

- nature
- cell discovery
- review articles
- article
Lysosome biology in autophagy
- Review Article
- Open access
- Published: 11 February 2020
Lysosome biology in autophagy
Cell Discovery volume 6, Article number: 6 (2020) Cite this article
Abstract
Autophagy is a major intracellular degradation system that derives its degradative abilities from the lysosome. The most well-studied form of autophagy is macroautophagy, which delivers cytoplasmic material to lysosomes via the double-membraned autophagosome. Other forms of autophagy, namely chaperone-mediated autophagy and microautophagy, occur directly on the lysosome. Besides providing the means for degradation, lysosomes are also involved in autophagy regulation and can become substrates of autophagy when damaged. During autophagy, they exhibit notable changes, including increased acidification, enhanced enzymatic activity, and perinuclear localization. Despite their importance to autophagy, details on autophagy-specific regulation of lysosomes remain relatively scarce. This review aims to provide a summary of current understanding on the behaviour of lysosomes during autophagy and outline unexplored areas of autophagy-specific lysosome research.
오토파지는
리소좀에서 분해 능력을 얻는
주요 세포 내 분해 시스템입니다.
가장 잘 연구된 오토파지의 형태는
이중막 오토파지를 통해
세포질 물질을 리소좀으로 전달하는 거대 오토파지입니다.
다른 형태의 오토파지,
즉 샤프론 매개 오토파지와 마이크로 오토파지는 리소좀에서
직접 발생합니다.
리소좀은
분해 수단을 제공하는 것 외에도
자가포식 조절에 관여하며,
손상되면 자가포식의 기질이 될 수 있습니다.
자가포식 과정에서
리소좀은 산성화 증가, 효소 활성 강화, 핵 주위 국소화 등
주목할 만한 변화를 보입니다.
리소좀의 자가포식에 대한 중요성에도 불구하고,
리소좀의 자가포식 특이적 조절에 대한 세부 사항은
상대적으로 부족합니다.
이 리뷰는
자가포식 중 리소좀의 거동에 대한
현재의 이해를 요약하고
자가포식 특이적 리소좀 연구의 미개척 분야를
개괄적으로 소개하는 것을 목표로 합니다
Similar content being viewed by others
The emerging mechanisms and functions of microautophagy
Article 12 September 2022
The mechanisms and roles of selective autophagy in mammals
Article 27 October 2022
Machinery, regulation and pathophysiological implications of autophagosome maturation
Article 23 July 2021
Introduction
Autophagy refers to a set of pathways by which cytoplasmic material is delivered into the lysosome for degradation (Fig. 1). Starvation and other threats to cellular homeostasis strongly induce autophagy to acquire nutrients by recycling non-essential material or to eliminate harmful material. It comes mainly in three forms: macroautophagy, chaperone-mediated autophagy (CMA), and microautophagy1. Central to all of them is the lysosome, the characteristically acidic organelle with over 60 luminal hydrolases and important cellular regulators2.
자가포식은
세포질 물질이 분해를 위해
리소좀으로 전달되는 일련의 경로를 말합니다(그림 1).
기아 및 기타 세포 항상성에 대한 위협은
자가포식이 비필수 물질을 재활용하여
영양분을 얻거나 유해한 물질을 제거하도록 강력하게 유도합니다.
오토파지는
주로 거시적 오토파지, 샤프론 매개 오토파지(CMA), 미세 오토파지1의
세 가지 형태로 이루어집니다.
이 모든 것의 중심에는
60개 이상의 루멘 가수 분해 효소와
중요한 세포 조절 인자가 있는 특징적인
산성 소기관인 리소좀이 있습니다2.
Fig. 1: Autophagy processes.
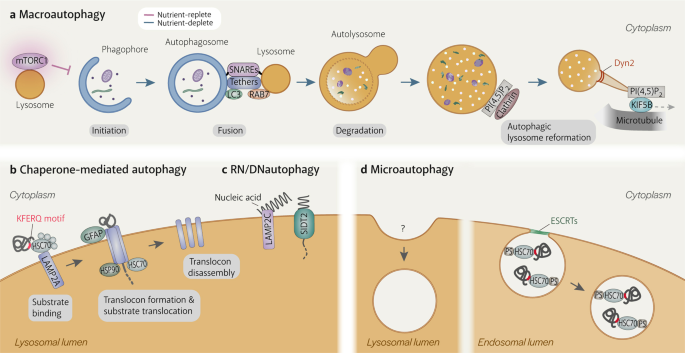
a Macroautophagy is the only autophagy process that involves another organelle, the autophagosome. It is induced when mTORC1 becomes inactivated upon dissociation from the lysosome. After the phagophore matures into a double-membraned autophagosome, the lysosome fuses with the outer autophagosomal membrane in a SNARE-dependent manner. Fusion is facilitated by tethering factors that bind to proteins on the autophagosome (e.g. LC3) and the lysosome (e.g. RAB7). Lysosomal enzymes degrade the inner autophagosomal membrane and sequestered material. Tubules extend from autolysosomes by KIF5B binding to clathrin-organised PI(4,5)P2 clusters on the autolysosomal membrane and moving away from the autolysosome on microtubules. The tubules are eventually cleaved from the autolysosome by Dyn2, generating new lysosomes. b Chaperone-mediated autophagy (CMA) involves the direct uptake of proteins with the KFERQ(-like) motif into lysosomes via a translocation complex consisting of LAMP2A monomers on the lysosomal membrane that is stabilised by GFAP and luminal HSP90. CMA substrates are delivered to LAMP2A by cytosolic HSC70 and other cytosolic chaperones. Substrate translocation is assisted by lysosomal HSC70. c RN/DNautophagy is the direct delivery of nucleic acids into lysosomes via the nucleic acid transporter, SIDT2. LAMP2C binds nucleic acids and potentially passes them to SIDT2 for translocation into the lysosomal lumen. d Microautophagy is the uptake of cytosolic material by invagination of the lysosomal membrane. Although it has been observed in lysosomes since the discovery of this organelle, mechanistic details are still scarce. Microautophagy in endosomes is more well-understood. Endosomal microautophagy substrates contain KFERQ(-like) motifs and are recognised by cytosolic HSC70 to be delivered to endosomes, where HSC70 binds to phosphatidylserine. Membrane deformation and eventually scission of intralumenal vesicle from the endosomal membrane are executed by the ESCRT machinery.
