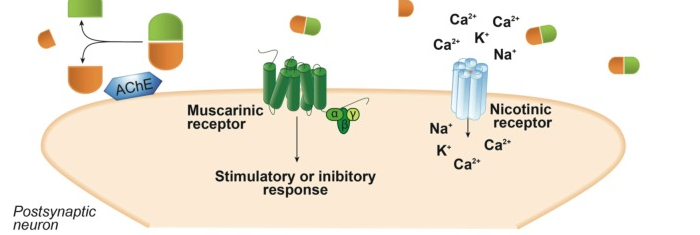
Schematic representation of biological aspects involving acetylcholine neurotransmission. Acetylcholine (ACh) is synthesized in the cytosol of cholinergic presynaptic neurons from choline and acetyl-coenzyme A (acetyl-CoA) by the enzyme choline acetyltransferase (ChAT) and is then transferred into synaptic vesicles by the vesicular acetylcholine transporter (VAChT). Depolarization of the presynaptic neuron promotes ACh exocytosis from synaptic vesicles into the synaptic cleft, where it can bind nicotinic or muscarinic receptors, leading to either a stimulatory or an inhibitory response. In the synaptic cleft, ACh is rapidly hydrolyzed by the enzyme acetylcholinesterase (AChE), releasing acetate and choline, which is reuptaken into the presynaptic cholinergic neuron by the high-affinity choline transporter (CHT1).
Alzheimer's Disease: Targeting the Cholinergic System
Talita H. Ferreira-Vieira, Isabella M. Guimaraes, [...], and Fabiola M. Ribeiro
Abstract
Acetylcholine (ACh) has a crucial role in the peripheral and central nervous systems. The enzyme choline acetyltransferase (ChAT) is responsible for synthesizing ACh from acetyl-CoA and choline in the cytoplasm and the vesicular acetylcholine transporter (VAChT) uptakes the neurotransmitter into synaptic vesicles. Following depolarization, ACh undergoes exocytosis reaching the synaptic cleft, where it can bind its receptors, including muscarinic and nicotinic receptors. ACh present at the synaptic cleft is promptly hydrolyzed by the enzyme acetylcholinesterase (AChE), forming acetate and choline, which is recycled into the presynaptic nerve terminal by the high-affinity choline transporter (CHT1). Cholinergic neurons located in the basal forebrain, including the neurons that form the nucleus basalis of Meynert, are severely lost in Alzheimer’s disease (AD). AD is the most ordinary cause of dementia affecting 25 million people worldwide. The hallmarks of the disease are the accumulation of neurofibrillary tangles and amyloid plaques.
However, there is no real correlation between levels of cortical plaques and AD-related cognitive impairment. Nevertheless, synaptic loss is the principal correlate of disease progression and loss of cholinergic neurons contributes to memory and attention deficits. Thus, drugs that act on the cholinergic system represent a promising option to treat AD patients.
1. ACETYLCHOLINE
1.1. Introduction
Acetylcholine (ACh) was the first neurotransmitter to be identified [1]. ACh is the neurotransmitter used by all cholinergic neurons, which has a very important role in the peripheral and central nervous systems. All pre- and post-ganglionic parasympathetic neurons and all pre-ganglionic sympathetic neurons use ACh as neurotransmitter. In addition, part of the post-ganglionic sympathetic neurons also uses ACh as neurotransmitter.
In the central nervous system (CNS), the cholinergic neurons are widely distributed [2, 3]. They are mainly found in the spinal cord, hindbrain, medial habenula, mesopontine region, basal forebrain, striatum, olfactory tubercle and islands of Cajella complex [2, 4-7]. Almost all regions of the brain are innervated by cholinergic neurons [3].
Given its widespread distribution in the brain, it is not surprising that cholinergic neurotransmission is responsible for modulating important neural functions. The cholinergic system is involved in critical physiological processes, such as attention, learning, memory, stress response, wakefulness and sleep, and sensory information. Experimental data using non-human primates and rodents have demonstrated that injuries introduced to basal forebrain cholinergic neurons that innervate the cortex lead to attention deficit [8, 9]. On the other hand, the facilitation of the cholinergic transmission by using the cholinesterase inhibitors can improve attention in humans [10, 11].
It has been demonstrated that the cholinergic system plays a role in the learning process [12, 13]. Moreover, published data indicate that ACh is involved in memory [14-16]. Further studies have demonstrated that endogenous acetylcholine is important for modulation of acquisition [17], encoding [18], consolidation [19], reconsolidation [20], extinction [21] and retrieval of memory [22]. The importance of the cholinergic neurons from the nucleus basalis of Meynert on memory is highlighted by the fact that the specific degeneration of these neurons takes place in Alzheimer’s disease (AD) and contributes to the memory loss exhibited by AD patients [23, 24].
