
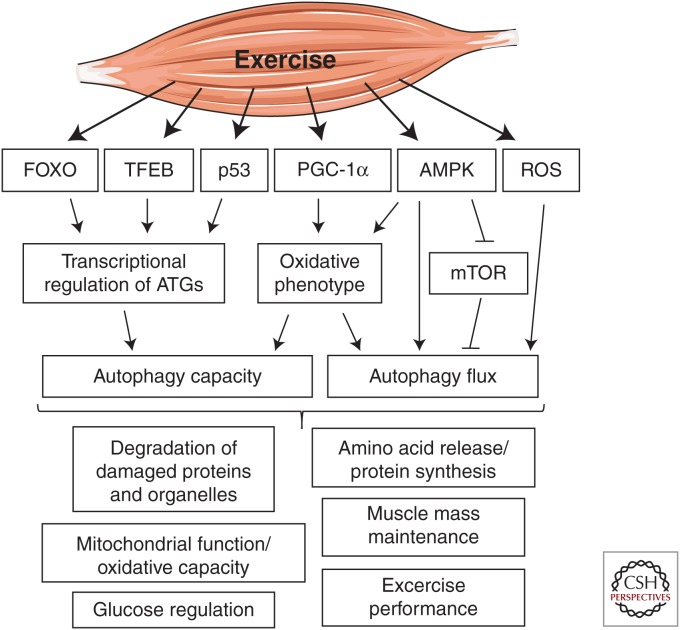
운동에 의한 골격근의 자가포식 활성화는
포크헤드 박스 O(FOXO) 전사인자 계열,
전사인자 EB(TFEB),
p53,
퍼옥시좀 증식인자 활성화 수용체 γ 보조인자 1α(PGC-1α)를 매개로 하는
자가포식 관련 유전자(ATG)의 전사 조절에 영향을 받아
자가포식 능력을 증가시키는 것으로 나타났습니다.
골격근의 산화적 표현형은
자가포식 능력/유량과 관련이 있으며
운동 반응성 5′ AMP 활성화 단백질 키나아제(AMPK)와
PGC-1α에 의해 긍정적으로 조절됩니다.
AMPK와 포유류 라파마이신 타겟(mTOR)은
각각 운동으로 인한 자가포식의
활성화제와 억제제 역할을 합니다.
운동으로 인한 활성 산소종(ROS)의 생성은
오토파지 활성화의 신호로
작용하는 것으로 보입니다.
운동의 자가포식에 의존하는 유익한 효과로는
산화적으로 손상된 단백질과
세포 소기관의 분해,
미토콘드리아 산화 능력 향상,
포도당 조절 개선,
단백질 합성,
근력 및 질량 보존,
지구력 운동 능력 향상 등이 있습니다.
결론
수많은 연구에서
운동에 반응하여 골격근에서
자가포식이 활성화된다는 증거가 제시되었습니다.
운동에 의한 자가포식 조절에는
자가포식 플럭스의 증가와
중요한 자가포식 유전자의 전사 활성화가 모두 포함되며,
그 결과 자가포식 능력이 향상될 수 있습니다.
또한
운동으로 유도된 자가포식은 운
동의 많은 유익한 효과를 매개하는 데
관여하는 것으로 보입니다.

Cold Spring Harb Perspect Med. 2017 Aug; 7(8): a029777.
doi: 10.1101/cshperspect.a029777
PMCID: PMC5538402
PMID: 28270532
Autophagy-Dependent Beneficial Effects of Exercise
Jens Frey Halling and Henriette Pilegaard
Author information Copyright and License information PMC Disclaimer
Abstract
Exercise has long been recognized as a powerful physiological stimulus for a wide variety of metabolic adaptations with implications for health and performance. The metabolic effects of exercise occur during and after each exercise bout and manifest as cumulative adaptive responses to successive exercise bouts. Studies on the beneficial effects of exercise have traditionally focused on the biosynthesis of metabolic proteins and organelles. However, the recycling of cellular components by autophagy has recently emerged as an important process involved in the adaptive responses to exercise. This review covers the regulation of autophagy by exercise, with emphasis on the potential autophagy-dependent beneficial effects of exercise.
