
https://jamanetwork.com/journals/jamaneurology/fullarticle/787860
Abstract
Parkinson disease (PD), the most common neurodegenerative movement disorder, is characterized by an extensive and progressive loss of dopaminergic neurons in the substantia nigra pars compacta. One of the pathological hallmarks of PD is the presence of Lewy bodies, intracellular inclusions of aggregated α-synuclein. Although the cause and pathogenesis of selective loss of dopamine neurons and the accumulation of α-synuclein in PD remain elusive, growing lines of evidence from environmental risk factors and early-onset genetics point to a convergence between energy metabolism and the disposal of damaged proteins in the development of PD. These findings suggest that impairments in mitochondrial and ubiquitin-proteasome system function can significantly contribute to the pathogenesis of PD. This review will summarize recent insights gained from genetic and environmental studies of PD that underscore this association.
초록
가장 흔한 신경 퇴행성 운동 장애인 파킨슨병(PD)은
흑질 흑질에서 도파민 신경세포가
광범위하고 점진적으로 손실되는 것이 특징입니다.
PD의 병리학적인 특징 중 하나는
응집된 α-시누클레인의 세포 내 내포물인
루이체가 존재한다는 것입니다.
https://www.nature.com/articles/nn.4660
도파민 뉴런의 선택적 손실과
PD에서
α-시누클레인 축적의 원인과 병인은 아직 밝혀지지 않았지만,
환경적 위험 요인과
초기 발병 유전학에서 점점 더 많은 증거가
PD 발병에서
에너지 대사와
손상된 단백질의 처리 사이의 수렴을 지적하고 있습니다.
이러한 연구 결과는
미토콘드리아 및
유비퀴틴-프로테아좀 시스템 기능의 손상이
pd의 발병에 크게 기여할 수 있음을 시사합니다.
이 리뷰에서는
이러한 연관성을 강조하는 PD에 대한
유전 및 환경 연구에서 얻은 최근의 통찰력을 요약합니다.
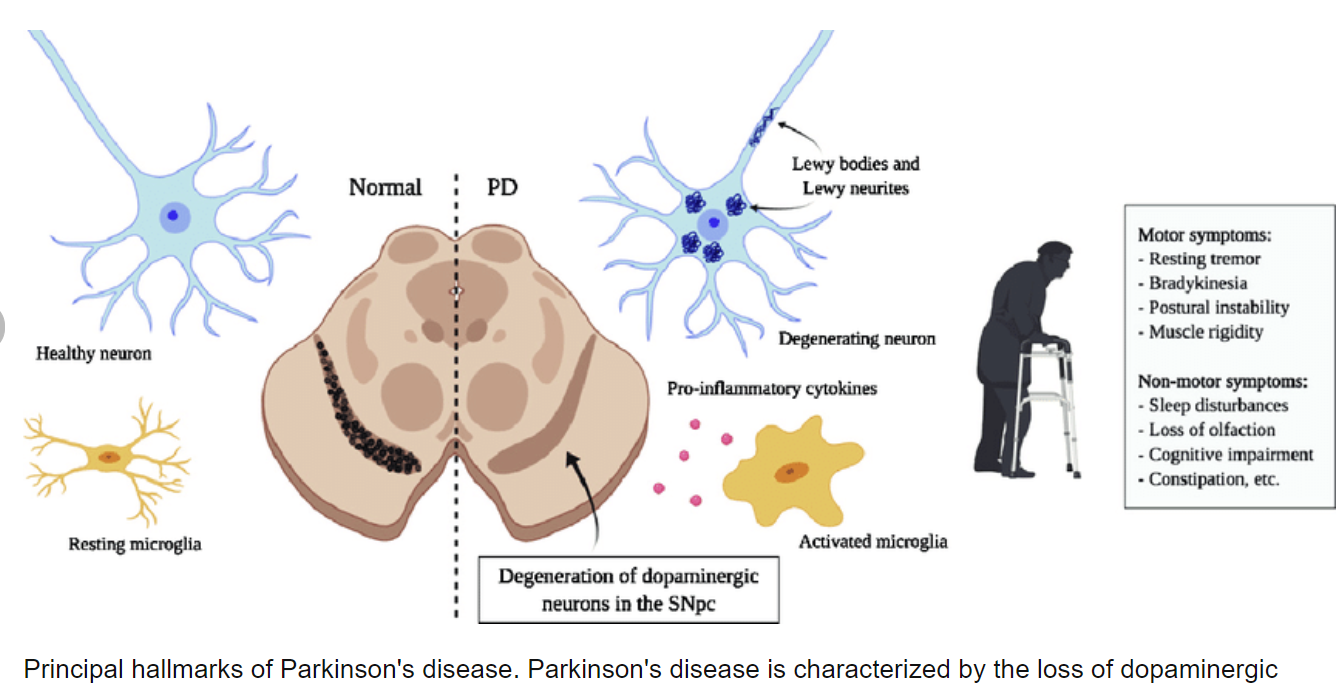
Parkinson disease (PD) is clinically defined as a neurological disorder characterized by 4 cardinal signs (resting tremor, bradykinesia, rigidity, and postural instability). Parkinson disease affects approximately 1% of the population by the age of 65 years, increasing to 4% to 5% of the population by the age of 85 years.1 The disease is chronic and progressive. Patients experience increasing difficulty in daily living functions along the course of the disease. Although environmental risk factors for PD have received significant interest, the importance of genetic factors underlying the likelihood of developing PD is increasingly recognized. Despite the overall rarity of the familial forms, which compose less than 10% of all cases, the identification of several genes causing early-onset PD (such as α-synuclein, UCHL1 [a ubiquitin carboxy-terminal hydrolase L1], parkin, DJ1[a parkin-associated protein involved with oxidative stress], and PINK1 [a putative serine threonine kinase]) has yielded crucial insights into the possible pathogenic mechanisms.1 Several other genes are associated with the parkinsonian phenotype, but usually additional clinical signs such as dementia, dystonia, or ataxia are evident on neurological examination.