Stress is another factor that can regulate ACh release in the forebrain and its function on the hypothalamic-pituitary-adrenal (HPA) system can modulate biological and emotional outcomes [25, 26]. In a work that evaluated the function of the cholinergic system in mediating the response to stress it was found that treatment of rats with the antagonist of the nicotinic receptor, mecamylamine, attenuates the activation of the HPA axis in response to a stressor agent [26]. Similarly, nicotine can mimic the ACh effects on the HPA axis by activating nicotinic receptors [27].
Another important function of the cholinergic system is to regulate the sleep cycle [28]. It has been demonstrated that stimulation of cholinergic neurons in precise regions of the brainstem can promote REM (rapid eye movement) sleep, dose-dependently [29]. Moreover, ACh has many modulatory functions in the sensory cortex [30-32]. ACh contributes to auditory synaptic transmission by facilitating thalamo-cortical communication [31]. In addition, ACh has a function in the organization and in the neuronal response to the visual cortex [33]. Recent evidences also suggest the involvement of ACh in adult neurogenesis [34].
1.2. Acetylcholine Synthesis
Cholinergic neurotransmission relies on proteins implicated in the synthesis, storage, transportation and degradation of its neurotransmitter, ACh (Fig. 11). ACh synthesis takes place in the cytoplasm of cholinergic neurons. The enzyme choline acetyltransferase (ChAT) can synthesize ACh from choline and acetyl-coenzyme A (acetyl-CoA). Following that, the neurotransmitter is transported by the vesicular acetylcholine transporter (VAChT) from the cytosol into synaptic vesicles [35, 36]. Acetyl-CoA is supplied by mitochondria and most of the choline comes from the diet. Although cholinergic neurons can synthesize choline, the de novo synthesis contributes only with a very small fraction of the total choline that is needed for ACh synthesis (see Fig. 11) [37, 38].
")
ChAT is a 69 kDa enzyme that is mainly localized in the cytoplasm of cholinergic nerve endings [39, 40]. The gene that encodes ChAT can generate multiple mRNAs by alternative splicing [41]. Two longer forms (74 and 82 kDa) of the ChAT protein, in addition to the 69 kDa ChAT, have also been detected in humans [41, 42]. It has been demonstrated that the 82 kDa ChAT is mainly found in the nuclear region when heterologously expressed in immortalized mammalian cells [43]. One main difference between 69 and 82 kDa ChAT is the number of functional nuclear localization signals (NLS). The cytosolic 69 kDa ChAT has only one NLS, although 82 kDa ChAT exhibits two NLS, which might underlie its retention in the cellular nucleus [44]. Moreover, expression of endogenous 82 kDa ChAT in the nuclear region of neurons has been detected by immunohistochemistry in human neuronal tissue [45].
ChAT activity is regulated by neuronal depolarization, influx of Ca2+ and phosphorylation of the enzyme by a wide variety of protein kinases [46-48]. Although ChAT has a vital function in the synthesis of ACh, it has been demonstrated that the enzyme is not the limiting step for ACh synthesis, as ChAT is present in kinetic excess [49, 50].
The genetic organization of VAChT and ChAT genes is unique, as the VAChT gene is nested in the first long intron of the ChAT gene [51-53]. This cholinergic gene locus has been detected in most species examined so far [53-56]. Some promoters are the same for the two genes, although there are other promoters that are specific for either VAChT or ChAT [57-59]. Thus, VAChT and ChAT can be regulated independently, which explains why these two proteins are differently expressed during development [60].
1.3. Acetylcholine Storage in Vesicles
ACh that is synthesized in the cytoplasm of cholinergic neurons is transported into synaptic vesicles by VAChT, which is located in the synaptic vesicle membrane (see Fig. 11). The gene encoding VAChT has been cloned and hydrophobic analysis indicates that the protein has twelve transmembrane domains [51, 53, 61, 62]. Each molecule of ACh transported by VAChT is in exchange for two vesicular protons, which leads to the fulfill of synaptic vesicles with the neurotransmitter [63, 64]. The vesicular transporter activity can be blocked by vesamicol, which is a non-competitive inhibitor [65, 66].
VAChT carboxyl-terminus has many motifs important for cellular trafficking and for its localization to synaptic vesicle membranes [67-70]. Interestingly, PKC can phosphorylate VAChT and regulate its vesicular localization [68, 71,72]. A VAChT knockdown mouse model, expressing about 68% less VAChT protein, shows major neuromuscular deficits. This data highlights the importance of the transporter to the peripheral nervous system. Furthermore, cognitive impairment can take place even due to a mild decrease in VAChT protein expression (about 45%) [73].