Autophagy is activated in skeletal muscle in response to exercise. This process may mediate many of the beneficial effects of exercise (e.g., improved oxidative capacity). Work to understand the molecular details is ongoing.
운동은
건강과 운동 능력에 영향을 미치는
다양한 신진대사 적응을 위한 강력한 생리적 자극으로
오랫동안 인식되어 왔습니다.
운동의 대사 효과는
각 운동 시합 중과 후에 발생하며
연속적인 운동 시합에 대한 누적된 적응 반응으로 나타납니다.
운동의 유익한 효과에 대한 연구는
전통적으로 대사 단백질과 세포 소기관의 생합성에 초점을 맞춰 왔습니다.
그러나
최근에는
자가포식에 의한 세포 구성 요소의 재활용이
운동에 대한 적응 반응에 관여하는
중요한 과정으로 부상하고 있습니다.
이 리뷰에서는
운동에 의한 자가포식의 조절을 다루며,
운동의 잠재적인 자가포식 의존적 유익한 효과에 중점을 두고 있습니다.
자가포식은
운동에 반응하여
골격근에서 활성화됩니다.
이 과정은
운동의 많은 유익한 효과(예: 항산화 능력 향상)를 매개할 수 있습니다.
분자적 세부 사항을 이해하기 위한 연구는 계속 진행 중입니다.
ADAPTATIONS TO EXERCISE TRAINING
Exercise training can induce multiple adaptations in skeletal muscle, the type of which depends on the mode of exercise. Hence, resistance exercise training is characterized by increased muscle fiber cross-sectional area (hypertrophy), whereas endurance exercise training is characterized by metabolic adaptations in skeletal muscle.
Resistance exercise training-induced increases in muscle mass contribute to increased muscle strength and muscle performance (Chesley et al. 1992; Biolo et al. 1995) with importance for both athletic ability and everyday physical function. This is, in part, facilitated by a net increase in myofilament proteins. In accordance, a single resistance exercise bout has been shown to increase protein synthesis (Chesley et al. 1992; Biolo et al. 1995; Phillips et al. 1997) for up to 48 hours involving increased translational initiation. Of notice is, however, that resistance exercise also increases protein degradation for up to 24 hours (Biolo et al. 1995; Phillips et al. 1997) underlining the importance of a well-regulated balance between protein synthesis and degradation in the regulation of muscle mass.
Endurance exercise training-induced metabolic adaptations in skeletal muscle contribute to increasing the maximal oxidative capacity as well as the metabolic efficiency of skeletal muscle with concomitant improvements in endurance performance and health. Endurance exercise training-induced increases in content and activity of proteins involved in oxidative metabolism in human and rodent skeletal muscle are well established (Holloszy 1967; Gollnick et al. 1972; Henriksson and Reitman 1977). These adaptations have, in general, been reported as a result of increased synthesis of metabolic proteins and investigations of the underlying mechanisms behind metabolic adaptations with exercise training have primarily focused on the regulation of protein synthesis. However, the original study by Henriksson and Reitman showing that the activity of oxidative enzymes in human skeletal muscle increased over the course of several weeks of endurance exercise training also showed that the gained enzyme activity was rapidly lost within 1–2 weeks of detraining (Henriksson and Reitman 1977). This underlines the high turnover of metabolic proteins in skeletal muscle. Furthermore, more recent evidence suggests that the regulation of protein removal by autophagy is of equal importance as increased synthesis of metabolic proteins for obtaining the full beneficial metabolic effects of endurance exercise training.
운동 훈련은
골격근에 여러 가지 적응을 유도할 수 있으며,
그 유형은 운동 방식에 따라 달라집니다.
따라서
저항 운동 훈련은
근섬유 단면적 증가(비대)가 특징인 반면,
지구력 운동 훈련은 골격근의 대사적 적응이 특징입니다.