매크로 오토파지는
다른 세포소기관인 오토파지좀이 관여하는
유일한 자가포식 과정입니다.
이는 리소좀에서
mTORC1이 분리되어
비활성화될 때 유도됩니다.
식세포가 이중막 자가포식체로 성숙하면
리소좀은 SNARE 의존적인 방식으로
외부 자가포식체 막과 융합합니다.
융합은 오토파지솜(예: LC3)과 리소좀(예: RAB7)의 단백질에 결합하는
테더링 인자에 의해 촉진됩니다.
리소좀 효소는
내부 자가포식체 막과 격리된 물질을 분해합니다.
세뇨관은 자가소체 막의 클라트린으로 구성된 PI(4,5)P2 클러스터에 결합한 KIF5B에 의해 자가소체에서 확장되어 미세소관에서 자가소체로부터 멀어집니다. 세뇨관은 결국 Dyn2에 의해 자가리소좀에서 절단되어 새로운 리소좀을 생성합니다.
b 샤페론 매개 자가포식(CMA)은
리소좀 막의 LAMP2A 단량체로 구성된 전위 복합체를 통해 KFERQ(- 유사) 모티프를 가진 단백질이
GFAP 및 루멘 HSP90에 의해 안정화된 리소좀으로 직
접 흡수되는 것을 포함합니다.
CMA 기질은 세포질 HSC70 및 기타 세포질 샤프론에 의해 LAMP2A로 전달됩니다. 기질 전위는 리소좀 HSC70의 도움을 받습니다.
c RN/DN 오토파지는
핵산 수송체 SIDT2를 통해 핵산을 리소좀으로 직접 전달하는 것입니다.
LAMP2C는 핵산과 결합하여 리소좀 루멘으로의 전위를 위해 잠재적으로 SIDT2로 전달합니다.
d 미세 오토파지는
리소좀 막의 침입을 통한 세포질 물질의 섭취입니다.
이 세포 소기관이 발견된 이후 리소좀에서 관찰되어 왔지만, 메커니즘에 대한 자세한 내용은 아직 밝혀지지 않았습니다. 엔도솜의 미세 오토파지는 더 잘 알려져 있습니다. 엔도솜의 미세 오토파지 기질은 KFERQ(- 유사) 모티프를 포함하고 있으며 세포질 HSC70에 의해 인식되어 엔도솜으로 전달되고, 여기서 HSC70은 포스파티딜세린과 결합합니다. 막 변형과 최종적으로 소포 내막에서 소포가 분리되는 것은 ESCRT 기계에 의해 실행됩니다.
While CMA and microautophagy take place directly on lysosomes (the former using a transmembrane protein translocation complex and the latter by membrane invagination), macroautophagy involves an additional organelle: the double-membraned autophagosome (Fig. 1a). Macroautophagy begins with the expansion of a piece of membrane, termed the phagophore, around cytoplasmic material that is targeted randomly or selectively with autophagy receptors. The expanding phagophore eventually resembles a sphere with a single opening, the sealing of which results in the autophagosome. Lysosomes fuse with the outer autophagosomal membrane (OAM), supplying acidic hydrolases that degrade the inner autophagosomal membrane (IAM) and sequestered material. The size of the autophagosome (~0.5–2 µm)3 enables macroautophagy to degrade material too large for CMA and microautophagy, which are restricted by the single-protein limitation of the translocation complex and the size of the lysosome (~0.5 µm)3, respectively. Protein aggregates, the ER, mitochondria, damaged lysosomes and bacteria are just a few of the targets of macroautophagy1.
In addition to serving as a source of degradative ability, lysosome is also involved in autophagy regulation, primarily through its relationship with the master kinase complex, mTORC14. The activity of mTORC1 directly reflects intracellular and extracellular nutrient levels. An abundance in nutrients or growth factor signalling prompts mTORC1 to localize onto lysosomes, where it becomes activated to initiate growth-promoting processes and suppress macroautophagy by inhibiting the autophagy initiation complex4 and the nuclear translocation of the transcription factor EB (TFEB), which governs the transcription levels of lysosomal and autophagy genes5,6,7. Conversely, starvation causes mTORC1 to dissociate from lysosomes, leading to the induction of macroautophagy4 and likely microautophagy8,9. mTORC1 does not stay inactivated; its reactivation is required to replenish the lysosomal pool during prolonged starvation10. Constant cross-talk between lysosomes and autophagy, in terms of fusion and regulation, underlies steady autophagic flux.
In this review, we aim to provide a summary of the changes that lysosomes undergo as essential agents of macroautophagy, CMA, microautophagy, and RN/DNautophagy. We also discuss how lysosomes end up as substrates of macroautophagy (lysophagy). Here, the term ‘lysosome’ refers to acidic organelles with degradative potential and a layer of glycosylation on the luminal side of its membrane. We focus mainly on findings from mammalian studies and discuss what is still missing from our understanding of autophagy-specific lysosome regulation.
CMA와 마이크로 오토파지는 리소좀에서 직접 일어나지만(전자는 막 통과 단백질 전위 복합체를 사용하고 후자는 막 침윤을 통해), 매크로 오토파지는 이중막 오토파지좀이라는 추가 소기관을 포함합니다(그림 1a). 거대 오토파지는 자가포식 수용체를 무작위로 또는 선택적으로 표적으로 삼는 세포질 물질 주위에 식세포라고 불리는 막 조각이 확장되는 것으로 시작됩니다. 확장된 식세포는 결국 하나의 입구가 있는 구와 비슷해지며, 이 구가 밀봉되면 오토파지좀이 됩니다. 리소좀은 외부 자가포식체막(OAM)과 융합하여 내부 자가포식체막(IAM)과 격리된 물질을 분해하는 산성 가수분해효소를 공급합니다. 오토파지의 크기(~0.5~2 µm)3는 전위 복합체의 단일 단백질 제한과 리소좀의 크기(~0.5 µm)3에 의해 각각 제한되는 CMA 및 마이크로 오토파지에 비해 너무 큰 물질을 분해할 수 있는 매크로 오토파지를 가능하게 합니다. 단백질 응집체, ER, 미토콘드리아, 손상된 리소좀 및 박테리아는 거대 오토파지의 표적 중 일부에 불과합니다1.
