
The concept of ‘idiopathic’ Parkinson’s disease (PD) as a single entity has been challenged with the identification of several clinical subtypes, pathogenic genes and putative causative environmental agents. In addition to classic motor symptoms, non-motor manifestations (such as rapid eye movement sleep disorder, anosmia, constipation and depression) appear at prodromic/premotor stage and evolve, along with cognitive impairment and dysautonomia, as the disease progresses, often dominating the advanced stages of the disease. The key molecular pathogenic mechanisms include α-synuclein misfolding and aggregation, mitochondrial dysfunction, impairment of protein clearance (associated with deficient ubiquitin-proteasome and autophagy-lysosomal systems), neuroinflammation and oxidative stress. The involvement of dopaminergic as well as noradrenergic, glutamatergic, serotonergic and adenosine pathways provide insights into the rich and variable clinical phenomenology associated with PD and the possibility of alternative therapeutic approaches beyond traditional dopamine replacement therapies.
One of the biggest challenges in the development of potential neuroprotective therapies has been the lack of reliable and sensitive biomarkers of progression. Immunotherapies such as the use of vaccination or monoclonal antibodies directed against aggregated, toxic α-synuclein.as well as anti-aggregation or protein clearance strategies are currently investigated in clinical trials.
The application of glucagon-like peptide one receptor agonists, specific PD gene target agents (such as GBA or LRRK2 modifiers) and other potential disease modifying drugs provide cautious optimism that more effective therapies are on the horizon. Emerging therapies, such as new symptomatic drugs, innovative drug delivery systems and novel surgical interventions give hope to patients with PD about their future outcomes and prognosis.
'특발성' 파킨슨병(PD)을 단일 질환으로 보는 개념은
여러 임상 아형, 병원성 유전자 및 추정 원인 환경 요인이 밝혀짐에 따라
도전받고 있습니다.
전형적인 운동 증상 외에도
비운동 증상(예: 급속 안구 운동 수면 장애, 무감각증, 변비 및 우울증)이
전구/전운동 단계에서 나타나며,
질환이 진행됨에 따라
인지 장애 및 자율 신경 이상과 함께 발전하여
종종 질환의 진행 단계를 지배하게 됩니다.

rapid eye movement sleep disorder, anosmia, constipation and depression
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8393958/
주요 분자 병원성 메커니즘에는
α-시누클레인 오접힘 및 응집,
미토콘드리아 기능 장애,
단백질 제거 장애(유비퀴틴-프로테아좀 및 오토파지-리소좀 시스템 결핍과 관련),
신경 염증 및 산화 스트레스가 포함됩니다.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6773515/

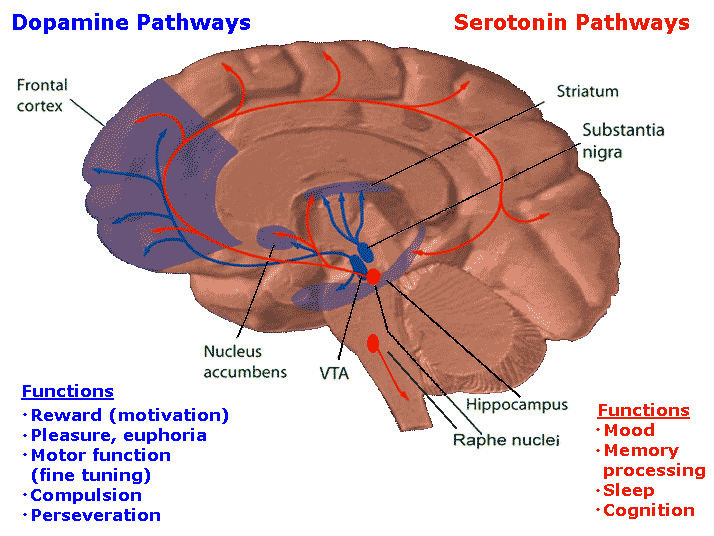
도파민성뿐만 아니라
노르아드레날린성, 글루탐산성, 세로토닌성 및 아데노신 경로의 관여는
PD와 관련된 풍부하고 다양한 임상 현상과
전통적인 도파민 대체 요법을 넘어서는
대체 치료 접근법의 가능성에 대한 통찰력을 제공합니다.
noradrenergic, glutamatergic, serotonergic and adenosine pathways

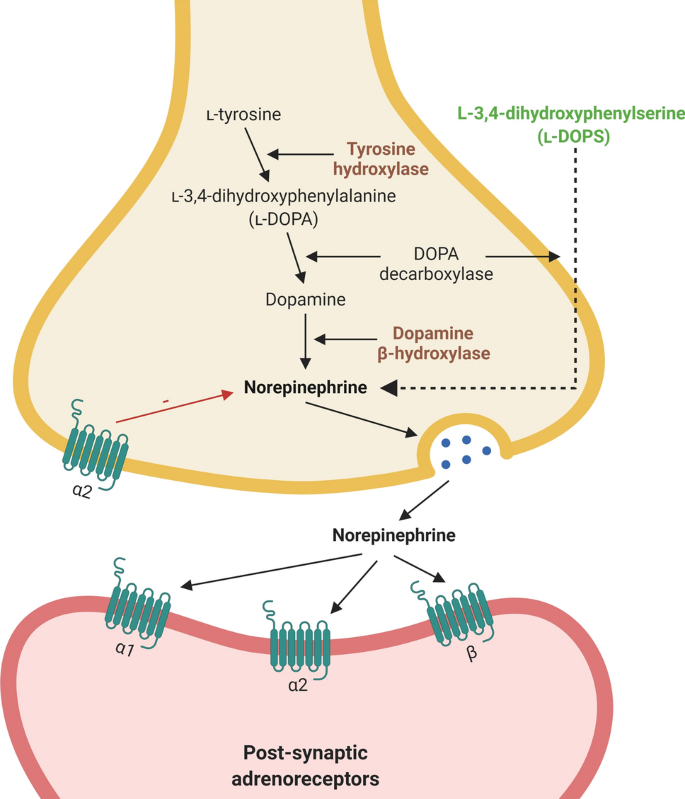
노르 아드레날린 시스템은
또한 도파민 성 변성에
항 염증 및 신경 보호 효과를 발휘하며
노르 아드레날린 손상은 결과적으로 파킨슨 질병의 진행을 조절
노르아드레날린성 핵은
뇌 전체에 광범위한 신경 분포를 제공하며
스트레스 반응, 감정 기억, 운동, 감각 및 자율 기능 조절에 관여하는 근본적인 신경 조절자 역할
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7034292/

https://link.springer.com/article/10.1007/s00401-020-02248-1




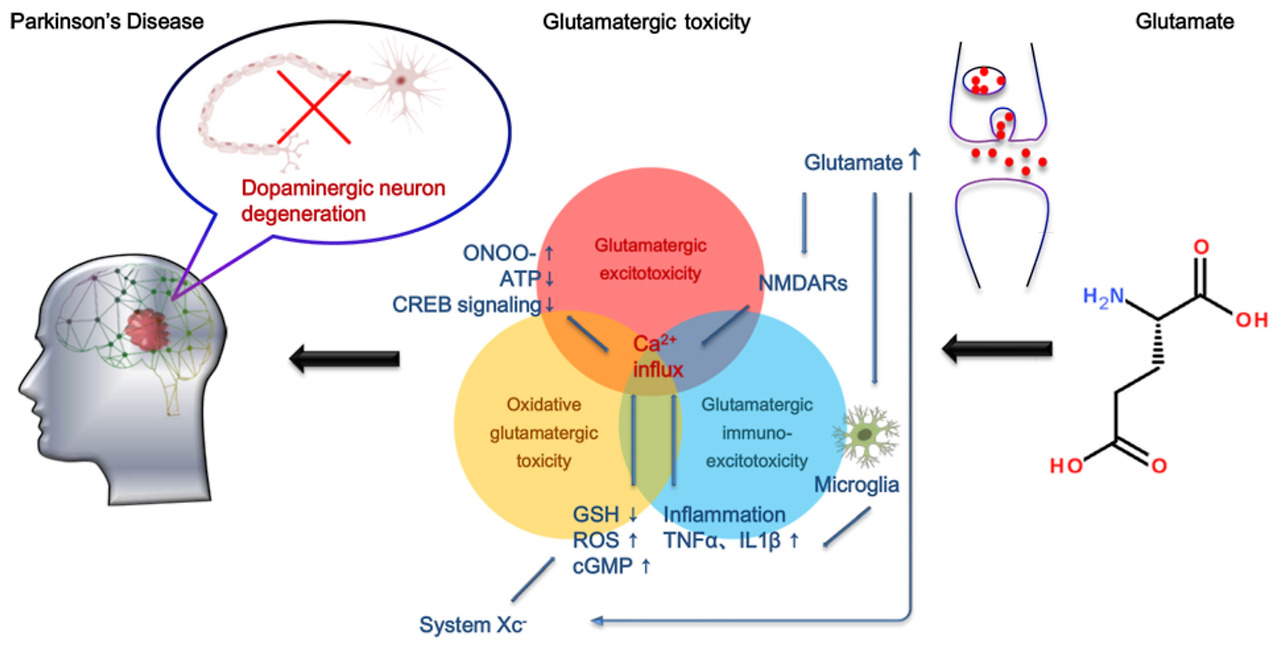
높은 수준의 세포 외 글루타메이트는
글루타메이트 수용체를 과활성화하여
중추 신경계에 흥분 독성 효과를 일으키므로
시냅스 틈새에서 글루타메이트를 적시에 제거하는 것이 필요
https://www.mdpi.com/1422-0067/25/5/3050
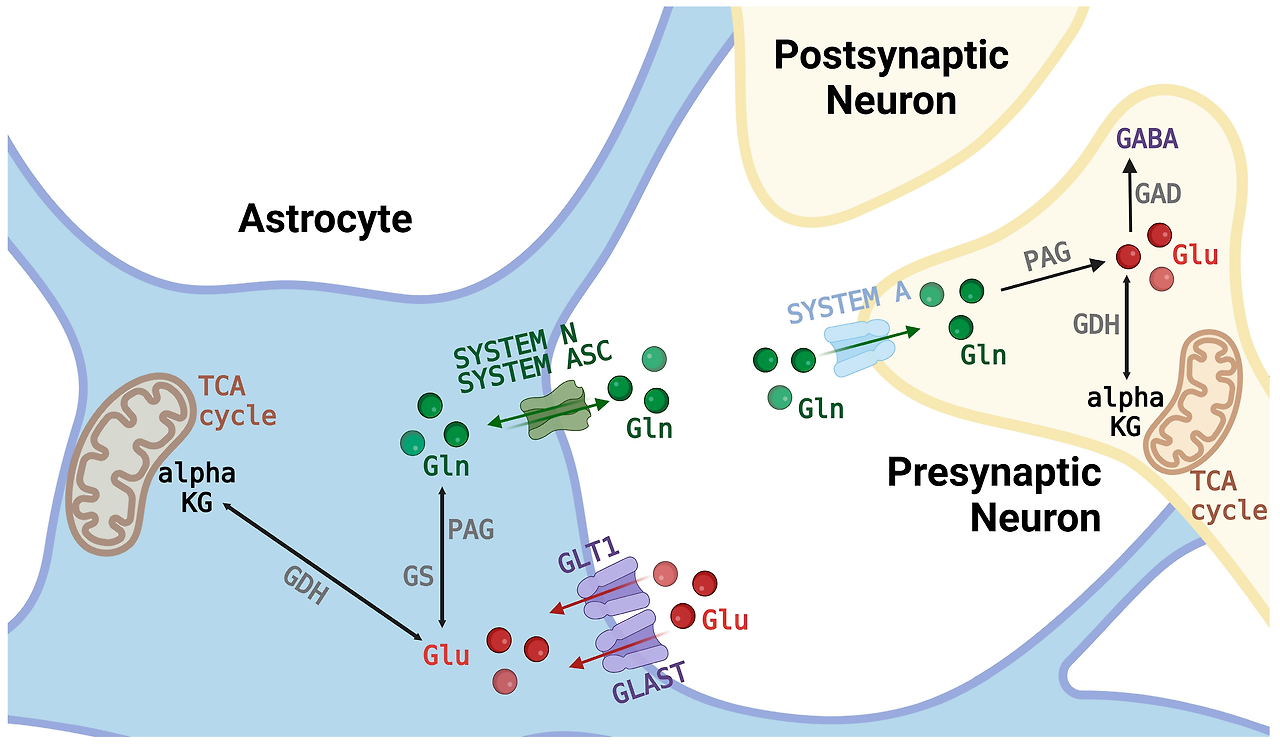


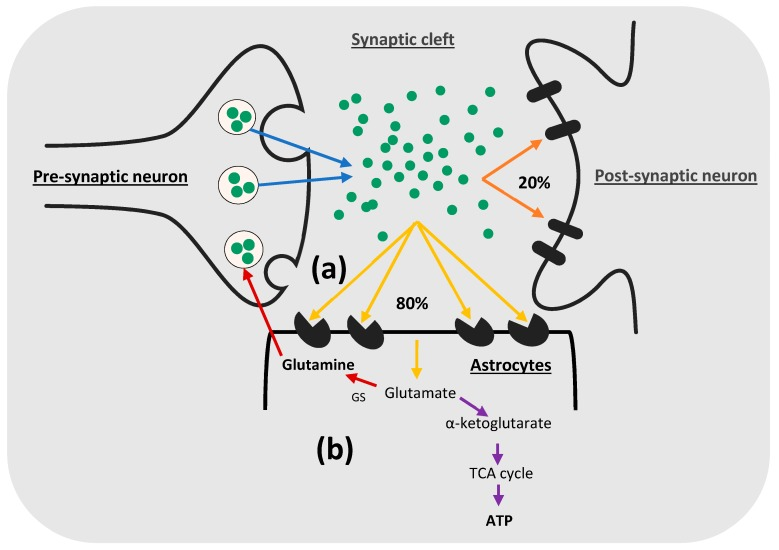

PD 및 LID 장애의 병태 생리학은
운동 및 비운동 장애를 일으킬 수 있는
세로토닌(5-HT) 뉴런의 기능 장애 활동과도 관련이 있는 것으로 나타났습니다.
5-HT 뉴런의 주요 집단은
거의 전체 신경축에 광범위한 신경 분포를 제공하고
뇌의 여러 기능을 제어하는 등쪽 시상핵(DRN)에 위치합니다.
DRN 5-HT 뉴런의 퇴화는
초기 PD에서 발생

비도파민성 치료제로서
아데노신 수용체 길항제를 사용한
전임상 및 임상 연구의 여러 노력에 대한 근거를 제시
잠재적인 신경 보호 치료법 개발의 가장 큰 난제 중 하나는
신뢰할 수 있고 민감한 진행 바이오마커가 부족하다는 점입니다.
응집된 독성 α-시누클레인을 겨냥한
백신 또는 단일 클론 항체 사용과 같은 면역 요법과
응집 방지 또는 단백질 제거 전략이
현재 임상 시험에서 연구되고 있습니다.
글루카곤 유사 펩타이드 1 수용체 작용제,
특정 PD 유전자 표적제(예: GBA 또는 LRRK2 조절제) 및
기타 잠재적 질병 조절 약물의 적용은
보다 효과적인 치료법이 곧 나올 것이라는 조심스러운 낙관론을 제시합니다.
The application of glucagon-like peptide one receptor agonists, specific PD gene target agents (such as GBA or LRRK2 modifiers) and other potential disease modifying drugs provide cautious optimism that more effective therapies are on the horizon
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6812410/
새로운 증상 치료제, 혁신적인 약물 전달 시스템, 새로운 외과적 개입과 같은 새로운 치료법은 PD 환자에게 향후 결과와 예후에 대한 희망을 줍니다.
Introduction
The clinical syndrome, described by James Parkinson in his 1817 ‘Essay on the shaking palsy’, and commonly referred to as ‘Parkinson’s disease’ (PD), is characterised by the cardinal features of rest tremor, bradykinesia, rigidity and postural instability, and a variety of other motor and non-motor symptoms.1–3 With ageing and increasing life span of the global population, age-related diseases like PD are receiving increased attention from the scientific community. Neurological disorders are now the leading source of disability in the world, and PD is the fastest growing of these disorders.4 The Global Burden of Disease Study estimates that the number of PD case will double from about 7 million in 2015 to about 13 million in 2040, suggesting a potential ‘PD Pandemic’.5 While this extrapolation based on future growth of population is just an estimate, it highlights the enormous burden that PD and related neurodegenerative conditions can pose for society.
Traditionally, the term ‘idiopathic’ PD has been used to describe the most common cause of parkinsonism in clinical practice. However, with the discovery of monogenic forms of PD (which may be clinically indistinguishable from the ‘idiopathic’ form), the clinical heterogeneity of the disease and the clinical overlap between PD dementia, dementia with Lewy bodies and other forms of parkinsonism, the nosology of PD classification needs to be continuously re-evaluated.6 7
1817년 제임스 파킨슨이
'떨림 마비에 관한 에세이'에서 기술한 임상 증후군으로
흔히 '파킨슨병'(PD)이라고 불리는 이 질환은
안정 시 떨림, 서동증, 경직 및 자세 불안정,
기타 다양한 운동 및 비운동 증상이 주요 특징입니다.1-3
세계 인구의 고령화와 수명 연장에 따라
PD와 같은 노화 관련 질환이
과학계의 주목을 받고 있습니다.