1.4. Acetylcholine Release and Inactivation
When cholinergic neurons are depolarized, ACh is exocytosed from synaptic vesicles and released into the synaptic cleft, where it can activate both muscarinic and nicotinic receptors. ACh present at the synaptic cleft is rapidly inactivated by the enzyme acetylcholinesterase (AChE), releasing choline and acetate [74]. Cholinergic neurons secrete AChE into the synaptic cleft, where the enzyme is normally associated with the plasma membrane (see Fig. 11) [75,76]. Neurotransmitters other than ACh are normally reuptaken into the presynaptic neuron and then inactivated by specific enzymes. Thus, the mechanism of ACh inactivation is unique. Each molecule of AChE can hydrolyze 5000 molecules of ACh per second, which makes AChE one of the most kinetically efficient enzymes known [77]. It has been demonstrated that AChE, in addition to metabolizing ACh, has a number of non-classical actions. For a review on non-classical actions of AChE, see [78].
AChE gene can generate multiple protein products due to mRNA splicing (Schumacher et al., 1988). These distinct transcripts exhibit unique expression in the various tissues, as well as different patterns of plasma membrane association [79-81]. The main AChE transcript is expressed in muscles and brain. Other transcripts of AChE are expressed in developing blood cells [81-83].
A number of compounds can inhibit AChE enzymatic activity. The organophosphate compounds are irreversible AChE inhibitors that are used as insecticides and nerve gases. For more information on organophosphate, see [84]. Following organophosphate exposure, AChE recovers very slowly. Thus, antidotes, including doxylamine, can be used to speed enzyme regeneration [85]. Moreover, AChE inhibitors that compete for the enzyme site, as succinylcholine, are used as anesthetic adjuvants since these drugs can promote neuromuscular blockage [86, 87]. Importantly, AChE competitive inhibitors are also the mostly wide used drugs to treat AD patients [88, 89]. These drugs will be further discussed in the section “4.2. Drugs to treat AD targeting the cholinergic system”.
1.5. Choline Reuptake
Choline that is released by ACh hydrolysis in the synaptic cleft is continuously reuptaken into the presynaptic cholinergic neuron by an active transport system (see Fig. 11) [90, 91]. Two choline transporters have been identified in neurons: a ubiquitous, low-affinity, sodium-independent transporter that can only be inhibited by high concentrations of hemicholinium-3 (HC-3) (Ki of about 50 μM), and a high-affinity, sodium-dependent, HC-3-sensitive (Ki of 10-100 nM) choline transporter (CHT1). CHT1 is mainly found in cholinergic neurons [92-94] and is responsible for supplying choline for the synthesis of ACh [95, 96]. The reuptake of choline is the rate-limiting step for ACh synthesis in most circumstances [95, 96]. Moreover, mice that display a disruption of CHT1 gene expression exhibit symptoms related to ACh deficit and inevitable death within an hour of birth [97].
The CHT1 gene was identified in 2000 (Okuda et al., 2000). The majority of the CHT1 proteins are localized to early endosomes and synaptic vesicles [98,99]. CHT1 localization in synaptic vesicles appears to be very pronounced, as 90% of CHT1 proteins present at the neuromuscular junction are found in the membrane of synaptic vesicles [100]. It has been demonstrated that CHT1 contains a dileucine-like motif in its carboxyl-terminus and that this motif is crucial for the constitutive internalization of the transporter on clathrin-coated pits [101]. Following internalization, the transporter is recycled back to the plasma membrane in a mechanism that can be modulated by depolarization in a Ca2+-dependent manner [102]. Interestingly, it has been demonstrated that an increase in neuronal firing can lead to an increase in choline uptake and in the synthesis of ACh [49, 50, 96, 103, 104]. It is possible that the presence of CHT1 in the membrane of synaptic vesicles and the consequent increase in CHT1 relocation to the plasma membrane following neuronal depolarization could explain why an increase in neuronal firing promotes increased choline reuptake and, thus, ACh synthesis [98-100]. In support to this hypothesis, rats that were exposed to attentional task-performing tests, which can increase cholinergic firing, exhibit augmented choline uptake and increased levels of CHT1 at the synaptic membrane in the right medial prefrontal cortex [105].
2. CHOLINERGIC NEURONS IN THE BRAIN
The localization of cholinergic neurons in the brain was possible by using anti-ChAT antibodies [106]. There are some cholinergic neurons in the CNS that are part of local circuits (interneurons) and others that are projection neurons, connecting different areas of the brain. Cholinergic interneurons are present in various brain substrates, including the caudate-putamen nucleus, nucleus accumbens, olfactory tubercle, and Islands of Calleja complex, where they have important modulatory actions. For example, the large size aspiny cholinergic interneurons located at the caudate-putamen are responsible for modulating the neuronal output from the basal ganglia [107, 108].