리소좀은 분해 능력의 원천이 될 뿐만 아니라 주로 마스터 키나아제 복합체인 mTORC14와의 관계를 통해 오토파지 조절에도 관여합니다. mTORC1의 활성은 세포 내 및 세포 외 영양소 수준을 직접적으로 반영합니다. 영양소 또는 성장 인자 신호가 풍부하면 mTORC1이 리소좀에 국한되어 활성화되어 성장 촉진 과정을 시작하고 오토파지 개시 복합체4와 리소좀 및 오토파지 유전자의 전사 수준을 조절하는 전사인자 EB(TFEB)의 핵 전위를 억제하여 거대 오토파지를 억제합니다5,6,7. 반대로, 기아는 mTORC1이 리소좀에서 분리되어 거대 오토파지4 및 미세 오토파지8,9를 유도하며, 장기간의 기아 동안 리소좀 풀을 보충하기 위해서는 mTORC1이 비활성화된 상태로 유지되지 않고 재활성화되어야 합니다10. 융합과 조절 측면에서 리소좀과 오토파지 사이의 지속적인 교차 작용은 꾸준한 오토파지 플럭스의 기초가 됩니다.
이 리뷰에서는 거대 오토파지, CMA, 미세 오토파지 및 RN/DN 오토파지의 필수 매개체로서 리소좀이 겪는 변화에 대해 요약하고자 합니다. 또한 리소좀이 어떻게 거대 오토파지(리소파지)의 기질이 되는지에 대해서도 설명합니다. 여기서 '리소좀'이란 분해 가능성이 있고 막의 내강 쪽에 당화 층이 있는 산성 소기관을 말합니다. 주로 포유류 연구 결과에 초점을 맞추고 자가포식 특이적 리소좀 조절에 대한 이해에서 아직 부족한 부분을 논의합니다.
Macroautophagy: autophagosome–lysosome fusion
A crucial step in macroautophagy is the autophagosome acquiring degradative enzymes by fusing with the lysosome (Fig. 1a). The high energy barrier of membrane fusion is overcome by the formation of a complex consisting of SNARE (soluble N-ethylmaleimide-sensitive factor attachment protein receptor) proteins embedded on either of the two membranes11. Autophagosome-lysosome fusion is executed by either of two SNARE complexes: STX17-SNAP29-VAMP7/VAMP812,13 or STX7-SNAP29-YKT64. SNARE complex formation is facilitated by tethering factors that hold the two vesicles close (Fig. 1a). For autophagosome-lysosome fusion, the HOPS complex14, PLEKHM115, and EPG516 play such a role by simultaneously interacting with proteins on both the autophagosomal membrane and the lysosomal membrane. PLEKHM1 binds to the lysosomal small GTPases, Arl8bGTP and RAB7GTP, while also binding to LC3 on the autophagosome15. Similarly, EPG5 binds to RAB7GTP and LC316. The HOPS complex has a more extensive reach, being able to interact with lysosomal Arl8bGTP17 and the autophagosomal Qa-SNARE STX17, either directly14 or via Pacer18,19.
STX17 was the first autophagosomal SNARE identified in mammals. It is precisely recruited to fully formed autophagosomes12,20, thereby avoiding potential complications that could arise from lysosomes fusing with phagophores (discussed later). The mechanism underlying STX17 recruitment and its timing is still unclear. At its C-terminus is a hairpin loop made from two transmembrane domains with glycine zipper motifs that allows STX17 to insert into the OAM7,9. The C-terminal region containing the transmembrane domains is sufficient for accurate autophagosomal targeting and hence may contain an amino acid sequence that can sense changes in the OAM during autophagosome formation12. Alternatively, the timing of STX17 recruitment may be enforced by other proteins. ULK1 when free from Ser-423 phosphorylation has been reported to recruit STX17 to autophagosomes, where STX17 then preferentially binds SNAP29, resulting in the dissociation of ULK121. STX17 has also been reported to bind directly to the autophagosomal protein, LC322. However, further analyses should be conducted to confirm whether the strict timing of STX17 recruitment can be established by these methods of recruitment. A highly effective inhibitor of STX17 recruitment that does not suppress autophagosome maturation has been reported23 but its mode of action is unknown.
While acute depletion of STX17 activity by siRNA treatment12 or drug inhibition23 suppresses autophagic flux, chronic deficiency of STX17 has little effect24. This finding led to the discovery of a second autophagosomal SNARE, YKT624, whose activity can compensate for STX17 deficiency. In mammalian cells, R-SNARE YKT6 forms a complex with Qa-SNARE STX7, and Qbc-SNARE SNAP29 (14A homologue was identified in Drosophila, in which YKT6 can replace VAMP7 to form a complex with Syx17 (the Drosophila homologue of STX17) and SNAP2925. In yeast, YKT6 is the sole autophagosomal SNARE26,27. Unlike STX17, YKT6 does not have transmembrane domains and must be modified with palmitoyl and farnesyl to associate with membranes24. YKT6 is also recruited to mature autophagosomes24, but the mechanism of this temporal regulation remains unknown.