신경계 질환은
현재 전 세계 장애의 주요 원인이며,
PD는 이러한 질환 중 가장 빠르게 증가하는 질환입니다.4
글로벌 질병 부담 연구(Global Burden of Disease Study)에 따르면
2015년 약 700만 명에서
2040년 약 1300만 명으로
두 배 증가할 것으로 추정되어
'PD 팬데믹' 가능성을 시사합니다.5
향후 인구 증가에 따른 추정치이지만,
PD 및 관련 신경 퇴행성 질환이 사회에 미칠 엄청난 부담을 강조하는 결과입니다.
전통적으로 '특발성' PD라는 용어는 임상에서 파킨슨병의 가장 흔한 원인을 설명하는 데 사용되어 왔습니다.
그러나
'특발성' 형태와 임상적으로 구별할 수 없는
단원성 형태의 PD가 발견되고,
질병의 임상적 이질성과 PD 치매,
루이소체 치매 및 다른 형태의 파킨슨병 간의 임상적 중복이 발견됨에 따라
PD 분류의 개념은 지속적으로 재평가될 필요가 있습니다.6 7
Historic milestones
Major milestones in PD etiopathogenesis include the identification of intracytoplasmic inclusion bodies (‘Lewy bodies’) as a pathologic hallmark by Frederick Lewy in 1912 and the discovery of dopamine deficiency and its involvement in the parkinsonian animal models. The pioneering work of Arvid Carlsson and Oleh Hornykiewicz starting in 1957 established the link between dopamine deficiency and PD. The latter was supported by the proof of concept demonstrating clinical rescue in the first trial in PD patients with intravenous levodopa in 1961 and the introduction of high dosage levodopa therapy by George Cotzias in 1967.8
In 1982, William Langston, a neurologist, described seven patients in the San Francisco Bay Area who were using ‘synthetic heroin’ and developed parkinsonian features.9 Subsequent investigations revealed the cause of this drug-induced parkinsonism, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine, which is toxic to substantia nigra dopaminergic neurons. The discovery had a remarkable impact on research into the etiopathogenesis of PD and experimental therapeutics, leading to drug trials in animal models and large-scale epidemiological studies on occupational exposure to potential toxins.
In 1996, Polymeropoulos and colleagues found genetic markers on chromosome 4q21-q23 to be linked to the PD phenotype in an Italian kindred and 3 Greek families with autosomal dominant PD, and the following year they reported a mutation in the α-synuclein gene (SNCA), highlighting for the first time that PD may have a genetic aetiology.10 11 This landmark discovery launched a highly productive period of successful gene hunting during which many more PD genes and genetic risk loci were identified. These findings facilitated the generation of genetic animal models which subsequently identified new therapeutic targets for clinical trials.7 12 13
역사적인 이정표
PD 병인의 주요 이정표로는
1912년 프레드릭 루이가
세포질 내 봉입체('루이체')를 병리학적 특징으로 규명하고
도파민 결핍과 파킨슨병 동물 모델에서의 관련성을 발견한 것을 들 수 있습니다.
1957년부터 시작된
아비드 칼슨과 올레 호니키에비츠의 선구적인 연구로
도파민 결핍과 PD 사이의 연관성이 확립되었습니다.
후자는 1961년
레보도파를 정맥 주사한 PD 환자를 대상으로 한
첫 번째 임상시험에서 임상적 완화를 입증한 개념 증명과
1967년 조지 코치아스가 고용량 레보도파 요법을 도입하면서 뒷받침되었습니다.8
1982년
신경과 전문의 윌리엄 랭스턴은 샌프란시스코 베이 지역에서
'합성 헤로인'을 사용하던 7명의 환자에게
파킨슨병 증상이 나타났다고 보고했습니다.9
이후 조사를 통해
약물로 인한 파킨슨병의 원인인
1-메틸-4-페닐-1,2,3,6-테트라하이드로피리딘이
흑질 도파민 신경세포에 독성이 있는 물질이라는 것이 밝혀졌습니다.
이 발견은
PD의 병인 연구와 실험적 치료법에 큰 영향을 미쳤으며,
동물 모델에서의 약물 실험과 잠재적 독소에 대한
직업적 노출에 대한 대규모 역학 연구로 이어졌습니다.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8752015/

1996년 폴리메로풀로스와 동료들은
상염색체 우성 PD를 가진 이탈리아 혈통과
그리스 3가족에서 PD 표현형과 연관된 염색체 4q21-q23의 유전자 마커를 발견했고,
이듬해에는
α-시누클린 유전자(SNCA)의 변이를 보고하여
처음으로 PD에 유전적 원인이 있을 수 있음을 강조했습니다.10 11
이 획기적인 발견을 시작으로
매우 생산적인 유전자 사냥의 시대가 열리고
더 많은 PD 유전자 및 유전 위험 유전자좌가 확인되었습니다.
이러한 발견은
이후 임상시험을 위한 새로운 치료 표적을 확인하는
유전적 동물 모델의 생성을 촉진했습니다.7 12 13
Clinical syndrome
The clinical criteria of the UK Parkinson’s Disease Society Brain Bank for probable PD require the presence of bradykinesia and one of the following features: rigidity, 4–6 Hz rest tremor, or postural instability; in addition, three supportive features are required.1 The International Parkinson’s and Movement Disorder Society (MDS) developed their own clinical diagnostic criteria that include (1) presence of parkinsonism (bradykinesia plus either rest tremor or rigidity); (2) absence of absolute exclusionary criteria, (3) supportive criteria and (4) no red flags.14 In addition to a variety of clinical rating scales, particularly the Unified Parkinson’s Disease Rating Scale (UPDRS) used to assess severity of the disease, reliable diagnostic, presymptomatic and progression biomarkers are being developed to support the diagnosis and to track the course of the disease.15
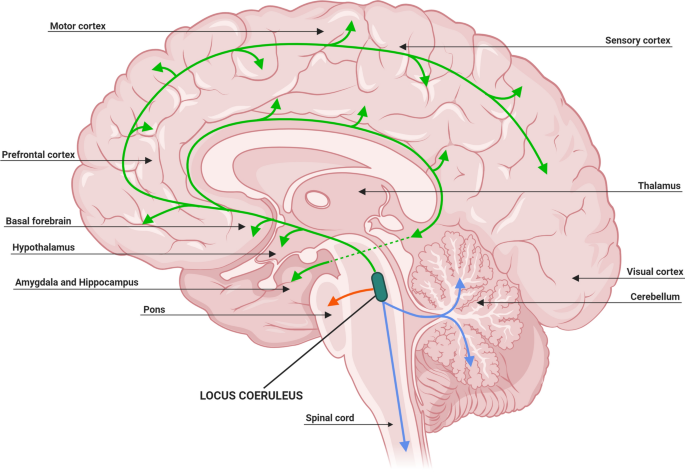
While the clinical syndrome of PD was initially attributed to basal ganglia dysfunction, human postmortem and animal model studies have subsequently shown that non-dopaminergic neurons in other brain regions (such as vagus dorsal motor nucleus, locus coeruleus and raphe nuclei) are also involved.16 These areas in the brain stem have been proposed to degenerate long before substantia nigra. Although this Braak hypothesis has been challenged,17 it is now well accepted that the involvement of non-dopaminergic pathways in the evolution of PD accounts for the increasingly recognised non-motor symptoms that adversely impact the quality of life of patients with PD.18–20 The involvement of noradrenergic, glutamatergic, serotonergic and adenosine pathways, among others, provides a biological basis for the various non-motor symptoms and suggests that modulation of these non-dopaminergic pathways can lead to alternative therapeutic approaches.21
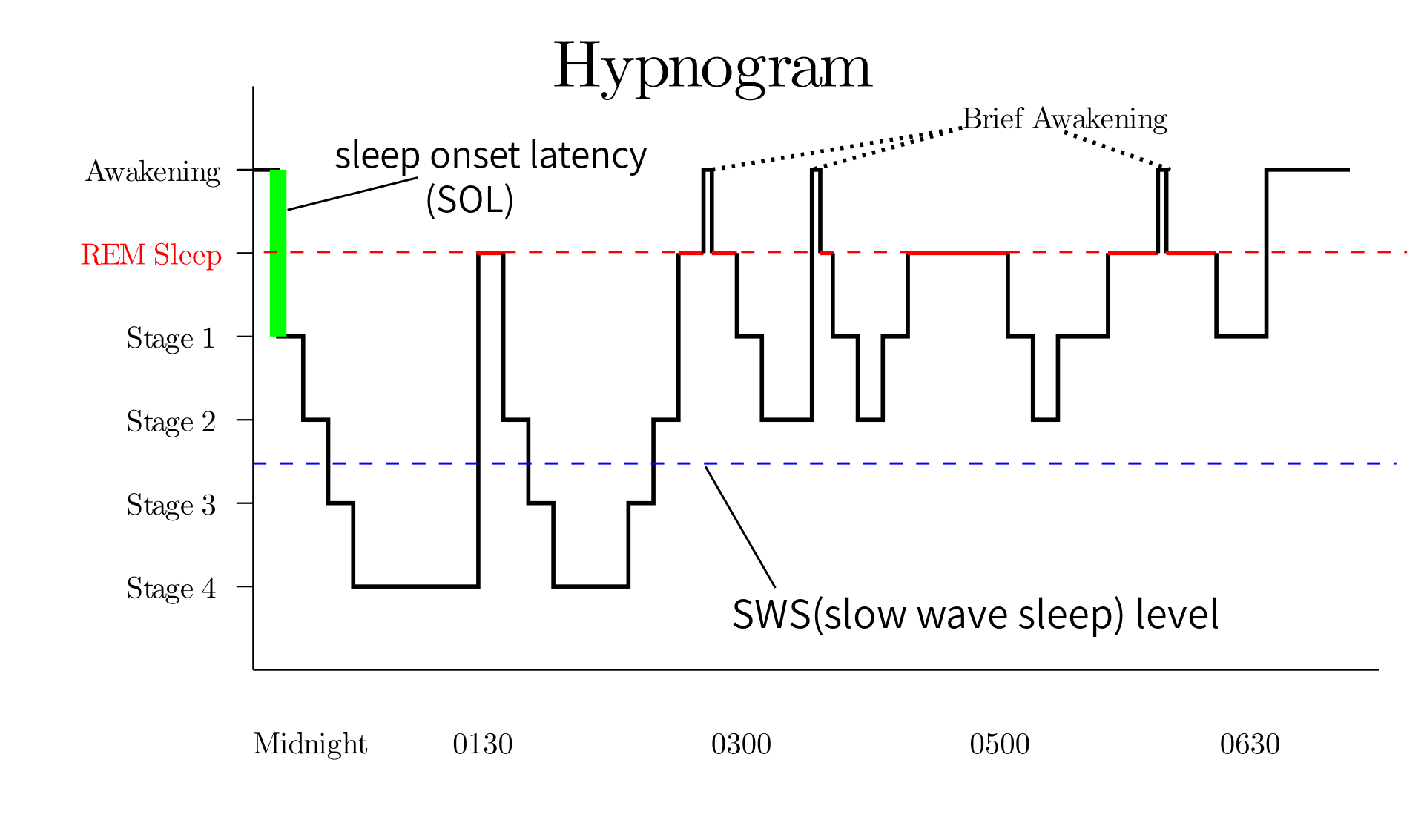
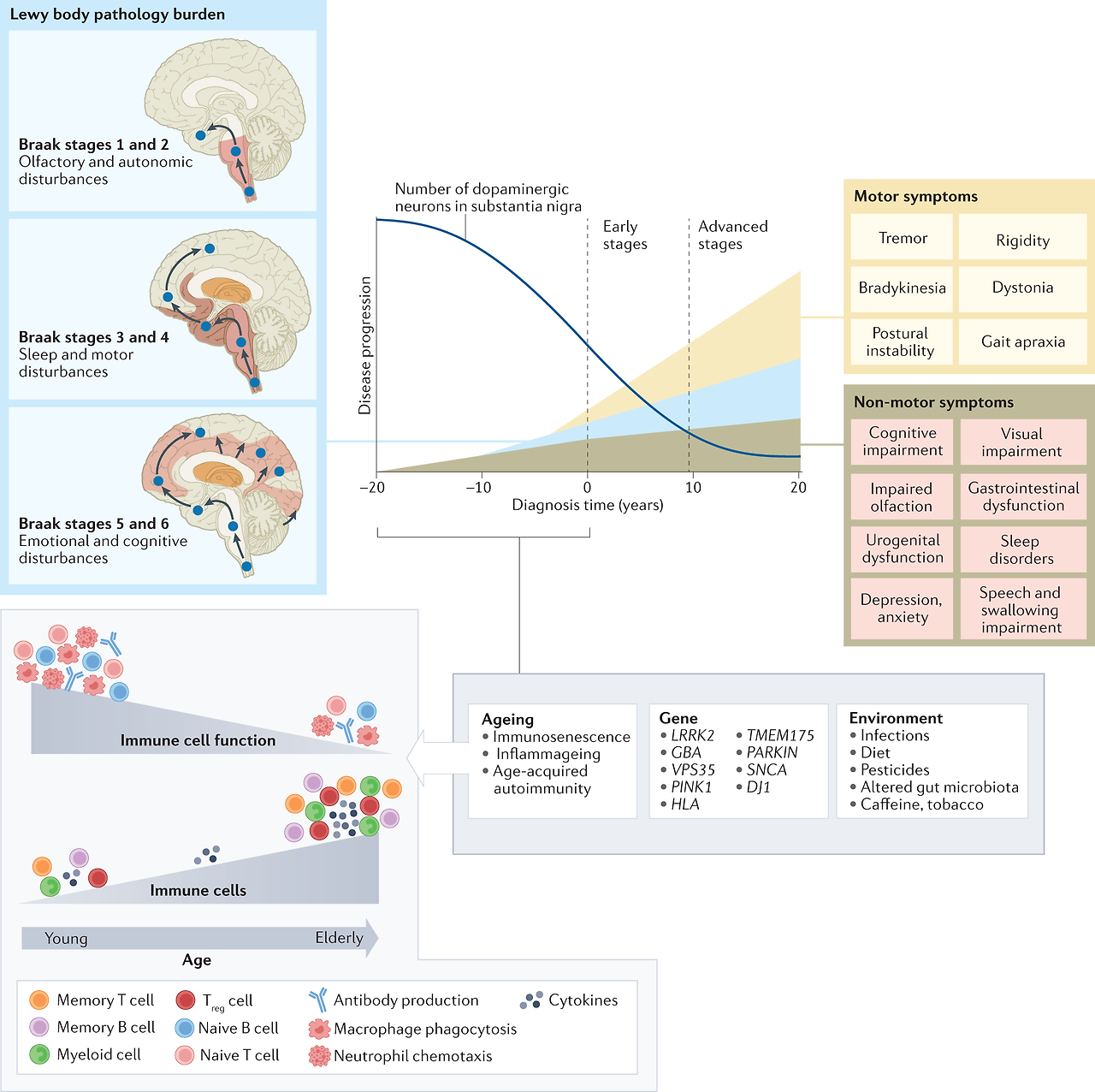
The term ‘prodromal’ PD refers to a phase (up to 15–20 years before onset of motor symptoms) during which clinical signs of disease are not evident but underlying neurodegeneration has started and progressed22 (figure 1). Clinical studies have shown that rapid eye movement sleep behaviour disorder (RBD), depression, olfactory dysfunction, constipation and autonomic dysfunction may be present during this period.23 24 The 2019 Movement Disorders Society diagnostic criteria for prodromic PD have added other new markers (such as diabetes mellitus and physical inactivity), facilitating a web-based calculation of prodromic risk.25 The list of potential clinical, biochemical, imaging and genetic risk markers will likely continue to increase in the future.
영국 파킨슨병 학회 뇌 은행의 임상 기준에 따르면
파킨슨병으로 추정되는 경우
서동증과 경직, 4~6Hz 안정 시 진전 또는 자세 불안정성 중 하나의 특징이 있어야 하며,
이 외에도 세 가지 보조적인 특징이 필요합니다.1
국제 파킨슨병 및 운동장애 학회(MDS)는
(1) 파킨슨병(서동증과 안정 시 진전 또는 경직 중 하나 이상),
(2) 절대 배제 기준의 부재,
(3) 지지 기준,
(4) 위험 신호 없음 등을 포함하는 자체 임상 진단 기준을 개발했습니다.14
다양한 임상 등급 척도, 특히 질병의 중증도를 평가하는 데 사용되는
통합 파킨슨병 등급 척도(UPDRS) 외에도
진단을 지원하고 질병의 경과를 추적하기 위해 신뢰할 수 있는
진단, 전조 증상 및 진행 바이오마커가 개발되고 있습니다.15
PD의 임상 증후군은 처음에는
기저핵 기능 장애에 기인했지만,
이후 인간 사후 및 동물 모델 연구에 따르면
다른 뇌 영역(미주 등 운동핵, 소교차핵 및 라페핵 등)의
비도파민성 신경세포도 관여하는 것으로 나타났습니다.16
뇌간의 이러한 영역은
흑질보다 훨씬 전에 퇴화하는 것으로 제안되어 왔습니다.
이 브라크 가설은 도전을 받아왔지만,17
이제는 PD의 진화에 비도파민성 경로의 관여가
PD 환자의 삶의 질에 악영향을 미치는
비운동 증상에 대해 점점 더 잘 인식되고 있습니다.18-20
특히
노르아드레날린, 글루탐산, 세로토닌 및 아데노신 경로의 관여는
다양한 비운동 증상에 대한 생물학적 기반을 제공하고
이러한 비도파민성 경로의 조절이 대체 치료 접근법으로 이어질 수 있음을 시사합니다.21
'전구' PD라는 용어는
질병의 임상 징후는 뚜렷하지 않지만
근본적인 신경 퇴행이 시작되고 진행되는 단계(운동 증상 발병 전 최대 15~20년)를 의미합니다22 (그림 1).