Projection cholinergic neurons connect two or more regions in both the peripheral and the CNS. In the CNS, there are a number of well-defined clusters of cholinergic neurons [109]. The cholinergic neurons found in the basal forebrain are the most well-known of the CNS cholinergic clusters [109-111]. In the substantia innominate of the basal forebrain it is located the nucleus basalis of Meynert with neurons projecting throughout the cortex and the amygdala. Cholinergic neurons present at the septum and the vertical limb of the diagonal band project mainly to the hippocampus. In addition, neurons located at the lateral part of the horizontal limb of the diagonal band nucleus project to the olfactory bulb [109, 112]. The cholinergic neuronal projections from the basal forebrain have unique roles in a wide variety of physiological functions and have an overall modulatory action on cortical functions [113-115]. Importantly, the neurons that form the nucleus basalis of Meynert undergo extensive degeneration in AD. The neurons present at the pedunculopontinus nucleus and laterodorsal tegmental nucleus in the brainstem project to the thalamus [109, 110]. These neurons, which project to both the cortex and spinal cord [116], have many important physiological roles, including the modulation of the sleep/awake cycle [28, 29, 117].
3. ACETYLCHOLINE RECEPTORS
ACh, which is a neurotransmitter with modulatory functions in the CNS, acts by activating its receptors and promoting either stimulation or inhibition, depending on receptor type and neuronal localization of the receptor. ACh receptors are classified, according to agonist selectivity and pharmacology, as either muscarinic or nicotinic receptors.
3.1. Nicotinic ACh Receptors
Nicotinic ACh receptors are ion gated receptor channels that are selective for cations (K+, Na+ and Ca2+) (see Fig. 11) [118-120]. Following nicotinic receptor activation, a rapid cellular response is generated. So far, nine nicotinic receptors were identified. Distinct types of nicotinic receptors are expressed in various structures, including the CNS, ganglions, and muscles. Nicotinic receptors are formed by five different subunits: α, β, δ, and γ (fetal) or ε (adult) [121]. Different combinations of such subunits form the various types of nicotinic receptors. Cholinergic nicotinic receptors expressed in muscle and ganglia are comprised of two α subunits plus each of the other three. On the other hand, neuronal nicotinic receptors are formed by the combination of only two types of subunits (α2-10 and β2-4) [122]. Moreover, the most widely expressed nicotinic subunit in the CNS is the α7 [123]. A great number of different types of nicotinic receptors can be generated by combining these different subunits. As a consequence, each type of nicotinic receptor may have distinct properties and functions [124].
In the peripheral nervous system, activation of nicotinic receptors leads to rapid synaptic transmission. However, in the CNS, these ACh receptors employ a modulatory influence, rather than excitation or inhibition [125]. There are a number of differences between peripheral and CNS nicotinic receptors. For example, nicotinic receptors expressed in the CNS are highly permeable to Ca2+, which is an important signaling molecule involved in the long-lasting modulatory effects of nicotinic receptors [125]. In the CNS, most of the nicotinic receptors are expressed at the presynaptic neuronal membrane and their main role is to regulate the release of neurotransmitters, whereas nicotinic receptors expressed in the peripheral nervous system are mainly post-synaptic [126-128]. Stimulation of nicotinic receptors present at the CNS presynaptic neuronal membrane leads to an increase in presynaptic Ca2+concentration, which may facilitate the release of a number of neurotransmitters, such as glutamate, GABA, dopamine, serotonin, norepinephrine, as well as ACh [129-134]. Thus, ACh can influence the strength and fidelity of various synapses and modulate overall CNS neurotransmission [135, 136]. Agonists of nicotinic ACh receptors can improve, while antagonists for the receptor impair, performance in cognitive tasks [137-139]. It has been demonstrated that the role of ACh in learning and memory seems to be related to the regulation of glutamatergic neurotransmission [127, 140]. Moreover, the interaction of the cholinergic and glutamatergic systems seems to be important not only for cognitive processes but also for neuroprotection, as it has been shown that nicotinic receptors agonists are neuroprotective in a mechanism that is Ca2+-dependent and that involves the glutamatergic system [141, 142].
Nicotinic receptor activity is strictly regulated. It has been demonstrated that phosphorylation of nicotinic receptors by protein kinase A, protein kinase C, and tyrosine kinase can regulate receptor activity [143-146]. Moreover, nicotinic receptors can also be regulated allosterically by Ca2+. Ca2+ ions can bind to the amino-terminal region of nicotinic receptors, which is extracellular, and promote potentiation of ACh-dependent currents in a voltage-insensitive manner [147]. On the other hand, when Ca2+ binds to the receptor intracellular sites, a voltage-dependent reduction in conductance takes place [148].