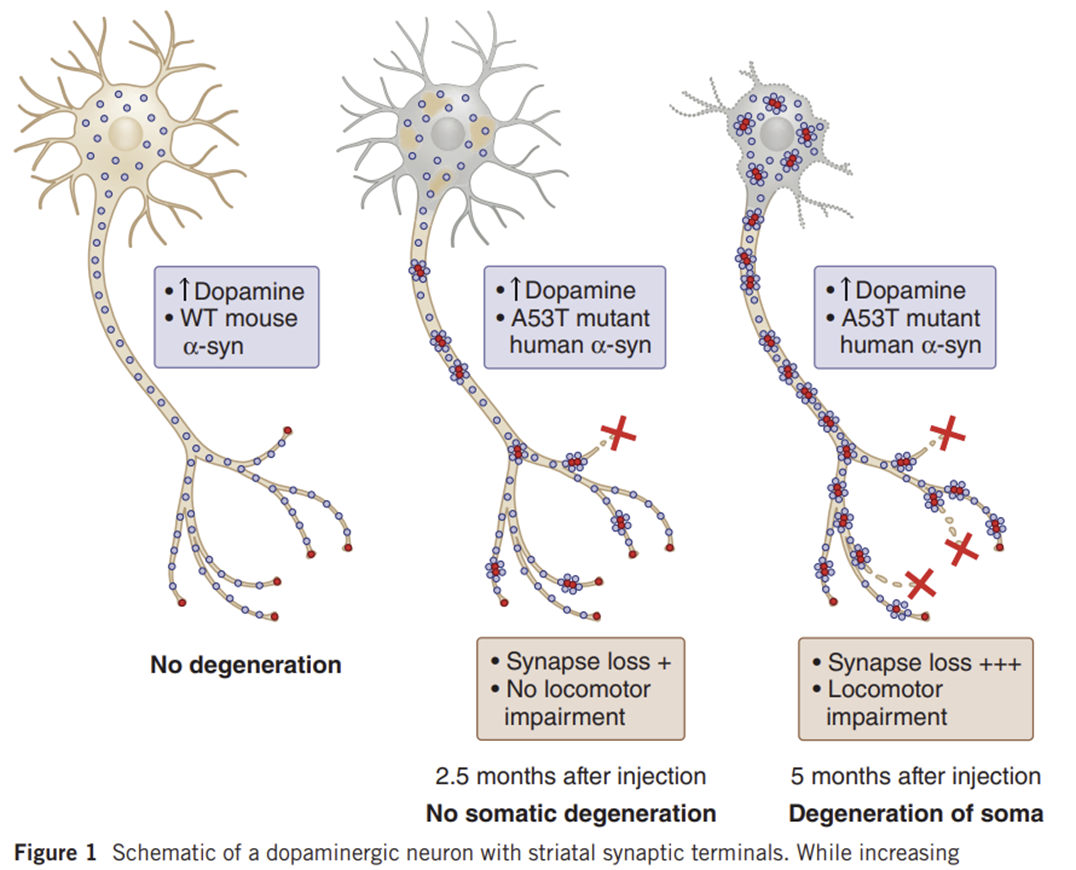
Parkinson disease is initially recognized by its clinical symptoms and ultimately diagnosed in postmortem analysis by the presence of Lewy bodies and the loss of dopaminergic neurons in the substantia nigra pars compacta.1 These neurons give rise to the nigrostriatal pathway, and, as a consequence of the neuronal loss, there is a depletion in striatal dopamine resulting in the clinical phenotype. Lewy bodies, found in significant quantities in PD brain specimens, consist of eosinophilic inclusions containing a loosely compacted core of aggregated α-synuclein and other proteins. Therefore, it was not surprising that the first genetic evidence to early-onset familial PD was linked to the α-synuclein gene (SYNCA-PARK1 locus). Linkage studies and subsequent mutation analysis of the SYNCA gene in the Contursi kindred and several Greek kindreds with autosomal dominant early-onset PD identified an autosomal dominant missense mutation (A53T).1 Subsequent to this initial finding, 2 additional missense mutations in SYNCA, A30P and E46K, were respectively identified in a German and a Spanish family with autosomal dominant early-onset PD.1,2 The role of α-synuclein as a key player in the pathogenesis of PD was further revealed when Singleton and colleagues3 discovered a genomic triplication of SYNCA in the original Spellman-Muenter-Iowa kindred, another autosomal dominant PD family; a concomitant increase in the soluble α-synuclein protein confirmed this finding. An important implication of this study was that the aberrant metabolism (protein mishandling) and increase of wild-type α-synuclein is likely to be a precipitating cause of dopaminergic neuronal loss in PD. While missense mutations in α-synuclein are likely to be pathogenic, it is unclear whether these mutations result in an altered functional protein or enhance the rate of cytoplasmic synuclein aggregation leading to disease.
