
Cold Spring Harb Perspect Biol. 2020 Mar; 12(3): a033993.
doi: 10.1101/cshperspect.a033993
PMCID: PMC6773515
PMID: 30936119
The Autophagy Lysosomal Pathway and Neurodegeneration
Author information Copyright and License information PMC Disclaimer
Abstract
The autophagy lysosomal pathway (ALP) is a major mechanism for degrading intracellular macromolecules. The catabolic products can then be used by the cell for energy or as building blocks to make other macromolecules. Since its discovery, a variety of cellular pathways have emerged that target components with varying specificity for lysosomal degradation. Under some circumstances, lysosomes may release their contents into the extracellular space where they may serve signaling or pathogenic functions. The ALP is active in healthy cells, and the level of activity can be regulated by nutrient-sensing and metabolic signaling pathways. The ALP is the primary pathway by which lipids and damaged organelles are degraded and may be the only pathway capable of degrading aggregated proteins. As such, there has been intense interest in understanding the role of the ALP in the accumulation of aggregated misfolded proteins characteristic of many of the major adult-onset neurodegenerative diseases. This review focuses on recent advances in our understanding of the ALP and its potential relationship to the pathogenesis and treatment of neurodegenerative diseases.
자가포식 리소좀 경로 autophagy lysosomal pathway (ALP)는
세포 내 거대 분자를 분해하는
주요 메커니즘입니다.
이 대사 산물은
세포에서 에너지로 사용되거나
다른 거대 분자를 만들기 위한 구성 요소로 사용될 수 있습니다.
리소좀이 발견된 이후,
리소좀 분해에 대한 다양한 특이성을 가진 성분을 표적으로 하는
다양한 세포 경로가 등장했습니다.
일부 상황에서 리소좀은
그 내용물을 세포 외 공간으로 방출하여
신호 전달 또는
병원성 기능을 수행할 수 있습니다.
ALP는
건강한 세포에서 활성화되며,
영양소 감지 및 대사 신호 전달 경로에 의해
활동 수준이 조절될 수 있습니다.
ALP는
지질과 손상된 세포 소기관이 분해되는 주요 경로이며
응집된 단백질을 분해할 수 있는 유일한 경로일 수 있습니다.
따라서
많은 주요 성인 발병 신경 퇴행성 질환의 특징인
응집된 잘못 접힌 단백질의 축적에서
ALP의 역할을 이해하는 데 관심이 집중되어 왔습니다.
이 리뷰에서는
ALP에 대한 최근의 이해와
신경 퇴행성 질환의 발병 및
치료와의 잠재적 관계에 대해 중점적으로 살펴봅니다.
HISTORY
The work of Drs. Christian de Duve and Yoshinori Ohsumi led to the discovery of the autophagy lysosomal pathway (ALP), and the contributions of each were recognized with the awards of the Nobel prize in physiology or medicine in 1974 and 2016, respectively. de Duve discovered the membrane-bound acidic compartment called the lysosome with biochemistry approaches (De Duve and Wattiaux 1966). Ohsumi discovered the core machinery responsible for autophagy by identifying the genes in yeast that are necessary for survival in the setting of caloric restriction (Klionsky et al. 2003; Tooze and Dikic 2016).
크리스찬 드 듀브(Christian de Duve) 박사와 요시노리 오스미(Yoshinori Ohsumi) 박사의 연구는
자가포식 리소좀 경로(ALP)의 발견으로 이어졌고,
각각 1974년과 2016년 노벨 생리의학상과 의학상을 수상하며 공로를 인정받았습니다.
드 듀브 박사는 생화학적인 접근법으로 리소좀이라는 막 결합 산성 구획을 발견했습니다(De Duve and Wattiaux 1966).
오스미는
칼로리 제한 환경에서
생존에 필요한 효모의 유전자를 확인하여
자가포식을 담당하는 핵심 메커니즘을 발견했습니다(Klionsky et al. 2003; Tooze and Dikic 2016).
LYSOSOME—THE HUB OF A PROTEOSTASIS NETWORK
The common way to portray the ALP is as a unidirectional pathway that begins with the de novo formation of double-membrane organelles called autophagophores and culminates in the fusion of autophagosomes and their contents with lysosomes to form autophagolysosomes, in which the contents of the autophagosomes are degraded. However, further study of the ALP has revealed multiple distinct pathways to the lysosome including nonspecific macroautophagy, substrate-specific forms of autophagy (e.g., mitophagy), chaperone-mediated autophagy (CMA), microautophagy, and micropinocytosis, to name a few (Fig. 1). One common feature of all these pathways is that degradation ultimately occurs in the lysosome. For this reason, it may be easier for the reader to understand the ALP using a hub and spoke model (Nixon et al. 2008; Perera and Zoncu 2016). Here, the hub, the lysosome, will be introduced first and then the different pathways into and out of the lysosome will be described.
리소좀 - 단백질 정체 네트워크의 허브
ALP를 묘사하는 일반적인 방법은
오토파지포어라고 하는
이중막 소기관의 신생 형성으로 시작하여
오토파지와 그 내용물이 리소좀과 융합하여
오토파지리소좀을 형성하는
단방향 경로로 마무리되며,
여기서 오토파지의 내용물이 분해되는 것으로
끝나는 것으로 설명합니다.
그러나
ALP에 대한 추가 연구를 통해
비특이적 거대 오토파지,
기질 특이적 형태의 오토파지(예: 미토파지),
샤프론 매개 오토파지(CMA),
미세 오토파지
및 미세 피노세포증 등
리소좀에 대한
여러 가지 경로가 밝혀졌습니다(그림 1).
nonspecific macroautophagy,
substrate-specific forms of autophagy (e.g., mitophagy),
chaperone-mediated autophagy (CMA),
microautophagy, and
micropinocytosis
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6304738/

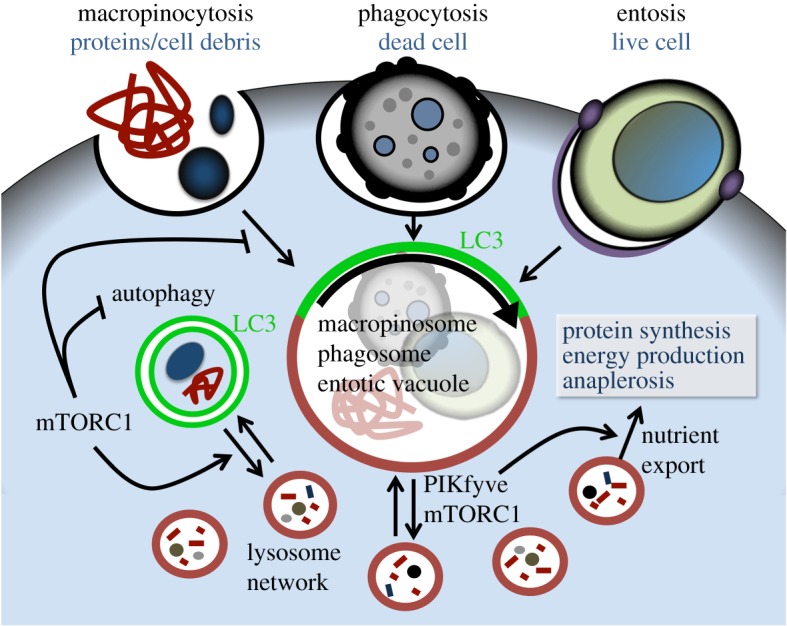

이러한 모든 경로의 공통적인 특징은 분해가 궁극적으로 리소좀에서 일어난다는 것입니다. 이러한 이유로 허브와 스포크 모델을 사용하면 독자가 ALP를 더 쉽게 이해할 수 있습니다(Nixon et al. 2008, Perera와 Zoncu 2016). 여기서는 허브인 리소좀을 먼저 소개한 다음 리소좀으로 들어오고 나가는 다양한 경로를 설
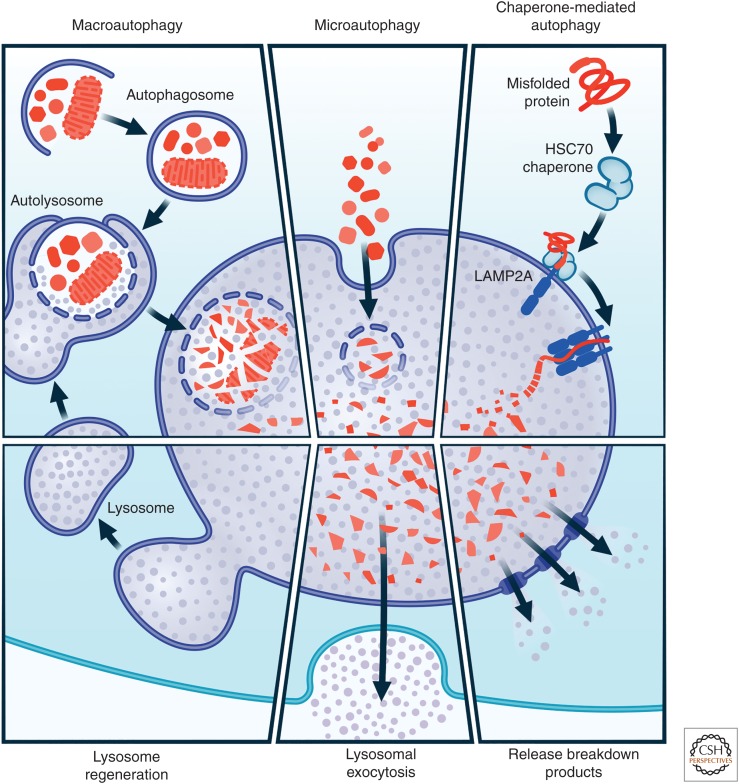
The autophagy lysosomal pathway.