Not only nicotine, but also DMPP (1,1-Dimethyl-4-phenylpiperazinium) and cystine can act as nicotinic receptor agonists. Agonist sensitivity is highly influenced by nicotinic receptor subunit composition [149]. Curare is the best known antagonist for nicotinic receptors, although this drug is not capable of blocking central nicotinic receptors. Despite the high number of nicotinic receptor antagonists that have been developed, specificity for the different types of CNS nicotinic receptors is still missing [150, 151].
3.2. Muscarinic ACh Receptors
Muscarinic receptors are G protein coupled receptors that are capable of modulating a wide variety of ion channels [152]. Genes for five isoforms of muscarinic receptors (M1-5) have been identified so far [153, 154]. M1, M3, and M5 are coupled to Gαq, whereas M2 and M4 are coupled to Gαi. Stimulation of Gαq coupled muscarinic receptors leads to activation of phospholipase C and formation of inositol phosphates and other second messengers, which can promote closure of K+ channels, thus facilitating cell excitability [155, 156]. Activation of Gαi coupled muscarinic receptors leads to inhibition of adenylyl cyclase and reduction of cyclic adenosine monophosphate (cAMP) levels, promoting inhibition of voltage-gated Ca2+channels and, thus, diminishing cell excitability [155, 157, 158]. Nevertheless, muscarinic receptor signaling is involved in the cross-talk with various other receptors, making the final outcome much more complex [152, 155, 159]. As a consequence, stimulation of ACh muscarinic receptors can promote the opening or closing of Ca2+, K+, or Cl- channels, which might facilitate either depolarization or hyperpolarization, depending on the cell type where these receptors are expressed [160-162]. In terms of synaptic localization, M1/M3 receptors are mainly located at the postsynaptic level [163-165]. On the other hand, M2/M4 receptors are generally located presynaptically, acting as autoreceptors and negatively regulating ACh release [163, 166] and/or as heteroreceptors modulating synaptic transmission by regulating Ca2+ channel activity [167, 168].
Stimulation of muscarinic receptors, as stated in the last paragraph, can lead to either excitation or inhibition. However, activation of these receptors more commonly produces excitation, especially in the cortex. There are many mechanisms underlying muscarinic-mediated excitation. Normally, excitation via these receptors involves suppression of K+ currents in postsynaptic sites. Activation of muscarinic receptors can block various types of K+ currents by inhibiting many resting and voltage-gated K+ channels [160, 169-171]. In addition to decreasing K+ currents, muscarinic receptors located at the presynaptic region can also inhibit GABAergic neurotransmission [172, 173]. On the other hand, stimulation of Gαq coupled muscarinic receptors generates intracellular second messengers that can facilitate N-methyl-D-aspartate (NMDA) currents [174]. Inhibition of GABAergic and stimulation of glutamatergic neuro-transmission will, then, result in excitation.
In general, inhibitory effects produced by muscarinic receptor activation are not very common in most areas of the CNS. It has been demonstrated that in certain regions of the cortex and the brainstem ACh can promote inhibition [158]. Nevertheless, it is debatable whether muscarinic-mediated inhibition is direct [175]. The proposed hypothesis is that muscarinic-mediated inhibition occurs via an increase in K+ currents [172], inhibition of glutamatergic excitatory post-synaptic potentials (EPSPs) [176], and decrease of Ca2+currents [177].
The first selective agonist identified for muscarinic receptors was muscarine and, so far, there is no agonist available with a specific selectivity for one particular subtype of muscarinic receptor [155]. The two best known muscarinic receptor antagonists are atropine and quinuclidinyl benzilate, which block all muscarinic receptors. However, more recently, a few subtype specific antagonists for muscarinic receptors have been developed [156, 157,178].