저항성 운동 훈련으로 인한 근육량 증가는
운동 능력과 일상적인 신체 기능 모두에 중요한
근력 및 근력 향상에 기여하며(Chesley 외. 1992; Biolo 외. 1995),
이는 운동 능력과 일상적인 신체 기능 모두에 중요합니다.
이는 부분적으로
근섬유 단백질의 순 증가에 의해 촉진됩니다.
따라서
한 번의 저항 운동으로
최대 48시간 동안 단백질 합성을 증가시키는 것으로 나타났습니다(체슬리 외. 1992; 비올로 외. 1995; 필립스 외. 1997).
그러나
주목할 점은
저항 운동도 최대 24시간 동안
단백질 분해를 증가시킨다는 사실(Biolo 외. 1995; Phillips 외. 1997)은
근육량 조절에서 단백질 합성과 분해 사이의 잘 조절된 균형이 중요하다는 것을 강조합니다.
지구력 운동 훈련으로 인한
골격근의 대사 적응은
최대 산화 능력과 골격근의 대사 효율을 증가시켜
지구력 수행 능력과 건강을 동시에 개선하는 데 기여합니다.
지구력 운동 훈련으로 인한
인간과 설치류 골격근의 산화 대사에 관여하는
단백질 함량과 활성의 증가는 잘 알려져 있습니다(Holloszy 1967; Gollnick 외. 1972; Henriksson and Reitman 1977).
이러한 적응은
일반적으로 대사 단백질의 합성 증가의 결과로 보고되어 왔으며,
운동 훈련에 따른 대사 적응의 기본 메커니즘에 대한 연구는
주로 단백질 합성 조절에 초점을 맞춰 왔습니다.
그러나
몇 주간의 지구력 운동 훈련을 통해
인간 골격근의 산화 효소 활성이 증가한다는 Henriksson과 Reitman의 초기 연구에 따르면
운동 중단 후 1~2주 이내에 증가된 효소 활성이
급격히 소실되는 것으로 나타났습니다(Henriksson and Reitman 1977).
이는 골격근에서 대사 단백질의 높은 회전율을 강조합니다.
또한,
최근의 증거에 따르면
지구력 운동 훈련의 유익한 대사 효과를 완전히 얻기 위해서는
대사 단백질의 합성 증가만큼이나
자가포식에 의한 단백질 제거 조절이 중요하다고 합니다.
MECHANISMS OF AUTOPHAGY
The term autophagy is derived from the ancient Greek words “auto” (self) and “phagein” (to eat). Autophagy is a conserved mechanism in which targeted cellular components are engulfed in maturing autophagosomes that subsequently fuse with lysosomes for hydrolytic degradation and recycling of proteins, membranes, and other cellular components.
Important knowledge has been gathered regarding key molecular mechanisms involved in initiation and execution of cellular recycling through autophagy, which have recently been comprehensively reviewed (Ktistakis and Tooze 2016). In brief, a central set of autophagy-related (ATG) proteins controlling autophagosome formation and elongation has been identified in yeast, most of which have been found to have one or more mammalian homologs. An essential step in autophagosome maturation is the recruitment of the ULK1 kinase complex (including ATG13, ATG101, and FIP200) and the class III PI 3-kinase (PI3K) complex (including VPS34, Beclin-1, ATG14, and p150) to the maturing autophagosome membrane (Fig. 1). The interaction between the ULK1 complex, LC3/GABARAP, and ATG5-12-16L1 at the autophagosome membrane increases the activity of ULK1 and sustains PI3K complex activity, which drives phosphatidylinositol 3-phosphate (PI3P) formation and promotes autophagosome maturation. Furthermore, ATG5-12-16L1-dependent phosphatidylethanolamine (PE) lipidation of LC3-I to LC3-II on the autophagosome membrane is crucial for autophagosome maturation (Fig. 1) (Mizushima and Komatsu 2011; Ktistakis and Tooze 2016). LC3-II and the LC3-II/LC3-I ratio are therefore widely used as markers for autophagosome content and autophagy flux, although interpretations of LC3-II as a general autophagy marker should be made cautiously (Mizushima and Yoshimori 2007; Rubinsztein et al. 2009; Klionsky et al. 2016). Although elevated LC3-II content can be interpreted as a marker of increased autophagosome content and therefore induction of autophagy, a reduction in LC3-II can, conversely, also be the result of increased autophagic flux, because LC3-II itself is degraded in the lysosome. Tracking of other proteins known to be degraded during autophagy can therefore be useful in interpreting alterations in autophagy (Klionsky et al. 2016).