The lysosome is the hub of a network of pathways that feed cargo into its lumen for degradation. These include pathways depicted here to deliver intracellular cargo such as macroautophagy, chaperone-mediated autophagy and microautophagy, as well as others that deliver extracellular cargo to the lysosome including endocytosis and micropinocytosis (see text for details). Cargo delivered to the lysosome can undergo degradation into molecular building blocks that can return to the cytoplasm to be catabolized further to supply cellular energy needs or to be reused in the synthesis of new macromolecules. Contents of lysosomes can also be extruded extracellularly by a Ca2+-dependent exocytic process. LAMP2A,
자가포식 리소좀 경로.
리소좀은
분해를 위해 화물물을
루멘으로 공급하는 경로 네트워크의 허브입니다.
여기에는
매크로 오토파지,
샤페론 매개 오토파지 및
마이크로 오토파지와 같은 세포 내화물을
리소좀으로 전달하는 경로와
엔도사이토시스 및 마이크로피노사이토시스 등
세포 외화물을 리소좀으로 전달하는 다른 경로가 포함됩니다(자세한 내용은 본문 참조).
리소좀으로 전달된 화물은
분자 구성 요소로 분해되어
세포질로 돌아가 세포에 필요한 에너지를 공급하거나
새로운 거대 분자의 합성에 재사용하기 위해 더 분해될 수 있습니다.
리소좀의 내용물은
Ca2+ 의존적 세포 외 배출 과정을 통해
세포 밖으로 배출될 수도 있습니다
Lysosome-associated membrane glycoprotein 2; HSC70, heat shock cognate 71.
Lysosomes are defined as membrane-bound organelles delimited by a single-lipid bilayer and characterized by an acidified milieu (pH ∼4.5). The membrane contains a variety of structural and signaling proteins, channels, transporters and trafficking, and fusion machinery. For example, the lysosomal membrane v-type ATPase (v-ATPase) pumps protons across the membrane and is the major mechanism for generating an acidic lumen and a pH gradient. Lysosomes are frequently located in the perinuclear area in the cell soma but undergo trafficking and fusion, mediated by a subset of membrane-associated RAB GTPases and SNARE proteins (Ao et al. 2014; Perera and Zoncu 2016). RAB5 and RAB7 function specifically to tether and dock endolysosomal membranes. LAMP1 makes up ∼50% of the lysosomal membrane protein, and it couples lysosomes to transport machinery.
Lysosomes contain approximately 60 soluble hydrolases, which are active at acidic pH, and carry out the degradation of macromolecules transported from the cytoplasm to the lumen of the organelle. Membranes and lipids are degraded within intralysosomal vesicles that contain specialized hydrolases and activator proteins suited to the task. Lipid droplets shuttled to lysosomes are hydrolyzed into free fatty acids and glycerol. Catabolites such as short peptides, amino acids, and other molecules are translocated from the lumen to the cytoplasm or other cellular compartments for the cell to use.
Lysosome biogenesis is coordinated by transcription factors that belong to the microphthalmia-transcription factor E family (MiT), including transcription factor EB (TFEB) and TFE3 (Settembre et al. 2011). They bind to a DNA response element motif known as the coordinated lysosomal expression and regulation (CLEAR) element within genes that encode proteins required for lysosomal biogenesis, autophagy, exocytosis, and endocytosis. In effect, CLEAR-dependent transcription promotes processes that lead to the isolation from the cytoplasm of unwanted cytoplasmic macromolecules, and their permanent removal via degradation and/or exocytosis (Decressac et al. 2013; Polito et al. 2014). TFEB and TFE3 are normally located in the cytoplasm, bound to the scaffold 14-3-3 protein owing to phosphorylation of two serines in the transcription factors by TFEB kinases such as ERK2 or mTORC1. In response to stresses such as starvation or lysosomal dysfunction, the lysosomal nutrient sensing (LYNUS) machinery, which sits on the cytoplasmic side of the lysosomal membrane, senses the nutrient content of the lysosome, and communicates the information to the nucleus (Alers et al. 2012; Inoki et al. 2012a; Lin et al. 2012; Settembre et al. 2013; Shanware et al. 2013). TFEB is dephosphorylated by mTOR, translocates to the nucleus, and drives the transcription of CLEAR-containing genes (Settembre et al. 2013). For example, TFEB induces expression of PPARα and PGC1α, which play a role in lipid oxidation and ketogenesis (Settembre et al. 2011, 2013; Tsunemi et al. 2012), emphasizing the tight integration of these catabolic processes. TFEB was also shown to be the main mediator of PGC1α-induced improvement in a mouse model of Huntington's disease (Tsunemi et al. 2012). Transcription of TFEB itself is controlled via an autoregulatory loop (Settembre et al. 2013).
Although it is well established that the protein composition of lysosomes evolves during biogenesis and maturation, an important unanswered question in the field is whether functionally important subpopulations of mature lysosomes exist within different cell types or even within the same cell. Lysosomes uniformly express LAMP-1, but other lysosome-associated proteins are expressed more variably. For example, LAMP-2A, which is required for CMA, is absent from ∼80% of lysosomes at the baseline (Cuervo et al. 1997; Kaushik and Cuervo 2012). It is unclear whether the heterogeneity reflects functionally distinct lysosomes in which production is regulated, or if it is related to stochastic variations in protein sorting or maturation. Lysosomes undergo “reformation” (Yu et al. 2010), suggesting that there is a lot of exchange between different endocytic compartments.
리소좀은
단일 지질 이중층으로 구분되고
산성화된 환경(pH ∼4.5)을 특징으로 하는
막 결합 소기관으로 정의됩니다.
이 막에는
다양한 구조 및 신호 단백질,
채널, 수
송체 및
이동체,
융합 기계가 포함되어 있습니다.
예를 들어,
리소좀 막의 v형 ATPase(v-ATPase)는 양성자를 막 전체로 펌핑하며
산성 루멘과 pH 구배를 생성하는 주요 메커니즘입니다.
리소좀은
세포질의 핵 주변 영역에 위치하는 경우가 많지만
막과 관련된 RAB GTPase 및 SNARE 단백질의 하위 집합을 매개로 이동과 융합을 거칩니다(Ao 등. 2014; Perera 및 Zoncu 2016).
RAB5와 RAB7은 특히
소포체 막을 묶고 고정하는 기능을 합니다.
LAMP1은 리소좀 막 단백질의 ∼50%를 구성하며,
리소좀을 운반 기계에 결합합니다.
리소좀에는
약 60개의 가용성 가수분해효소가 포함되어 있으며,
산성 pH에서 활성화되어
세포질에서 세포소기관 내강으로 운반되는
거대 분자의 분해를 수행합니다.
세포막과 지질은
해당 작업에 적합한 특수 가수분해 효소 및
활성화 단백질을 포함하는
소포체 내에서 분해됩니다.
리소좀으로 운반된 지질 방울은
유리 지방산과 글리세롤로
가수분해됩니다.
짧은 펩타이드, 아미노산 및 기타 분자와 같은
이화 물질은
세포가 사용할 수 있도록
루멘에서 세포질 또는 다른 세포 구획으로 이동합니다.
리소좀 생성은
전사인자 EB(TFEB) 및
TFE3를 포함하여
미세 전사인자 E 계열(MiT)에 속하는
전사인자에 의해 조정됩니다(Settembre 외. 2011).
이들은
리소좀 생물 생성,
자가포식,
세포외이동,
세포내이식에 필요한 단백질을 암호화하는
유전자 내에서 조정된 리소좀 발현 및 조절(CLEAR) 요소로 알려진
DNA 반응 요소 모티프에 결합합니다.
실제로 CLEAR 의존적 전사는
원치 않는 세포질 거대 분자를 세포질에서 분리하고
분해 및/또는 세포외 분해를 통해
영구적으로 제거하는 과정을 촉진합니다(Decressac 외. 2013; Polito 외. 2014).
TFEB와 TFE3는 일반적으로 세포질에 위치하며 ERK2 또는 mTORC1과 같은 TFEB 키나제에 의한 전사인자의 두 세린의 인산화로 인해 스캐폴드 14-3-3 단백질에 결합됩니다.