4. ALZHEIMER’S DISEASE
4.1. Alzheimer’s Disease and the Cholinergic System
Cholinergic neurotransmission has been implicated in a number of disease states. Because ACh has an important role in cognitive processes, the cholinergic system is pointed as an important factor in many forms of dementia, including AD [179, 180]. Deficits in the cholinergic transmission can potentially influence all aspects of cognition and behavior, including cortical and hippocampal processing information [181]. Disruption of cholinergic inputs to the cortex can impair attention and the use of instructive cues needed for decision-making related to ongoing behavior [182]. Moreover, it has been shown that blocking CA3 cholinergic receptors impairs the encoding of information and memory [183]. In addition to cognitive alterations, psychiatric symptoms are frequently observed in AD patients, including apathy and depression [184]. It has been postulated that the loss of cholinergic neurons and the consequent impairment in dopaminergic transmission could be the main factors underling AD-related psychiatric symptoms [184, 185]. Corroborating this hypothesis, it has been demonstrated that dopamine efflux is increased in the nucleus accumbens of M4 knockout mice [186]. One possible explanation for these findings is that cholinergic neuronal projections from the laterodorsal tegmental nucleus and pedunculopontinus nucleus to the nucleus accumbens can regulate dopamine release through M4 autoreceptors [187, 188]. Interestingly, the nucleus accumbens can project to the nucleus basalis and control cholinergic neurotransmission [189]. Further highlighting the importance of the cholinergic system in the CNS, cholinergic neuronal loss, especially in the basal forebrain, occurs not only in AD, but also in Parkinson’s disease [190,191], Down syndrome [192], amyotrophic lateral sclerosis [193, 194], progressive supranuclear palsy [195, 196], and olivopontocerebellar atrophy [197]. Moreover, Huntington’s disease appears to be associated with decreased ChAT activity [198, 199].
AD, the most common cause of dementia, was first described more than 100 years ago [200]. Nowadays, about 24 million people worldwide suffers from some form of dementia and the global prevalence of dementia is expected to double every 20 years, reaching 42 million people in 2020 and 81 million people in 2040 [201]. AD corresponds to 60-80% of all cases of dementia, mainly affecting people over 65 years of age, and leading to inevitable death in about seven years following disease onset [202]. AD is a progressive neurodegenerative disease, with cognitive decline gradually worsening over the years. Impairment of short term memory is one of the first symptoms that AD patients experience. Patients worsen gradually and start to exhibit other symptoms, including long-term memory loss, confusion, disturbances in language and sleep-wake cycle, mood swings, loss of body functions, and death [203]. The rapid progression of the disease has a great impact for the affected individual, their family members, caregivers and society [204].
The pathological hallmarks of AD are the accumulation of neurofibrillary tangles and amyloid plaques [205]. Histopathological analysis reveals, in both humans and animal models of Alzheimer's disease, the presence of amyloid plaques and neurofibrillary tangles in several brain regions, especially in the basal forebrain, frontal lobe [206], hippocampus and cerebral cortex [207-209]. Amyloid plaques are formed from β-amyloid peptides (Aβ), which are produced by the enzymatic cleavage of the amyloid precursor protein (APP) [210-212]. However, there is no strict correlation between number of cortical plaques and cognitive decline in AD, indicating that other factors might have a role in disease progression [213, 214]. Neurofibrillary tangles are formed due to hyperphosphorylation and oligomerization of tau, a microtubule-associated protein [215, 216]. Interestingly, a post mortem human brain study found an important association between neocortical neurofibrillary tangle density and dementia severity in patients with AD [217].
It has been demonstrated that cholinergic synapses are particularly affected by Aβ oligomers early neurotoxicity [218, 219] and that synaptic loss is the major correlate of cognitive impairment [213, 220]. In fact, the memory deficit and neuropathology severity observed in patients exhibiting AD highly correlate with changes in hippocampal synaptic transmission, especially with changes in synaptophysin expression, a presynaptic vesicle protein [220-222]. Electrophysiological recordings from the hippocampus of mutant APP mice also showed important deficits (a 40% loss) in basal synaptic transmission [223, 224]. Cholinergic neurons located at the nucleus basalis of Meynert are the main neurons affected in AD [23, 24, 219]. The loss of the nucleus basalis cholinergic neurons in AD patients is severe: from about 500,000 in the healthy adult to less than 100,000 in patients displaying advanced AD [225]. Moreover, it has been demonstrated that ChAT transcription is severely diminished in the remaining cholinergic neurons, which leads to decreased ChAT activity and progression of dementia [179, 226, 227]. All these cholinergic alterations that take place in AD closely correlates with impaired attention and memory observed in patients [179, 228]. Furthermore, some evidences also suggest the involvement of AChE in the pathogenesis of AD [229-231]. A molecular modeling study showed that AChE interacts with the Aβ peptide and promotes amyloid fibril formation. In this work, kinetic analyses revealed that a structural motif in AChE (a hydrophobic sequence of 35 resides peptides) was able to promote amyloid formation and its incorporation into the growing Aβ-fibrils [232]. Others found around the β-amyloid plaques in the neocortex and hippocampus of aged transgenic Tg2576 mice a strong staining for AchE, which was accompanied by local distortions of the cholinergic fiber. In addition, quantitative autoradiography assay revealed that choline uptake was reduced in the hippocampus of these animals and that the expression of muscarinic and nicotinic cholinergic receptors was diminished [233]. Based on these and other evidences, three different cholinesterase inhibitors, donepezil, galantamine, and rivastigmine, have been used to treat AD with relative success [234, 235].