임상 연구에 따르면 이 시기에는
안구 운동 수면 행동 장애(RBD),
우울증,
후각 기능 장애,
변비 및 자율 기능 장애가 나타날 수 있습니다.23 24
2019년 운동 장애 학회의 전구기 PD 진단 기준에는
다른 새로운 마커(당뇨병 및 신체 비활동성 등)가 추가되어
웹 기반 전구기 위험 계산이 용이해졌습니다.25
잠재적인 임상, 생화학, 영상 및 유전 위험 마커 목록은
향후 계속 늘어날 가능성이 높습니다.
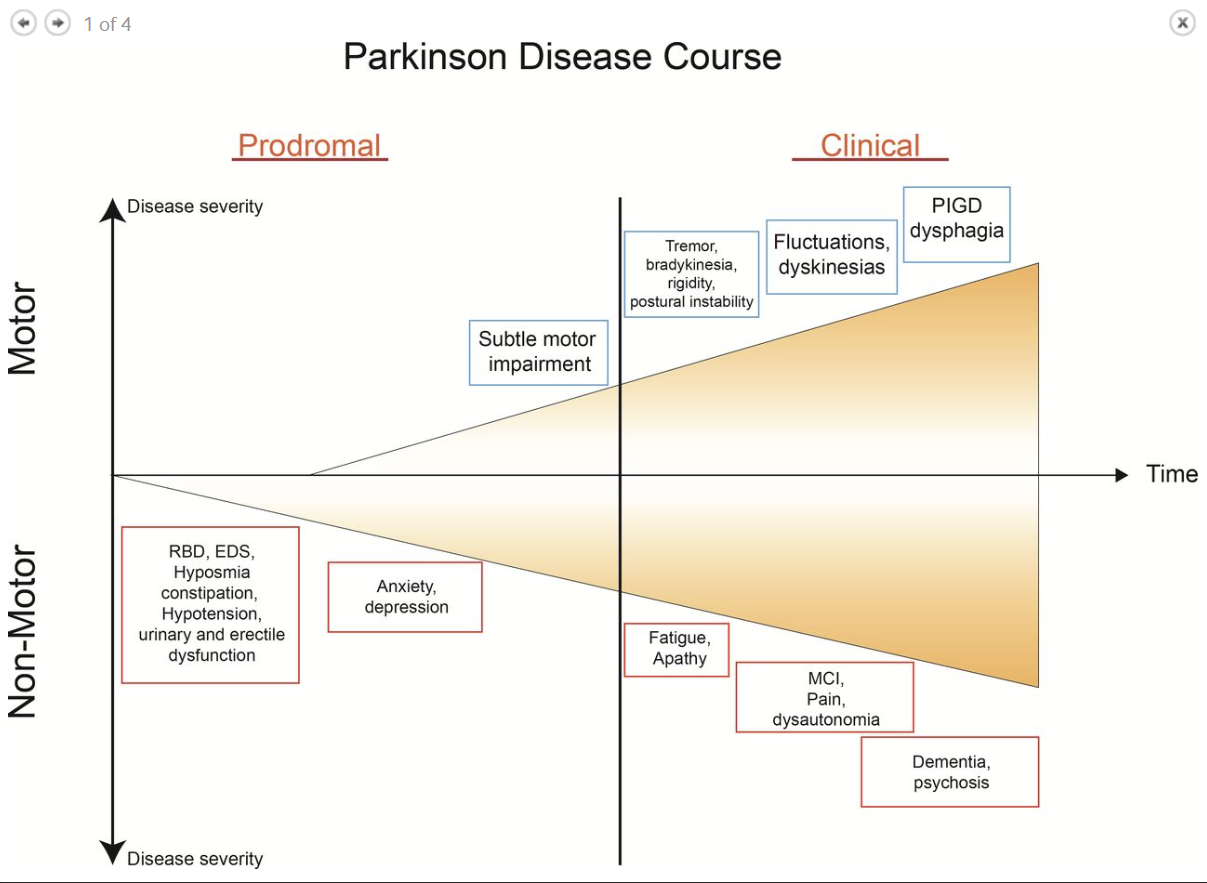
Figure 1
Course of PD from prodromal phase to clinical phase, including levodopa-related complications. PD, Parkinson’s disease; PIGD, postural-instability-gait-disorder; RBD, rapid eye movement sleep behaviour disorder.
Etiology
The relative contribution of genes and environmental/lifestyle factors in pathogenesis of PD has been debated. With median age at onset at 60 years, age is the single most important risk factor for PD.26 27 The frequency appears higher in men compared with women (ratio ranges from 1.3 to 2.0) but the incidence may be influenced by differences in prevalence of variables such as cigarette smoking behaviour, use of postmenopausal hormones and caffeine intake (see section on lifestyle and protective factors).26 Like in other neurodegenerative diseases, age-related biological dysfunction including telomere dysfunction, genomic instability, epigenetic changes, ubiquitin-proteasome and autophagy-lysosomal system, and mitochondrial defects, may underpin and facilitate neuronal demise.28 29
Subtypes of PD have been proposed, categorising patients according to distinct clinical clusters, such as tremor-dominant and postural-instability-gait-disorder (PIGD) subtypes.30–33 Many studies have found that the PIGD phenotype is characterised by more severe disease manifestation and more rapid progression than the tremor-dominant for of PD. It has been suggested the clinical subtypes both determine the phenotype and natural progression/prognosis and also reflect underlying and distinct pathogenic mechanisms. This concept, however, has been challenged because motor subtypes are not fixed but change with progression of the disease and with treatment.34–36
pd의 발병에 있어
유전자와 환경/생활습관 요인의 상대적 기여도에 대한 논의가 계속되고 있습니다.
발병 연령의 중앙값은 60세로,
나이가 PD의 가장 중요한 단일 위험 요인입니다.26 27
여성에 비해 남성에서
발병 빈도가 더 높지만(비율 범위는 1.3~2.0),
흡연 행위,
폐경 후 호르몬 사용 및 카페인 섭취와 같은 변수의
유병률 차이에 영향을 받을 수 있습니다(라이프스타일 및 보호 요인 섹션 참조). 26
다른 신경 퇴행성 질환과 마찬가지로
텔로미어 기능 장애,
게놈 불안정성,
후성유전학적 변화,
유비퀴틴-프로테아좀 및 자가포식-리소좀 시스템,
미토콘드리아 결함 등
노화와 관련된 생물학적 기능 장애는
신경세포 사멸을 뒷받침하고 촉진할 수 있습니다.28 29


떨림 우세형 및
자세 불안정 보행 장애(PIGD) 아형과 같은
뚜렷한 임상 군집에 따라 환자를 분류하는
파킨슨병의 하위 유형이 제안되었습니다.30-33
tremor-dominant and
postural-instability-gait-disorder (PIGD) subtypes
많은 연구에 따르면
PIGD 표현형은 떨림 우세형보다
더 심각한 질병 증상과 더 빠른 진행이 특징입니다.
임상 아형이 표현형과
자연 진행/예후를 결정하고
근본적이고 뚜렷한 병원성 메커니즘을 반영한다고 제안되었습니다.
그러나
운동 아형은
고정된 것이 아니라
질병의 진행과 치료에 따라 변화하기 때문에
이 개념은 도전받고 있습니다.34-36
Environmental risk factors
The potential cause and effect relationship between etiologic factors and disease has been traditionally explored through clinical association studies using a cross-sectional (hospital and community-based) or prospective (population-based) methodology. Several risk factors have been implicated including pesticide and heavy metal exposure, rural living, agricultural occupation, traumatic head injury, history of melanoma, consumption of dairy products, type 2 diabetes mellitus (reduced by the use of antidiabetic drugs), among many others26 (figure 2). Although these links are supported by underling biological plausibility, a number of the observations cannot be consistently replicated. A recent meta-analysis which involved both quantitative and qualitative analyses of various environmental exposures suggests a lack of robust consistency in some of these associations (such as rural living, well-water consumption, farming and pesticide exposure).37 While other meta-analyses reaffirmed a positive association with pesticide exposure,38 others found lack of support for a link with traumatic head injury.39 Due to several challenges and inherent limitations, it is not surprising that such epidemiological studies sometimes give conflicting results.
병인 요인과 질병 사이의 잠재적인 인과 관계는
전통적으로 횡단면(병원 및 지역사회 기반) 또는
전향적(인구 기반) 방법론을 사용한
임상 연관성 연구를 통해 탐구되어 왔습니다.
살충제 및 중금속 노출, 농촌 거주, 농업 직업,
외상성 두부 손상, 흑색종 병력,
유제품 섭취, 제2형 당뇨병(항당뇨병제 사용으로 감소) 등
이러한 연관성은
생물학적 타당성을 바탕으로 뒷받침되지만,
관찰 결과 중 상당수는 일관되게 재현할 수 없습니다.
다양한 환경 노출에 대한 양적 및 질적 분석을 모두 포함한 최근 메타 분석에 따르면 이러한 연관성 중 일부(농촌 거주, 우물물 소비, 농업 및 살충제 노출 등)에서 강력한 일관성이 부족하다고 합니다.37 다른 메타 분석에서는 살충제 노출과 긍정적인 연관성을 재확인한 반면,38 다른 연구에서는 외상성 머리 손상과의 연관성을 뒷받침하지 못했습니다.39 몇 가지 과제와 내재된 한계로 인해 이러한 역학 연구에서 때로 상충되는 결과가 나오는 것은 놀라운 일이 아닙니다.
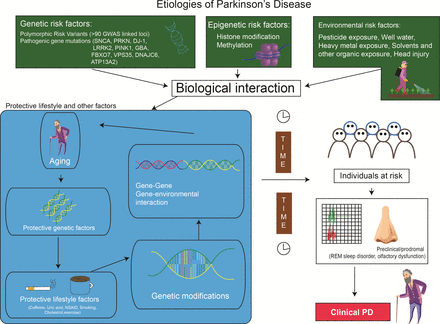
Figure 2
Etiologies of PD: biologic interaction between genetic, epigenetic and environmental factors. PD, Parkinson’s disease; REM, rapid eye movement.
Lifestyle and other protective factors
Cigarette smoking and caffeine consumption are the two most consistent protective factors associated with a reduced risk of PD.26 37 Other reported associations include higher serum urate, ibuprofen use and exercise, among others.26 The negative association between cigarette smoking and PD is most intriguing. This inverse relationship is not easily explained, but some have suggested that PD-related cautious personality (avoidance trait) predisposes some individuals to quitting neuroprotective smoking as the biological mechanism involved in PD.40 The other hypothesis links nicotine to dopaminergic neuronal protection since it has been shown to stimulate the release of dopamine in the striatum and preserve dopaminergic function in experimental models. It is also possible that there are other unidentified neuroprotective components in cigarette smoke.
The relative risk reduction of PD among caffeine drinkers is between 0.5 and 0.8 and, similar to smoking, a dose-dependent effect has been consistently demonstrated in most studies.26 Caffeine, an antagonist of adenosine A2a receptor, has been postulated to exert neuroprotective role by blocking this receptor. In addition to caffeine, it is possible that antioxidants present in some beverages (such as tea) may contribute to a protective effect among black tea drinkers, independent of caffeine.
Uric acid, a product of purine metabolism is an antioxidant with radical scavenger properties. A meta-analysis involving 13 studies has demonstrated that serum uric acid is lower in PD compared with controls, with the same pattern in those with more severe diseases compared with early stage PD.41 However, the Copenhagen General Population Study and some other studies have shown no causal relationship, suggesting that some unknown confounders exist.42 Ibuprofen has been suggested to lower PD risk though the association with other non-steroidal anti-inflammatory drugs has not been consistent.26 Statin use and lipid levels have also been extensively studied. However, the relative role of hydrophilic and hydrophobic statins, types of lipid levels and their specific interactions have not been conclusively addressed and methodological differences make it difficult to draw definitive conclusions.43
흡연과 카페인 섭취는
PD 위험 감소와 관련된
가장 일관된 두 가지 보호 요인입니다.26 37
기타 보고된 연관성으로는
혈청 요산염 증가,
이부프로펜 사용 및
운동 등이 있습니다.26
흡연과 PD 사이의 부정적인 연관성은
가장 흥미롭습니다.
이러한 역관계는 쉽게 설명할 수 없지만,
일부에서는 PD와 관련된 신중한 성격(회피 특성)이
PD와 관련된 생물학적 기전으로
일부 개인이 신경 보호 흡연에 취약하다고 제안했습니다.40
다른 가설은
실험 모델에서
니코틴이
도파민의 방출을 자극하고
도파민 기능을 보존하는 것으로 나타났기 때문에
니코틴과 도파민 신경 보호와의 연관성이 있다는 가설입니다.
담배 연기에는
확인되지 않은
다른 신경 보호 성분이 있을 가능성도 있습니다.
카페인을 마시는 사람의 PD의 상대적 위험 감소는
0.5에서 0.8 사이이며,
흡연과 마찬가지로
대부분의 연구에서 용량 의존적 효과가 일관되게 입증되었습니다.26
아데노신 A2a 수용체의 길항제인
카페인은
이 수용체를 차단하여
신경 보호 역할을 하는 것으로 가정하고 있습니다.
카페인 외에도
일부 음료(예: 홍차)에 함유된 항산화 물질이
카페인과 무관하게 홍차를 마시는 사람들의 신경 보호 효과에 기여할 수 있습니다.
퓨린 대사의 산물인
요산은
라디칼 제거 기능이 있는
항산화제입니다.
13개의 연구가 포함된 메타 분석에 따르면,
혈청 요산은 대조군에 비해 PD에서 더 낮으며,
초기 PD에 비해 더 심각한 질병을 가진 사람들에게서
동일한 패턴이 나타납니다.41
https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0173731


그러나
코펜하겐 일반 인구 연구와 일부 다른 연구에서는
인과 관계가 없는 것으로 나타나 알려지지 않은
교란 요소가 존재함을 시사합니다.42
이부프로펜은
다른 비스테로이드 항염증제와의 연관성은 일관되지 않았지만
PD 위험을 낮추는 것으로 제안되었습니다.26
스타틴 사용과 지질 수치도
광범위하게 연구되어 왔습니다.
그러나
친수성 및 소수성 스타틴의 상대적 역할,
지질 수치의 유형 및 특정 상호 작용은 결
정적으로 다루어지지 않았으며
방법론적 차이로 인해 확실한 결론을 내리기 어렵습니다.43
Genetics
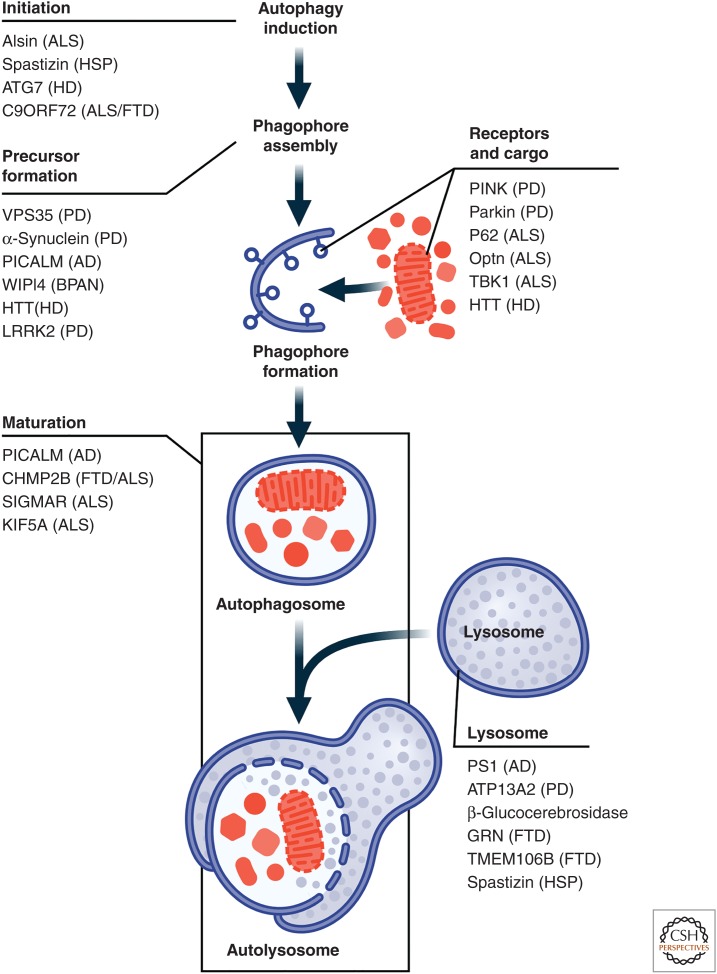
Early twin studies and the identification of several families exhibiting a Mendelian inheritance pattern (dominant and recessive) provided evidence for genetic causes of PD which culminated in the discovery of the first PD-related gene, α-synuclein (SNCA), in 1997.10 A year later, mutation in Parkin (PRKN), linked to autosomal recessive form of PD, was identified.44 The nomenclature of assigning a ‘PARK’ number to these genes has been confusing and we, therefore, prefer the classification proposed by the International Parkinson and Movement Disorders Society using the gene names.6
With the advancement of genetic techniques and population studies, including genome-wide association studies (GWAS), over 20 monogenic forms of PD have been described and over 100 loci have been identified as risk factors for PD.44–47 It is daunting to decipher clinically useful information from the huge amount of published clinical and genetic data that may help differentiate the various forms of genetic parkinsonism.47 Here, we highlight some of the distinguishing features of the more important monogenetic PD disorders.