Parkin mutations (PARK2) cause a form of autosomal recessive juvenile PD (ARJP)4; however, patients with ARJP are atypical in clinical presentation (dystonia is common) and in most cases there is absence of Lewy bodies.5 Parkin is a ubiquitin E3 ligase that functions to prepare substrates for protein degradation mediated through the ubiquitin-proteasome system (UPS). Initially, much effort was focused on the relationship between parkin and the putative substrates that might occur in Lewy bodies (ie, α-synuclein) because the accumulation of these substrates could be potentially toxic to cells. Our insight into the role of parkin in PD was changed when it was shown that fruit fly models of the disease, made by inactivation of parkin, had little effect on dopaminergic neurons, whereas muscle mitochondria seemed compromised.6 In support of this finding, knocking out the gene in mice caused several physiological changes but no neuronal loss in the substantia nigra, despite the severe loss of dopamine neurons observed in ARJP. Moreover, steady-state levels of the known parkin substrates were surprisingly unaltered in the parkin knockout mouse.7,8 Two-dimensional gel electrophoresis followed by mass spectrometry analysis revealed a decrease in the abundance of several proteins involved in mitochondrial function or in the protection from oxidative stress and age-dependent increase in oxidative damage in the substantia nigra of parkin knockout compared with wild-type mice.7 In another parkin knockout mouse model, the loss of locus coaeruleus neurons and a marked reduction of the norepinephrine-dependent startle response were observed.8 The physiological changes in this model have yet to be addressed. Last, parkin was shown to protect against ceramide, an agent that induces mitochondria-dependent apoptosis by reducing mitochondrial swelling and cytochrome c release in vitro. Protection was blocked in the presence of a proteasomal inhibitor, suggesting that the protective action of parkin requires its ubiquitin E3 ligase activity and is mediated through the UPS.7
In addition to parkin, mutations in 2 other genes were recently identified that were associated with mitochondrial impairment. Bonifati and colleagues9 identified a rare form of ARJP caused by mutations in the DJ1 gene (PARK7). The investigators found a large deletion comprising exons 1 to 5 and a point mutation (L166P) in one Dutch and one Italian family, respectively. The L166P missense mutation has been shown to alter the stability and function of the DJ1 protein. Although the exact role of DJ1 remains unclear, wild-type and mutant DJ1 differed in the colocalization to the mitochondria in transfected cells.9 DJ1 overexpression protects against mitochondrial damage and oxidative stress whereas mutant DJ1 (and DJ1 knockdown experiments) sensitizes cells to these effects.7,9
Valente and colleagues10 recently identified a familial, early-onset, autosomal recessive form of parkinsonism caused by mutations in a putative mitochondrial protein kinase named PINK1 (phosphatase and tensin homolog–induced kinase–PARK6). PINK1 colocalizes to the mitochondria, where the protein is believed to have a role in maintaining mitochondrial homeostasis. Mutant PINK1 increases cell susceptibility to stress conditions, inducing mitochondrial dysfunction and apoptosis.10 Whether PINK1 or DJ1 has potential substrates and/or interactors has not been addressed. Given the convergence of function between these early-onset autosomal recessive genes, it is tempting to speculate whether parkin, DJ1, and PINK1 interact at the functional level. To our knowledge, there is no pathological confirmation of PD for either DJ1 or PINK1 families (because of lack of postmortem tissue). It will be interesting to see whether patients with these mutations present with Lewy body inclusions or present like patients with ARJP. The early-onset genetics brings together a common and recurring theme—mitochondrial impairment, oxidative stress, and proteotoxic stress are critical factors in the pathogenesis of PD. Proteasomal activity is highly dependent on adenosine triphosphate production, and inhibition of either mitochondrial or proteasomal function can lead to PD-like pathological features in experimental models. For example, administration of environmental toxins that disrupt mitochondrial complex I activity, such as N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (a derivative of a synthetic opiate) and rotenone (a pesticide), induce PD-like symptoms. Rats treated with rotenone show a selective loss of dopaminergic neurons and the formation of Lewy bodies. Similarly, administration of proteasomal inhibitors induces PD-like symptoms in rodents. McNaught and colleagues11 have examined the effects of inhibiting the UPS in rats by administering synthetic and naturally occurring proteasome inhibitors. Within 2 weeks after receiving the proteasome inhibitors, the rats began to show clinical signs that were remarkably similar to PD patients, including bradykinesia, rigidity, and tremor; brain specimens from these animals had selective neuronal loss and Lewy body–like inclusions in the substantia nigra, locus coaeruleus, and other areas typically affected in PD patients. These findings place mitochondrial impairment, oxidative stress, and proteasome dysfunction at center stage in the pathogenesis of genetic and environmental forms of PD. Environmental factors, such as exposure to toxins that have been linked to PD, seem to be more important in late-onset forms of the disease whereas in early-onset PD genetic factors assume predominant importance. Nevertheless, similar elements seem to link these pathogenic factors to a convergent mechanism of cell death. Do events that affect mitochondrial energy production and decrease resistance to oxidative stress result in UPS dysfunction?
파킨슨병(PD)은
임상적으로 4가지 주요 증상(안정 시 진전, 서동증, 경직, 자세 불안정)을 특징으로 하는
신경학적 장애로 정의됩니다.
파킨슨병은 65세까지
인구의 약 1%에서 발병하며
85세까지 인구의 4~5%로 증가합니다.1
이 질환은 만성적이고 진행성입니다.
환자는 질병이 진행됨에 따라
일상 생활 기능에 점점 더 어려움을 겪게 됩니다.