Besides recruitment, the SNAREs involved in autophagosome-lysosome fusion are also subjected to other means of regulation. SNAP29 modified with O-linked N-acetylglucosamine28 and STX17 phosphorylated on its N-terminal domain13 cannot be incorporated into the SNARE complex. STX17 may also be suppressed by the ubiquitin conjugation enzyme BRUCE as STX17-positive autophagosomes accumulate in BRUCE-deficient cells29. Since BRUCE interacts with both STX17 and SNAP2929, it might interfere with STX17-SNAP29 binding on the autophagosome. On the other hand, VAMP7 competes with its SNARE-deficient isoform, VAMP7B, for incorporation into the SNARE complex. VAMP7 is favoured when VAMP7B is bound to DIPK2A30. When formed, the STX17-SNAP29-VAMP7 bundle must be stabilised by EPG516 and ATG14L31. The YKT6-containing SNARE complex is less well-studied. In addition to molecular and genetic studies, structural information on both autophagosome-lysosome SNARE complexes will provide invaluable insights into the regulation of autophagosome-lysosome fusion.
The efficiency of autophagosome-lysosome fusion is also sensitive to the types and levels of particular phosphatidylinositol (PI) phosphates in the autophagosomal and lysosomal membranes. So far shown to be important are the reduction of PI(3,5)P2, production of PI4P, and suppression of PI(4,5)P2 appearance on either or both membranes. PI(3,5)P2 competes with actin for binding to cortactin on lysosomes and thus prevents the formation of stable actin filaments, which is crucial for efficient fusion. INPP5E dephosphorylates PI(3,5)P2 to PI3P, which allows cortactin to bind to actin32. Nevertheless, INPP5E activity must be restrained as PI(3,5)P2 must be present to activate TRPML1, the primary Ca2+ channel in the lysosomal membrane33. Although not yet directly demonstrated to be required for autophagosome-lysosome fusion, TRPML1 activity on lysosomes is still important for fusion as it contributes to the perinuclear localization of lysosomes34 and general lysosomal homeostasis33. Concurrently, PI4P is already present or being generated on both autophagosomal and lysosomal membranes35,36. The exact function of PI4P on the autophagosomal membrane is unclear but is proposed to be required for the association of fusion-promoting factors35. This has been shown for the lysosomal membrane, where the deliberate conversion of PI4P to PI(4,5)P2 causes the dissociation of RAB7 and its associated fusion-promoting effectors, including PLEKHM136. Furthermore, reduced PI4P levels on the lysosomal membrane leads to tubulation37, which would likely hinder fusion. Eventually, PI4P is converted to PI(4,5)P2 but this occurs strictly after fusion38,39 as its premature appearance releases fusion-promoting factors from the lysosomal membrane36 in addition to inhibiting TRPML1 activity39,40. The appearance of PI(4,5)P2 is one of the steps the autolysosome undergoes to regenerate lysosomes, a process called autophagic lysosome reformation (ALR; described later)41.
Lysosomes fusing with spherical but unclosed phagophores has been observed in cells with defective autophagosome closure resulting from a deficiency in ATG conjugation proteins20 or the ESCRT-III subunit CHMP2A42,43. Degradation of the IAM is considerably delayed in such cells20, which would cause autophagic flux to stall and futile depletion of the lysosomal pool. Moreover, leaving lysosomal enzymes in the intermembrane space of autolysosomes runs the risk of them damaging the membrane and leaking into the cytoplasm. The many layers of regulation set upon SNARE recruitment, SNARE complex formation, and lipid composition ensure that autophagosome-lysosome fusion occurs only when the time is right.
Macroautophagy/autophagy: degradation of the inner autophagosomal membrane and autophagic substrates
Degradation within autolysosomes starts with disruption of the IAM (Fig. 1a). In the vacuole of budding yeast, Atg15 was identified as the enzyme responsible for degrading the IAM (i.e. the membrane of autophagic bodies in the vacuole)44,45. An in vitro study found Atg15 to be a phospholipase that prefers phosphatidylserine46. The unidentified mammalian IAM lipase(s) might function similarly. In both organisms, the outer membrane (vacuolar membrane in yeast and OAM in mammalian cells) is spared from degradation despite being exposed to the IAM-degrading enzyme(s). The mechanism enabling resistance is unknown. One hypothesis is that the inner leaflet of the OAM lacks the substrates for the lipase, which is the mechanism proposed for the yeast vacuolar membrane against Atg15 activity46. Another hypothesis is that the OAM inherits membrane-protecting properties from the lysosomal membrane after fusion. This is supported by the observation of LAMP1, a lysosomal membrane protein, being present in the IAM of phagophores in CHMP2A-depleted cells42. As aforementioned, the IAM of phagophores is not readily degraded even after exposure to lysosomal enzymes20. However, the mechanism of enzymatic resistance is likely more complex since the IAM of phagophores can eventually be degraded, which is speculated to occur following autophagosomal closure20. The act of separating the phagophore membrane into the IAM and OAM during autophagosomal closure might confer different properties to the membranes, including the ability to resist degradation.
Lysosomal enzymes gain access to autophagic substrates after IAM degradation (Fig. 1a). More than 60 lysosomal hydrolases2 work in unison to digest the sequestered material, ranging from nucleic acids to bacteria1. Most of these enzymes have acidic pH optima47, making their function reliant on efficient acidification of autolysosomes. Poor lysosomal acidification is often attributed as the cause of impaired autophagy in diseases that are not apparently related to autophagy proteins48,49,50. Re-acidifying lysosomes by treatment with acidic nanoparticles48, drugs51 or by mTOR inhibition52 has been shown to restore autophagic flux, highlighting the importance of optimal enzymatic function.
The fate of catabolites generated from the degradation of autophagic substrates is poorly understood. It is widely accepted that they are exported from the lysosomes through numerous transporters on the lysosomal membrane and reused by the cell40. The activity of most transporters varies according to membrane voltage or intralysosomal proton levels40, which would make them reliant on V-ATPase activity. This is suggested by the finding that V-ATPase inhibition resulted in the accumulation of non-essential amino acids from a study on lysosomal metabolomics53. However, the same study also showed that V-ATPase inhibition did not affect the efflux of most essential amino acids, which was instead found to be regulated by mTORC1 activity in an SLC38A9-dependent manner53. Hence, catabolite efflux from lysosomes may be subjected to several regulatory mechanisms that are not just based on lysosomal membrane properties. These mechanisms are still mostly unclear, especially with respect to lipid egress. NPC1, NPC254 and LIMP255 have been identified to transport cholesterol from the lysosomal lumen to the lysosomal membrane but little is known about the transport of other lipid products. As indicated by recent studies, lipids may be transferred from lysosomal membranes to other organellar membranes via membrane contact sites56. Lipid egress should be tightly regulated to prevent the lysosomal membrane from losing lipids essential to its function. Since the release of catabolites from lysosome is essential for the cell to adapt to starvation, further investigations should be conducted, particular for catabolites besides amino acids and cholesterol, to gain a complete understanding of this process.