초기 쌍둥이 연구와 멘델 유전 패턴(우성 및 열성)을 보이는 여러 가족을 확인함으로써 PD의 유전적 원인에 대한 증거가 제공되었고, 1997년 최초의 PD 관련 유전자인 α-시누클레인(SNCA)이 발견되었습니다.10 1년 후, 상 염색체 열성 형태의 PD와 관련된 파킨(PRKN)의 돌연변이가 확인되었습니다.44 이러한 유전자에 'PARK' 번호를 부여하는 명명법은 혼란스러웠기 때문에 국제 파킨슨병 및 운동 장애 학회에서 유전자 이름을 사용하여 제안한 분류를 선호합니다.6
유전 기법과 게놈 전체 연관성 연구(GWAS)를 포함한 인구 연구의 발전으로 20가지 이상의 단일 유전자 형태의 파킨슨병이 기술되었고 100개 이상의 유전자좌가 파킨슨병의 위험 요인으로 확인되었습니다.44-47 다양한 형태의 유전성 파킨슨병을 감별하는 데 도움이 되는 방대한 양의 임상 및 유전 데이터에서 임상적으로 유용한 정보를 해독하는 것은 어려운 일입니다.47 다음은 보다 중요한 단일 유전자 PD 장애의 특징 중 일부를 강조하는 내용입니다.
PARK-SNCA (PARK1)
Although SNCA mutations are a rare cause of PD, the pivotal role of α-synuclein in the pathogenesis of PD is now clearly recognised.16 17 48–50 A small protein, α-synuclein (140 amino acids) is involved in (1) vesicle trafficking; (2) vesicle docking and priming; (3) vesicle fusion and neurotransmitter release and (4) axonal transport, but its function in normal brain is not fully understood. Overexpression of α-synuclein in transgenic mice can cause levodopa-responsive motor impairment and nigral degeneration. The protein’s toxicity has been demonstrated with excessive amounts of wild-type (multiplication), pathogenic mutations and modification by dopamine (toxic interactions between α-synuclein oligomers and lipids). Importantly, α-synuclein (non-soluble, aggregated, fibrillar form) is a major component of Lewy bodies and Lewy neurites, the pathological hallmarks of PD. Many studies have shown that α-synuclein, pathology spreads from peripheral nervous system and olfactory bulb and then propagates from caudal brainstem rostrally (see below discussion of Braak hypothesis and staging).
Despite the rarity of SNCA mutations, the discovery of whole gene duplication, triplication and quadriplication provides considerable insights into the underlying pathogenesis involving SNCA protein and also supports earlier observations that SNCA promoter polymorphic variant increases risk in sporadic PD. SNCA triplication is associated with early onset disease (compared with duplication carriers) and cognitive impairment suggesting a gene dosage effect.
SNCA 돌연변이가 파킨슨병의 드문 원인이지만,
파킨슨병 발병에서 α-시누클레인의 중추적인 역할은
이제 명확하게 인식되고 있습니다.16 17 48-50
작은 단백질인 α-시누클레인(140 아미노산)은
(1) 소포 이동,
(2) 소포 도킹 및 프라이밍,
(3) 소포 융합 및 신경 전달물질 방출,
(4) 축삭 수송에 관여하지만 정상 뇌에서의 기능은 완전히 이해되지 않고 있습니다.
형질전환 마우스에서 α-시누클레인의 과발현은
레보도파 반응성 운동 장애와
흑질 퇴행을 유발할 수 있습니다.
이 단백질의 독성은
과도한 양의 야생형(증식),
병원성 돌연변이 및 도파민에 의한 변형(α-시누클레인 올리고머와 지질 간의 독성 상호작용)을 통해
입증되었습니다.
중요한 것은
α-시누클레인(비용해성, 응집된 원섬유 형태)이
루이체와 루이 뉴라이트의 주요 구성 요소이며,
이는 PD의 병리학적인 특징이라는 점입니다.
많은 연구에 따르면
α-시누클레인은 말초 신경계와 후각 전구에서 병리가 퍼진 다음
꼬리 뇌간에서 등쪽으로 전파되는 것으로 나타났습니다(아래 Braak 가설 및 병기 논의 참조).
SNCA 돌연변이는 드물지만
전체 유전자 복제, 삼중화 및 사중화의 발견은
SNCA 단백질과 관련된 근본적인 병인에 대한 상당한 통찰력을 제공하며
SNCA 프로모터 다형성 변이가
산발성 PD의 위험을 증가시킨다는 이전의 관찰을 뒷받침합니다.
SNCA 삼중화는
조기 발병 질환(중복 보인자와 비교) 및
인지 장애와 관련이 있으며,
이는 유전자 용량 효과를 시사합니다.
PARK-Parkin (PARK2)
Parkin (PRKN) is the most common autosomal recessive PD-related gene; compound heterozygotes for PRKN account for nearly 50% of patients with early onset PD.7 44 The disease may present with dystonic gait, leg tremor at rest and on standing, cervical dystonia, dopa-responsive dystonia. freezing, festination, retropulsion, marked sleep benefit, hyperreflexia, ataxia, peripheral neuropathy and dysautonomia. Although usually symmetrical, it may be rarely present as hemiparkinsonism-hemiatrophy. Excellent levodopa response is typically complicated by early development of levodopa-induced dyskinesia. At autopsy, there is typically loss of neurons in substantia nigra pars compacta, but the dorsal tier is well preserved and Lewy bodies are rarely present.
파킨(PRKN)은
가장 흔한 상염색체 열성 PD 관련 유전자로,
PRKN의 복합 이형접합체는
초기 발병 PD 환자의 거의 50%를 차지합니다.7 44
이 질환은
근긴장성 보행,
안정 시 및 서 있을 때의
다리 떨림, 경부 근긴장 이상증, 도파 반응성 이상증, 동
결, 축제, 역추진, 뚜렷한 수면 효과, 과다 반사, 운동 실조증, 말초 신경증 및 이상 자율 감각증 등을 나타낼 수 있습니다.
대개 대칭적으로 나타나지만
드물게 반파킨슨증-조혈 위축증이 나타날 수 있습니다.
우수한 레보도파 반응은
일반적으로 레보도파 유발 운동 이상증의 조기 발생으로 인해 복잡해집니다.
부검 시 일반적으로 흑질 흑질의 신경세포 손실이 있지만
등쪽 층은 잘 보존되어 있고
루이체는 거의 존재하지 않습니다(중복 운반자 포함) 및 인지 장애는 유전자 투여 효과를 시사합니다.
PARK-LRRK2 (PARK8)
LRRK2 is the most common autosomal dominant PD-related gene and a common mutation (G2019S) has been identified in both familial and sporadic PD with age-dependent penetrance.7 47 G2019S mutation accounts for 1%–3% of sporadic PD and 3%–4% familial PD in most Caucasian populations and up to 40% in North African Berbers, Iberian and Ashkenazi Jews populations.7 47 51 This mutation is largely absent in Asians who have a 5%–10% carrier rate of the ethnic specific coding risk variants (G2385R and R1628P).7 12 47 51 Most LRRK2 carriers are of late onset simulating typical PD, and clinically indistinguishable from non-carriers, but seem to have more benign course, manifested chiefly by PIGD phenotype, with less RBD and relatively preserved olfaction. Atypical features include orthostatic hypotension, dementia, hallucinations, corticobasal syndrome and primary progressive aphasia. Pathology is quite heterogeneous; it may or not include Lewy bodies and may overlap with synucleinopathies and tauopathies.
A large protein (2527 amino acids), also referred to as ‘dardarin’ (meaning tremor), LRRK2 is involved in vesicular trafficking, autophagy, protein synthesis and cytoskeletal function; it also interacts with mitochondrial proteins and may be involved in immune system. LRRK2 is highly expressed in the medium-sized spiny neurons of the striatum; also in macrophages and microglia suggesting an involvement in inflammatory pathways. Mutational hotspots are mainly in the functional domains (Kinase and Roc-Cor) suggesting a dysregulation of the kinase and GTPase activities, with a toxic gain of function as a possible underpinning mechanism.7 12
LRRK2는 가장 흔한 상염색체 우성 PD 관련 유전자이며 가족성 및 산발성 PD 모두에서 연령에 따른 침투력을 가진 공통 돌연변이(G2019S)가 확인되었습니다.7 47 대부분의 백인 인구에서 산발성 PD의 1~3%, 가족성 PD의 3~4%를 차지하고 북아프리카 베르베르, 이베리아 및 아시케나지 유대인 인구에서는 최대 40%까지 발생합니다.7 12 51 47 51 이 돌연변이는 인종 특이적 코딩 위험 변이(G2385R 및 R1628P)의 보인자 비율이 5~10%인 아시아인에서는 거의 나타나지 않습니다.7 12 47 51 대부분의 LRRK2 보인자는 전형적인 PD와 유사한 후기 발병이며 임상적으로 비보유자와 구별되지 않지만 주로 PIGD 표현형으로 나타나는 양성 경과가 더 많으며 RBD가 적고 상대적으로 후각이 보존되는 것처럼 보입니다. 비정형적인 특징으로는 기립성 저혈압, 치매, 환각, 피질 기저 증후군 및 원발성 진행성 실어증이 있습니다. 병리학은 매우 이질적이며 루이체를 포함하거나 포함하지 않을 수 있으며 시뉴클레인 병증 및 타우 병증과 겹칠 수 있습니다.
'다다린'(떨림을 의미)이라고도 하는 대형 단백질(2527개 아미노산)인 LRRK2는 소포 이동, 자가포식, 단백질 합성 및 세포 골격 기능에 관여하며 미토콘드리아 단백질과 상호작용하고 면역 체계에도 관여할 수 있습니다. LRRK2는 선조체의 중간 크기의 가시 뉴런과 대식세포 및 미세아교세포에서 높게 발현되어 염증 경로에 관여할 수 있음을 시사합니다. 돌연변이 핫스팟은 주로 기능 영역(키나아제 및 Roc-Cor)에 있으며, 이는 키나아제 및 GTPase 활성의 조절 장애를 시사하며, 독성 기능 증가가 가능한 기저 메커니즘으로 추정됩니다.7 12

PARK-GBA
Glucocerebrosidase (GBA) gene, located on chromosome 1q21, encodes the lysosomal enzyme glucocerebrosidase that decomposes glucocerebroside into glucose and ceramide and plays an important role in sphingolipid degradation. Homozygous or compound heterozygous mutations of this gene are linked to Gaucher’s disease, the most prevalent lysosomal storage disorder. Due to low glucocerebrosidase enzymatic activity, Gaucher’s disease is associated with elevated serum chitotriosidase and glucocerebroside accumulation in the spleen, liver and bone marrow, and an increased risk of PD. Heterozygous, homozygous or compound heterozygous mutations of the GBA gene represent the single most important genetic risk factor of PD in the general population, conferring more than five times increased risk of PD.52 53 Common pathogenic variants include p.N370S, p.E326K and p.T369 M with effect sizes between 2.6 and 0.9 year reduction in age-at-onset. GBA mutations are found in 10% of sporadic PD and in over 40% of familial PD in Ashkenazi Jewish patients.54 Genetic modifiers of GBA-associated PD are being investigated in several large GWAS and other studies.55 PARK-GBA has a younger age at onset, higher prevalence of cognitive impairment and of RBD than in typical PD (in non-carriers). It has been postulated that loss-of-function of glucocerebrosidase leads to impaired lysosomal enzyme function followed by α-synuclein accumulation and aggregation. Indeed, postmortem studies of brains from PD patients with GBA mutations show Lewy bodies in cortical areas in addition to the classic PD pathology.
The rest of the pathogenic autosomal dominant (VPS35, EIF4G1, DNAJC13, CHCHD2) and autosomal recessive (PINK1, DJ1, ATP13A2, GIGYF2, PLA2G6, FBXO7, DNJAC6, SYNJ1, VPS13C) genes are quite rare and are often manifested by atypical features.3 4 40 With the easy access to direct consumer genetic testing kits and whole exome and genome sequencing services, the reported variants of uncertain significance are often difficult to interpret.56 57
1q21 염색체에 위치한 글루코세레브로시다아제(GBA) 유전자는 글루코세레브로사이드를 포도당과 세라마이드로 분해하는 리소좀 효소 글루코세레브로시다아제를 암호화하며 스핑고지질 분해에 중요한 역할을 합니다. 이 유전자의 동형접합 또는 복합 이형접합 돌연변이는 가장 흔한 리소좀 저장 장애인 고셔병과 관련이 있습니다. 글루코세레브로시다아제 효소 활성도가 낮기 때문에 고셔병은 비장, 간, 골수에서 혈청 키토트리오시다아제 및 글루코세레브로사이드 축적 증가와 PD 위험 증가와 관련이 있습니다. GBA 유전자의 이형접합, 동형접합 또는 복합 이형접합 돌연변이는 일반 인구에서 PD의 가장 중요한 단일 유전적 위험 요인으로, PD 위험을 5배 이상 증가시킵니다.52 53 일반적인 병원성 변이로는 p.N370S, p.E326K 및 p.T369 M이 있으며 발병 연령 감소 효과 크기는 2.6년에서 0.9년 사이입니다. GBA 돌연변이는 산발성 파킨슨병의 10%와 아시케나지 유대인 환자의 가족성 파킨슨병의 40% 이상에서 발견됩니다.54 GBA 관련 파킨슨병의 유전적 변형자는 여러 대규모 GWAS 및 기타 연구에서 조사되고 있습니다.55 PARK-GBA는 일반적인 파킨슨병(비보유자)보다 발병 연령이 더 젊고 인지 장애 및 RBD 유병률이 더 높습니다. 글루코세레브로시다아제의 기능 상실은 리소좀 효소 기능 장애와 α-시누클레인 축적 및 응집으로 이어진다는 가설이 제기되었습니다. 실제로 GBA 돌연변이가 있는 PD 환자의 뇌를 사후에 연구한 결과, 전형적인 PD 병리 외에도 피질 영역에서 루이체가 발견되었습니다.
나머지 병원성 상염색체 우성(VPS35, EIF4G1, DNAJC13, CHCHD2) 및 상염색체 열성(PINK1, DJ1, ATP13A2, GIGYF2, PLA2G6, FBXO7, DNJAC6, SYNJ1, VPS13C) 유전자는 매우 드물고 종종 비정형적인 특징으로 나타납니다.3 4 40 소비자가 직접 유전자 검사 키트와 전체 엑솜 및 게놈 시퀀싱 서비스를 쉽게 이용할 수 있게 되면서, 보고된 중요도가 불확실한 변이는 종종 해석하기 어려운 경우가 많습니다.56 57
Pathophysiologic mechanisms
It is well recognised in human postmortem studies that PD patients have neuronal loss in the substantia nigra par compacta, locus ceruleus and other neuronal populations.58 The Braak hypothesis suggests that the early pathological changes occur in the medulla oblongata and olfactory bulb (Braak stages 1 and 2) before advancing rostrally to substantia nigra and midbrain (Braak stages 3 and 4) by which time clinical symptoms and signs are likely to be present; in late stages, the cortical regions eventually become affected (Braak stages 5 and 6).
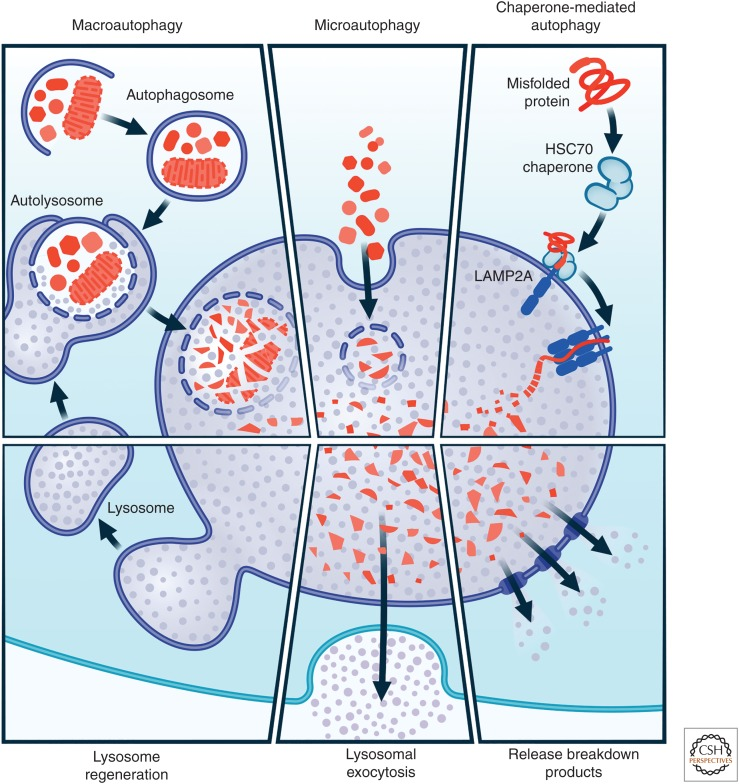
It is beyond the scope of this review to describe in detail the various possible pathophysiologic mechanisms. However, regardless of the underpinning etiologies (environmental, genetic or other risk factors), several key molecular events and hallmarks have been consistently reported in human postmortem tissues, in vitro human cells lines, human brain organoids and animal models (1A). These include α-synuclein misfolding and aggregation, mitochondrial dysfunction, impairment of protein clearance (involving key ubiquitin-proteasome and autophagy-lysosomal systems), neuroinflammation and oxidative stress (figure 3). These major molecular and cellular hallmarks are often associated with many other interlinked events including vesicular transport disruption, loss of microtubular integrity, neuronal excitotoxicity, disruption of trophic factors, iron metabolic pathway dysregulation, endoplasmic reticulum impairment, poly (ADP-ribose) polymerase and other enzymatic activation, among several others. Axonal mitochondria are particularly vulnerable and their dysfunction can contribute to impaired axonal transport and some have suggested that distal axons in the striatum may be the initial site of neurodegeneration in PD.59 Essentially, all of these mechanisms potentially promote programmed cell death (apoptosis) or necrosis. As cellular processes are dynamic and neurodegeneration occurs over a prolonged period of insults/stresses and with various compensatory mechanisms at play, it is impossible to determine with any certainty if these pathways work independently or converge to a single route to neuronal death. It is more likely that the various pathophysiologic processes intersect with each other, resulting in a viscous cascade of insults and ultimately irreversible cellular damage.