파킨슨병의 환경적 위험 요인이 큰 관심을 받고 있지만,
파킨슨병 발병 가능성의 근간이 되는 유전적 요인의 중요성이 점점 더 인식되고 있습니다.
전체 사례의 10% 미만을 차지하는 가족형은
전반적으로 드물지만,
초기 발병 PD를 유발하는 여러 유전자
(α-시누클레인,
UCHL1 [유비퀴틴 카르복시 말단 가수분해효소 L1],
파킨,
DJ1[산화 스트레스와 관련된 파킨 관련 단백질],
PINK1 [추정 세린 트레오닌 키나제])가
확인되면서
가능한 발병 메커니즘에 대한 중요한 통찰을 얻게 되었습니다.1
α-synuclein,
UCHL1 [a ubiquitin carboxy-terminal hydrolase L1],
parkin,
DJ1[a parkin-associated protein involved with oxidative stress], and
PINK1 [a putative serine threonine kinase
파킨슨병 표현형과 관련된 다른 여러 유전자가 있지만,
일반적으로 신경학적 검사에서
치매, 근긴장 이상 또는 운동 실조증과 같은 추가적인 임상 징후가 나타납니다.
파킨슨병은 처음에는 임상 증상으로 인식되며,
사후 분석에서 루이체가 존재하고
흑질 흑질에서 도파민 신경세포가 소실되어 궁극적으로 진단됩니다.1
이러한 신경세포는
흑질질 경로를 생성하고,
신경세포 소실의 결과로
선조체 도파민이 고갈되어 임상 표현형이 발생합니다.
PD 뇌 표본에서 상당량 발견되는 루이체는
응집된 α-시누클레인 및
기타 단백질의 느슨하게 압축된 코어를 포함하는 호산구 내포물로 구성됩니다.
따라서
조기 발병 가족성 PD에 대한 최초의 유전적 증거가
α-시누클레인 유전자(SYNCA-PARK1 유전자좌)와 관련이 있다는 것은 놀라운 일이 아니었습니다.
상염색체 우성 조기 발병 PD를 가진 콘투르시 혈통과 여러 그리스 혈통의 SYNCA 유전자에 대한 연관성 연구 및 후속 돌연변이 분석에서 상염색체 우성 미센스 변이(A53T)가 확인되었습니다.1 이 초기 발견에 이어, 독일과 스페인의 상염색체 우성 조기 발병 PD 가족에서 각각 A30P와 E46K라는 2개의 추가 SYNCA 미센스 변이가 확인되었습니다.1,2 싱글턴과 동료3가 또 다른 상염색체 우성 PD 가족인 스펠만-뮤엔터-아이오와 혈통에서 SYNCA의 게놈 3배 증가를 발견하고 가용성 α-시누클레인 단백질의 수반 증가를 확인하면서 PD 발병의 핵심 요소로서 α-시누클레인의 역할이 더욱 밝혀졌습니다.
이 연구의 중요한 시사점은
비정상적인 대사(단백질의 잘못된 처리)와
야생형 α-시누클레인의 증가가
PD에서 도파민성 신경세포 손실의 촉발 원인일
가능성이 높다는 것입니다.
α-시누클레인의 미센스 돌연변이는 병원성이 있을 가능성이 있지만, 이러한 돌연변이가 기능 단백질의 변형을 초래하는지 아니면 세포질 시누클레인 응집 속도를 높여 질병을 유발하는지는 불분명합니다.
파킨 돌연변이(PARK2)는
상염색체 열성 청소년 PD(ARJP)4의 한 형태를 유발하지만,
ARJP 환자는 임상 양상이 비정형적이며(근긴장이상증이 흔함)
대부분의 경우 루이체가 없습니다.5
파킨은
유비퀴틴-프로테아좀 시스템(UPS)을 통해 매개되는
단백질 분해를 위한 기질을 준비하는 기능을 하는
유비퀴틴 E3 리가제(ubiquitin E3 ligase)입니다.
https://www.nature.com/articles/35086067

초기에는
파킨과 루이체에서 발생할 수 있는 추정 기질(즉, α-시누클레인)의 관계에 많은 노력이 집중되었는데,
이러한 기질의 축적이 세포에 잠재적으로 독성을 일으킬 수 있기 때문입니다.
파킨의 비활성화에 의해 만들어진 초파리 질병 모델이
도파민 신경세포에는 거의 영향을 미치지 않는 반면
근육 미토콘드리아는 손상된 것으로 나타났을 때
PD에서 파킨의 역할에 대한 우리의 통찰력은 바뀌었습니다.6
이 발견을 뒷받침하기 위해
생쥐에서 유전자를 제거하면 ARJP에서 관찰된
도파민 신경세포의 심각한 손실에도 불구하고
여러 생리적 변화가 발생했지만 실질핵에서는
신경세포 손실이 나타나지 않았습니다.
또한, 알려진 파킨 기질의 정상 상태 수준은
파킨 녹아웃 마우스에서 놀랍게도 변하지 않았습니다.7,8
2차원 젤 전기영동과 질량 분석 분석 결과, 야생형 마우스에 비해 파킨 녹아웃 마우스에서 미토콘드리아 기능 또는 산화 스트레스에 대한 보호와 연령 의존적 산화 손상 증가에 관련된 여러 단백질의 양이 감소한 것으로 나타났습니다.8,10 파킨 녹아웃의 실질 흑질은 나이에 따라 산화적 손상이 증가했습니다. 또 다른 파킨 녹아웃 생쥐 모델에서는 시교차상핵 뉴런의 손실과 노르에피네프린 의존성 놀람 반응의 현저한 감소가 관찰되었습니다.8 이 모델의 생리적 변화는 아직 밝혀지지 않았습니다.