Macroautophagy: autophagic lysosome reformation
During prolonged macroautophagy, persistent autophagosome-lysosome fusion results in most, if not all, lysosomes being incorporated into autolysosomes10. Besides lysosomal biogenesis, the cell replenishes its lysosome stores by autophagic lysosome reformation (ALR), a process by which lysosomes are regenerated from autolysosomes during prolonged starvation and other lysosome-depleting circumstances57,58. Without ALR, the cell struggles to adapt to starvation and becomes more susceptible to cell death57.
ALR begins with the reactivation of mTORC110,57, initiated by lysosomal calcium-based negative feedback59 as well as increased amino acid levels in the cytosol60,61 and the lysosomes62. The link between mTORC1 reactivation and ALR initiation is not known but may be the phosphorylation of UVRAG by reactivated mTOR. Phosphorylated UVRAG activates the class III PI 3-kinase VPS34 to generate PI3P on autolysosomes, whose levels may determine rate of tubulation57. PI3P is also implicated in the recruitment of spastizin and spatacsin, two proteins of unknown function but have been reported to be essential for autolysosomal tubule formation63. RAB7 must also be removed from autolysosomes before ALR can take place10. RAB7 enforces lysosomal association to dynein for perinuclear localization which facilitates autophagosome-lysosome fusion64. Post-fusion, autolysosomes might dispense with dynein and instead associate with kinesin, which drives autolysosomal tubulation.
Tubule formation requires the conversion of autolysosomal PI4P to PI(4,5)P2 by the PI4P 5-kinases, PIP5K1A and PIP5K1B. Clathrin binds PI(4,5)P2 via AP2 and organises PI(4,5)P2 into clusters on the autolysosomal membrane41. Tubules are generated by kinesin motor protein KIF5B41,65 binding to the PI(4,5)P2 clusters and presumably pulling on the autolysosomal membrane while moving away on microtubules65. Tubulation is facilitated by WHAMM-mediated actin formation at the autolysosome core and at the base of the tubules66. It is unclear what prevents the autolysosome core from moving with KIF5B; it may be held in place by actin66 or by a dynein-based counterforce as a balance between dynein-driven and kinesin-driven movement has been reported to be important for tubulation34,67,68. This balance is proposed to be maintained by the lysosomal Ca2+ channel TRPML1, which has also been implicated in scission of the tubules34.
During tubulation, the movement of lysosomal luminal contents is restricted to prevent them from entering the tubules and potentially disrupting the membrane57. This is achieved by an unidentified mechanism dependent on optimal levels of PI4P37 and PI3P57. The autolysosomal tubules are eventually severed by the GTPase Dynamin 2 (Dyn2) powered by hydrolysis of GTP69. In Dyn2-depleted cells, electron-dense tubules extending from autolysosomes were observed69, suggesting that lysosomal enzymes are only weakly retained in the autolysosomal core. The new lysosomes derived from the severed tubules eventually become acidic and capable of hydrolysis10, perhaps by transiently fusing with late endosomes or mature lysosomes70.
Autophagy regulation by lysosomes
Starvation-induced inactivation of mTORC1 is one of the main inducers of autophagy (except perhaps for CMA). When the cell has sufficient levels of nutrients, mTORC1 is recruited to lysosomes by a complex composed of Rag-GTPases. The Rag complex is in turn tethered to the lysosomal membrane via another multi-subunit complex called Ragulator that interacts with the lysosomal V-ATPase and the amino acid transporter SLC38A971,72. Both the Rag complex and Ragulator must be in their ‘active’ conformations71,72 and located in RAB7-free microdomains on the lysosomal membrane73 to recruit mTORC1. When on the lysosomal membrane, mTORC1 is activated by GTP-bound Rheb. Activated mTORC1 suppresses macroautophagy by phosphorylating ULK1 and ATG13 of the autophagy initiation complex, preventing its activation71,72.
mTORC1 activation is regulated by nutrient levels in the cytosol and the lysosome71,72. Cytosolic nutrient levels are detected by protein sensors that inform Rag and Ragulator conformations and in turn determine whether mTORC1 is recruited to lysosomes for activation71,72. A drop in nutrient levels turns off the mechanism to recruit mTORC1, resulting in mTORC1 inactivation. Autophagy is initiated and lysosomes begin receiving large numbers of macromolecules. Within lysosomes, some macromolecules may trigger signalling that promotes autophagic flux, such as mitochondrial DNA and its induction of TLR9 signalling39. The macromolecules are gradually broken into their constituents, such as amino acids that would be used in the synthesis of essential proteins. However, amino acid efflux during starvation requires mTORC1 reactivation53, which is achieved by the lysosomal V-ATPase strengthening Ragulator-Rag interaction in response to the rise in intralysosomal amino acid levels and enabling mTORC1’s lysosomal recruitment62. Amino acid efflux is amplified when free arginine in the cytosol and lysosomal lumen activates SLC38A9, an amino acid transporter and another positive regulator of mTORC1 activity74,75. mTORC1 reactivation also initiates ALR, replenishing the lysosomal pool (10; see previous section). Should intracellular nutrient levels remain low, mTORC1 will become inactivated again and the cycle will continue till starvation is resolved.
Lysosomal activation during autophagy
Autophagic flux during starvation is supported by elevated lysosomal activity. Starvation-induced inactivation of mTORC1 removes its suppression on TFEB, which then translocates to the nucleus, where it upregulates the transcription of lysosomal and autophagy genes, supporting the production of lysosomes and autophagosomes5,6,7. Lysosomes associate with dynein instead of kinesin to move to the perinuclear region, where most autophagosome-lysosome fusion occurs64. Perinuclear lysosomes are more acidic76,77,78,79, which enhances enzymatic function80 to efficiently degrade autophagic substrates.