오토파지라는
용어는 고대 그리스어 '오토'(스스로)와 '파지인'(먹다)에서 유래했습니다.
오토파지는
표적 세포 성분이 성숙된 오토파지솜에 포획된 후 리소좀과 융합하여
단백질, 세포막 및 기타 세포 성분을
가수분해하고
재활용하는
보존된 메커니즘입니다.
자가포식을 통한 세
포 재활용의 시작과 실행에 관여하는
주요 분자 메커니즘에 관한 중요한 지식이 최근 종합적으로 검토되고 있습니다(Ktistakis와 Tooze 2016).
간단히 말해,
효모에서 자가포식체 형성과 연장을 조절하는
자가포식 관련(ATG) 단백질의 중심 세트가 확인되었으며,
이들 대부분은 하나 이상의 포유류 동족체가 있는 것으로 밝혀졌습니다.
오토파지솜 성숙의 필수 단계는
ULK1 키나아제 복합체(ATG13, ATG101, FIP200 포함)와
클래스 III PI 3-kinase(PI3K) 복합체(VPS34, Beclin-1, ATG14, p150 포함)를
성숙 오토파지솜 막에 모집하는 것입니다(그림 1).
자가포식체 막에서
ULK1 복합체, LC3/GABARAP, ATG5-12-16L1의 상호작용은
ULK1의 활성을 증가시키고
포스파티딜이노시톨 3-포스페이트(PI3P) 형성을 유도하고
자가포식체 성숙을 촉진하는 PI3K 복합체의 활성을 유지합니다.
또한,
오토파지솜 막에서 LC3-I에서
LC3-II로의 ATG5-12-16L1 의존 포스파티딜에탄올아민(PE) 지질화는
오토파지솜 성숙에 매우 중요합니다(그림 1)
(Mizushima and Komatsu 2011, Ktistakis and Tooze 2016).
따라서
LC3-II와 LC3-II/LC3-I 비율은
오토파지의 함량과 오토파지 플럭스의 마커로 널리 사용되지만,
일반적인 오토파지 마커로서 LC3-II를 해석하는 것은
신중하게 이루어져야 합니다(미즈시마 및 요시모리 2007; 루빈슈타인 등 2009; 클리온스키 등 2016).
LC3-II 함량 증가는
자가포식소체 함량 증가와
이에 따른 자가포식 유도의 마커로 해석될 수 있지만,
반대로
LC3-II의 감소는
리소좀에서 LC3-II 자체가 분해되기 때문에
자가포식 플럭스 증가의 결과일 수도 있습니다.
따라서
자가포식 과정에서 분해되는 것으로
알려진 다른 단백질을 추적하는 것은
자가포식의 변화를 해석하는 데 유용할 수 있습니다(Klionsky 등. 2016).
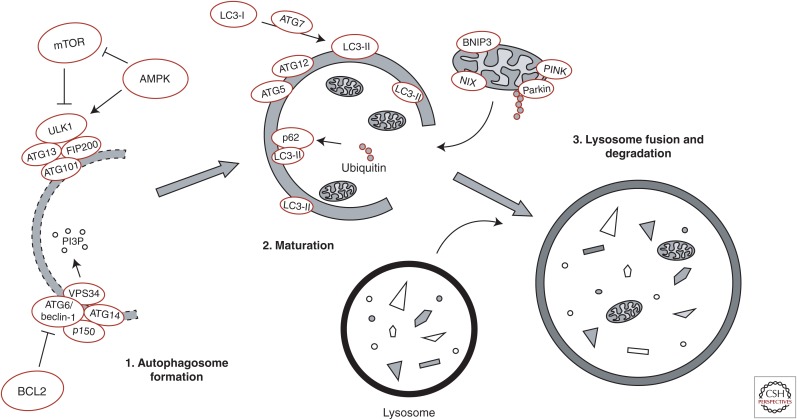
Overview of basic molecular mechanisms involved in initiation of autophagy.