기아 또는 리소좀 기능 장애와 같은 스트레스에 반응하여
리소좀 막의 세포질 쪽에 있는
리소좀 영양소 감지(LYNUS) 기계가
리소좀의 영양소 함량을 감지하고
그 정보를 핵에 전달합니다(Alers 외. 2012, Inoki 외. 2012a, Lin 외. 2012, Settembre 외. 2013, Shanware 외. 2013).
TFEB는
mTOR에 의해 탈인산화되고
핵으로 이동하여 CLEAR 함유 유전자의 전사를 유도합니다(Settembre et al. 2013).
예를 들어,
TFEB는
지질 산화와 케톤 생성에 중요한 역할을 하는
PPARα와 PGC1α의 발현을 유도하여(세템브레 외. 2011, 2013; 츠네미 외. 2012),
이러한 대사 과정이 긴밀하게 통합되어 있음을 강조합니다(세템브레 외. 2013).
또한, 헌팅턴병 마우스 모델에서 TFEB는 PGC1α에 의한 개선의 주요 매개체임이 밝혀졌습니다(츠네미 외. 2012). TFEB 자체의 전사는 자가 조절 루프를 통해 제어됩니다(Settembre et al. 2013).
리소좀의 단백질 구성이
생물 발생과 성숙 과정에서 진화한다는 것은 잘 알려져 있지만,
이 분야에서 중요한 미해결 질문은
성숙한 리소좀의 기능적으로 중요한 하위 집단이
다른 세포 유형 또는 심지어 같은 세포 내에 존재하는지 여부입니다.
리소좀은 LAMP-1을 균일하게 발현하지만, 다른 리소좀 관련 단백질은 더 다양하게 발현됩니다. 예를 들어, CMA에 필요한 LAMP-2A는 기준선에서 리소좀의 ∼80%에 존재하지 않습니다(Cuervo et al. 1997; Kaushik and Cuervo 2012). 이질성이 생산이 조절되는 기능적으로 구별되는 리소좀을 반영하는지, 아니면 단백질 분류 또는 성숙의 확률적 변화와 관련이 있는지는 불분명합니다. 리소좀은 "변형"(Yu et al. 2010)을 겪는데, 이는 서로 다른 세포 내 구획 사이에 많은 교류가 있음을 시사합니다.
PATHWAYS TO THE LYSOSOME—MACROAUTOPHAGY
Macroautophagy is the major intracellular pathway by which intracellular cargo are delivered to the lysosome (Biswas et al. 2008). The complex biochemistry of the pathway is excellently reviewed elsewhere (Klionsky 2005, 2007; Yamamoto and Yue 2014; Bingol 2018). Induction is governed by the Ulk1 complex, which induces the nucleation of the isolation membrane (IM) or phagophore. Then, the Beclin1-Atg14L-Vps34 lipid kinase complex catalyzes the production of PI3P at the IM, resulting in the recruitment of PI3P-binding proteins such as WIPI-1, -2, and DFCP1 (Ferguson et al. 2009). The sites of autophagy induction and the source of membranes has been controversial (Mari et al. 2011); endoplasmic reticulum (ER) and ER-mitochondrial contacts are likely sources but others have been suggested (Axe et al. 2008; Itoh et al. 2008). The IM expands and envelops the cargo in a process that requires Atg9 and two ubiquitin-like conjugation systems, which trigger the covalent attachment of the lipid phosphatidylethanolamine (PE) to Atg8 or its mammalian homolog, microtubule-associated protein 1 light chain 3 (LC3) (Nakatogawa et al. 2007; Fujita et al. 2008; Sou et al. 2008). Embedded into the autophagosome membrane via the PE moiety, LC3-II serves as an adaptor protein that binds cargo being engulfed by the autophagosome through other adaptor proteins described below (Tan et al. 2008; Tung et al. 2010). The mature autophagosome fuses with lysosomes by a mechanism that likely depends on SNAREs to form autophagolysosomes, exposing cargo to the lysosomal degradation machinery for digestion (Jäger et al. 2004; Stroikin et al. 2004; Cai et al. 2010; Yuzaki 2010; Itoh et al. 2011; Hyttinen et al. 2013; Albanesi et al. 2015; Murrow et al. 2015). Interestingly, although LC3 remains the most specific known marker of autophagosomes, LC3-independent forms of autophagy have been described (Kuma et al. 2007; Szalai et al. 2015; Engedal and Seglen 2016).
Initially, macroautophagy was thought to be nxxxxonselective, with phagophores scooping up and engulfing cytoplasm and cargo that happen to be in the neighborhood. Subsequently, a large number of adaptor proteins have been discovered, including p62/SQSTM1 (Sequestosome 1), NBR1, Nix, NDP52, Alfy/WDFY3, and OPTN (optineurin), which play a role in targeting specific cargo for autophagic clearance including protein aggregates, mitochondria, other damaged organelles, etc. (Fortun et al. 2003; Komatsu et al. 2007, 2010; Farré et al. 2008; Geisler et al. 2010; Brady et al. 2011; Matsumoto et al. 2011; Lim et al. 2015). Many of these adaptors contain ubiquitin- and LC3-interacting regions and often undergo oligomerization, which increases the avidity of their binding to cargo. In some cases, binding is regulated by clinically relevant modulators of these adaptor proteins such as TBK1, which phosphorylates OPTN (Wild et al. 2011). Defining the extent of substrate specificity and the associated mechanisms of regulation are a major focus of ongoing research in the field (Anding and Baehrecke 2017).
리소좀-대식작용으로 가는 경로
매크로 오토파지는
세포 내 화물이 리소좀으로 전달되는 주요 세포 내 경로입니다(Biswas 등. 2008).
이 경로의 복잡한 생화학은
다른 곳에서 훌륭하게 검토되었습니다(Klionsky 2005, 2007; Yamamoto and Yue 2014; Bingol 2018).
유도는 분리막(IM) 또는 식세포의 핵 형성을 유도하는 Ulk1 복합체의 지배를 받습니다. 그런 다음 Beclin1-Atg14L-Vps34 지질 키나아제 복합체가 IM에서 PI3P 생성을 촉매하여 WIPI-1, -2, DFCP1과 같은 PI3P 결합 단백질의 모집을 초래합니다(Ferguson et al. 2009). 자가포식 유도 부위와 막의 출처는 논란의 여지가 있습니다(Mari et al. 2011). 소포체(ER)와 ER-미토콘드리아 접촉이 유력한 출처이지만 다른 출처도 제안되었습니다(Axe et al. 2008; Itoh et al. 2008). IM은 Atg9와 두 개의 유비퀴틴 유사 접합 시스템을 필요로 하는 과정에서 화물을 확장하고 감싸며, 이는 지질 포스파티딜에탄올아민(PE)을 Atg8 또는 포유류 동족체인 미세소관 관련 단백질 1 경쇄 3(LC3)에 공유 결합을 촉발합니다(Nakatogawa 등 2007; Fujita 등 2008; Sou et al. 2008). PE 모이티를 통해 오토파지솜 막에 내장된 LC3-II는 아래에 설명된 다른 어댑터 단백질을 통해 오토파지솜에 포획되는 화물과 결합하는 어댑터 단백질 역할을 합니다(Tan et al. 2008; Tung et al. 2010). 성숙한 오토파지좀은 SNARE에 의존하는 메커니즘에 의해 리소좀과 융합하여 오토파지리소좀을 형성하고 소화를 위해 화물을 리소좀 분해 기계에 노출시킵니다(Jäger 외. 2004; Stroikin 외. 2004; Cai 외. 2010; Yuzaki 2010; Itoh 외. 2011; Hyttinen 외. 2013; Albanesi 외. 2015; Murrow 외. 2015). 흥미롭게도 LC3가 오토파지좀의 가장 구체적인 마커로 알려져 있지만, LC3와 무관한 형태의 오토파지가 기술되었습니다(Kuma 외. 2007; Szalai 외. 2015; Engedal and Seglen 2016).
처음에 거대 오토파지는 비선택적인 것으로 여겨졌으며, 포식자가 주변에 있는 세포질과 운반물을 퍼서 삼키는 것으로 생각되었습니다. 이후 단백질 응집체, 미토콘드리아, 기타 손상된 소기관 등 자가포식 제거를 위해 특정화물을 표적으로 삼는 역할을 하는 p62/SQSTM1(시퀘스토솜 1), NBR1, Nix, NDP52, Alfy/WDFY3, OPTN(옵티뉴린) 등 수많은 어댑터 단백질이 발견되었습니다. (Fortun 외. 2003; Komatsu 외. 2007, 2010; Farré 외. 2008; Geisler 외. 2010; Brady 외. 2011; Matsumoto 외. 2011; Lim 외. 2015). 이러한 어댑터의 대부분은 유비퀴틴 및 LC3와 상호 작용하는 영역을 포함하고 있으며 종종 올리고머화를 거쳐 화물과의 결합에 대한 열성을 증가시킵니다. 경우에 따라 결합은 OPTN을 인산화하는 TBK1과 같은 이러한 어댑터 단백질의 임상적으로 관련된 조절 인자에 의해 조절됩니다(Wild et al. 2011). 기질 특이성의 정도와 관련 조절 메커니즘을 정의하는 것은 이 분야에서 진행 중인 연구의 주요 초점입니다(Anding and Baehrecke 2017).