Although starvation-induced lysosomal activation is mainly attributed to mTORC1 inhibition, certain findings indicate that autophagy proteins may also be required. A study on the relationship between lysosomes and autophagy found that lysosomes in cells without ATG5 or ATG7 (members of the ATG conjugation system) failed to acidify and showed no enhancement in enzymatic activity in response to starvation or mTORC1 inhibition despite TFEB activity being unaffected81. Consistent with this is the finding that amino acid starvation-induced V-ATPase assembly is independent of mTORC1 activity74, suggesting that lysosomal activation, at least in terms of acidification, is not regulated by mTORC1 activity and may be linked to autophagosome formation. However, acidification of lysosomes in normal cells was not observed after initiating mTORC1-independent autophagy by trehalose treatment81. Furthermore, a separate study observed acidification of lysosomes in ATG5-deficient cells starved of amino acids and serum77. The effect of autophagosome formation on lysosomal function should be further investigated.
Quality control of lysosomes by lysophagy
Despite being fortified with the glycocalyx, a 5–12 nm-thick layer of sugar residues on the luminal side of its membrane proteins82, the lysosomal membrane remains susceptible to damage by various stressors such as drug-mediated/disease-related lysosomotropism, the loss of stabilising proteins, and trapped infectious agents83. When the lysosomal membrane is breached, lysosomal function is lost. Moreover, lysosomal contents are released into the cytoplasm, resulting in damage to cytoplasmic components and, ultimately, cell death83.
Lysophagy is the engulfment of damaged lysosomes by autophagosomes with the aim of limiting the spread of damage (Fig. 2). It is employed when ESCRT-mediated repair, the first line of defence, proves to be insufficient84,85. Although direct evidence is still lacking, lysophagy most likely targets severely damaged lysosomes whose membranes no longer act as barriers against free movement of proteins and other material. This is indicated by observations of autophagy-promoting proteins and modifications on the luminal side of the lysosomal membrane and that such proteins (e.g. ubiquitin ligases86,87,88) appear on damaged lysosomes after ESCRT recruitment84,85.
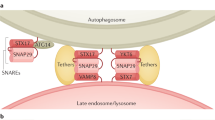
Fig. 2: Mechanisms of autophagy machinery recruitment to damaged lysosomes.
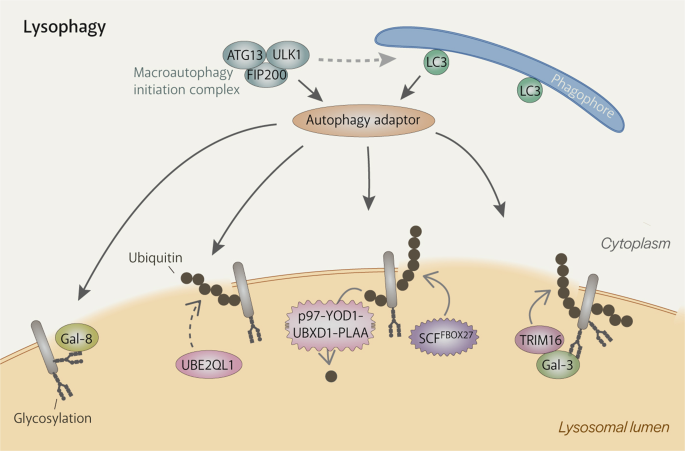
Extensive damage to the lysosomal membrane allows cytosolic proteins to pass through freely, including glycan-binding galectins and ubiquitin ligases. Damaged lysosomes are heavily ubiquitinated, which is carried out by ubiquitylation enzymes such as UBE2QL1 (an E2 enzyme), TRIM16 (an E3 ligase) and SCFFBOX27 (an E3 ligase). K48-linked ubiquitin chains are removed by the p97-YOD1-UBXD1-PLAA complex to emphasise the presence of K63-linked chains, which are preferred by the autophagy machinery. Autophagy adaptors bind either directly to galectins (e.g. NDP52) or to ubiquitin (e.g. p62, OPTN, TAX1BP1). They then recruit the autophagy machinery, including the initiation complex, and serve to promote the formation of the autophagosome specifically around the damaged lysosome.
The autophagy machinery is primarily recruited by ubiquitination of damaged lysosomes89,90. Membrane damage exposes the otherwise hidden glycocalyx, which recruits galectins (Gals). Amongst them is Gal-3, which draws the E3 ligase TRIM16 into the lumen of damaged lysosomes. TRIM16 then mediates ubiquitination of the damaged lysosomes (actual targets are still unidentified) and also recruits upstream autophagic factors, ULK1, Beclin1 and ATG16L191. Another E3 ligase involved in lysophagy was identified as the SKP1-CUL1-F-box protein 27 (SCFFBXO27) ubiquitin ligase complex88, which can directly bind to the exposed glycocalyx and associate with the damaged membrane via myristoylated FBXO27. SCFFBXO27 ubiquitinates SNARE proteins and lysosomal membrane proteins88. Cells deficient in either E3 ligase experienced impaired ubiquitination and lysophagy88,91 but still had residual ubiquitination that may have been produced by the other E3 ligase or other unidentified E3 ligases. The latter is more likely as both E3 ligases were not found to function downstream of UBE2QL1, which was identified to be a lysophagy-mediating E2 ligase86. UBE2QL1 enters the lumen of damaged lysosomes by an unknown mechanism and mediates mostly K48-linked ubiquitination of the luminal ends of lysosomal membrane proteins86. UBE2QL1 activity is important for recruitment of the autophagy receptors, p62 and TAX1BP1, and p9786. The latter is part of a complex with YOD1, UBXD1 and PLAA, that removes K48-linked ubiquitin and thus emphasises the presence of K63-linked ubiquitin, which is preferred by the autophagy machinery87,92. The profile of the ubiquitination substrates on damaged lysosomes and the types of ubiquitin linkage utilised in lysophagy are still unclear.