The UNC-51-like kinase 1 (ULK1) kinase complex (including autophagy-related genes (ATG1)3, ATG101, and FAK family kinase-interacting protein of 200 kDa (FIP200)) and the class III PI3-kinase complex (including the catalytic vacuolar protein sorting mutant 34 (VPS34) and the regulatory ATG6/Beclin-1, p150, and ATG14) are stabilized on a phagophore membrane and generate a pool of phosphatidylinositol-3-phosphate (PI3P), which is essential for autophagosome formation. The 5′ AMP-activated protein kinase (AMPK) and mammalian target of rapamycin (mTOR) act as opposing regulators of ULK1 complex activity through stimulatory and inhibitory phosphorylation of ULK1, respectively, whereas AMPK inhibits mTOR activity. Disassociation of ATG6/Beclin-1 and B-cell lymphoma 2 (BCL2) is required for the assembly of the class III PI3-kinase complex. ATG7 regulates the recruitment of cytosolic LC3-I to the maturing autophagosome membrane, where ATG5-ATG12-dependent lipidation forms membrane-bound LC3-II. The autophagy adaptor protein p62 links ubiquitinated substrates with LC3-II. Selective autophagic clearance of mitochondria (mitophagy) is regulated by the kinase PTEN-induced putative kinase 1 (PINK1), which is stabilized on depolarized mitochondrial membranes and recruits the E3 ubiquitin ligase parkin to mitochondria. Parkin regulates ubiquitination of mitochondrial membrane proteins, thereby targeting mitochondria for degradation. The mitophagy receptors BCL2/adenovirus E1B 19-kDa protein-interacting protein 3 (BNIP3) and NIP3-like protein X (NIX) present on mitochondrial membranes can directly interact with LC3-II to induce mitophagy. Finally, the mature autophagosomes fuse with lysosomes causing hydrolytic degradation of autophagosomal cargo.
오토파지의 시작에 관여하는 기본적인 분자 메커니즘에 대한 개요.
UNC-51 유사 키나아제 1(ULK1) 키나아제 복합체(자가포식 관련 유전자(ATG1)3, ATG101, 200kDa의 FAK 계열 키나아제 상호 작용 단백질(FIP200) 포함)와 클래스 III PI3 키나아제 복합체(촉매성 액포 단백질 분류 변이 34(VPS34) 및 조절 ATG6/Beclin-1, p150 및 ATG14)는
식세포막에서 안정화되어
자가포식체 형성에 필수적인
포스파티딜이노시톨-3-인산(PI3P) 풀을 생성합니다.
5′ AMP 활성화 단백질 키나아제(AMPK)와
라파마이신의 포유류 표적(mTOR)은
각각 ULK1의 자극 및 억제 인산화를 통해
ULK1 복합체 활성의 상반된 조절자 역할을 하는 반면,
AMPK는 mTOR 활성을 억제합니다.
클래스 III PI3-키나아제 복합체의 조립을 위해서는 ATG6/Beclin-1과 B세포 림프종 2(BCL2)의 해리가 필요합니다. ATG7은 성숙한 자가포식체 막에 세포질 LC3-I의 모집을 조절하며, ATG5-ATG12 의존성 지질화는 막 결합 LC3-II를 형성합니다. 오토파지 어댑터 단백질 p62는 유비퀴틴화된 기질과 LC3-II를 연결합니다. 미토콘드리아의 선택적 자가포식 제거(미토파지)는 탈분극화된 미토콘드리아 막에서 안정화되고 E3 유비퀴틴 리가제 파킨을 미토콘드리아로 모집하는 키나아제 PTEN 유도 추정 키나아제 1(PINK1)에 의해 조절됩니다.