PATHWAYS TO THE LYSOSOME—MICROAUTOPHAGY, MULTIVESICULAR BODIES, AND PINOCYTOSIS
Another path to lysosomal degradation is microautophagy (Sahu et al. 2011; Shpilka and Elazar 2011) and the closely related endosomal microautophagy (Bingol 2018). These relatively less studied pathways begin with the invagination of the lysosomal membrane. Eventually, the invaginations, which contain contents from the cytoplasm, pinch off to form vesicles in the lysosomal lumen (Shpilka and Elazar 2011). The degree of substrate specificity and relative importance of microautophagy for physiology remains unclear.
리소좀-미세 자가포식, 다소포체 및 피노세포증으로 가는 경로
리소좀 분해의
또 다른 경로는
마이크로 오토파지(Sahu 외. 2011; Shpilka와 Elazar 2011)와 밀접하게 관련된
엔도솜 마이크로 오토파지(Bingol 2018)입니다.
상대적으로 덜 연구된 이러한 경로는
리소좀 막의 침입으로 시작됩니다.
결국
세포질의 내용물을 포함하는 침윤은
리소좀 내강에 소포를 형성하기 위해 꼬집어 들어갑니다(Shpilka and Elazar 2011).
기질 특이성의 정도와 생리학에서
미세 오토파지의 상대적 중요성은 아직 불분명합니다.
PATHWAYS TO THE LYSOSOME—CHAPERONE-MEDIATED AUTOPHAGY
CMA is a pathway to translocate polypeptides with the KFERQ motif directly from the cytoplasm into the lumen of the lysosome (Olson et al. 1991; Kiffin et al. 2004). Translocation is mediated by LAMP-2A and HSP8A, a member of the HSP70 family. Interestingly, the KFERQ motif is found in ∼30% of the proteome and in many proteins implicated in neurodegenerative diseases including α-synuclein (Olson et al. 1991; Cuervo et al. 2004; Martinez-Vicente et al. 2008). Importantly, and unlike macroautophagy, proteins evidently need to be translocated into the lysososme as monomers, so CMA may be less effective than autophagy at degrading aggregated protein. Nevertheless, induction of CMA has been shown to accelerate the clearance of some disease-causing proteins (Massey et al. 2006; Bauer et al. 2010; Wang et al. 2010b), which presumably reduces their propensity to accumulate and aggregate.
리소좀-샤페론 매개 자가포식 경로
CMA는
KFERQ 모티프를 가진 폴리펩타이드를
세포질에서 리소좀 내강으로 직접 이동시키는 경로입니다(Olson 외. 1991; Kiffin 외. 2004).
전위는 HSP70 계열의 구성원인 LAMP-2A와 HSP8A에 의해 매개됩니다. 흥미롭게도 KFERQ 모티브는 단백질체의 ∼30%에서 발견되며 α-시누클레인을 포함한 신경 퇴행성 질환과 관련된 많은 단백질에서 발견됩니다(Olson 외. 1991; Cuervo 외. 2004; Martinez-Vicente 외. 2008). 중요한 것은 거대 오토파지와 달리 단백질은 단량체로서 리소좀으로 이동해야 하므로 CMA는 응집된 단백질을 분해하는 데 오토파지보다 덜 효과적일 수 있다는 점입니다. 그럼에도 불구하고 CMA의 유도는 일부 질병을 유발하는 단백질의 제거를 가속화하는 것으로 나타났습니다(Massey 외. 2006; Bauer 외. 2010; Wang 외. 2010b), 이는 아마도 축적 및 응집 성향을 감소시키는 것으로 추정됩니다.
OTHER PATHS TO THE LYSOSOME
Autophagy, microautophagy, and CMA are pathways to convey cytoplasmic contents to the lysosomal lumen for degradation. Additional pathways, including macropinocytosis, phagocytosis, and endocytosis can bring extracellular material to the lysosome for degradation (Binotti et al. 2015). In cancer cells, macropinocytosis may be a critical way to take up macromolecules for metabolic support, but relatively less is known about the role and importance of macropinocytosis in neurons. In the context of neurodegenerative disease, these pathways may be important routes by which extracellular aggregation-prone proteins enter neurons and transmit proteinopathy between neighboring cells. An important future direction will be to better understand mechanisms by which misfolded proteins are taken up by neurons and how they get access to the cytosol to template and propagate misfolded protein conformations.
자가포식,
마이크로오토파지,
CMA는
세포질 내용물을 분해하기 위해
리소좀 내강으로 운반하는 경로입니다.
대식세포증, 식세포증, 엔도사이토시스 등의 추가 경로는
세포 외 물질을 리소좀으로 가져와 분해할 수 있습니다(Binotti 등. 2015).
암세포에서 대식세포증은 대사 지원을 위해 거대 분자를 흡수하는 중요한 방법일 수 있지만, 신경세포에서 대식세포증의 역할과 중요성에 대해서는 상대적으로 덜 알려져 있습니다. 신경 퇴행성 질환의 맥락에서 이러한 경로는 세포 외 응집성 단백질이 뉴런으로 들어가 이웃 세포 간에 단백질 병증을 전달하는 중요한 경로가 될 수 있습니다. 향후 중요한 방향은 잘못 접힌 단백질이 뉴런에 의해 흡수되는 메커니즘과 잘못 접힌 단백질 형태를 형성하고 전파하기 위해 세포질에 접근하는 방법을 더 잘 이해하는 것입니다.
PATHWAYS FROM THE LYSOSOME
Cargo delivered to the lysosome can be degraded into substituent molecular building blocks, then released into the cytoplasm to be metabolized or reused in the synthesis of new macromolecules. However, lysosomes or autolysosomes can also translocate to the plasma membrane, and fuse and release their contents into the extracellular space (Takenouchi et al. 2009; Settembre et al. 2013). This process of lysosomal exocytosis is believed to begin with activation of the lysosome, causing kinesin-associated lysosomes to be trafficked to the plasma membrane (Perera and Zoncu 2016). Ca2+ efflux from the lysosome mediated by the TRPML1 channel is likely critical for triggering SNARE-mediated plasma membrane fusion (Xu and Ren 2015; Perera and Zoncu 2016). TRPML1 gating is modulated by lysosomal pH and by phosphoinositides, such as PI(3,5)P2, PI(4,5)P2 , PI(3,4)P2, and PI(3,4,5)P2. Interestingly, lysosomal Ca2+ release and exocytosis may be triggered in neurons by back-propagating action potentials, and exocytosed proteolytic enzymes may play a role in synaptic plasticity (Padamsey et al. 2017). Fe2+ reportedly accumulates in lysosomes, likely as a consequence of degrading Fe2+-containing cellular proteins, and abnormal neuronal accumulation of Fe2+ is a feature of neurodegenerative diseases (Kwan et al. 2012; Wang et al. 2016; van Duijn et al. 2017). Whether that is so because of lysosomal dysfunction is not known. How lysosomal exocytosis is related to other release mechanisms, including exosomes, is also not fully understood (Baixauli et al. 2014; Poehler et al. 2014).
리소좀의 경로
리소좀으로 전달된 화물은 치환 분자 구성 요소로 분해된 다음 세포질로 방출되어 대사되거나 새로운 거대 분자의 합성에 재사용될 수 있습니다. 그러나 리소좀 또는 자가 리소좀은 원형질막으로 이동하여 내용물을 융합하여 세포 외 공간으로 방출할 수도 있습니다(타케노우치 외. 2009; 세템브레 외. 2013). 이러한 리소좀 세포외 유출 과정은 리소좀의 활성화로 시작되어 키네신 관련 리소좀이 혈장막으로 이동하는 것으로 여겨집니다(Perera and Zoncu 2016). TRPML1 채널을 매개로 한 리소좀의 Ca2+ 유출은 SNARE 매개 혈장막 융합을 촉발하는 데 중요할 수 있습니다(Xu and Ren 2015, Perera and Zoncu 2016). TRPML1 게이트는 리소좀 pH와 PI(3,5)P2, PI(4,5)P2 , PI(3,4)P2 및 PI(3,4,5)P2와 같은 포스포이노시타이드에 의해 변조됩니다. 흥미롭게도 리소좀 Ca2+ 방출과 세포외 이동은 역전파 활동 전위에 의해 뉴런에서 유발될 수 있으며, 세포외 이동된 단백질 분해 효소는 시냅스 가소성에 역할을 할 수 있습니다(Padamsey et al. 2017). Fe2+는 리소좀에 축적되는 것으로 알려졌는데, 이는 Fe2+ 함유 세포 단백질 분해의 결과일 수 있으며, 비정상적인 신경세포의 Fe2+ 축적은 신경 퇴행성 질환의 특징입니다(Kwan 외. 2012; Wang 외. 2016; van Duijn 외. 2017). 이것이 리소좀 기능 장애 때문인지 여부는 알려지지 않았습니다. 리소좀 세포 외 배출이 엑소좀을 포함한 다른 방출 메커니즘과 어떻게 관련되어 있는지도 완전히 이해되지 않았습니다(Baixauli 등. 2014; Poehler 등. 2014).