4.2. Drugs to Treat AD Targeting the Cholinergic System
Cholinesterase inhibitors can increase ACh levels in the synaptic cleft and partially ameliorate cognitive symptoms, enhance quality of life and diminish caregiver burden for patients with mild to severe AD [234, 235]. Nevertheless, these drugs have positive effects for only a short period of time, about 1 to 3 years, and cannot alter disease progression [235]. It has been demonstrated that treatment with donepezil is associated with significant cognitive and functional benefits over the course of 12 months in patients with moderate to severe AD [236]. In a double-blind study designed to assess patients survival with mild to moderately severe AD, the long-term treatment (2 years) with galantamine significantly reduced mortality and cognition decline, besides improving daily live activities in mild to moderate AD patients [237]. Rivastigmine has also been used for AD treatment due to its ease of use (transdermal patch) and good tolerability by patients [238, 239]. Despite the slight variations among these medications, there is no evidence of any difference among them with respect to efficacy. However, the donepezil appears to be associated with less adverse effects when compared to rivastigmine [240].
Although cholinesterase inhibitors are not very effective in slowing AD progression, other drugs targeting the cholinergic system might still produce promising results [234, 235, 241]. It has been demonstrated that M1 muscarinic receptors coupling to G-proteins is impaired in the neocortex of AD patients and that the extent of M1/G-protein uncoupling is related to the severity of cognitive symptoms in AD [242]. In addition, activation of muscarinic receptors can shift the APP processing towards the nonamyloidogenic pathway [243, 244]. Based on these findings and many other studies, M1 acetylcholine receptor agonists are emerging as an important therapeutic tool to treat AD. Interestingly, M1 receptor signaling affects several of AD major hallmarks, including cholinergic deficit, cognitive dysfunction, and tau and Aβ pathologies [245-247]. It has been shown that activation of M1 receptors decreases tau hyperphosphorylation via activation of PKC and inhibition of GSK-3β [247, 248]. Moreover, the M1 agonist AF267B can rescue the cognitive impairment and decrease Aβ42 and tau abnormalities in the cortex and hippocampus of a mouse model of AD [247]. Moreover, AF267B treatment leads to an increase in alpha secretase, which is an enzyme that can prevent the production of Aβ peptide [247]. Clinical trials are currently being hold to test this compound. In addition to M1 agonists, M1 allosteric modulators have also been proposed as potential pharmacological tools to treat AD, as they can decrease Aβ production [249]. Moreover, the use of M2 antagonists, such as SCH-57790 and SC-72788, can lead to blockage of M2-mediated inhibition of presynaptic release of ACh, which can activate M1 and nicotinic receptors, ameliorating cognitive impairment in AD [250]. In addition, activation of M2 receptors can cause an increase in Aβ production [251]. It has been shown that activation of nicotinic receptors can promote nonamyloidogenic processing of APP, increase ACh release and ameliorate cognition deficit [252]. However, further studies have demonstrated that nicotinic receptor activation can lead to an increase in Aβ-mediated tau phosphorylation [252]. Therefore, modulation of different cholinergic receptors might play different roles in AD pathology.
In a large-scale placebo-controlled clinical trial, it was demonstrated that xanomeline, a muscarinic agonist with reasonable selectivity for M1/M4 receptors, exhibits a positive effect in minimizing, in a dose-dependent manner, cognitive and psychiatric symptoms in AD, including memory deficit, mood disturbance, agitation and hallucinations [253]. Moreover, the discovery of M1/M4 positive allosteric modulators have also provided a new approach that holds great potential for the treatment of CNS disorders. The pharmacological characterization of these allosteric agonists demonstrated that these drugs bind to an allosteric site, increasing affinity of the receptor for ACh, without, however, binding or modulating other mAChR subtypes [254,255]. For example, it has been shown that the benzyl quinolone carboxylic acid (BQCA), which is an M1-selective allosteric agonist, is effective in increasing spontaneous excitatory postsynaptic currents in the medial prefrontal cortex (mPFC) pyramidal cells and improving memory in a transgenic mouse model of AD [254]. Regarding M4 selective allosteric agonists, VU0010010 and VU0152100 can induce a selective depression of transmission at excitatory synapses in the hippocampus and reverse amphetamine-induced hyperlocomotion in rats, respectively, suggesting that M4 PAMs can have an antipsychotic effect [256, 257].