파킨은
미토콘드리아 막 단백질의 유비퀴틴화를 조절하여
미토콘드리아를 표적으로 삼아 분해합니다.
미토콘드리아 막에 존재하는
미토파지 수용체 BCL2/아데노바이러스 E1B 19-kDa 단백질 상호작용 단백질 3(BNIP3) 및
NIP3 유사 단백질 X(NIX)는 LC3-II와
직접 상호작용하여
미토파지를 유도할 수 있습니다.
마지막으로
성숙한 오토파지는
리소좀과 융합하여
오토파지 화물의 가수분해 분해를 일으킵니다.
Selective Autophagy
Although autophagy can function in a nxxxxonselective manner, for example to provide substrates during energy deprivation, mechanisms for selective clearance of specific organelles and proteins have been identified. The autophagy adaptor protein p62 contains a carboxy-terminal ubiquitin-associated domain and directly interacts with LC3-II, thereby facilitating the incorporation of ubiquitinated substrates into autophagosomes (Fig. 1). Because p62 is degraded during autophagy, changes in p62 protein content have together with LC3 lipidation been used as a surrogate marker for autophagy flux (Klionsky et al. 2016).
An emerging autophagy-dependent mechanism with potential implications for exercise training adaptations is the autophagic clearance of damaged mitochondria termed mitophagy and a molecular machinery that serves to target mitochondria for degradation has been discovered. The kinase PTEN-induced putative kinase 1 (PINK1) is stabilized on depolarized mitochondrial membranes, for example, in response to oxidative stress, and recruits the E3 ubiquitin ligase parkin to mitochondria. On activation, parkin ubiquitinates several mitochondrial membrane proteins, thereby targeting mitochondria for degradation. Furthermore, specific mitophagy receptors such as BCL2/adenovirus E1B 19-kDa protein-interacting protein 3 (BNIP3) and NIP3-like protein X (NIX) present on mitochondrial membranes can directly interact with LC3-II to induce mitophagy (Fig. 1) (Youle and Narendra 2011).
선택적 자가포식
자가포식은
에너지 부족 시 기질을 제공하는 등
비선택적인 방식으로 기능할 수 있지만,
특정 세포소기관과 단백질의 선택적 제거 메커니즘이 확인되었습니다.
오토파지 어댑터 단백질 p62는
카르복시 말단 유비퀴틴 관련 도메인을 포함하고
LC3-II와 직접 상호작용하여
유비퀴틴화된 기질을 오토파지솜에 통합하는 것을 촉진합니다(그림 1).
p62는
자가포식 과정에서 분해되기 때문에
p62 단백질 함량의 변화는 LC3 지질화와 함께
자가포식 플럭스의 대리 마커로 사용되어 왔습니다(Klionsky et al. 2016).
운동 훈련 적응에 잠재적인 영향을 미치는
새로운 자가포식 의존 메커니즘은
미토파지라고 하는 손상된 미토콘드리아의 자가포식 제거이며,
미토콘드리아 분해를 표적으로 하는
분자 기계가 발견되었습니다.
예를 들어
산화 스트레스에 반응하여
탈분극된 미토콘드리아 막에서
키나아제 PTEN 유도 추정 키나아제 1(PINK1)이 안정화되고
E3 유비퀴틴 리가제 파킨을
미토콘드리아로 모집합니다.
활성화되면
파킨은 여러 미토콘드리아 막 단백질을 유비퀴틴화하여
미토콘드리아를 표적으로 삼아 분해합니다.
또한
미토콘드리아 막에 존재하는
BCL2/아데노바이러스 E1B 19-kDa 단백질 상호작용 단백질 3(BNIP3) 및
NIP3 유사 단백질 X(NIX)와 같은
특정 미토파지 수용체는
LC3-II와 직접 상호작용하여
미토파지를 유도할 수 있습니다(그림 1)(Youle and Narendra 2011).