ALP IN THE CONTEXT OF THE PROTEOSTASIS NETWORK
It is important to remember that the ALP does not function in isolation. It exists within an elaborate proteostasis network and signaling system that enables cells to sense and respond to metabolic changes (Crighton et al. 2006; Maiuri et al. 2010; He et al. 2012). Although the ALP plays an important role in the turnover of proteins, other pathways including the unfolded protein response (UPR) and the ubiquitin proteasome system (UPS) are also critical (Webb et al. 2003; Keller et al. 2004; Kabuta et al. 2006; Wang et al. 2009, 2010a; Benbrook and Long 2012; Wang and Mandelkow 2012; Lee et al. 2013), and there is abundant evidence that there is cross talk between these pathways (Bernales et al. 2006; Pandey et al. 2007; Matus et al. 2008; Kirkin et al. 2009; Rouschop et al. 2010; Watanabe et al. 2010; Senft and Ronai 2015). For example, inhibitors of the UPR are capable of inducing autophagy, perhaps because they lead to ER-associated degradation and the extrusion of aggregated protein to the cytoplasm that must be cleared by autophagy (Høyer-Hansen and Jäättelä 2007; Hetz et al. 2009; Suh et al. 2012). Cross talk and redundancy between pathways probably explain how cells and organisms can survive at least some significant impairments in specific components of the proteostasis network.
ALP는 단독으로 기능하지 않는다는 점을 기억하는 것이 중요합니다. ALP는 세포가 대사 변화를 감지하고 이에 반응할 수 있도록 하는 정교한 단백질 정체 네트워크와 신호 체계 내에 존재합니다(Crighton 외. 2006; Maiuri 외. 2010; He 외. 2012). ALP는 단백질의 턴오버에 중요한 역할을 하지만, 펼쳐진 단백질 반응(UPR) 및 유비퀴틴 프로테아좀 시스템(UPS)을 포함한 다른 경로도 중요합니다(Webb et al. 2003; Keller et al. 2004; Kabuta et al. 2006; Wang et al. 2009, 2010a; Benbrook and Long 2012; Wang and Mandelkow 2012; Lee et al. 2013), 이러한 경로 간에 상호 작용이 있다는 증거가 풍부합니다(Bernales et al. 2006; Pandey et al. 2007; Matus et al. 2008; Kirkin et al. 2009; Rouschop et al. 2010; Watanabe et al. 2010; Senft and Ronai 2015). 예를 들어, UPR의 억제제는 자가포식을 유도할 수 있는데, 이는 아마도 ER과 관련된 분해와 자가포식에 의해 제거되어야 하는 응집된 단백질의 세포질로의 유출을 유도하기 때문입니다(Høyer-Hansen and Jäättelä 2007; Hetz et al. 2009; Suh et al. 2012). 경로 간의 상호 작용과 중복성은 아마도 세포와 유기체가 단백질 정체 네트워크의 특정 구성 요소에 적어도 일부 심각한 손상이 발생하더라도 생존할 수 있는 방법을 설명할 수 있을 것입니다.
ALP IN NEURODEGENERATIVE DISEASE—DO NEURONS MANAGE AUTOPHAGY DIFFERENTLY?
The genetics of autophagy were elucidated in yeast, and the majority of autophagy studies involve the use of nonneuronal cells (Kim and Klionsky 2000), raising the question of whether neuronal autophagy is unique (Larsen and Sulzer 2002; Boland and Nixon 2006; Moruno Manchon et al. 2016). Because neurons are postmitotic, they have lost one important mechanism to clear long-lived proteins—cell division and dilution (Eden et al. 2011). Neurons also have an elaborated morphology, with long dendrites and axons that can be a meter long or longer, and therefore must manage the degradation of cargo at distant sites (Shen and Ganetzky 2009; Bowling and Klann 2014; Tang et al. 2014). The cell biology of the autophagy pathways in neurons was initially characterized by Holzbaur and colleagues, who showed that autophagosomes are formed constitutively in axon terminals, and are trafficked to the cell body (Maday and Holzbaur 2014). En route, the protein composition gradually resembles mature lysosomes and the intralumenal pH drops. Eventually, in the soma, they fuse with lysosomes to form autophagolysosomes, and the degradation of their contents ensues.
In addition, neurons in the central nervous system are substantially buffered by astrocytes from large variations in nutrients that can occur elsewhere in the body and that can induce starvation-dependent activation of mTOR-dependent autophagy. Indeed, a number of studies have suggested that mTOR-dependent autophagy may be regulated in neurons differently than in nonneuronal cells (Boland et al. 2008). For example, inhibition of mTOR with rapamycin or sirolimus induces autophagy less effectively in neurons compared with nonneuronal cells (Tsvetkov et al. 2010; Krüger et al. 2012). Rapamycin binds FKB12, a nonobligate component of the mTORC1 complex, but not the mTORC2 complex, and one study has suggested that differences in the TORC complexes in neurons could contribute to the differences in reported efficacy of rapamycin (Roscic et al. 2011). There is also evidence that subtypes of neurons may differ in their dependence on autophagy (Hara et al. 2006; Komatsu et al. 2006; Friedman et al. 2012).
자가포식의 유전학은 효모에서 밝혀졌고, 대부분의 자가포식 연구는 비신경세포를 사용하며(Kim and Klionsky 2000), 신경세포의 자가포식이 독특한지에 대한 의문이 제기되었습니다(Larsen and Sulzer 2002; Boland and Nixon 2006; Moruno Manchon 외. 2016). 뉴런은 세포사멸 후이기 때문에 수명이 긴 단백질을 제거하는 중요한 메커니즘인 세포 분열과 희석 기능을 상실했습니다(Eden 외. 2011). 뉴런은 또한 길이가 1미터 이상 되는 긴 수상돌기와 축삭돌기를 가진 정교한 형태를 가지고 있기 때문에 먼 곳에서 화물의 분해를 관리해야 합니다(Shen and Ganetzky 2009; Bowling and Klann 2014; Tang et al. 2014). 뉴런에서 자가포식 경로의 세포 생물학은 처음에는 Holzbaur와 동료들에 의해 특징지어졌는데, 이들은 자가포식체가 축삭 말단에서 구성적으로 형성되어 세포체로 운반된다는 것을 보여주었습니다(Maday와 Holzbaur 2014). 그 과정에서 단백질 구성은 점차 성숙한 리소좀과 유사해지고 내강 내 pH가 떨어집니다. 결국 체세포에서는 리소좀과 융합하여 자가포식소좀을 형성하고 그 내용물의 분해가 이어집니다.
또한 중추 신경계의 뉴런은 신체의 다른 곳에서 발생할 수 있는 영양소의 큰 변화로부터 성상 세포에 의해 상당히 완충되며, 이는 기아에 의존하는 mTOR 의존성 자가포식의 활성화를 유도할 수 있습니다. 실제로 많은 연구에서 신경세포에서는 mTOR 의존성 자가포식이 비신경세포와 다르게 조절될 수 있다고 제안했습니다(Boland 등. 2008). 예를 들어, 라파마이신 또는 시롤리무스로 mTOR을 억제하면 비신경세포에 비해 신경세포에서 자가포식이 덜 효과적으로 유도됩니다(Tsvetkov 등. 2010; Krüger 등. 2012). 라파마이신은 mTORC1 복합체의 비필수 성분인 FKB12와 결합하지만 mTORC2 복합체에는 결합하지 않으며, 한 연구에 따르면 뉴런에서 TORC 복합체의 차이가 라파마이신의 효능 차이에 기여할 수 있다고 합니다(Roscic et al. 2011). 또한 뉴런의 하위 유형에 따라 자가포식에 대한 의존도가 다를 수 있다는 증거도 있습니다(Hara 외. 2006; Komatsu 외. 2006; Friedman 외. 2012).
ALP IN NEURODEGENERATIVE DISEASE—WHAT IS THE EVIDENCE?
A common thread cutting across multiple adult-onset neurodegenerative diseases, including Alzheimer's disease (AD), Parkinson's, Huntington disease, amyotrophic lateral sclerosis (ALS), and frontotemporal dementia, is the abnormal deposition of misfolded aggregated protein(s) (Friedman et al. 2012). Given the critical role of autophagy in the clearance of aggregated protein, this could indicate that autophagy dysfunction is a common mechanism in neurodegenerative disease.