마지막으로,
파킨은 시험관 내에서
미토콘드리아의 부종과
시토크롬 C 방출을 감소시켜
미토콘드리아 의존적 세포 사멸을 유도하는
세라마이드에 대해 보호하는 것으로 나타났습니다.
프로테아좀 억제제가 있는 경우 보호 효과가 차단되었으며,
이는 파킨의 보호 작용이
유비퀴틴 E3 리가제 활성을 필요로 하며 UPS를 통해 매개된다는 것을 시사합니다.7
파킨 외에도 최근 미토콘드리아 손상과 관련된 두 가지 다른 유전자의 돌연변이가 확인되었습니다. 보니파티와 동료들9은 DJ1 유전자(PARK7)의 돌연변이로 인해 발생하는 희귀한 형태의 ARJP를 확인했습니다. 연구진은 각각 네덜란드와 이탈리아의 한 가족에서 엑손 1~5와 점 돌연변이(L166P)로 구성된 대규모 결실을 발견했습니다. L166P 미센스 돌연변이는 DJ1 단백질의 안정성과 기능을 변화시키는 것으로 나타났습니다. DJ1의 정확한 역할은 아직 불분명하지만, 야생형과 돌연변이 DJ1은 감염된 세포에서 미토콘드리아와의 공동화에서 차이가 있었습니다.9 DJ1 과발현은 미토콘드리아 손상과 산화 스트레스로부터 보호하는 반면 돌연변이 DJ1(및 DJ1 녹다운 실험)은 이러한 효과에 세포를 민감하게 만듭니다.7,9
발렌테와 동료들10은 최근 PINK1(포스파타제 및 텐신 동족 유도 키나아제-PARK6)이라는 추정 미토콘드리아 단백질 키나아제의 돌연변이로 인한 가족성 조기 발병 상염색체 열성 형태의 파킨슨병을 확인했습니다. PINK1은 미토콘드리아에 국소화되며, 이 단백질은 미토콘드리아 항상성 유지에 중요한 역할을 하는 것으로 알려져 있습니다. 돌연변이 PINK1은 스트레스 조건에 대한 세포 감수성을 증가시켜 미토콘드리아 기능 장애와 세포 사멸을 유도합니다.10 PINK1 또는 DJ1에 잠재적인 기질 및/또는 상호작용자가 있는지는 아직 밝혀지지 않았습니다. 이러한 조기 발병 상염색체 열성 유전자 간의 기능 융합을 고려할 때 파킨, DJ1 및 PINK1이 기능 수준에서 상호 작용하는지 여부를 추측할 수 있습니다. 우리가 아는 한, DJ1 또는 PINK1 계열에 대한 PD의 병리학적 확인은 없습니다(사후 조직이 부족하기 때문에). 이러한 돌연변이를 가진 환자에게 루이소체 내포물이 나타나는지 또는 ARJP 환자처럼 나타나는지 확인하는 것은 흥미로울 것입니다. 초기 발병 유전학은 미토콘드리아 손상, 산화 스트레스, 단백질 독성 스트레스가 루게릭병 발병에 중요한 요소라는 공통적이고 반복적인 주제를 담고 있습니다. 프로테아좀 활성은 아데노신 삼인산 생성에 크게 의존하며, 미토콘드리아 또는 프로테아좀 기능을 억제하면 실험 모델에서 PD와 유사한 병리학적 특징이 나타날 수 있습니다. 예를 들어, N-메틸-4-페닐-1,2,3,6-테트라하이드로피리딘(합성 아편유도체)과 로테논(살충제)과 같이 미토콘드리아 복합체 I의 활동을 방해하는 환경 독소를 투여하면 PD 유사 증상이 유발될 수 있습니다. 로테논을 투여한 쥐는 도파민 신경세포가 선택적으로 소실되고 루이체가 형성되는 것으로 나타났습니다. 마찬가지로 프로테아좀 억제제를 투여하면 설치류에서 PD 유사 증상이 유발됩니다. 맥노트와 동료들11은 합성 및 자연 발생 프로테아좀 억제제를 투여하여 쥐의 UPS를 억제하는 효과를 조사했습니다.
프로테아좀 억제제를 투여한 지 2주 이내에 쥐는 서동증, 경직, 진전 등 PD 환자와 매우 유사한 임상 증상을 보이기 시작했으며, 이 동물의 뇌 표본에서는 선택적 신경세포 소실과 흑질, 소교세포 및 기타 PD 환자에서 일반적으로 영향을 받는 부위에서 루이체와 유사한 내포물이 발견되었습니다.
이러한 연구 결과는
미토콘드리아 손상,
산화 스트레스,
프로테아좀 기능 장애가
유전적 및 환경적 형태의 파킨슨병 발병의 중심 단계에 있음을 시사합니다.
PD와 연관된 독소 노출과 같은 환경적 요인은
후기 발병 형태의 질병에서 더 중요한 것으로 보이는 반면,
조기 발병 PD에서는 유전적 요인이 가장 중요한 것으로 추정됩니다.
그럼에도 불구하고 유사한 요소들이 이러한 병원성 요인과 세포 사멸의 수렴 메커니즘을 연결하는 것으로 보입니다.