Acute Exercise-Induced Autophagy
The ability of a single endurance exercise bout to induce an autophagic response in skeletal muscle was first described in 1984. Thus, using electron microscopy (EM), the formation of autophagic vacuoles was detected in mouse muscle fibers 2 to 7 days after an extremely strenuous (9 h) treadmill running bout (Salminen and Vihko 1984). However, the notion that exercise may stimulate autophagic removal of cellular components has only recently received widespread attention following reports that LC3-II as well as autophagosome content detected by EM was increased in mouse muscle after a single bout of treadmill running (Grumati et al. 2011). This finding has since been supported by several other studies in rodents (He et al. 2012; Jamart et al. 2012b; Pagano et al. 2014; Saleem et al. 2014; Fritzen et al. 2015; Liu et al. 2015; Vainshtein et al. 2015b; Halling et al. 2016). However, the autophagic response in human muscle to acute endurance exercise seems to be more complex. Hence, ultra-endurance running has been shown to potently increase LC3-II content (Jamart et al. 2012b), whereas exercise bouts of lower duration have been reported to lower the content of LC3-II (Fritzen et al. 2015; Moller et al. 2015; Schwalm et al. 2015). Moreover, some studies have observed unchanged skeletal muscle LC3-II content in humans following a single cycling exercise bout (Masschelein et al. 2014; Tachtsis et al. 2016). In addition, reduced p62 content following endurance exercise has been reported in both mouse (He et al. 2012; Pagano et al. 2014) and human (Schwalm et al. 2015) skeletal muscle, although others have observed unchanged p62 content after a single endurance exercise bout (Jamart et al. 2012b; Fritzen et al. 2015; Moller et al. 2015; Halling et al. 2016; Tachtsis et al. 2016). Altogether, there is robust evidence suggesting that autophagy is stimulated in skeletal muscle in response to acute endurance exercise. However, the exact mechanism behind induction of autophagy may depend on factors such as intensity/duration of exercise (Schwalm et al. 2015), feeding (Jamart et al. 2013), and timing of muscle sampling (Halling et al. 2016), which may contribute to the variability of the observed autophagic responses to acute endurance exercise.
Several studies have also examined the effects of strength/resistance exercise on regulation of autophagy. It has been shown that LC3-II protein and GABARAP mRNA were reduced from 3 to 24 hours after a single session of resistance exercise in both young and aged human muscle, which was interpreted as decreased activation of autophagy (Fry et al. 2013). Furthermore, it has been shown that the decreased LC-II content following resistance exercise was dependent on post-exercise intake of essential amino acids (Glynn et al. 2010), suggesting that resistance exercise-induced regulation of autophagy is dependent on nutrient availability. However, the apparent modulation of autophagy by resistance exercise in combination with amino acid intake was not associated with changes in either overall protein synthesis or protein breakdown at 2 hours post-exercise (Glynn et al. 2010). This indicates that autophagy-mediated release of amino acids is not involved in anabolic/catabolic processes in the early recovery period. The reported lowering of LC3-II and LC3-II/I content in human skeletal muscle during the early recovery period after a resistance exercise bout has been recapitulated in several recent studies (Dickinson et al. 2016; Francaux et al. 2016; Smiles et al. 2016). This may suggest a deactivation of autophagy during recovery from resistance exercise, assuming that LC3-II/I is a valid marker for autophagic flux. In contrast, others have reported no change (Smiles et al. 2015) or increase (Ogborn et al. 2015) in autophagy markers at various timepoints after resistance exercise. However, as previously described, LC3-II itself is degraded when autophagosomes fuse with lysosomes suggesting that, in certain cases, a reduction in LC3-II content can actually be the result of increased autophagic flux (Mizushima and Yoshimori 2007; Rubinsztein et al. 2009). It is, therefore, noteworthy that a recent study showed resistance exercise-induced activation of chaperone-assisted selective autophagy and LC3 puncta formation in human skeletal muscle up to 24 hours into recovery (Ulbricht et al. 2015), similar to that observed after treadmill running in mice (Grumati et al. 2011; He et al. 2012). Therefore, it is uncertain how resistance exercise affects autophagic flux. An approach that may be used to address this is colchicine-induced blockade of lysosomal degradation as previously used to determine autophagic flux in mice in response to endurance exercise (Vainshtein et al. 2015b). However, rodent models of resistance exercise may not be comparable with humans, underlined by the observation that “resistance exercise” did not affect LC3-II levels in rat skeletal muscle (Ogasawara et al. 2016). On the other hand, the activity of VPS34, a component of the PI3K complex, has been shown to be increased following high-resistance electrically stimulated contraction of rat skeletal muscle (MacKenzie et al. 2009). This suggests that autophagy is activated during recovery from resistance exercise similarly to endurance exercise, although further studies are required to directly assess the effects of resistance exercise on autophagic flux in human muscle.