That autophagy dysfunction may be a major mechanism in neurodegenerative disease is further suggested from the established role of mitochondrial damage and associated deficits in bioenergetics or excessive reactive oxygen species in these conditions (Beal et al. 2006; Lezi and Swerdlow 2012; Ryan et al. 2015; Wang 2017). Because autophagy is the principal pathway by which cells remove catastrophically damaged mitochondria (Youle and Narendra 2011), it stands to reason that impairment in this process may result in abnormal accumulation of deleterious mitochondria (Dagda et al. 2008; Narendra et al. 2008; Ferree et al. 2013; Lemasters 2014; Lazarou et al. 2015; Chen et al. 2017; Jinn et al. 2017).
More broadly, the link between autophagy and aging may be an important consideration (Meléndez et al. 2003; Bergamini et al. 2004; Rubinsztein et al. 2011; Inoue et al. 2012b; Carnio et al. 2014). Aging is the number-one risk factor for neurodegenerative diseases, and is associated with down-regulation of autophagy in the brain (Lipinski et al. 2010). Conversely, induction of autophagy has been shown in model organisms to increase their longevity (Simonsen et al. 2008; Zheng et al. 2010; Pyo et al. 2013). More importantly, autophagy induction is associated with an increase in their health span and quality of life, suggesting that the promotion of autophagy may confer a more fundamental improvement in quality of the function of biological systems that could bring broad benefits (Cuervo et al. 2005; Levine et al. 2011; Oka et al. 2012; Kang et al. 2015).
Beyond the intuitive appeal of the pathway as a therapeutic target (Levine and Kroemer 2008; Nixon 2013), there is a remarkable genetic association between neurodegenerative disease and autophagy. Mutations in many different genes associated with different steps in autophagy have been linked to AD, Parkinson's, or Huntington's disease, as well as ALS, frontotemporal dementia, and others (Fig. 2; Table 1) (Kegel et al. 2000; Petersén et al. 2001; Skibinski et al. 2005; Ramirez et al. 2006; Cheung and Ip 2009; Ju et al. 2009; Montie et al. 2009; Winslow et al. 2010; Dehay et al. 2012; Lucin et al. 2013; Ochaba et al. 2014; Wong and Holzbaur 2014; Martin et al. 2015; Ciura et al. 2016; Sellier et al. 2016; Sullivan et al. 2016; Manzoni 2017; Fujikake et al. 2018), indicating that mutations and variants in genes associated with autophagy are sufficient to cause neurodegenerative disease in some cases. Indeed, mutations in some genes critical for lysosome function lead to lysosomal storage disorders that cause neurological symptoms in childhood (Nixon et al. 2008). The extent to which a mismatch between the production and autophagic clearance of misfolded proteins exists in the much more common cases of idiopathic or sporadic AD and Parkinson's disease or ALS is not known (Ahmed et al. 2012). However, recent studies in patients with AD and the dynamics of amyloid β turnover suggest the intriguing possibility that genetic forms of AD may cause an overproduction of misfolded proteins, whereas idiopathic forms may be associated with reduced clearance, with all forms of AD associated with a fundamental mismatch between production and clearance, and the same may hold for other common but idiopathic forms of neurodegenerative disease (Mawuenyega et al. 2010).
알츠하이머병(AD),
파킨슨병,
헌팅턴병,
근위축성 측색 경화증(ALS),
전측두엽 치매 등
여러 성인 발병 신경 퇴행성 질환의 공통점은
잘못 접힌 응집 단백질의 비정상적인 침착입니다(Friedman 등. 2012).
응집 단백질의 제거에서
자가포식의 중요한 역할을 고려할 때,
이는 자가포식 기능 장애가
신경 퇴행성 질환의 일반적인 메커니즘임을 나타낼 수 있습니다.
자가포식 기능 장애가
신경 퇴행성 질환의 주요 기전일 수 있다는 것은
미토콘드리아 손상과
생체 에너지 또는
과도한 활성 산소 종의 생체 에너지 결핍과 관련된 역할이
확립된 데서 더욱 시사됩니다(Beal 외. 2006; Lezi and Swerdlow 2012; Ryan 외. 2015; Wang 2017).
자가포식은
세포가 치명적으로 손상된 미토콘드리아를 제거하는 주요 경로이므로(Youle and Narendra 2011),
이 과정의 장애는 해로운 미토콘드리아의 비정상적인 축적을 초래할 수 있다는 것은 당연한 일입니다(Dagda 외 2008, Narendra 외 2008, Ferree 외 2013, Lemasters 2014, Lazarou 외 2015, Chen 외 2017, Jinn 외 2017).
보다 광범위하게는
오토파지와 노화 사이의 연관성도
중요한 고려사항이 될 수 있습니다(Meléndez 외. 2003; Bergamini 외. 2004; Rubinsztein 외. 2011; Inoue 외. 2012b; Carnio 외. 2014).
노화는
신경 퇴행성 질환의 가장 큰 위험 요인이며,
뇌의 자가포식 조절 저하와 관련이 있습니다(Lipinski 등. 2010).
반대로,
모델 유기체에서 자가포식을 유도하면
수명이 늘어나는 것으로 나타났습니다(Simonsen 외. 2008, Zheng 외. 2010, 표 외. 2013).
더 중요한 것은
자가포식 유도가 건강 수명과 삶의 질 증가와 관련이 있다는 것으로,
자가포식의 촉진이 생물학적 시스템의 기능의 질을 보다 근본적으로 개선하여 광범위한 이점을 가져올 수 있음을 시사합니다(Cuervo et al. 2005; Levine et al. 2011; Oka et al. 2012; Kang et al. 2015).
치료 표적으로서
이 경로의 직관적인 매력 외에도(Levine and Kroemer 2008; Nixon 2013),
신경 퇴행성 질환과 오토파지 사이에는
놀라운 유전적 연관성이 존재합니다.
오토파지의 여러 단계와 관련된 다양한 유전자의 돌연변이는
루게릭병, 전두측두엽 치매뿐만 아니라 AD, 파킨슨병, 헌팅턴병과도 관련이 있습니다(그림 2, 표 1)(Kegel 외. 2000, Petersén 외. 2001, Skibinski 외. 2005, Ramirez 외. 2006, Cheung and Ip 2009, Ju 외. 2009, Montie 외. 2009, Winslow et al. 2009; 윈슬로우 등 2010; 데하이 등 2012; 루신 등 2013; 오차바 등 2014; 웡과 홀츠바우어 2014; 마틴 등 2015; 시우라 등 2016; 셀리에 등 2016; 설리반 등 2016; 만조니 2017; 후지케 등 2018),
오토파지와 관련된 유전자의 변이 및 변이가
신경 퇴행성 질환을 유발하기에 충분하다는 것을 시사합니다.
실제로 리소좀 기능에 중요한
일부 유전자의 돌연변이는
어린 시절에 신경 증상을 유발하는 리소좀 저장 장애로 이어집니다(Nixon 외. 2008).
특발성 또는 산발성 AD와 파킨슨병 또는
ALS의 훨씬 더 일반적인 경우에서
잘못 접힌 단백질의 생산과
자가포식 제거 사이의 불일치가 어느 정도 존재하는지는 알려져 있지 않습니다(Ahmed 외. 2012).
그러나
최근 AD 환자에 대한 연구와
아밀로이드 β 회전율의 역학에 따르면
유전적 형태의 AD는 잘못 접힌 단백질의 과잉 생산을 유발할 수 있는 반면
특발성 형태는 제거율 감소와 관련이 있을 수 있으며,
모든 형태의 AD는 생산과 제거 사이의 근본적인 불일치와 관련이 있을 수 있다는
흥미로운 가능성을 제시하고 있으며,
다른 흔하지만 특발성 형태의 신경 퇴행성 질환도 마찬가지입니다(Mawuenyega et al. 2010).
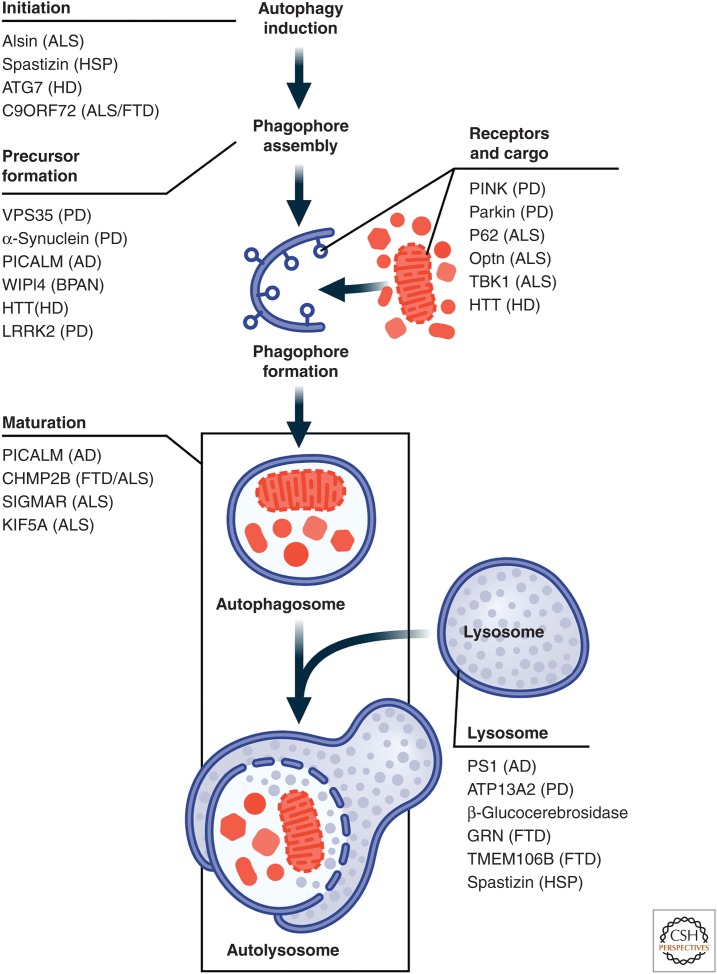
Genetic links between autophagy and neurodegenerative disease.