미토콘드리아 에너지 생산에 영향을 미치고
산화 스트레스에 대한 저항력을 감소시키는 사건이
UPS 기능 장애를 초래할까요?
Methods
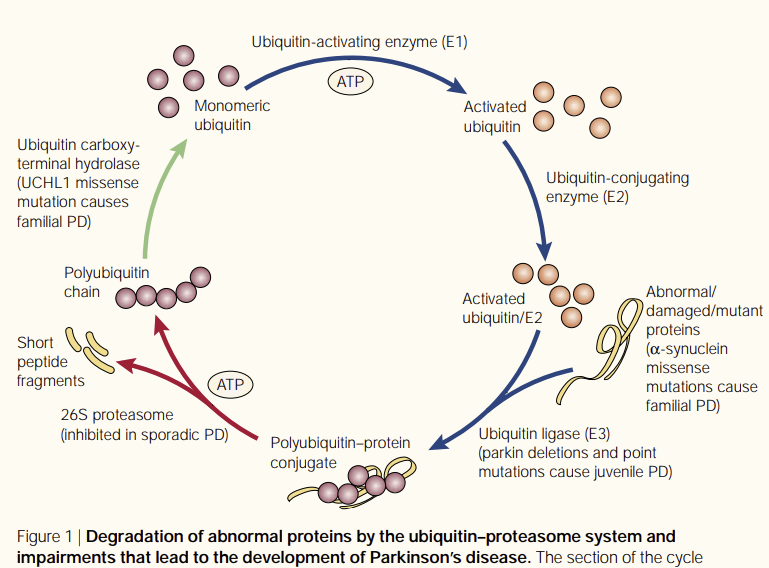
Considerable evidence suggests the neuronal damage that occurs in early-onset PD may be caused by impairment in the UPS. One influential hypothesis in PD is that subsets of neurons are susceptible to failure in proteasome-mediated protein turnover. It has been previously reported that overexpression of mutant α-synuclein increases sensitivity to proteasome inhibitors by decreasing proteasome function. In addition, mutant α-synuclein also causes selective toxic reaction to catecholaminergic neurons in primary mesencephalic cultures, an effect that can be mimicked by the application of proteasome inhibitors.12 As previously noted, McNaught and colleagues11 provided compelling in vivo data that inhibition of the UPS resulted in the clinical symptoms and pathological features of PD.
Despite this evidence and the existence of excellent mouse models for PD, in vivo assessment of the involvement of the UPS and analysis of its activity remain major challenges. Specifically, our understanding of the link between proteolysis and disease would benefit from the ability to follow the activity of the UPS in affected brain tissues before and after administration of disease modifiers (eg, N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine and epoxomicin) or in the presence of specific genetic abnormalities (eg, α-synuclein mutations). We and other groups have started to address the question of proteasomal impairment in neurodegenerative disease by developing transgenic mouse lines that allow the UPS to be monitored in vivo.5 These mice use similar constructs; they express a green fluorescent protein backbone fused with a proteasome substrate. Impairment of the UPS using pharmacological manipulation leads to an accumulation of the reporter that can be conveniently assessed by fluorescent intensity. These novel mouse models are powerful research tools that will eventually allow investigators to directly test the relationship between mitochondrial activity and proteasomal activity as it relates to the development of PD. The results from these studies will hopefully lead to a better understanding of the pathogenic cascade and provide a rationale for development of therapeutic agents.
방법
초기 파킨슨병에서 발생하는 신경세포 손상은
UPS의 손상으로 인해 발생할 수 있다는
상당한 증거가 있습니다.
PD의 영향력 있는 가설 중 하나는
뉴런의 하위 집합이
프로테아좀 매개 단백질 턴오버에 장애를 일으키기 쉽다는 것입니다.
이전에
돌연변이 α-시누클레인의 과발현은
프로테아좀 기능을 감소시켜
프로테아좀 억제제에 대한 민감성을 증가시킨다고 보고된 바 있습니다.
또한 돌연변이 α-시누클레인은
원발성 중뇌 배양에서 카테콜아민성 뉴런에 선택적 독성 반응을 일으키는데,
이는 프로테아좀 억제제의 적용으로 모방할 수 있는 효과입니다.12
앞서 언급했듯이 McNaught와 동료들은11 UPS를 억제하면 PD의 임상 증상 및 병리학적인 특징이 발생한다는 강력한 생체 내 데이터를 제공했습니다.
이러한 증거와 PD에 대한 우수한 마우스 모델의 존재에도 불구하고,
UPS의 관여에 대한 생체 내 평가와 그 활성 분석은 여전히 주요 과제로 남아 있습니다.
특히,
단백질 분해와 질병 사이의 연관성을 이해하려면
질병 조절제(예: N-메틸-4-페닐-1,2,3,6-테트라하이드로피리딘 및 에폭소미닌)의 투여 전후 또는
특정 유전적 이상(예: α-시누클린 변이)이 있는 경우 영향을 받은 뇌 조직에서
UPS의 활동을 추적할 수 있는 능력이 있어야 할 것입니다.
저희와 다른 그룹은 생체 내에서 UPS를 모니터링할 수 있는 형질전환 마우스 라인을 개발하여 신경 퇴행성 질환의 프로테아좀 손상 문제를 해결하기 시작했습니다.5 이 마우스는 유사한 구조를 사용하며 프로테아좀 기질과 융합된 녹색 형광 단백질 백본을 발현합니다. 약리학적인 조작을 통해 UPS를 손상시키면 형광 강도로 편리하게 평가할 수 있는 리포터가 축적됩니다. 이 새로운 마우스 모델은 연구자들이 PD 발병과 관련된 미토콘드리아 활동과 프로테아좀 활동 간의 관계를 직접 테스트할 수 있는 강력한 연구 도구입니다. 이러한 연구 결과는 병원성 캐스케이드에 대한 이해를 높이고 치료제 개발의 근거를 제공할 수 있을 것으로 기대됩니다.