Another possible approach to treat AD patients could be drugs targeting CHT1, since choline uptake by cholinergic neurons is the rate-limiting step for ACh production. It has been shown that changes in the expression, trafficking or activity of CHT1 could directly affect choline uptake and, consequently, cholinergic neurotransmission [95, 96]. As mentioned in the topic “Choline reuptake”, a good strategy to increase choline reuptake, and thus ACh synthesis, could be through increasing neuronal firing, which consequently could increase CHT1 relocation to the plasma membrane [98-100]. Thus, drugs that could increase cholinergic neuronal firing, perhaps by acting on ion channels that regulate neurotransmitter release, have the potential to increase CHT1 plasma membrane expression and, as a result, ACh synthesis. In fact, it has been shown that APP interacts with CHT1 proteins increasing their endocytosis from the cell surface [258] and that mice that display disruption of CHT1 gene expression exhibit symptoms related to ACh deficit [97]. Therefore, drugs capable of increasing CHT1 at the cell surface hold potential as future therapy for AD.
Another therapeutic strategy that has been investigated to rescue neuronal cell death is memantine. Memantine is a FDA approved drug to treat AD patients, which is an uncompetitive (channel blocking) NMDA receptor antagonist, that can provide beneficial effects in patients with AD by protecting the cholinergic neurons of excitotoxic destruction [259, 260]. In fact, memantine is able to reduce the excitotoxicity in AD [261]. Furthermore, it has also been proposed that the nerve growth factor (NGF) has the potential to preserve cholinergic neurons [262]. Clinical trials indicate that NGF can rescue cholinergic neurons and stabilize the rate of cognitive decline in AD patients [263, 264]. Moreover, previous studies have shown that treatment with NGF can counteract cholinergic atrophy and memory deficits in aged rats [265].
The current treatments for AD patients provide only symptomatic relief and are not very efficient. Thus, a novel approach for AD treatment using animal models has been investigated [266]. Immunization of transgenic mice with a synthetic Aβ peptide (ative immunization) has shown satisfactory results in minimizing cognitive dysfunction [267], including rescuing short-term memory deficit [268] and reducing amyloid deposition in the brain of both young and old mice [269]. In humans, vaccination with anti-Aβ antibody (passive immunization) has also shown promising results in improving cholinergic transmission and attenuating memory deficits associated with early AD [270]. Some of these vaccines are facing clinical trials [241]. Although both, active [271] and passive [272] immunization, present promising results, it is important to consider the risks involved in AD immunization, including the development of autoimmune disorders [273].
CONCLUSION
The cholinergic system plays an important role in memory and attention and the loss of cholinergic neurons from the nucleus basalis of Meynert that takes place in the AD patient’s brain appears to be a very important factor contributing to AD memory deficit. There are only four drugs that have been approved for treating AD patients so far, including the cholinesterase inhibitors - donepezil, galantamine, and rivastigmine – and memantine, which is an NMDA receptor antagonist. However, none of them are disease-modifying drugs. Therapeutic interventions aiming to replenish lost neurons and/or avoid neuronal death have the potential to modify AD progression. Based on this idea, new drugs are being tested aiming to diminish Aβ production, increase trophic factors and decrease glutamate excitotoxicity. Such therapies have the potential to rescue cholinergic neuronal death, as well as to avoid the loss of cortical and hippocampal neurons, preventing AD progression.
ACKNOWLEDGEMENTS
T.H.F.V., I.M.G., F.R.S. and F.M.R. were responsible for manuscript design, literature review and paper writing. This work was supported by FAPEMIG (PPM-00015-13) and CNPq (400133/2014-8 and 442077/2014-9) grants to F.M.R. T.H.F.V. is the recipient of a CNPq postdoctoral fellowship.
CONFLICT OF INTEREST
The authors declare that there is no conflict of interests regarding the publication of this paper.
LIST OF ABBREVIATIONS
- ACh
- = acetylcholine
- AChE
- = acetylcholinesterase
- AD
- = Alzheimer’s disease
- APP
- = amyloid precursor protein
- Aβ
- = β-amyloid protein peptides
- ChAT
- = choline acetyltransferase
- CHT1
- = high-affinity choline transporter
- CNS
- = central nervous system
- DMPP
- = 1,1-Dimethyl-4-phenylpiperazinium
- EPSP
- = excitatory post-synaptic potential
- HC-3
- = hemicholinium-3
- HPA
- = hypothalamic-pituitary-adrenal
- NGF
- = Nerve growth factor
- NMDA
- = N-methyl-D-aspartate receptor
- VAChT
- = vesicular acetylcholine transporter
Article information
REFERENCES
demented and nondemented persons aged 80 years and older. 1993. [PubMed] [CrossRef]