Mutations in many genes that encode proteins that play a role in the autophagy lysosomal pathway (ALP) lead to neurodegenerative disease syndromes in humans, indicating that impairment of the ALP can be sufficient to produce neurodegenerative disease. Some examples are shown in this figure. AD, Alzheimer's disease; ALS/FTD, amyotrophic lateral sclerosis/frontotemporal dementia; BPAN, β-propeller protein-associated neurodegeneration; CMT2, Charcot-Marie-Tooth disease 2; HD, Huntington's disease; HSP, hereditary spastic paraplegia; PD,
오토파지와 신경 퇴행성 질환 사이의 유전적 연관성.
자가포식 리소좀 경로(ALP)에서 역할을 하는 단백질을 암호화하는
많은 유전자의 돌연변이는
인간에게 신경 퇴행성 질환 증후군을 유발하며,
이는 ALP의 손상만으로도
신경 퇴행성 질환이 발생할 수 있음을 나타냅니다.
이 그림에는 몇 가지 예가 나와 있습니다. AD, 알츠하이머병; ALS/FTD, 근위축성 측삭 경화증/전두측두엽 치매; BPAN, β-프로펠러 단백질 관련 신경 퇴행; CMT2, 샤르코-마리-투스병 2; HD, 헌팅턴병; HSP, 유전성 경련 마비증; PD,
Parkinson's disease.
Table 1.
Autophagy lysosomal pathway and Parkinson's disease–associated genes
Genetic locusGenePossible roles in autophagy
| PARK1 | SNCA (synuclein) | Blocks chaperone-mediated autophagy (CMA); may promote Atg9 mislocalization; vesicle fusion |
| PARK2 | Parkin | Ubiquitin ligase that tags mitochondria for binding of adaptor proteins for mitophagy |
| PARK6 | PINK1 | Ubiquitin kinase whose accumulation on mitochondrial membranes is a marker for recruitment of Parkin to mediate mitophagy |
| PARK 7 | DJ-1 | Works in parallel with PINK1/Parkin to play a role in mitophagy |
| PARK8 | LRRK2 | Kinase for key Rab proteins that may play a role in endosomal/lysosomal trafficking |
| PARK9 | P-type ATPase ATP13A2 | Essential for the maintenance of lysosomal pH and autophagosome–lysosome fusion |
| PARK14 | PLA2G6 | May play a role mediating mTOR-independent autophagy |
| PARK15 | FBOX7 | Mitophagy |
| PARK17 | Vps35 (vacuolar sorting-associated protein 35) | Mutant Vps35 may mislocalize Atg9, leading to impaired autophagy |
| PARK19 | DNAJC6 | Endocytosis and endocytic trafficking |
| PARK20 | SYNJ1 | Endosomal trafficking and autophagy |
| PARK21 | DNAJC13 | Endocytosis |
| Unassigned | GBA | Lysosomal function, synuclein metabolism |
A number of studies have sought to further determine whether brains of patients with neurodegenerative disease exhibit morphological abnormalities that indicate autophagic malfunction. Although widespread abnormalities have been reported, the nature of the morphological changes appear to be, at least partly, disease-specific (Anglade et al. 1997; Sikorska et al. 2004; Nixon 2007; Boland et al. 2008; Chu et al. 2009; Crews et al. 2010; Lucin et al. 2013; Yamamoto and Yue 2014; Martin et al. 2015; Fujikake et al. 2018). Some of these differences have been highlighted in excellent recent reviews (Nixon 2013; Yamamoto and Yue 2014). That different neurodegenerative diseases exhibit sometimes striking differences in patterns of autophagy-associated cytopathology underscores the importance of defining the underlying disease-associated mechanisms. This knowledge will help guide approaches to the development of effective therapeutics and for stratifying patient populations to do sensitive clinical trials.
Based in part on these lines of reasoning, many groups have found evidence in nonhuman models of neurodegenerative disease that autophagy modulation accelerates clearance of disease-causing proteins (Ravikumar et al. 2002; Sarkar et al. 2007a; Dolan and Johnson 2010; Tsvetkov et al. 2010; Wang et al. 2010a; Roscic et al. 2011; Congdon et al. 2012; Kruger et al. 2012; Barmada et al. 2014; Polito et al. 2014; Marrone et al. 2018) and improves disease phenotypes (Ravikumar et al. 2004; Jia et al. 2007; Sarkar et al. 2007b; Hetz et al. 2009; Montie et al. 2009; Spencer et al. 2009; Bauer et al. 2010; Rodríguez-Navarro et al. 2010; Tsvetkov et al. 2010; Watanabe et al. 2010; Schaeffer et al. 2012; Castillo et al. 2013; Decressac et al. 2013; Barmada et al. 2014; Höllerhage et al. 2014; Polito et al. 2014). It is worth noting that some important limitations apply to many of these studies. The assays available to measure autophagy induction are limited in their sensitivity. In general, these assays depend on making inferences about flux based on snapshots of levels of pathway intermediates, which can be prone to misinterpretation, even failing to distinguish upstream autophagy induction with downstream blockade (Klionsky et al. 2011, 2012, 2016). One assay that avoids this pitfall is the optical pulse labeling approach, which monitors the dynamic clearance of an autophagy substrate fused to a photo-switchable protein in live cells, allowing clearance to be calculated directly (Barmada et al. 2014). Assays in vivo are even more limited (Mizushima et al. 2004), making it difficult to accurately assess the effects of putative autophagy inducers, and complicating any correlations seen with disease phenotypes. Very few studies that have reported positive effects of autophagy induction on disease-associated phenotypes have done appropriate pharmacokinetic/pharmacodynamic studies. Without a direct demonstration that the intervention is, in fact, inducing autophagy, the exact relationship is difficult to determine.
Some studies have reported that interventions designed to modulate autophagy flux have unexpected effects (Zhang et al. 2011). Maniatis and colleagues tested whether genetic inhibition of the autophagy pathway would worsen disease-associated phenotypes in a mouse model of ALS based on mutations in superoxide dismutase 1 (Rudnick et al. 2017). They specifically knocked out Atg7, a gene required for autophagy, in motor neurons early in development in mice expressing a transgene encoding the ALS-associated mutant SOD1G93A. Early on, denervation and behavioral deficits were accelerated compared with SOD1G93A mice in which Atg7 was intact. But later, they observed that the loss of Atg7 was associated with less interneuron and astrocyte pathology and an extension of life span.
What could be happening? One possibility is that autophagy may play multiple roles in disease that differ depending on the stage of disease progression. For example, perhaps the initial and somewhat expected worsening of deficits in motor unit function and motor neuron-dependent phenotypes could result from an acceleration in pathogenesis in motor neurons. But Cleveland and colleagues have shown by selectively reducing mutant SOD1 expression in a cell-type specific manner that motor neurons, astrocytes, microglia, and oligodendrocytes each contribute to different extents and in different ways to deficits exhibited by murine models of ALS (Ilieva et al. 2009). The exact nature and mechanisms of the cell-nonautonomous contributions to ALS pathogenesis still need to be worked out. But it is conceivable that if disease begins in motor neurons and if propagation of disease to neighboring nonneuronal cells depends on the release of misfolded proteins by lysosomal exocytosis, then blocking autophagy might actually mitigate the cell-nonautonomous component of disease at the expense of worsening the cell-autonomous component. It is interesting to note that whereas Maniatis and colleagues reported almost no effects of knocking out Atg-7 on baseline motor neuron structure or function (Rudnick et al. 2017), Komatsu found that disruption of Atg-7 and autophagy throughout the brain caused significant neurodegeneration (Komatsu et al. 2006).
But the explanation could be even more complicated. Others have shown that germline mutations that are designed to block specific arms of the protein homeostasis pathway, such as autophagy, are sufficient to induce neurodegeneration (Komatsu et al. 2005, 2006; Hara et al. 2006). Presumably, germline mutations in the autophagy pathway must induce widespread adaptive remodeling of other arms of the proteostasis network to compensate, possibly even in a complicated cell-specific way, which could confound the simple interpretation of the role of autophagy in disease. To what extent does the loss of autophagy at an early stage in development instruct us on the role of autophagy impairment in the aged brain when many neurodegenerative diseases develop? Answers to these questions will be critical for shaping our thinking about pathogenesis and treatment.