Relevance to the study of neuroscience
The active turnover of damaged cellular proteins is of major importance in the intracellular environment. By using the methods herein and other cellular and biochemical techniques, questions regarding the involvement of genetic and environmental triggers can be identified. A specific facet of cell death of interest is to monitor the UPS through the progression of a disease. Abnormal and misfolded proteins are degraded primarily by the UPS in an adenosine triphosphate–dependent manner5 (Figure). Impaired UPS activity, caused by depressed mitochondrial metabolism or through direct inhibition of UPS components, may potentially result in the accumulation of toxic proteins that contribute to the pathogenesis of PD. It has long been suggested that exposure to environmental toxins plays a role in the development of PD; the finding that exposure to rotenone and epoxomicin results in Lewy body inclusions in rodents bolsters this observation. The UPS itself may be inhibited by the accumulation of aggregation-prone proteins,5 including α-synuclein.12 Lewy bodies could inhibit the UPS by saturating and/or sequestering one or more molecular chaperones and/or ubiquitin ligases required for UPS function or by interaction with the proteasome. There is scientific evidence to support either of these hypotheses. Indeed, parkin, the molecular chaperones Hsp70/Hsp90, and proteasome subunits colocalize in Lewy bodies and mitochondrial proteins (eg, cytochrome c).13 Whether protein aggregation is a cause or a consequence of neurotoxicity remains an unanswered question. Nevertheless, a decline in UPS activity due to environmental or genetic reasons may explain the irreversible and precipitous loss of dopamine neurons that characterizes PD. These results are provocative because they suggest that proteasome inhibition might be a common link between the different early-onset genetic and environmental triggers of PD.
신경과학 연구와의 관련성
손상된 세포 단백질의 활발한 회전율은
세포 내 환경에서 매우 중요합니다.
여기서의 방법과 기타 세포 및 생화학 기술을 사용하여
유전적 및 환경적 유발 요인의 관여에 관한 질문을 확인할 수 있습니다.
관심 있는 세포 사멸의 특정 측면은 질병의 진행을 통해
UPS를 모니터링하는 것입니다.
비정상적이고 잘못 접힌 단백질은
주로 UPS에 의해
아데노신 삼인산 의존적인 방식으로 분해됩니다5 (그림).
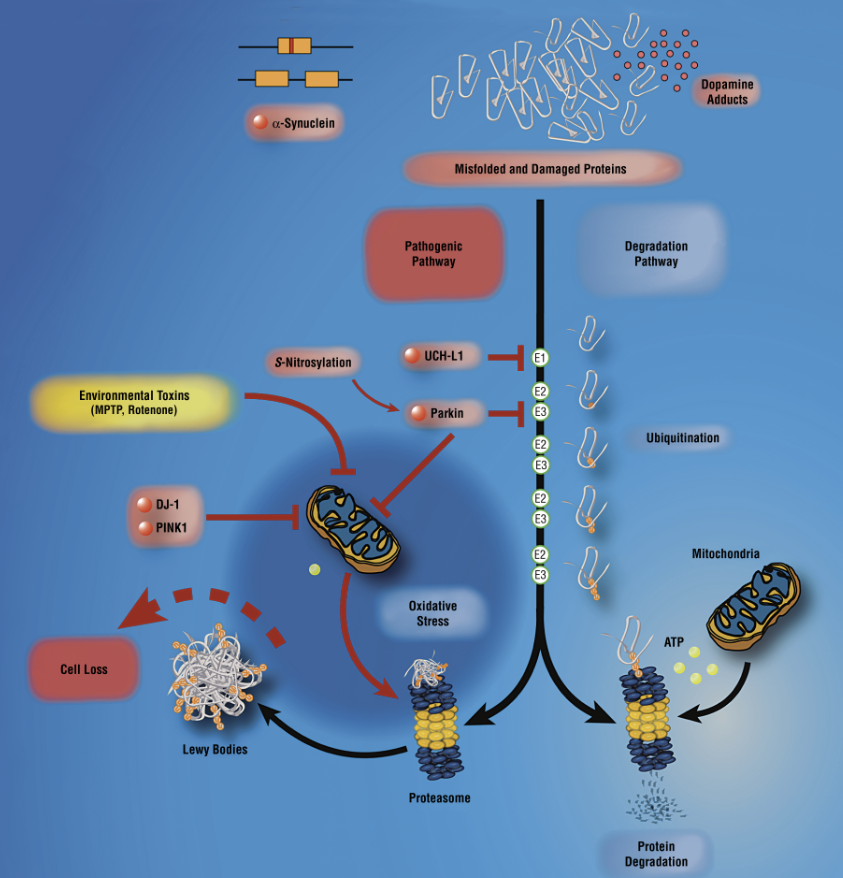
미토콘드리아 대사가 저하되거나
UPS 성분의 직접적인 억제를 통해 UPS 활동이 손상되면
잠재적으로 PD의 발병에 기여하는
독성 단백질이 축적될 수 있습니다.