인간 사후 연구에서 PD 환자는
흑질상핵, 세쿨루스 및 기타 신경세포 집단에서
신경세포 손실이 있다는 것이 잘 알려져 있습니다.58
브라크 가설에 따르면
초기 병리학적인 변화는
흑질과 중뇌(브라크 1, 2단계)로 진행하기 전에
흑질 수질과 후각구(브라크 3, 4단계)에서 발생하며,
이때 임상 증상과 징후가 나타날 가능성이 높고
말기에는 결국 피질 영역(브라크 5, 6단계)이 영향을 받는다고 합니다.
가능한 다양한 병리 생리학적 메커니즘을 자세히 설명하는 것은 이 리뷰의 범위를 벗어납니다. 그러나 근본적인 원인(환경적, 유전적 또는 기타 위험 요인)에 관계없이 인간 사후 조직, 시험관 내 인간 세포주, 인간 뇌 오가노이드 및 동물 모델에서 몇 가지 주요 분자적 사건과 특징이 일관되게 보고되고 있습니다(1A).
여기에는
α-시누클레인 오접힘 및 응집,
미토콘드리아 기능 장애,
단백질 제거 장애(주요 유비퀴틴-프로테아좀 및 오토파지-리소좀 시스템 관련),
신경 염증 및 산화 스트레스 등이 포함됩니다(그림 3).
이러한 주요 분자 및 세포적 특징은
소포체 수송 장애,
미세소관 완전성 상실,
신경세포 흥분 독성,
영양 인자 장애,
철 대사 경로 조절 장애,
소포체 손상,
폴리(ADP-리보스) 중합효소 및 기타 효소 활성화 등
여러 가지 다른 상호 연관된 사건과 관련이 있는 경우가 많습니다.
축삭 미토콘드리아는 특히 취약하며 기능 장애는 축삭 수송 장애에 기여할 수 있으며 일부에서는 선조체의 원위 축삭이 PD에서 신경 퇴행의 초기 부위일 수 있다고 제안했습니다.59 기본적으로 이러한 모든 메커니즘은 잠재적으로 프로그램된 세포 사멸(아포토시스) 또는 괴사를 촉진할 수 있습니다.
세포 과정은
역동적이고
신경 퇴화는
장기간의 모욕/스트레스와 다양한 보상 메커니즘이 작용하면서 발생하므로
이러한 경로가 독립적으로 작동하는지 또는
신경 세포 사멸의 단일 경로로 수렴하는지 확실하게 판단하는 것은 불가능합니다.
다양한 병리 생리학적 과정이 서로 교차하여
점성이 있는 연속적인 모욕과
궁극적으로 돌이킬 수 없는
세포 손상을 초래할 가능성이 더 높습니다.
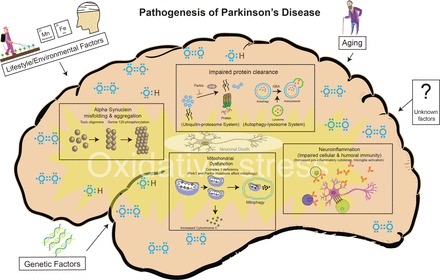
Figure 3
Pathogenesis of PD: a variety of cellular mechanisms on the background of oxidative stress, coupled with again, lifestyle/environmental and genetic factors contribute to the PD-related neurodegeneration. PD, Parkinson’s disease.
We highlight some of the key research pathophysiologic mechanistic insights that may have potential therapeutic implications.
PD의 발병 기전: 산화 스트레스의 배경에 대한 다양한 세포 메커니즘과 다시, 생활 습관/환경 및 유전적 요인이 결합하여 PD 관련 신경 퇴화에 기여합니다. PD, 파킨슨병.
잠재적인 치료적 영향을 미칠 수 있는 주요 연구 병리 생리학적 메커니즘에 대한 통찰력을 강조합니다.
α-시누클레인은 기본적으로 펼쳐져 있으며 특정 생화학적 상호 작용에 따라 3차 구조를 채택합니다. 이 단백질의 비정상적인 응집은 도파민성 뉴런에 독성이 있어 PD와 관련된 신경 퇴행을 유발하는 것으로 밝혀졌습니다. 산화 스트레스, PD 유전자 돌연변이 및 과발현은 α-시누클레인의 형태 변화와 응집에 영향을 미칠 수 있습니다. α-시누클레인은 실험 조건에 따라 다양한 형태/종으로 존재하며, 올리고머 및 피브릴 종의 상대적 독성에 대한 논란이 있습니다.16 이러한 종 중 일부는 신경 염증 반응을 활성화하고 더 중요한 것은 α-시누클레인 병리를 세포에서 세포로 '시드'하고 확산시킬 수 있습니다. 이러한 관찰은 발현 억제부터 올리고머 종 생성 및 세포 전달 감소에 이르는 치료적 접근법의 기초를 제공하며, 이러한 전략 중 일부는 현재 진행 중인 임상 시험으로 전환되었습니다.- PD 환자에서 미토콘드리아 복합체 1 활성의 감소가 발견되었으며, 이 억제제(예: 로테논)를 사용하면 실험적 PD 모델에서 미토콘드리아 손상(사이토크롬 c 방출, 카스파제 캐스케이드 활성화 및 최종 세포 사멸과 같은 미토콘드리아 잠재력 감소)을 일으키는 것으로 밝혀졌습니다. 마찬가지로 미토파지 장애를 포함한 미토콘드리아 기능 장애의 특징은 Parkin, PINK1, DJ1 (7,12,58)과 같은 특정 PD 관련 유전자의 해로운 영향의 결과로 확인되었습니다. 연구자들은 또한 미토콘드리아 손상이 산화 도파민 축적과 글루코세레브로시다아제 감소를 촉진하여 도파민이 α-시누클린 축적과 리소좀 손상 사이의 공통된 연관성이 있음을 보여주었습니다.59 이러한 연구는 향후 미토콘드리아 표적 및 항산화 치료 접근법에 대한 원동력을 제공할 수 있을 것입니다.
- PD 환자에서는 염증성 사이토카인의 증가와 면역 세포 집단(단핵구 및 그 전구체 등)의 변화를 포함하여 선천성 및 적응성 면역 반응 이상이 모두 강조되었습니다.60 이는 자가 면역 질환과 PD 사이의 연관성을 보여주는 임상 연관성 연구, 분자 영상에서 염증 세포 활성화(예: 미세아교세포)의 증거 및 실험적 PD 모델에서 신경 염증의 특징을 통해 더욱 뒷받침되고 있습니다.
- 미주신경이 응집된 α-시누클레인이 위장관에서 하부 뇌간으로 전달되는 '고속도로' 역할을 하는 장-뇌 연결이 PD 발병에 기여하는 요인이라는 증거가 점점 더 많이 나오고 있습니다.61 장과 뇌의 상호작용은 PD 환자에게서 분리한 α-시누클레인을 형질전환 α-시누클레인 생쥐에 이식했을 때 운동 결함이 발생하고 항생제 치료로 일부 결함이 회복되었으며 미생물 재식민화가 병리 생리를 악화시킨 실험에 의해 뒷받침됩니다. 또한, 여러 연구에 따르면 질 절제술과 충수 절제술이 파킨슨병 발병 위험을 잠재적으로 줄일 수 있는 것으로 나타났습니다. PD의 병원성 메커니즘으로서 α-시누클레인 응집과 중추신경계로의 확산을 유발하는 데 있어 위장 미생물과 장내 미생물총, 감염 및 염증의 역할을 더 명확히 규명하기 위해서는 추가 연구가 필요합니다.
https://www.nature.com/articles/s41577-022-00684-6

Treatment
PD is a complex neurodegenerative disorder with a broad spectrum of motor and non-motor features that require individualised therapeutic approach. Clinical trials designed to provide evidence-based data must both include a well-defined population of patients and controls and should also utilise the most objective, reliable and validated tools to assess the effects of the therapeutic intervention. Although a variety of clinical rating scales and other instruments have been utilised in assessing response to various therapies, the UPDRS is used most frequently as the primary outcome measure in various clinical trials.62
An overview of medical and surgical therapeutic options for patients with PD in various stages of their disease is highlighted in table 1 and figure 4. In addition to conventional therapies, we also provide insights into evidence-based63 as well as emerging and experimental therapeutics of PD.
파킨슨병은
광범위한 운동 및 비운동 특징을 가진 복잡한 신경 퇴행성 질환으로,
개별화된 치료적 접근이 필요합니다.
근거 기반 데이터를 제공하기 위해 고안된 임상시험은 잘 정의된 환자군과 대조군을 모두 포함해야 하며, 치료적 개입의 효과를 평가하기 위해 가장 객관적이고 신뢰할 수 있으며 검증된 도구를 활용해야 합니다. 다양한 치료법에 대한 반응을 평가하는 데 다양한 임상 평가 척도 및 기타 도구가 활용되고 있지만, 다양한 임상시험에서 1차 결과 측정으로 가장 많이 사용되는 것은 UPDRS입니다.62
표 1과 그림 4에는 질병의 다양한 단계에 있는 다발성 경화증 환자를 위한 의학적 및 수술적 치료 옵션에 대한 개요가 나와 있습니다. 또한 기존 치료법 외에도 근거 기반63 및 새로운 실험적 치료법에 대한 인사이트를 제공합니다.
Table 1
Drugs used in the treatment of Parkinson’s disease and levodopa-related complications
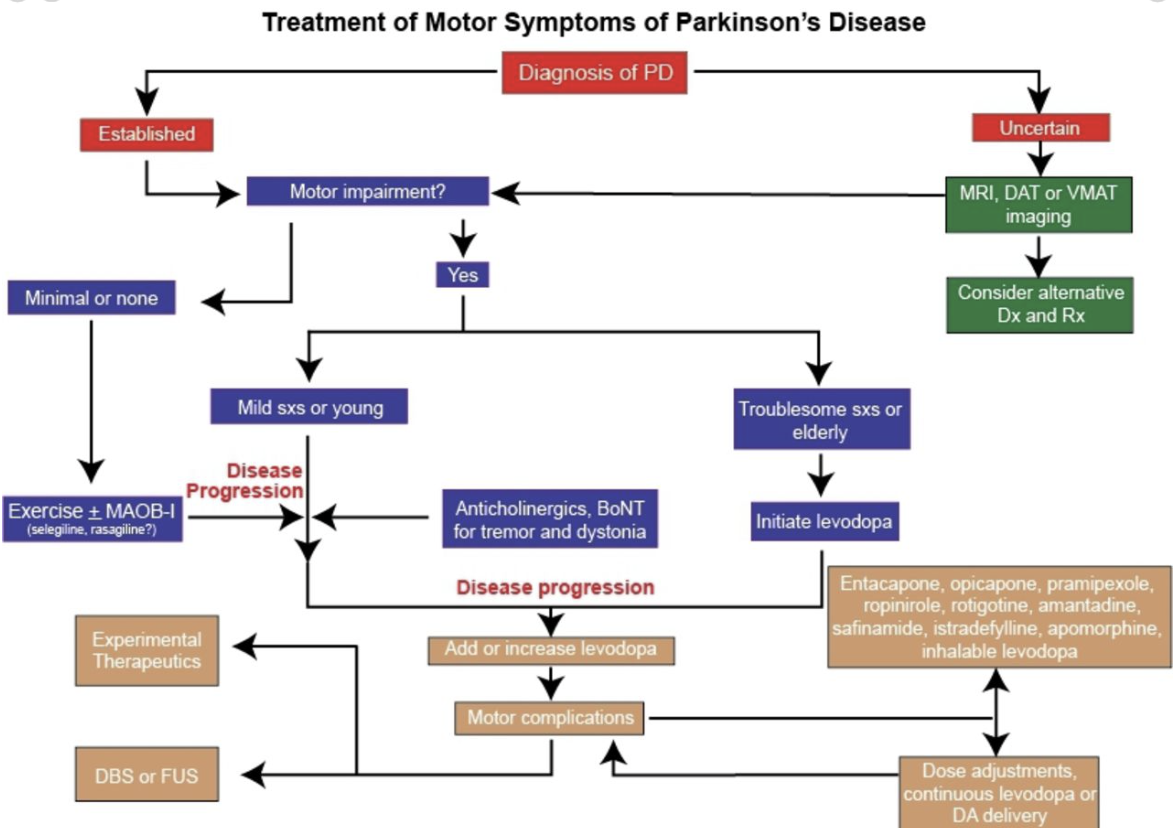
Figure 4
Algorithm for the treatment of motor symptom of PD. DBS, deep brain stimulation; MAOB, imonoamine-oxidase type B inhibitor;
PD, Parkinson’s disease.
Neuroprotective or disease-modifying therapies
In order to consider disease-modifying therapies, it is critical to recognise the variable slopes of progression in patients with PD, reflecting the clinical (and pathological) heterogeneity of the disease.64 An increasing understanding of etiopathogenesis of PD has led to hypotheses about potential neuroprotective strategies that, when applied early (perhaps even in the prodromal phase), may favourably alter the progression of the disease.64 However, double-blind placebo-controlled trials of potential disease-modifying therapies have been thus far disappointing64. The first such trial, DATATOP (Deprenyl and Tocopherol Antioxidative Therapy of Parkinsonism), randomised patients with early PD to treatment with selegiline (selective monoamine oxidase, MAO-B inhibitor or monoamine oxidase inhibitors (MAOI)), tocopherol (vitamin E), or both, and followed them until their disability was severe enough to require levodopa.65 Although the group randomised to selegiline had a delay in reaching the endpoint, the interpretation of the study was confounded by the drug’s mild symptomatic antiparkinsonian and antidepressant properties, as well as the potential effects of its amphetamine metabolites. Another MAOI, rasagiline, has been shown to have modest symptomatic benefit,66 but its effect on disease progression is uncertain. In a delayed-start design trial,67 used to assess the potential disease-modifying effects of rasagiline (ADAGIO (Attenuation of Disease Progression with Azilect Given Once-Daily)), 1176 patients with early untreated PD were randomised into four treatment groups: 1 or 2 mg/day, early-start vs delayed treatment). While the 1 mg dose group showed improvement in total UPDRS score and slower slope of progression compared with placebo at the end of 9 months, there was no observable benefit with the 2 mg dose. Because of the confounding symptomatic effect and lack of long-term benefits of early start rasagiline, this drug cannot be recommended as a disease-modifying treatment.
Development of neuroprotective strategies has been challenging, partly because of lack of reliable and sensitive biomarkers of progression15 and yet incomplete understanding of the pathogenesis of the disease. One of the most exciting developments of potential neuroprotective or disease-modifying therapies is the use of α-synuclein monoclonal antibodies to minimise accumulation and spread of aggregated, toxic, α-synuclein.68–70 Other antisynuclein strategies currently in development include active immunisation against synuclein, antiaggregation drugs, certain Abelson (c-Abl) kinase inhibitors, such as Nilotinib and K0706 and strategies designed to increase clearance.71 Given the history of failed ‘neuroprotective trials’, we should temper our expectations that safe and effective disease-modifying drugs will be approved in the near future.2 72
In addition, a variety of other approaches, such as the use of glucagon-like peptide 1 receptor agonists, are being explored as potential disease-modifying strategies. Furthermore, specific GBA or LRRK2 modifiers, such as ambroxol hydrochloride and DNL201 or DNL151, respectively, are currently tested in genetically defined parkinsonian populations.73 74
신경 보호 또는 질병 조절 요법
질병 수정 요법을 고려하기 위해서는
파킨슨병 환자의 임상적(및 병리학적) 이질성을 반영하여
질병의 다양한 진행 경사를 인식하는 것이 중요합니다.64
파킨슨병의 병인 기전에 대한 이해가 증가하면서
조기에(아마도 전구기에도) 적용하면
질병의 진행을 유리하게 바꿀 수 있는
잠재적 신경 보호 전략에 대한 가설이 제기되었습니다.64
그러나 잠재적 질병 수정 요법에 대한 이중 맹검 위약 대조 시험은 지금까지 실망스러운 결과를 가져왔습니다64.
첫 번째 임상시험인 D
ATATOP(파킨슨병의 데프레닐 및 토코페롤 항산화 치료)에서는
초기 파킨슨병 환자를 무작위로 셀레길린(선택적 모노아민 산화효소, MAO-B 억제제 또는 모노아민 산화효소 억제제(MAOI)), 토코페롤(비타민 E) 또는 두 가지 치료제로 분류하여 레보도파가 필요할 정도로 장애가 심해질 때까지 추적 관찰했습니다.65
셀레길린에 무작위로 배정된 그룹은 종료점에 도달하는 데 시간이 지연되었지만, 이 약물의 경미한 항파킨슨 및 항우울 효과와 암페타민 대사 산물의 잠재적 효과로 인해 연구 해석이 혼란스러웠습니다. 또 다른 MAOI인 라사길린은 약간의 증상 개선 효과가 있는 것으로 나타났지만,66 질병 진행에 미치는 영향은 불확실합니다. 지연 시작 설계시험67에서 라사길린의 잠재적인 질병 조절 효과를 평가하는 데 사용된 임상시험(ADAGIO(1일 1회 아질렉트 투여를 통한 질병 진행의 완화))에서는 치료 경험이 없는 초기 PD 환자 1176명을 하루 1 또는 2mg, 조기 시작 대 지연 치료의 네 가지 치료 그룹으로 무작위 배정했습니다.) 1mg 투여군은 9개월 후 위약에 비해 총 UPDRS 점수가 개선되고 진행 속도가 느려진 반면, 2mg 투여군에서는 관찰할 수 있는 이점이 나타나지 않았습니다. 라사길린 조기 시작의 혼란스러운 증상 효과와 장기적인 이점의 부족으로 인해 이 약은 질병 조절 치료제로 권장할 수 없습니다.