ALP AS A POTENTIAL THERAPEUTIC TARGET FOR NEURODEGENERATIVE DISEASE
The evidence suggesting that autophagy is a therapeutic target is intriguing and warrants further investigation. However, while it has been shown that it can be safe to chronically target a major protein homeostasis pathway (e.g., the drug bortezomib, which inhibits the UPS, is a clinically approved treatment for multiple myeloma), there have as yet been no clinical trials of autophagy modulators in humans, and there are multiple caveats to consider.
The mechanisms by which disease-associated genetic mutations in the autophagy pathway cause neurodegeneration is a focus of ongoing research (Nixon 2006), and is critical to the design of therapeutic strategies. Deficits that attenuate the pathway might respond to interventions that stimulate the pathway to restore normal levels of activity (Bose et al. 2011), but that depends on the step in the process that is impaired and the nature of the impairment. For example, mutations in CHMP2B, which plays a role in autophagosome/lysosome fusion (Lu et al. 2013), cause frontotemporal dementia (Skibinski et al. 2005). If disease-causing mutations block that step completely, therapeutically stimulating autophagy might be detrimental, because induced autophagosomes would accumulate without a path to clear them.
Unlike the UPS, autophagy is active basally and can be induced normally by caloric restriction, which can occur during fasting (Alirezaei et al. 2010). Chronic caloric restriction in nonhuman organisms has generally been associated with positive outcomes including increases in longevity and resistance to disease (Bergamini et al. 2007). However, the mechanism by which caloric restriction induces autophagy is thought to be via the inhibition of the mTOR kinase (Balgi et al. 2009; Inoki et al. 2012a). Whether mTOR could be a safe drug target is controversial (Zhang et al. 2011) because mTOR regulates other critical biological pathways in addition to autophagy (Inoki et al. 2012a), such as protein translation (King et al. 2008), and the mTOR inhibitor rapamycin is an immunosuppressant. mTOR inhibitors would be expected to have activity in the periphery, and it has been difficult to develop brain penetrant inhibitors. Partly for these reasons, a focus of the field has been on the development of autophagy inducers that act by mTOR-independent mechanisms (Høyer-Hansen and Jäättelä 2007; Høyer-Hansen et al. 2007; Williams et al. 2008; Lipinski et al. 2010; Decuypere et al. 2011; Bootman et al. 2017).
One additional concern with targeting this pathway has been the potential that systemic induction of autophagy might increase the risk of developing cancer (Shintani and Klionsky 2004; Amaravadi et al. 2007; Kimmelman 2011; Lock et al. 2011; Rosenfeldt et al. 2013; Cui et al. 2014; Rao et al. 2014; Galluzzi et al. 2015; Lévy et al. 2015; Perera et al. 2015; White 2015). Several studies suggest that cancers, particularly as they form tumors that outstrip their blood and nutrient supply, up-regulate the autophagy pathway, possibly in conjunction with macropinocytosis, to enable the cells to endure stress, scavenge nutrients, survive, and proliferate (Qu et al. 2003; Amaravadi and Thompson 2007; Mathew et al. 2007; Guo et al. 2011; Lopez et al. 2011; Yang et al. 2011, 2014, 2016; Amaravadi and Debnath 2014; Karsli-Uzunbas et al. 2014). It is important to note that although autophagy induction might enhance the growth of a preexisting cancer by these mechanisms, there is also evidence that autophagy can function to suppress the initial development of cancer (Liang et al. 1999; Yue et al. 2003; Takamura et al. 2011; Wu et al. 2012; Guo et al. 2013). Interestingly, Levine and colleagues developed a cell-permeant version of beclin, an autophagy-inducing gene, and showed that chronic administration did not significantly increase the incidence of tumors in mice. In the end, it will be critical to do careful preclinical safety studies for any potential autophagy inducer before first-in-human trials. In addition, the field would benefit tremendously from the development of target-engagement biomarkers for autophagy to guide dosing regimens and to ensure that any clinical trial of a putative autophagy inducer is, in fact, inducing autophagy at the doses tested.
SUMMARY AND CONCLUSIONS
Previously relegated to a narrow role as the cell's trash bin, the lysosome has gained new prominence as an organelle centrally positioned to send and receive critical signals and to regulate cellular metabolism as well as catabolism of macromolecules and other organelles. Autophagy is one of several cellular pathways to deliver intracellular cargo to lysosomes for degradation, a process that depends in many cases on special sets of adaptor proteins that are specific for certain cargoes.
Autophagy may have particular importance for neurodegenerative disease given its demonstrated capacity to metabolize aggregated proteins, damaged organelles linked to neurodegenerative disease such as mitochondria, and its close association with aging, the most important risk factor for neurodegenerative diseases. The myriad mutations in genes with established or apparent roles in autophagy, which result in neurodegenerative disease underscore the conclusion that deficits in autophagy are sufficient to cause neurodegenerative disease and suggest that lysosomal dysfunction may be a common thread that even extends to the more common idiopathic forms of adult-onset neurodegenerative disease.
Nevertheless, it remains unclear whether modulation of autophagy and lysosomal function will be a successful strategy to treat one or more neurodegenerative diseases. Neuropathology studies support the conclusion that dysfunction in the ALP is a common feature of neurodegenerative diseases. However, the ALP is complex and our understanding of the nature of the impairments remains limited in most cases, making it uncertain whether the same type of intervention is likely to work in all diseases and at all stages of those disease. Some of these questions may be difficult to fully address in nonhuman models, given the poor predictive value many of these models have for results in human clinical trials. Frustratingly, knowledge of druggable targets in the pathway remains limited, and the throughput and resolution of current autophagy assays pose challenges for the effective prosecution of potential lead optimization programs. Finally, there is the question of whether and how modulation of the ALP could be achieved safely in humans. Good pharmacodynamic markers for the ALP would be invaluable to enable investigators to test the hypothesis in humans that modulation of the autophagy pathway could be a safe and effective treatment for neurodegenerative diseases.
ACKNOWLEDGMENTS
We wish to thank Kathryn Claiborn and Kelley Nelson for editorial assistance and Giovanni Maki for assistance with graphics. We regret our inability to cite many outstanding publications of our colleagues simply because of space limitations. This work was made possible with support from Grants RF1AG058476, R37NS101966, and P01AG54407.
Footnotes
Editors: Richard I. Morimoto, F. Ulrich Hartl, and Jeffery W. Kelly
Additional Perspectives on Protein Homeostasis available at www.cshperspectives.org
REFERENCES
- Ahmed I, Liang Y, Schools S, Dawson VL, Dawson TM, Savitt JM. 2012. Development and characterization of a new Parkinson's disease model resulting from impaired autophagy. J Neurosci 32: 16503–16509. 10.1523/jneurosci.0209-12.2012 [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Albanesi J, Wang H, Sun HQ, Levine B, Yin H. 2015. GABARAP-mediated targeting of PI4K2A/PI4KIIα to autophagosomes regulates PtdIns4P-dependent autophagosome-lysosome fusion. Autophagy 11: 2127–2129. 10.1080/15548627.2015.1093718 [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Alers S, Löffler AS, Wesselborg S, Stork B. 2012. Role of AMPK-mTOR-Ulk1/2 in the regulation of autophagy: Cross talk, shortcuts, and feedbacks. Mol Cell Biol 32: 2–11. 10.1128/MCB.06159-11 [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Alirezaei M, Kemball CC, Flynn CT, Wood MR, Whitton JL, Kiosses WB. 2010. Short-term fasting induces profound neuronal autophagy. Autophagy 6: 702–710. 10.4161/auto.6.6.12376 [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Amaravadi R, Debnath J. 2014. Mouse models address key concerns regarding autophagy inhibition in cancer therapy. Cancer Discov 4: 873–875. 10.1158/2159-8290.CD-14-0618 [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Amaravadi RK, Thompson CB. 2007. The roles of therapy-induced autophagy and necrosis in cancer treatment. Clin Cancer Res 13: 7271–7279. 10.1158/1078-0432.CCR-07-1595 [PubMed] [CrossRef] [Google Scholar]
- Amaravadi RK, Yu D, Lum JJ, Bui T, Christophorou MA, Evan GI, Thomas-Tikhonenko A, Thompson CB. 2007. Autophagy inhibition enhances therapy-induced apoptosis in a Myc-induced model of lymphoma. J Clin Invest 117: 326–336. 10.1172/JCI28833 [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Anding AL, Baehrecke EH. 2017. Cleaning house: Selective autophagy of organelles. Dev Cell 41: 10–22. 10.1016/j.devcel.2017.02.016 [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Anglade P, Vyas S, Javoy-Agid F, Herrero MT, Michel PP, Marquez J, Mouatt-Prigent A, Ruberg M, Hirsch EC, Agid Y. 1997. Apoptosis and autophagy in nigral neurons of patients with Parkinson's disease. Histol Histopathol 12: 25–31. [PubMed] [Google Scholar]