환경 독소에 대한 노출이
PD 발병에 영향을 미친다는 사실은
오래 전부터 제안되어 왔으며,
설치류에서 로테논과 에폭소미닌에 노출되면 루이체 내포물이 생성된다는 사실은
이러한 관찰을 뒷받침하는 결과입니다.

루이체는 α-시누클레인을 포함하여 응집되기 쉬운
단백질5의 축적에 의해 UPS 자체가 억제될 수 있습니다.12
루이체는
UPS 기능에 필요한 하나 이상의 분자 샤프론 및/또는 유비퀴틴 리가제 또는
프로테아좀과의 상호작용을 통해
포화 및/또는 격리함으로써 UPS를 억제할 수 있습니다.
이러한 가설 중 하나를 뒷받침하는 과학적 증거가 있습니다. 실제로 파킨, 분자 샤프론인 Hsp70/Hsp90, 프로테아좀 하위 유닛은 루이체와 미토콘드리아 단백질(예: 사이토크롬 C)에 공동 분포합니다.13 단백질 응집이 신경 독성의 원인인지 결과인지는 아직 밝혀지지 않은 의문입니다.
그럼에도 불구하고
환경적 또는 유전적 이유로 인한
UPS 활동의 감소는
PD의 특징인 도파민 뉴런의 비가역적이고
급격한 손실을 설명할 수 있습니다.
이러한 결과는
프로테아좀 억제가
PD의 다양한 초기 발병 유전적 및 환경적 유발 요인 사이의 공통된 연결고리일 수 있음을 시사하기 때문에
도발적인 결과입니다.
Relevance to the practice of neurology
In many diseases, genetic testing offers the physician the ability to verify a differential diagnosis and select an appropriate route of treatment based on an individual’s genetic profile. In PD, accurate diagnosis of the disease cannot be confirmed only by clinical examination because different mutations (ie, SYNCA and parkin) may confer similar behavioral phenotypes but have different underlying neuropathological features. However, the use of genetic screening in individuals with a family history of PD should be carefully evaluated because it is not understood how most of these mutations can cause PD. For instance, a growing body of evidence suggests that, for the autosomal recessive genes, the presence of a single mutation in 1 of the 2 alleles may confer increased susceptibility to PD. Many patients have been described as having single heterozygous mutations in the parkin, DJ1, or PINK1 gene. In these cases, the failure to detect a second mutation may have been an artifact of the screening procedure or a result of a genomic rearrangement of the other copy of the gene. However, a more parsimonious hypothesis is that a loss of a single copy of one of these genes results in haploinsufficiency, acting as a susceptibility factor toward PD.8 Given the finding that these genes only confer susceptibility to PD, epigenetic causes are likely significant contributing factors. Factors such as smoking, geographic area, family history, exposure to exogenous toxins and agents (N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine, pesticides, and infection agents), and aging may greatly influence the propensity for an individual to develop PD, potentially because of effects on other underlying biochemical processes. While there seem to be multiple causes of PD, recent lines of evidence suggest that the mechanisms and potential treatments of this disease are converging on potential pathological processes found in the interplay between the UPS and mitochondria.
신경과 진료와의 관련성
많은 질병에서 유전자 검사는
의사가 감별 진단을 확인하고 개인의 유전자 프로필에 따라
적절한 치료 경로를 선택할 수 있는 능력을 제공합니다.
파킨슨병에서는
서로 다른 돌연변이(예: SYNCA 및 파킨)가
유사한 행동 표현형을 보일 수 있지만
근본적인 신경 병리학적 특징이 다르기 때문에
임상 검사만으로는 정확한 진단을 확인할 수 없습니다.
그러나 이러한 돌연변이의 대부분이
어떻게 PD를 유발할 수 있는지 이해되지 않았기 때문에
PD 가족력이 있는 개인에게 유전자 검사를 사용하는 것은 신중하게 평가해야 합니다.
예를 들어,
상염색체 열성 유전자의 경우,
2개의 대립 유전자 중 하나에 단일 돌연변이가 존재하면
PD에 대한 감수성이 높아질 수 있다는 증거가 점점 더 많이 제시되고 있습니다.
많은 환자가
파킨, DJ1 또는 PINK1 유전자에
단일 이형접합 돌연변이가 있는 것으로 알려져 있습니다.
이러한 경우,
두 번째 돌연변이를 발견하지 못한 것은
선별 절차의 부작용이거나 다른 유전자 사본의 게놈 재배열의 결과일 수 있습니다.
그러나 보다 근사한 가설은
이러한 유전자 중 하나의 단일 사본이 손실되면
일배체결핍이 발생하여 PD에 대한 감수성 요인으로 작용한다는 것입니다.8
이러한 유전자가
PD에 대한 감수성만 부여한다는 사실을 고려할 때
후성 유전적 원인이 중요한 기여 요인일 가능성이 높습니다.
흡연, 지역, 가족력,
외인성 독소 및 약제(N-메틸-4-페닐-1,2,3,6-테트라하이드로피리딘, 살충제 및 감염제) 노출,
노화와 같은 요인은
다른 근본적인 생화학적 과정에 영향을 미치기 때문에
개인의 PD 발병 성향에 큰 영향을 미칠 수 있습니다.
N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine, pesticides, and infection agents
PD의 원인은 여러 가지가 있는 것으로 보이지만,
최근의 증거에 따르면 이 질환의 메커니즘과 잠재적 치료법은
UPS와 미토콘드리아 사이의 상호작용에서 발견되는 잠재적 병리 과정에 수렴하고 있습니다.