신경 보호 전략의 개발은 부분적으로는 신뢰할 수 있고 민감한 진행 바이오마커가 부족하고15 질병의 발병 기전에 대한 이해가 불완전하기 때문에 어려운 과제였습니다. 잠재적인 신경 보호 또는 질병 수정 치료법 중 가장 흥미로운 개발 중 하나는 응집된 독성 α-시누클레인의 축적과 확산을 최소화하기 위해 α-시누클레인 단일 클론 항체를 사용하는 것입니다.68 -70 현재 개발 중인 다른 항시누클레인 전략으로는 시누클레인에 대한 활성 면역, 항응집 약물, 특정 아벨슨(c-Abl) 키나제 억제제(예: 닐로티닙 및 K0706), 클리어런스 증가 전략 등이 있습니다.71 '신경 보호 시험'의 실패 역사를 고려할 때 가까운 미래에 안전하고 효과적인 질병 조절 약물이 승인될 것이라는 기대는 줄여야 합니다.2 72
또한 글루카곤 유사 펩타이드 1 수용체 작용제의 사용과 같은 다양한 다른 접근법이 잠재적인 질병 조절 전략으로 모색되고 있습니다. 또한, 암브록솔 염산염과 DNL201 또는 DNL151과 같은 특정 GBA 또는 LRRK2 조절제는 현재 유전적으로 정의된 파킨슨병 환자 집단에서 테스트되고 있습니다.73 74
Symptomatic treatment of motor symptoms
Levodopa
A majority of patients with PD require levodopa therapy within 2 years of symptom onset. Levodopa, the most effective drug in the treatment of PD, is almost always combined with carbidopa or benserazide, aromatic acid decarboxylase inhibitors that prevent its peripheral metabolism and markedly reduce the risk of nausea. Increasing the ratio of carbidopa:levodopa from the current standard 1:4 has been shown to increase on time without dyskinesia and reduce off time.75
The global antiparkinsonian efficacy of levodopa is so predictable that a positive therapeutic response is used to support the diagnosis of PD. Adverse effects of levodopa include nausea and vomiting, orthostatic hypotension, sedation, confusion, sleep disturbance, hallucinations and dyskinesias. There are many different types of dyskinesia but peak-dose chorea or stereotypy and wearing off dystonia are most common.76 About half of the patients experience wearing off, and a third experience dyskinesias within 2 years after initiation of levodopa therapy. Latency from ingestion of levodopa to observable therapeutic benefit can be shortened by taking levodopa on an empty stomach (if tolerated without nausea), avoiding or reducing protein intake, or by crushing the levodopa tablet and mixing it with a carbonated beverage.
Because of a concern about development of levodopa-related motor complications, many patients and physicians are reluctant to initiate levodopa therapy even though the patients experience troublesome symptoms. This is especially true in patients with young-onset PD who are more likely to develop motor fluctuations and dyskinesia early in the course of levodopa treatment.77 78 While the treatment of PD must be individualised and tailored to the needs of each patient, this ‘levodopa phobia’ unfortunately and unnecessarily may delay needed and effective relieve of PD-related motor symptoms. Furthermore, there is no evidence from animal or human studies that levodopa accelerates disease progression or that delaying initiation of levodopa delays onset of dyskinesia. Indeed, a 9-month study, called the earlier versus later L-dopa trial, compared 150, 300 and 600 mg daily doses of levodopa with placebo found no evidence of levodopa toxicity, although 16.5% of the patients in the 600 mg group developed dyskinesia.79 Furthermore, a more recent multicenter double-blind placebo-controlled delayed-start trial using carbidopa:levodopa in early PD showed no significant change in rate of progression between early or delayed-start groups suggesting that levodopa does not have disease modifying effect.80
In patients with short duration of response to levodopa, fractionation of total daily dose is usually the initial strategy in an attempt to smooth out fluctuations and prevent wearing off symptoms (figure 4). The duration of benefit from each dose of levodopa may be enhanced by blocking dopamine metabolism with MAOIs or catechol-O-methyl transferase inhibitors (COMTIs), or by adding dopamine agonists or extended release preparations of amantadine (see below).
Different formulations of levodopa have been developed or are in development to provide more desirable delivery that avoids or prevents levodopa-related complications (table 1) 7. Studies have shown that carbidopa/levodopa extended-release (IPX066) as 95, 145, 195 and 255 mg capsules is useful in patients who continue to have motor fluctuations despite high frequency levodopa,81 though the decrease in daily ‘off’ time of about an hour is modest82 and may be comparable to other add-on oral therapies. Other formulations of levodopa that have been found effective in smoothing out motor fluctuations include continuous intrajejunal infusion of levodopa-carbidopa intestinal gel.83 The efficacy of continuous infusion formulation is comparable to subthalamic nucleus (STN) deep brain stimulation (DBS) surgery though only the latter treatment led to improvement in the duration and disability of levodopa-related dyskinesias.84 Another approach being investigated in patients with severe motor fluctuations, involves a soluble carbidopa/levodopa formulation that is continuously delivered subcutaneously via less invasive subcutaneous route that allows for a stable levodopa level over 72 hours.85 Although the preliminary data are encouraging in regard to motor fluctuations and dyskinesias, but the formation of cutaneous nodules may present a major limitation to future adaptation of this therapy.
A formulation of levodopa, approved by the US Food and Drug Administration (FDA) in 2018, as a ‘rescue’ from off periods is inhalable levodopa powder without carbidopa.86 The drug is supplied in cartons containing a 3-part inhaler and two 42 mg levodopa capsules in blister packaging. The drug must be actively inhaled and is, therefore, often associated with troublesome cough. Once patients are able to tolerate it, they can inhale it up to five times per day as needed and expect a reversal of parkinsonian symptoms within 10–30 min. The time of onset is comparable to dissolvable benserazide:levodopa formulation87 but slower than subcutaneous or sublingual apomorphine (7–10 min)88 (table 1).
레보도파
대부분의 파킨슨병 환자는
증상 발현 후 2년 이내에 레보도파 치료를 받아야 합니다.
PD 치료에 가장 효과적인 약물인
레보도파는
거의 항상 말초 대사를 막고
메스꺼움의 위험을 현저히 감소시키는
방향족산 탈카르복실효소 억제제인
카비도파 또는 벤데라자이드와 병용합니다.
카비도파:레보도파의 비율을 현재 표준인 1:4에서 증가시키면
운동 이상증 없이 온타임은 증가하고
오프타임은 감소하는 것으로 나타났습니다.75
레보도파의 전 세계적인 항파킨슨 효능은
매우 예측 가능하기 때문에 긍
정적인 치료 반응은 PD 진단을 뒷받침하는 데 사용됩니다.
레보도파의 부작용으로는
메스꺼움과 구토,
기립성 저혈압,
진정,
혼란,
수면 장애,
환각 및
운동 이상증 등이 있습니다.
운동 이상증에는 다양한 유형이 있지만
최고 용량 무도병 또는
근긴장 이상증과 탈진 운동 이상증이 가장 흔합니다.76
환자의 약 절반이 탈진 운동 이상증을 경험하며,
레보도파 치료 시작 후 2년 이내에
1/3이 운동 이상증을 경험합니다.
레보도파를
공복에 복용하거나(메스꺼움 없이 견딜 수 있는 경우),
단백질 섭취를 피하거나 줄이거나,
레보도파 정제를 분쇄하여 탄산음료와 혼합하여 복용하면
레보도파 섭취 후 관찰 가능한 치료 효과까지의 지연 시간을 단축할 수 있습니다.
레보도파 관련 운동 합병증 발생에 대한 우려로 인해
많은 환자와 의사는 환자가 골치 아픈 증상을 경험하더라도
레보도파 치료를 시작하기를 꺼립니다.
이는 특히
레보도파 치료 초기에
운동 변동과 운동 이상증이 발생할 가능성이 높은
젊은 발병 PD 환자의 경우 더욱 그렇습니다.77 78
PD 치료는
각 환자의 필요에 따라 개별화되고
맞춤화되어야 하지만,
이러한 '레보도파 공포증'은 불행히도
필요하고 효과적인 PD 관련 운동 증상 완화를
불필요하게 지연시킬 수 있습니다.
또한,
레보도파가 질병의 진행을 가속화하거나
레보도파 투여를 늦추면
운동 이상증의 발병이 지연된다는 동물 또는 인간 연구 결과는 없습니다.
실제로 초기 대 후기 L-도파 시험이라고 불리는 9개월간의 연구에서 레보도파를 매일 150, 300, 600mg씩 투여한 그룹과 위약을 비교한 결과, 600mg 그룹 환자의 16.5%에서 이상운동증이 발생했지만 레보도파 독성에 대한 증거는 발견되지 않았습니다.79 또한, 최근 다기관 이중맹검 위약 대조 지연 시작 시험에서 초기 파킨슨병 환자에서 카비도파:레보도파를 사용한 결과 조기 또는 지연 시작 그룹 간에 진행 속도에 큰 변화가 없는 것으로 나타나 레보도파가 질병 수정 효과가 없음을 시사했습니다.80
레보도파에 대한 반응 기간이 짧은 환자의 경우,
변동성을 완화하고
증상 소실을 방지하기 위해
일반적으로 일일 총 용량을 분할 투여하는 것이 초기 전략입니다(그림 4).
각 레보도파 용량에 따른 효과의 지속 기간은
MAOI 또는
카테콜-O-메틸 전이효소 억제제(COMTI)로
도파민 대사를 차단하거나
도파민 작용제 또는
아만타딘 서방형 제제를 추가함으로써 연장될 수 있습니다(아래 참조).
레보도파 관련 합병증을 피하거나 예방하는
보다 바람직한 전달을 제공하기 위해
다양한 제형의 레보도파가 개발되었거나 개발 중입니다(표 1) 7.
연구에 따르면
95, 145, 195, 255mg 캡슐의 카비도파/레보도파 서방형(IPX066)은
고빈도 레보도파 투여에도 불구하고
운동 변동이 지속되는 환자에게 유용하며,81
일일 '오프' 시간 감소는 약 1시간으로 크지 않고82
다른 추가 경구 치료제와 비슷할 수 있습니다.
운동 변동을 완화하는 데 효과적인 것으로 밝혀진 다른 제형의 레보도파에는 레보도파-카르비도파 장내 겔의 지속적인 관절 내 주입이 있습니다.83 지속적인 주입 제형의 효과는 시상하핵(STN) 심부 뇌 자극(DBS) 수술과 비슷하지만 후자의 치료만이 레보도파 관련 이상운동증의 지속 기간과 장애를 개선하는 것으로 나타났습니다.84 운동 변동이 심한 환자를 대상으로 연구 중인 또 다른 접근법은 덜 침습적인 피하 경로를 통해 지속적으로 피하로 전달되어 72시간 동안 레보도파 수치를 안정적으로 유지하는 수용성 카비도파/레보도파 제형입니다.85 예비 데이터는 운동 변동 및 운동 이상증과 관련하여 고무적이지만 피부 결절의 형성은 향후 이 치료법의 적응에 큰 제한이 될 수 있습니다.
2018년 미국 식품의약국(FDA)에서 휴지기 '구제'로 승인한 레보도파 제형은 카비도파가 없는 흡입용 레보도파 분말입니다.86 이 약은 3분할 흡입기와 42mg 레보도파 캡슐 2개가 블리스터 포장된 상자에 담겨 공급됩니다. 이 약은 적극적으로 흡입해야 하므로 종종 기침과 관련된 문제가 발생할 수 있습니다. 환자가 내성이 생기면 필요에 따라 하루에 최대 5회까지 흡입할 수 있으며, 10~30분 이내에 파킨슨병 증상이 회복될 것으로 기대할 수 있습니다. 발병 시간은 용해성 벤데라지드:레보도파 제형87과 비슷하지만 피하 또는 설하 아포모르핀(7~10분)88보다는 느립니다(표 1).
Other drugs
Besides levodopa, there are many other types of medications available for the treatment of PD-related motor symptoms: anticholinergics, amantadine, MAOIs, COMTIs, dopamine agonists and istradefylline.77
Anticholinergics
Anticholinergics, such as trihexyphenidyl and benztropine, antagonise the effects of acetylcholine at muscarinic receptors postsynaptic to striatal interneurons. They are predominantly used to reduce tremor and have no effect on bradykinesia. Antagonism of acetylcholine can be associated with a variety of adverse effects such as cognitive impairment, confusion, hallucination, blurred vision, dry mouth, constipation and urinary retention. These side effects limit the usefulness of anticholinergics in the treatment of PD.
기타 약물
레보도파 외에도
항콜린제, 아만타딘, MAOI, COMTI, 도파민 작용제, 이스트레이드필린 등
다양한 종류의 약물이 PD 관련 운동 증상 치료에 사용할 수 있습니다.77
항콜린제
트리헥시페니딜 및 벤즈트로핀과 같은 항콜린제는
시냅스 후 선조체 뉴런에 대한 무스카린 수용체에서
아세틸콜린의 효과를 길항합니다.
이들은 주로 떨림을 줄이는 데 사용되며
서동증에는 영향을 미치지 않습니다.
아세틸콜린의 길항작용은
인지 장애, 혼란, 환각, 시야 흐림, 구강 건조, 변비 및 소변 정체와 같은
다양한 부작용과 관련될 수 있습니다.
이러한 부작용은
PD 치료에서
항콜린제의 유용성을 제한합니다.
Antiglutamatergics
Glutamate mediates neurotransmission of most excitatory synapses and is vital for normal physiologic function of the brain. Amantadine (originally developed as an anti-influenza drug) is currently the main drug used in the treatment of levodopa-related dyskinesia. Besides its antiglutamatergic effect (presumably as glutamate/NMDA receptor antagonist), amantadine has also been thought to stimulate the release of endogenous dopamine stores, block reuptake of dopamine from the synaptic cleft and have anticholinergic properties. Extended release formulation of amantadine (ADS-5102 or Amantadine ER), administered before bedtime, has been found to improve both dyskinesia and also motor fluctuations. In one double-blind, placebo-controlled trial involving 126 patients, Amantadine ER significantly decreased mean off time and increased mean on time without troublesome dyskinesia.89 The most common side effects were visual hallucinations, peripheral oedema and dizziness. Amantadine ER is available as 68.5 and 137 mg capsules. Another formulation of amantadine, Osmolex ER (available as 129, 193 and 258 mg tablets), in contrast to Amantadine ER, delivers amantadine throughout the day (median Tmax 7.5 hours; half-life 16 hours). When administered in the morning, it reaches highest plasma levels during waking hours and are lowest during the night. Amantadine is contraindicated in patients with renal impairment.
항글루타메이트제
글루타메이트는
대부분의 흥분성 시냅스의 신경 전달을 매개하며
뇌의 정상적인 생리적 기능에 필수적입니다.
아만타딘(원래 항인플루엔자 약으로 개발됨)은
현재 레보도파 관련 운동 이상증 치료에 주로 사용되는 약물입니다.
아만타딘은
항글루타메이트 효과(글루타메이트/NMDA 수용체 길항제로 추정) 외에도
내인성 도파민 저장소의 방출을 자극하고
시냅스 틈새에서 도파민의 재흡수를 차단하며
항콜린성을 갖는 것으로 여겨져 왔습니다.
취침 전에 투여하는
아만타딘 서방형 제제(ADS-5102 또는
아만타딘 ER)는 운동 이상증과 운동 변동을
모두 개선하는 것으로 밝혀졌습니다.
126명의 환자를 대상으로 한 이중 맹검, 위약 대조 시험에서 아만타딘 ER은 운동 이상증 없이 평균 취침 시간을 유의하게 감소시키고 평균 취침 시간을 증가시켰습니다.89
가장 흔한 부작용은
시각 환각,
말초 부종 및 어지럼증이었습니다.
아만타딘 ER은 68.5mg 및 137mg 캡슐로 제공됩니다. 아만타딘의 또 다른 제형인 오스모렉스 ER(129, 193, 258mg 정제)은 아만타딘 ER과 달리 하루 종일 아만타딘을 전달합니다(T맥스 중앙값 7.5시간, 반감기 16시간). 아침에 투여하면 깨어 있는 시간 동안 혈장 농도가 가장 높고 밤에는 가장 낮습니다.
아만타딘은
신장 장애가 있는 환자에게는
금기입니다.
Monoamine oxidase inhibitors
Although selegiline and rasagiline are most frequently used in early, mild PD, these MAOIs are also effective in patients with moderately advanced PD with levodopa-related motor complications. Another MAOI, safinamide, administered once daily (50–100 mg/day), has been found to increase mean on time without troublesome dyskinesia and reduce daily and morning off times.90 Safinamide is both a reversible MAOI and it also reduces neuronal dopamine reuptake and blocks voltage-dependent activated sodium channel and intracellular calcium entry thus reducing neuronal glutamate release.