Although the upstream autophagy factor ATG16L1 can directly recognise ubiquitin90, the autophagy machinery is mainly recruited by the binding of autophagy receptors to ubiquitin. p6286,87,88,89,90, NDP5293, TAX1BP186 and OPTN94 are autophagy receptors that have ubiquitin-binding domains95 and known to localize to damaged lysosomes. Autophagy receptors bind autophagic substrates and LC3 on the phagophore at the same time, encouraging the phagophore to expand around autophagic substrates95. Recent work has shed new light on the role of autophagy receptors. NDP52 was discovered to be able to interact with subunits of the autophagy initiation complex, FIP20096 and ULK197, and TANK-binding kinase 1 (TBK1)96,97 and thus specifically initialise autophagosome formation around damaged lysosomes. p62 can also recruit FIP200 to ubiquitin condensates and probably does the same during lysophagy98.
Lysophagy is supported by mTORC1 inactivation, the resulting TFEB activation, and AMPK activation, which are mediated by galectins99. Gal-8 interacts with the Ragulator-Rag signalling machinery to cause mTORC1 dissociation and subsequent inactivation while Gal-9 recruits TAK1, which activates AMPK by phosphorylation99. Activated AMPK then phosphorylates ULK1 and ATG13 of the autophagy initiation complex, enhancing autophagic activity and thus lysosome clearance99.
Chaperone-mediated autophagy
Chaperone-mediated autophagy (CMA) is the direct translocation of protein substrates from the cytosol into the lysosomal lumen mediated by LAMP2A100, one of the three splice variants of the LAMP2 gene101. The generation of mice with a liver-specific deficiency of LAMP2A revealed that CMA is important for liver metabolism102 and increased CMA activity has been observed in response to a variety of conditions such as starvation, hypoxia, and oxidative stress103.
CMA substrates are delivered to lysosomes by HSC70, a cytosolic chaperone. HSC70 binds to a five amino acid-long motif, KFERQ or a variation of which, on the CMA substrate and brings it to LAMP2A on the lysosomal membrane (Fig. 1b). Both HSC70 and the CMA substrate then associate with the cytosolic region of LAMP2A, triggering the formation of multimeric LAMP2A complex104,105. Exactly how multimerisation of LAMP2A, a single-pass transmembrane protein, results in a transmembrane pore has yet to be determined. Multimerisation can only occur in cholesterol-poor regions of the lysosomal membrane106 and the resulting complex has to be stabilised by another lysosomal membrane protein, GFAP107, and luminal HSP90104 before it can translocate CMA substrates. The translocation channel of the complex is only wide enough to accommodate proteins that have been unfolded by HSC70 and several other chaperones in the cytosol108,109. Translocation is assisted by HSC70 in the lysosomal lumen110. After the substrate reaches the lysosomal lumen, substrate-free cytosolic HSC70 on the lysosomal membrane surface disperses the LAMP2A complex104. Since LAMP2A is the defining factor of CMA103, a full characterization of this protein, including structural studies of full-length LAMP2A and the translocation complex, would provide a significant advancement to current understanding of CMA.
While it is generally accepted that the rate of CMA is regulated by the levels of LAMP2A and its multimerisation efficiency103, the signalling upstream remains mostly unclear. Unlike other autophagy processes, mTORC1 does not regulate CMA111. mTORC2, however, influences the rate of LAMP2A multimerisation by activating Akt, which then phosphorylates GFAP, preventing it from stabilising LAMP2A complexes107. During prolonged starvation, Akt is inactivated by the phosphatase PHLPP1, leading to higher levels of GFAP that can associate with LAMP2A complexes112. The phosphatase for GFAP, if there is one, has not been identified. As mTORC2 and Akt levels on CMA-active lysosomes during prolonged starvation stay relatively stable, translocation complex formation depends mainly on PHLPP1’s recruitment to the lysosome112. The signal for recruitment of PHLPP1 and how CMA is activated only after prolonged starvation are two of the many unanswered questions on the regulation of CMA.
RN/DNautophagy
RN/DNautophagy (RDA) refers to the autophagic pathway by which nucleic acids are taken up directly by lysosomes for degradation (Fig. 1c). Its discovery began with the finding that LAMP2C was capable of binding RNA and DNA113,114. Subsequently, it was shown that isolated lysosomes could take up nucleic acids and that LAMP2-deficient lysosomes were less efficient in doing so113,114. Although LAMP2B can also bind nucleic acids113,115, its affinity for nucleic acids is much weaker than that of LAMP2C113,114,115. LAMP2C was thus named the first RDA receptor113,114.
The observation that LAMP2-deficient lysosomes had decreased but remaining RDA activity113,114 prompted the search for other RDA receptors. This led to the identification of SIDT2116,117, a putative double-stranded RNA transporter previously reported to localize to lysosomes118. SIDT2 is able to independently transport nucleic acids across the lysosomal membrane116,117 unlike LAMP2C, whose inability to multimerise renders it incapable of doing so119. Hence, SIDT2 is regarded to be the more important of the two116. LAMP2C can interact with SIDT2116, suggesting that it might pass its bound DNA or RNA to SIDT2 for delivery into lysosomes, but this has yet to be demonstrated. Furthermore, whether SIDT2 displays substrate selectivity is still unknown. By contrast, LAMP2C has been shown to prefer guanine-rich sequences120. Studies outside of the autophagy field have reported that SIDT2 exports viral RNA from lysosomes into the cytoplasm121 and that it has sodium ion transporter activity122. Whether these functions are related to RDA should be investigated.
The physiological relevance of RDA might involve the degradation of unwanted nucleic acids (e.g. viral DNA and mitochondrial DNA) as indicated by the increased mortality rates experienced by SIDT2-deficient mice post-viral infection121. However, this same study reported an accumulation of RNA in lysosomes and that the function of SIDT2 is to export RNA from lysosomes into the cytosol121. Additionally, several studies characterizing SIDT2-knockout mice have reported defects in insulin secretion123,124, hepatic lipid metabolism125,126 and autophagic flux127, which do not seem to involve nucleic acid degradation but should be investigated to clarify the physiological role of RDA.