모노아민 산화효소 억제제
셀레길린과 라사길린은
초기 경증 파킨슨병에 가장 많이 사용되지만,
이 MAOI는 레보도파 관련 운동 합병증을 동반한
중등도 진행성 파킨슨병 환자에게도 효과적입니다.
또 다른 MAOI인 사피나미드는
1일 1회(50-100 mg/일) 투여하면 문제가 되는
운동 이상증 없이 평균 온타임을 증가시키고
일일 및 아침 휴면 시간을 줄이는 것으로 밝혀졌습니다.90
사피나미드는
가역적인 MAOI이며
신경세포 도파민 재흡수를 감소시키고
전압 의존적으로 활성화된 나트륨 채널과
세포 내 칼슘 유입을 차단하여
신경세포 글루타메이트 방출도 감소시킵니다.
Dopamine agonists
Dopamine receptor agonists stimulate dopamine receptors (G protein-coupled, two major families, D2-like (D1 and D5) and D1-like (D2, D3 and D4)) and when introduced early in the course of PD treatment, they delay levodopa-related complications such as motor fluctuations and dyskinesias. But evidence is lacking to support the hypothesis that early introduction of dopamine agonists slows progression of the disease or even improves long-term quality of life. Common non-ergot dopamine agonists used in clinical practice include pramipexole, ropinirole, rotigotine and apomorphine. Dopamine agonists can be used as monotherapy for motor symptoms or as an adjunct therapy when the symptoms are not sufficiently controlled by levodopa or when motor fluctuations are present.91 In an open-label randomised trial involving 1620 newly diagnosed PD patients, those who were assigned to levodopa alone reported better mobility scores compared with those on dopamine agonists or MAOBI.92 An earlier 5-year study showed that those on ropinirole has less dyskinesias compared with those on levodopa.93
The most common side effects of dopamine agonists include orthostatic hypotension, sleepiness, hallucinations and leg oedema. In addition, these drugs have been linked to relatively high frequency a variety of behavioural problems that include pathological gambling, compulsive shopping and eating, hypersexuality and other impulse-control disorders (ICD).94 Patients with PD who experience ICD seem to have a variety of associated psychiatric symptoms, such as psychosis, interpersonal sensitivity, obsessive-compulsive symptoms and depression and seem to be prone to dopamine dysregulation syndrome, an addictive behaviour associated with excessive use of dopaminergic medications.73 Patients and their care givers must be counselled about the risk of ICD before treatment initiation and at each visit, as many patients may not be forthcoming about these behaviours.95
Because of relatively high frequency of adverse effects, particularly ICD, the role of dopamine agonists has changed over the recent decades. This class of drugs is now primarily used in early treatment of PD before initiating levodopa and in patients with motor fluctuations in order to prolong the response to levodopa. In addition to two orally administered non-ergoline formulations, pramipexole and ropinirole, rotigotine is available as a patch. Apomorphine, a nonergoline dopamine agonist, is water soluble and lipophilic and is therefore suitable for intravenous, subcutaneous, sublingual, intranasal or transdermal administration.88 . Apomorphine, when administered via subcutaneous injection, may provide a rapid rescue from hypomobility end-of-dose wearing off or unpredictable on/off episodes, typically observed in advanced PD. In a study of 109 patients who were randomly assigned to receive apomorphine sublingual film or placebo, there was a significant and clinically meaningful improvement in MDS–UPDRS part 3 within 30 min, 31% of 54 patients receiving apomorphine sublingual film discontinued treatment because of oropharyngeal side effects.96 In another randomised study, apomorphine infusion (mean dose 4.68 mg/hour) reduced ‘off’ time of 2.5 hours/day compared with 0.6 hours/day with placebo in PD patients with motor fluctuations despite optimal oral or transdermal therapy.97
도파민 수용체 작용제는
도파민 수용체(G 단백질 결합, 두 가지 주요 계열, D2 유사(D1 및 D5) 및 D1 유사(D2, D3 및 D4))를 자극하며, PD 치료 초기에 도입하면 운동 변동 및 운동 이상증과 같은 레보도파 관련 합병증을 지연시킵니다.
그러나
도파민 작용제의 조기 도입이
질병의 진행을 늦추거나 장기적인 삶의 질을 향상시킨다는 가설을 뒷받침하는 증거는 부족합니다.
임상에서 사용되는 일반적인 비 에르고트 도파민 작용제에는 프라미펙솔, 로피니롤, 로티고틴 및 아포모르핀이 있습니다. 도파민 작용제는 운동 증상에 대한 단독 요법으로 사용하거나 레보도파로 증상이 충분히 조절되지 않거나 운동 변동이 있는 경우 보조 요법으로 사용할 수 있습니다.91 새로 진단받은 1620명의 PD 환자를 대상으로 한 공개 라벨 무작위 시험에서 레보도파 단독 투여군은 도파민 작용제 또는 MAOBI 투여군에 비해 운동성 점수가 더 좋았습니다.92 이전의 5년 연구에 따르면 로피니롤을 사용한 환자는 레보도파를 사용한 환자보다 운동 이상증이 적었습니다.93
도파민 작용제의 가장 흔한 부작용으로는 기립성 저혈압, 졸음, 환각, 다리 부종 등이 있습니다. 또한 이러한 약물은 병적 도박, 강박적 쇼핑 및 식사, 과잉 성욕 및 기타 충동 조절 장애(ICD)를 포함한 다양한 행동 문제와 비교적 높은 빈도로 연관되어 있습니다.94 ICD를 경험하는 PD 환자는 정신병, 대인 관계 민감성, 강박 증상 및 우울증과 같은 다양한 관련 정신과 증상을 보이며 도파민 약물의 과도한 사용과 관련된 중독성 행동인 도파민 조절 장애 증후군에 걸리기 쉬운 것으로 보입니다.73 많은 환자들이 이러한 행동에 대해 솔직하게 말하지 않을 수 있으므로 치료 시작 전과 방문 시마다 환자와 간병인에게 ICD 위험에 대해 상담해야 합니다.95
특히 ICD의 부작용 빈도가 상대적으로 높기 때문에 도파민 작용제의 역할은 최근 수십 년 동안 변화해 왔습니다. 이 계열의 약물은 현재 레보도파를 시작하기 전 PD의 초기 치료와 레보도파에 대한 반응을 연장하기 위해 운동 변동이 있는 환자에게 주로 사용됩니다. 로티고틴은 두 가지 경구용 비에르고린 제형인 프라미펙솔과 로피니롤 외에도 패치제로도 제공됩니다. 비 에르고린 도파민 작용제인 아포모르핀은 수용성 및 친유성이므로 정맥, 피하, 설하, 비강 내 또는 경피 투여에 적합합니다.88. 아포모르핀은 피하 주사를 통해 투여할 경우, 일반적으로 진행성 파킨슨병에서 관찰되는 저운동성 투약 말기 마모 또는 예측할 수 없는 온/오프 에피소드로부터 신속하게 환자를 구할 수 있습니다. 아포모르핀 설하 필름 또는 위약을 무작위로 배정받은 109명의 환자를 대상으로 한 연구에서 아포모르핀 설하 필름을 투여받은 54명의 환자 중 31%가 구강인두 부작용으로 인해 치료를 중단한 가운데 30분 이내에 MDS-UPDRS 파트 3에서 유의미하고 임상적으로 의미 있는 개선이 나타났습니다.96 또 다른 무작위 연구에서 아포모르핀 주입(평균 용량 4.68 mg/시간)은 최적의 경구 또는 경피 치료에도 불구하고 운동 변동이 있는 PD 환자에서 위약의 하루 0.6시간에 비해 하루 2.5시간의 '오프' 시간을 감소시켰습니다.97
Catechol-O-methyl transferase inhibitors
COMTIs (entacapone, tolcapone and opicapone) block degradation of peripheral levodopa and tolcapone in addition blocks central degradation of levodopa and dopamine, increasing central levodopa and dopamine levels. Hepatotoxicity associated with tolcapone has limited its use. Triple-combination therapy containing levodopa (50, 75, 100, 125, 150 and 200 mg), carbidopa and entacapone (Stalevo) is available but often denied by third-party payers. Opicapone, a novel COMTI, administered once-daily (50 mg), has been found to significantly reduce off time.98 99 The primary role of COMTIs is to prolong the effects of levodopa and, therefore, they are useful as adjunctive drugs for patients who experience levodopa-related motor fluctuations. COMTIs are generally well tolerated, but besides increasing levodopa-related dyskinesias, they may cause nausea, postural hypotension, diarrhoea and orange discoloration of urine. There is no evidence that COMTIs prevent or delay the onset of levodopa-related motor complications.
카테콜-O-메틸 전이효소 억제제
COMTI(엔타카폰, 톨카폰 및 오피카폰)는 말초 레보도파의 분해를 차단하고 톨카폰은 레보도파와 도파민의 중추 분해를 차단하여 중추 레보도파와 도파민 수치를 증가시킵니다. 톨카폰과 관련된 간독성으로 인해 톨카폰의 사용이 제한되었습니다. 레보도파(50, 75, 100, 125, 150, 200mg), 카르비도파, 엔타카폰(스탈레보)을 포함하는 3중 병용 요법을 사용할 수 있지만 제3자 지불자가 거부하는 경우가 많습니다. 새로운 COMTI인 오피카폰은 1일 1회(50mg) 투여하면 휴약 시간을 크게 줄이는 것으로 밝혀졌습니다.98 99 COMTI의 주요 역할은 레보도파의 효과를 연장하는 것이므로 레보도파 관련 운동 변동을 경험하는 환자에게 보조 약물로 유용합니다. COMTI는 일반적으로 내약성이 좋지만 레보도파 관련 운동 이상증을 증가시키는 것 외에도 메스꺼움, 자세 저혈압, 설사 및 소변의 주황색 변색을 유발할 수 있습니다. COMTI가 레보도파 관련 운동 합병증의 발병을 예방하거나 지연시킨다는 증거는 없습니다.
Adenosine A2 receptor antagonist
In 2019, the FDA approved istradefylline (Nourianz), an adenosine A2 receptor antagonist, as adjunctive treatment for levodopa/carbidopa in patients with PD experiencing off episodes.100 Available as 20 and 40 mg tablets, the drug provides a modest benefit in patients with levodopa-related motor fluctuations. It is generally well tolerated, but has been reported to cause or increase dyskinesia, dizziness, constipation, nausea, hallucinations and insomnia.
아데노신 A2 수용체 길항제
2019년에 FDA는 아데노신 A2 수용체 길항제인 이스트라데필린(누리안즈)을 레보도파/카비도파의 보조 치료제로 승인했습니다.100 20mg 및 40mg 정제로 제공되는 이 약은 레보도파 관련 운동 변동이 있는 환자에서 약간의 이점을 제공합니다. 일반적으로 내약성이 좋지만 운동 이상증, 현기증, 변비, 메스꺼움, 환각 및 불면증을 유발하거나 증가시키는 것으로 보고되었습니다.
Symptomatic treatment of levodopa-resistant and non-motor symptoms
Levodopa-resistant symptoms
There are many levodopa-resistant motor symptoms such as dysarthria and dysphagia, freezing of gait, postural instability and dysautonomia. Freezing, sudden immobility of the feet when attempting to walk, often associated with falls, may be seen in either the off or the on period. Although off-period freezing may improve with optimisation of medications, on-period freezing is usually resistant to pharmacologic treatment.101 102
Physical therapy, including strategies that utilise sensory cues, such as stepping over a horizontal laser beam, may be helpful.103 Dysarthria and dysphagia are often treated by speech and voice therapists. Injection of botulinum toxin has been found to be effective in controlling high-amplitude rest and postural hand tremor which may be resistant to levodopa.104 Botulinum toxin may be also beneficial in the treatment of a variety of other non-levodopa responsive parkinsonian symptoms such as blepharospasm, apraxia of eyelid opening, anterocollis, camptocormia, bruxism, sialorrhea, seborrhea, hyperhidrosis, overactive bladder and constipation.105
레보도파 내성 운동 증상으로는
구음 장애 및 연하 장애,
보행 정지,
자세 불안정 및
자율 신경 이상증 등이 있습니다.
걸으려고 할 때 갑자기 발이 얼어붙고 움직이지 않는 증상은
종종 낙상과 관련이 있으며,
휴지기 또는 휴지기 모두에서 나타날 수 있습니다.
오프 기간의 동결은 약물을 최적화하면 개선될 수 있지만, 온 기간의 동결은 일반적으로 약물 치료에 내성이 있습니다.101 102
수평 레이저 빔을 밟는 것과 같이 감각 신호를 활용하는 전략을 포함한 물리 치료가 도움이 될 수 있습니다.103 구음 장애와 연하 장애는 언어 및 음성 치료사가 치료하는 경우가 많습니다. 보툴리눔 독소 주사는 레보도파에 내성이 생길 수 있는 고진폭 휴식 및 자세성 손 떨림을 조절하는 데 효과적인 것으로 밝혀졌습니다.104 보툴리눔 독소는 안검 경련, 눈꺼풀떨림증, 전안검하수, 캄프토코미아, 구안와사, 지루, 다한증, 과민 방광 및 변비와 같은 다양한 기타 레보도파 비반응성 파킨슨 증상 치료에도 도움이 될 수 있습니다.105
Non-motor symptoms
It is well recognised that non-motor symptoms comprise an important component of the clinical spectrum of PD even though most of them present with motor symptoms initially. These non-motor symptoms include depression, anxiety, apathy, psychosis, impulse control dysfunction, cognitive impairment, dementia, autonomic dysfunction (drooling orthostatic hypotension, urinary retention/incontinence, erectile dysfunction, gastrointestinal dysfunction excessive sweating), insomnia, RBD, olfactory dysfunction, pain and fatigue.106 Non-motor symptoms can affect quality of life, even more than motor problems. RBD, olfactory dysfunction and gastrointestinal dysfunction may precede motor symptoms. The Movement Disorders Society study group conducted a detailed review of available therapies for non-motor symptoms in PD.106
Here we highlight treatment of some of the common non-motor symptoms. Donepezil and rivastigmine (cholinesterase inhibitors) and memantine (NMDA receptor antagonist) provide modest benefit in patients with PD-associated dementia. Hallucinations, often associated with PD dementia and/or triggered by anti-PD drugs, usually improve with atypical antipsychotics such as quetiapine and clozapine which, in contrast to other antipsychotics (dopamine receptor blockers), have a relatively low risk of exacerbating parkinsonism. Pimavanserin, a non-dopaminergic and selective serotonin inverse agonist with high affinity at the 5-HT2A receptor, has been approved by the FDA in 2016 for the treatment of hallucinations and delusions associated with PD.107 It is available as a 10 mg tablet or 34 mg capsule.
Cholinesterase inhibitors, in addition to improving cognitive function, may reduce hallucinations, improve postural stability and might even reduce the frequency of falls in some patients.108 Sleep disorders should be addressed by strategies designed to improving sleep hygiene, and if needed, supplemented by hypnosedatives, tricyclic antidepressants, mirtazapine, trazodone, quetiapine or nighttime dopaminergic therapy.109 Excessive daytime sleepiness may respond to methylphenidate, modafinil or armodafinil.
Treatment of dysautonomia associated with PD is beyond the scope of this article but readers are referred to an excellent review of this topic.110 Orthostatic hypotension can be managed conservatively with salt supplementation, fludrocortisone, midodrine and droxidopa. Urological medications, such as migrabegron, and botulinum toxin injections into the bladder wall may improve bladder dysfunction. Dietary changes along with medications such as linaclotide and lubiprostone may improve constipation.111
Finally, the important role of physical, occupational and speech/voice therapy coupled with regular exercise programme cannot be overemphasised.112 The effectiveness of home-based and remotely supervised aerobic exercise in reducing the off-state MDS–UPDRS score was demonstrated in a double-blind, randomised clinical trial.113 One meta analysis of eight prospective studies with 2192 PD patients with a mean follow-up of 12 years demonstrated that high or moderate (but not light) physical activity reduced the risk of PD, with the association stronger in men than women.114 The PD risk reduction was between 10% and 17% for every each 10 metabolic equivalent of task-hours/week increase in high or moderate physical activity.
파킨슨병 환자 대부분이 초기에 운동 증상을 보이지만 비운동 증상도 파킨슨병의 임상 스펙트럼에서 중요한 부분을 차지한다는 사실은 잘 알려져 있습니다. 이러한 비운동 증상에는 우울증, 불안, 무관심, 정신병, 충동 조절 기능 장애, 인지 장애, 치매, 자율 기능 장애(침 흘림 기립성 저혈압, 요실금/요폐, 발기부전, 위장 기능 장애 과도한 발한), 불면증, RBD, 후각 기능 장애, 통증 및 피로가 포함됩니다.106 비운동 증상은 운동 문제보다 삶의 질에 더 큰 영향을 미칠 수 있습니다. RBD, 후각 기능 장애 및 위장 기능 장애가 운동 증상보다 먼저 나타날 수 있습니다. 운동 장애 학회 연구 그룹은 파킨슨병의 비운동 증상에 대해 사용 가능한 치료법에 대한 자세한 검토를 수행했습니다.106
여기에서는 일반적인 비운동 증상 중 일부에 대한 치료법을 강조합니다. 도네페질과 리바스티그민(콜린에스테라아제 억제제)과 메만틴(NMDA 수용체 길항제)은 PD 관련 치매 환자에게 약간의 혜택을 제공합니다. PD 치매와 관련이 있거나 항PD 약물에 의해 유발되는 환각은 일반적으로 다른 항정신병약물(도파민 수용체 차단제)과 달리 파킨슨병을 악화시킬 위험이 상대적으로 낮은 케티아핀 및 클로자핀과 같은 비정형 항정신병약물을 사용하면 개선됩니다. 5-HT2A 수용체에 대한 친화력이 높은 비도파민성 및 선택적 세로토닌 역작용제인 피마반세린은 2016년 FDA에서 파킨슨병과 관련된 환각 및 망상 치료제로 승인되었습니다.107 10mg 정제 또는 34mg 캡슐로 제공됩니다.