Microautophagy
Microautophagy refers to the process whereby lysosomes directly engulf cytosolic material by membrane invaginations (Fig. 1d). Although over 50 years have passed since it was first described128, little is known about the molecular machinery and regulation of microautophagy in mammals. This is mainly due to the difficulty in observing membrane invaginations in the small lysosomes of mammalian cells and also to the lack of robust assays to specifically measure the rate of microautophagy.
In contrast to the lysosome setup in mammalian cells, yeast cells typically have one large degradative ‘lysosome’, called the vacuole, whose size makes microautophagy easier to observe and studies with yeast cells have yielded several critical findings revealing the scope of microautophagy. Proteins and organelles were found to be targeted by vacuolar microautophagy and substrates differ according to the cell’s condition. Peroxisomes were found to be eliminated by microautophagy in yeast when methanol is replaced by glucose as an energy source129,130. Nutrient deprivation induces microautophagy of portions of the nucleus via nucleus-vacuole junctions131 and lipid droplets132. The ER is taken up during ER stress133. Studies with yeast have also determined that microautophagy is mediated by the ESCRT machinery134 and regulated by TORC1 activity135. The importance of GTP availability, membrane fluidity and membrane potential was also discovered, pointing to the existence of unidentified factors136.
On the mammalian front, microautophagy was recently discovered to occur on endosomes137. Termed endosomal microautophagy (eMI), substrates are either randomly or selectively taken up into endosomes. eMI substrates have KFERQ(-like) motifs and are delivered to endosomes by HSC70, reminiscent of CMA (137; see previous section) (Fig. 1d). However, eMI requires neither LAMP2A nor substrate unfolding137. As LAMP2A is found only in the genomes of mammals and birds, eMI might have emerged in other organisms to eliminate KFERQ-containing proteins8. Like multivesicular body formation and vacuolar microautophagy in yeast, membrane invagination in eMI is executed by the ESCRT machinery137 and partly HSC70138 which can deform membrane upon its binding to phosphatidylserine137,139 (Fig. 1d). After being incorporated into intraluminal vesicles, eMI substrates can be degraded within endosomes or lysosomes137. They can even be secreted out of the cell140. A similar process has also been found in fission yeast141.
Although the regulation of mammalian eMI is still mostly unknown, some hints can be derived from findings obtained from studies with Drosophila8. Drosophila eMI can be induced by starvation in a manner involving TOR (homologous to mTOR) inactivation8. Mammalian eMI may also be subjected to mTORC1-mediated regulation as it could be strongly induced by rapamycin treatment9. Like mammalian eMI, proteins of the ATG conjugation system are not involved in Drosophila eMI8,137. Further investigation revealed that ATG1 and ATG13, components of the macroautophagy initiation complex, are essential for Drosophila eMI8, which suggests that eMI and macroautophagy are regulated by the same upstream factors. Further indication of cross-talk between eMI and macroautophagy comes from the discovery that macroautophagy receptors are rapidly degraded by eMI during the first few hours of starvation in mammalian cells142.
eMI has been postulated to be the primary microautophagy pathway8,137, but the possibility of a lysosome-based microautophagy pathway still cannot be discounted. Although endosomes isolated from VPS4-depleted cells (and thus incapable of eMI) barely contain typical microautophagy substrates (cyclophilin, GAPDH and aldose), lysosomes from these cells have increased levels of the same proteins compared to those from normal cells137. On a related note, GAPDH puncta were still observed in cells depleted of both LAMP2A and TSG101 (a component of ESCRT-I)143 and could represent lysosomes. Although upregulation of CMA could explain the former observation and macroautophagy for the latter, they could also be due to lysosomes directly engulfing proteins for degradation as seen from earlier studies where isolated lysosomes were shown to be able to take up material such as Percoll particles144 and ferritin144,145.
There is still much to learn about microautophagy. Remaining questions include how it is regulated, what factors are involved, whether substrates are taken up specifically, whether membrane proteins are actively excluded (which has been demonstrated to occur for the V-ATPase in yeast microautophagy146 and indicated by the poor particle density of intravacuolar tubules147), and the extent of its physiological significance.
Conclusion
Despite the fact that the lysosome is essential to autophagy, it has been mostly relegated to a role secondary to the autophagosome in studies on macroautophagy (the most well-characterized form of autophagy). Lysosomal function is intricately linked with that of autophagy: autophagic dysfunction is often caused by defective lysosomal activity as exemplified by the phenotypes of lysosomal storage diseases148. And yet, autophagy-related lysosomal defects are rarely characterized in detail. The extent of interdependency between autophagy machinery and lysosomal activation during starvation is also unclear. Even changes that occur to the lysosomal membrane and lumen during autophagy have only been partially described. Furthermore, microautophagy is a research field that is mostly unexplored. Increasing efforts to understand the lysosome is necessary to achieve a complete picture of autophagy.
리소좀이 오토파지에 필수적이라는 사실에도 불구하고, 리소좀은 오토파지의 가장 특징적인 형태인 거대 오토파지에 대한 연구에서 대부분 오토파지에 부차적인 역할로 치부되어 왔습니다. 리소좀 기능은 오토파지의 기능과 복잡하게 연결되어 있습니다. 오토파지 기능 장애는 종종 리소좀 저장 질병의 표현형에서 예시되는 것처럼 리소좀 활동의 결함으로 인해 발생합니다148. 그러나 자가포식 관련 리소좀 결함은 자세히 밝혀진 바가 거의 없습니다. 기아 시 자가포식 기계와 리소좀 활성화 사이의 상호 의존성 정도도 불분명합니다. 자가포식 과정에서 리소좀 막과 루멘에 일어나는 변화조차도 부분적으로만 설명되어 있습니다. 게다가 미세 오토파지는 대부분 미개척 연구 분야입니다. 자가포식에 대한 완전한 그림을 얻기 위해서는 리소좀을 이해하려는 노력이 더 많이 필요합니다,
References