콜린에스테라아제 억제제는 인지 기능 개선 외에도 환각을 줄이고 자세 안정성을 개선하며 일부 환자의 낙상 빈도를 줄일 수 있습니다.108 수면 장애는 수면 위생을 개선하기 위한 전략으로 해결해야 하며, 필요한 경우 최면제, 삼환계 항우울제, 미르타자핀, 트라조돈, 케티아핀 또는 야간 도파민 치료로 보완할 수 있습니다.109 과도한 주간 졸음은 메틸페니데이트, 모다피닐 또는 아르모다피닐에 반응할 수 있습니다.
PD와 관련된 자율신경실조증 치료는 이 글의 범위를 벗어나지만 이 주제에 대한 훌륭한 리뷰를 참조하시기 바랍니다.110 기립성 저혈압은 소금 보충, 플루드로코르티손, 미도드린 및 드록시도파를 사용하여 보존적으로 관리할 수 있습니다. 미그라베그론과 같은 비뇨기과 약물과 방광벽에 보툴리눔 독소 주사로 방광 기능 장애를 개선할 수 있습니다. 리나클로타이드 및 루비프로스톤과 같은 약물과 함께 식이 요법을 바꾸면 변비가 개선될 수 있습니다.111
마지막으로, 규칙적인 운동 프로그램과 함께 물리, 작업 및 언어/음성 치료의 중요한 역할은 아무리 강조해도 지나치지 않습니다.112 가정 기반 및 원격 감독 유산소 운동의 효과는 이중 맹검, 무작위 임상 시험에서 오프 상태 MDS-UPDRS 점수를 줄이는 데 입증되었습니다.113 평균 12년간 추적 관찰한 2192명의 PD 환자를 대상으로 한 8건의 전향적 연구 메타 분석에 따르면, 고강도 또는 중등도 신체 활동이 PD 위험을 감소시키며, 여성보다 남성에서 그 연관성이 더 강한 것으로 나타났습니다.114 고강도 또는 중등도 신체 활동이 주당 10시간 증가할 때마다 PD 위험 감소율은 10%에서 17% 사이로 나타났습니다.
Surgical treatment
Deep brain stimulation
Despite optimal medical therapy, many patients with moderate to advanced disease have a poor quality of life because of fluctuating response, troublesome dyskinesia or levodopa-unresponsive symptoms. Ablative surgical approaches such as stereotactic destruction of physiologically defined overactive brain nuclei (thalamotomy, pallidotomy) have been largely replaced by DBS using implanted pulse generators. The chief advantage of DBS over ablative lesioning is that the stimulation parameters can be customised to the needs of the patient in order to optimise the benefits. Thalamic DBS is most frequently used to control high-amplitude tremor in patients with essential tremor, but STN or globus pallidus interna (GPi) are the most frequent targets for DBS treatment of patients with PD with disabling tremor and/or levodopa-related motor complications. To address the question whether optimal medical therapy or DBS provides more robust improvement, 255 patients at seven Veterans Affairs and six university hospitals were enrolled in a randomised controlled trial designed to compare the effects of DBS (STN, n=60; or GPi, n=61) and ‘best medical therapy’ (n=134) after 6 months of treatment.115 Patients treated with DBS gained a mean of 4.6 hours/day of on time without troubling dyskinesia, compared with 0 hours/day for patients who received best medical therapy (p<0.001). Furthermore, motor function improved by five or more points on the motor UPDRS in 71% of DBS and 32% of medical therapy patients. This was accompanied by improvements in the majority of PD-related health-related quality of life measures and only minimal decrement in neurocognitive testing. The overall risk of experiencing a serious adverse event, however, was 3.8 times higher in the DBS than in the medical therapy group (40% vs 11%).
The relative efficacy of STN and GPi as therapeutic targets has been debated since the advent of DBS.3 The Veterans Affairs Cooperative Study investigated STN and GPi DBS outcomes after 24 months in 299 patients, and there were no differences in mean changes in the motor (part III) UPDRS between the two targets.116 Patients undergoing STN required a lower dose of DAs than those undergoing pallidal stimulation (p=0.02), and visuomotor processing speed declined more after STN than after GPi stimulation (p=0.03). On the other hand, there was worsening of depression after STN DBS, but mood improved after GPi DBS (p=0.02). Slightly more than half of the patients experienced serious adverse events, but there was no difference in the frequency of these events between the two groups. Based on these and other studies, there is emerging evidence that GPi DBS may be particularly suitable for patients who may have troublesome dyskinesias as well as mild cognitive or behavioural impairment, whereas bilateral STN DBS may be the surgical choice for patients who are cognitively intact but in whom reduction in levodopa dosage is the primary goal. Compared with GPi, STN DBS seems to have a greater beneficial impact on off periods but is more likely associated with adverse effects such as ICD.
One common clinical question is whether DBS surgery at an earlier stage of PD or at a younger age can lead to similar positive outcome.3 In a randomised trial involving 251 relatively young (mean age 52 years) PD with early motor complications, STN DBS plus medical therapy was compared with medical therapy alone. The quality of life scores mean score improved by 7.8 points in DBS group compared with 0.2 point worsening in medical group. Motor disability, activities of daily living, levodopa-related motor complications were better in the surgery group.117 When considering early DBS, it is important to first optimise medical treatment, including strategies to improve motor fluctuations and dyskinesia and to consider the risks of surgery and other factors.118 119
While DBS is a proven effective therapeutic strategy, its success depends on the appropriate selection of patients and the experience and skill of the stereotactic surgeon in order to optimise the results and minimise complications. Advances in DBS technology, such as the use of adaptive stimulation, improving connectivity, directional stimulation and an exploration of for new targets will likely continue to improve.120 121
뇌심부 자극술
최적의 의학적 치료에도 불구하고 중등도에서 진행된 질환을 가진 많은 환자는 반응의 변동, 번거로운 운동 이상증 또는 레보도파에 반응하지 않는 증상으로 인해 삶의 질이 떨어집니다. 생리적으로 정의된 과민성 뇌핵의 정위적 파괴(시상 절개술, 구개 절개술)와 같은 절제적 수술 접근법은 이식형 펄스 발생기를 사용하는 DBS로 대부분 대체되었습니다. 절제적 병변 제거술에 비해 DBS의 가장 큰 장점은 환자의 필요에 따라 자극 파라미터를 맞춤화하여 효과를 최적화할 수 있다는 점입니다. 본태성 진전 환자의 고진폭 진전을 조절하기 위해 시상 DBS가 가장 많이 사용되지만, 장애성 진전 및/또는 레보도파 관련 운동 합병증을 동반한 본태성 진전 환자의 DBS 치료에는 STN 또는 내부 구상체(GPi)가 가장 빈번한 표적입니다. 최적의 의학적 요법 또는 DBS 중 어느 것이 더 강력한 개선 효과를 제공하는지 알아보기 위해 7개 보훈병원과 6개 대학병원에서 255명의 환자를 무작위 대조 시험에 등록하여 6개월 치료 후 DBS(STN, n=60; 또는 GPi, n=61)와 '최선의 의학적 요법'(n=134)의 효과를 비교했습니다.115 DBS로 치료받은 환자들은 문제가 되는 이상운동증 없이 하루 평균 4.6시간의 온타임이 증가한 반면, 최선의 의학적 요법을 받은 환자들은 0시간/일이었습니다(p<0.001). 또한, DBS 환자의 71%와 의학적 치료 환자의 32%에서 운동 기능이 운동 UPDRS에서 5점 이상 개선되었습니다. 이는 대부분의 파킨슨병 관련 건강 관련 삶의 질 측정 항목의 개선과 함께 신경인지 검사에서 최소한의 감소만을 동반했습니다. 그러나 심각한 부작용이 발생할 위험은 전반적으로 DBS 그룹이 의료 요법 그룹보다 3.8배 더 높았습니다(40% 대 11%).
치료 표적으로서 STN과 GPi의 상대적 효능은 DBS의 등장 이후 논의되어 왔습니다.3 재향군인회 협력 연구에서는 299명의 환자를 대상으로 24개월 후 STN과 GPi DBS 결과를 조사한 결과, 두 표적 간 운동(파트 III) UPDRS의 평균 변화에는 차이가 없었습니다.116 STN을 받은 환자는 팔라이드 자극을 받은 환자보다 더 적은 용량의 DA가 필요했으며(p=0.02), 시각 운동 처리 속도는 GPi 자극 후보다 STN 후 더 많이 감소했습니다(p=0.03). 반면, 우울증은 STN DBS 후 악화되었지만, 기분은 GPi DBS 후 개선되었습니다(p=0.02). 환자의 절반 이상이 심각한 부작용을 경험했지만, 두 그룹 간 부작용 발생 빈도에는 차이가 없었습니다. 이러한 연구와 다른 연구를 바탕으로, GPi DBS는 문제가 되는 운동 이상증과 경미한 인지 또는 행동 장애가 있는 환자에게 특히 적합할 수 있는 반면, 양측성 STN DBS는 인지에는 문제가 없지만 레보도파 용량 감소가 주된 목표인 환자에게 수술적 선택이 될 수 있다는 새로운 증거가 나오고 있습니다. GPi에 비해 STN DBS는 휴지기에는 더 유익한 영향을 미치는 것으로 보이지만 ICD와 같은 부작용과 관련이 있을 가능성이 더 높습니다.
한 가지 일반적인 임상적 질문은 파킨슨병 초기 단계 또는 더 어린 나이에 DBS 수술을 해도 비슷한 긍정적인 결과를 얻을 수 있는지 여부입니다.3 초기 운동 합병증이 있는 비교적 젊은(평균 연령 52세) 251명을 대상으로 한 무작위 시험에서 STN DBS와 의료 요법을 병행한 치료와 의료 요법만 시행한 치료가 비교되었습니다. 삶의 질 점수의 평균 점수는 DBS 그룹에서 7.8점 개선된 반면, 의료 요법 그룹에서는 0.2점 악화되었습니다. 운동 장애, 일상생활 활동, 레보도파 관련 운동 합병증은 수술 그룹에서 더 좋았습니다.117 조기 DBS를 고려할 때는 먼저 운동 변동과 이상 운동증을 개선하는 전략을 포함하여 의료 치료를 최적화하고 수술 위험과 기타 요인을 고려하는 것이 중요합니다.118 119
DBS는 효과적인 치료 전략으로 입증되었지만, 결과를 최적화하고 합병증을 최소화하기 위해서는 적절한 환자 선택과 정위 외과의의 경험과 숙련도에 따라 성공 여부가 결정됩니다. 적응 자극의 사용, 연결성 개선, 방향성 자극, 새로운 표적에 대한 탐색과 같은 DBS 기술의 발전은 계속 개선될 것입니다.120 121
Focused ultrasound
Unilateral focused ultrasound lesioning of the STN or thalamus (in tremor-dominant forms of PD) has been found to be beneficial in some patients, particularly if the symptoms are markedly asymmetric.89 Finally, spinal cord stimulation is increasingly being explored in patients with PD who are most troubled by their gait disorder.122
Cell replacement therapies
The outcomes of prior fetal tissue-derived cell transplants in PD have been variable. Although some transplanted patients showed some initial improvement, many developed ‘off’ dyskinesias despite robust graft survival.123 The presence of troubling dyskinesias in some patients, ethical concerns and the restricted availability of the tissues limited the clinical applicability of fetal transplantation.
Advances in the generation dopaminergic neurons from somatic human cells and improvement in the efficacy of differentiation protocols have led to a resurgence of cell transplantation in PD.124–127 Human embryonic stem cell lines and somatic cells which can be converted into authentic midbrain dopaminergic cells that satisfy good manufacturing practice grade criteria can be generated in unlimited amounts for clinical application.124–127
In 2020 a single case report of a patient with PD who was implanted with induced pleuripotent stem cells derived from his own fibrobalsts has generated controversy because of questionable scientific and ethical issues.127 Separately, a clinical trial involving 12 PD patients was conducted in Australia using parthenogenetic stem cells (derived from chemically induced unfertilized oocytes).128 There has been no report of any adverse side effects. The trial has been completed and results are expected in first half of 2020. The TRANSEURO is an open-label multicentre European study using human fetal dopamine cells and will be completed in 2021.129 Eleven patients have received transplantation in this observational study that recruited younger early PD. Although stem cell-derived cells have advantages over fetal cells, including near-unlimited availability, several critical issues, including potential tumorigenicity, immunosuppressive therapies, off-target effects, techniques of surgical delivery devices must be addressed before these approaches become clinically available.130
Finally, surgical delivery of gene therapy is an emerging area of experimental therapeutics. In phase 1 study, 15 patients with moderately advanced PD underwent MRI-guided delivery of adeno-associated viral vector serotype-2 encoding the complementary DNA for the enzyme, aromatic L-amino acid decarboxylase (VY-AADC01) into the putamen.131 This resulted in up to 42% coverage of the putamen and up to 79% corresponding increases in enzyme activity assessed by PET. There were dose-related improvements in clinical outcomes, including increases in patient-reported ON-time without troublesome dyskinesia and quality of life at 12 months. A phase 2 trial, randomised, placebo surgery controlled, double-blinded, multicentre, phase 2 clinical trial, evaluating the efficacy and safety of VY-AADC02 in advanced Parkinson’s disease patients with motor fluctuations, is currently being conducted in multiple centres.
In conclusion, much progress has been made in the understanding the etiopathogenesis of PD and in the symptomatic treatment of PD-related symptoms. However, currently there are no effective neuroprotective or disease modifying therapies that would slow the progression of the disease. The physical, mental, social and economic burden of PD is daunting and continues to be the most challenging therapeutic hurdle, especially in the advanced stages of the disease.
이전에 태아 조직 유래 세포 이식을 받은 파킨슨병 환자의 결과는 다양했습니다. 일부 이식 환자들은 초기에 어느 정도 호전을 보였지만, 많은 환자들이 이식 생존율이 높았음에도 불구하고 운동 이상증이 발생했습니다.123 일부 환자에서 문제가 되는 운동 이상증, 윤리적 우려, 조직의 제한된 가용성으로 인해 태아 이식의 임상 적용 가능성이 제한적이었습니다.
인간 체세포로부터 도파민 신경세포를 생성하는 기술이 발전하고 분화 프로토콜의 효능이 개선되면서 PD에서 세포 이식이 다시 부활했습니다.124-127 우수 제조 관리 기준 등급을 충족하는 진정한 중뇌 도파민 세포로 전환할 수 있는 인간 배아 줄기세포주 및 체세포는 임상 적용을 위해 무제한으로 생성할 수 있습니다.124-127
2020년에 자신의 섬유막에서 유래한 유도만능줄기세포를 이식받은 PD 환자의 사례 보고 한 건이 과학적, 윤리적 문제로 인해 논란을 일으켰습니다.127 이와는 별도로, 호주에서 12명의 PD 환자를 대상으로 한 임상시험이 (화학적으로 유도된 미수정 난자세포에서 유래한) 만능줄기세포를 사용하여 실시되었습니다.128 부작용에 대한 보고는 없었습니다. 임상시험은 완료되었으며 2020년 상반기에 결과가 나올 예정입니다. 인간 태아 도파민 세포를 사용하는 공개 라벨 다기관 유럽 연구인 TRANSEURO는 2021년에 완료될 예정입니다.129 이 관찰 연구에서 11명의 환자가 이식을 받았으며, 젊은 초기 PD를 모집했습니다. 줄기세포 유래 세포는 거의 무제한에 가까운 가용성 등 태아 세포에 비해 장점이 있지만 잠재적 종양원성, 면역 억제 요법, 표적 이탈 효과, 외과적 전달 장치 기술 등 몇 가지 중요한 문제를 해결해야 이러한 접근법이 임상적으로 이용 가능하기 전에 해결해야 합니다.130
마지막으로, 유전자 치료의 외과적 전달은 실험적 치료의 새로운 영역입니다. 1상 연구에서 중등도 진행성 PD 환자 15명을 대상으로 효소의 상보적 DNA를 코딩하는 아데노 관련 바이러스 벡터 혈청형-2, 방향족 L-아미노산 탈카르복실효소(VY-AADC01)를 퍼타멘에 MRI 유도 하에 전달했습니다.131 그 결과 퍼타멘의 최대 42% 범위와 PET로 평가한 효소 활성의 최대 79% 증가가 나타났습니다. 12개월 후 환자가 보고한 정시 투약률과 삶의 질이 개선되는 등 용량과 관련된 임상 결과가 개선되었으며, 이는 문제가 되는 운동 이상증 없이 정시 투약률이 증가한 것으로 나타났습니다. 운동 동요가 있는 진행성 파킨슨병 환자를 대상으로 VY-AADC02의 효능과 안전성을 평가하는 무작위, 위약 수술 대조, 이중 맹검, 다기관, 2상 임상 시험이 현재 여러 센터에서 진행 중입니다.
결론적으로, 파킨슨병의 병인 기전에 대한 이해와 파킨슨병 관련 증상에 대한 대증적 치료에는 많은 진전이 있었습니다. 그러나 현재로서는 질병의 진행을 늦출 수 있는 효과적인 신경 보호 또는 질병 수정 치료법은 없습니다. 파킨슨병의 신체적, 정신적, 사회적, 경제적 부담은 매우 크며, 특히 질병의 진행 단계에서 가장 어려운 치료 장애물로 남아 있습니다.