급성 운동 유발 자가포식 작용
골격근에서
자가포식 반응을 유도하는
단일 지구력 운동 시합의 능력은
1984년에 처음 설명되었습니다.
따라서
전자 현미경(EM)을 사용하여
극도로 격렬한(9시간) 러닝머신 달리기 시합 후
2~7일 후에 마우스 근육 섬유에서
자가포식 액포의 형성이 감지되었습니다(Salminen and Vihko 1984).
그러나
운동이
세포 성분의 자가포식 제거를 자극할 수 있다는 개념은
최근에야 트레드밀 달리기를 한 번 한 후
마우스 근육에서 LC3-II와 EM으로 검출된
오토파지솜 함량이 증가했다는 보고 이후 널리 주목을 받았습니다(Grumati 등. 2011).
이 발견은 이후 설치류를 대상으로 한 여러 다른 연구에서도 뒷받침되었습니다(He 외. 2012, Jamart 외. 2012b, Pagano 외. 2014, Saleem 외. 2014, Fritzen 외. 2015, Liu 외. 2015, Vainshtein 외. 2015b, Halling 외. 2016).
그러나
급성 지구력 운동에 대한
인간 근육의 자가포식 반응은
더 복잡한 것으로 보입니다.
따라서
초지구력 달리기는
LC3-II 함량을 강력하게 증가시키는 것으로 나타난 반면(Jamart 외. 2012b),
지속 시간이 짧은 운동은
LC3-II 함량을 낮추는 것으로 보고되었습니다(Fritzen 외. 2015; Moller 외. 2015; Schwalm 외. 2015).
또한 일부 연구에서는
한 번의 사이클링 운동 시합 후
사람의 골격근 LC3-II 함량이 변하지 않는 것으로 관찰되었습니다(Masschelein 등. 2014; Tachtsis 등. 2016).
또한,
지구력 운동 후 마우스(He 외. 2012; Pagano 외. 2014)와
사람(Schwalm 외. 2015) 골격근에서
모두 p62 함량 감소가 보고되었지만,
한 번의 지구력 운동 시합 후에도
p62 함량이 변하지 않는 것으로 관찰된 사례도 있습니다(Jamart 외. 2012b; Fritzen 외. 2015; Moller 외. 2015; Halling 외. 2016; Tachtsis 외. 2016).
전체적으로,
급성 지구력 운동에 반응하여
골격근에서 자가포식이 자극된다는 강력한 증거가 있습니다.
그러나
자가포식 유도의 정확한 메커니즘은
운동의 강도/기간(Schwalm 외. 2015),
먹이(Jamart 외. 2013),
근육 샘플링 시기(Halling 외. 2016) 등의 요인에 따라 달라질 수 있으며,
이는 급성 지구력 운동에 대한 관찰된 자가포식 반응의 변동성에 기여할 수 있습니다.
근력/저항 운동이
자가포식 조절에 미치는 영향도
여러 연구에서 조사되었습니다.
젊은 사람과 노인의 근육에서
한 번의 저항 운동 후 3시간에서 24시간 사이에
LC3-II 단백질과 GABARAP mRNA가 감소하는 것으로 나타났는데,
이는 자가포식의 활성화가 감소한 것으로 해석되었습니다(Fry 등. 2013).
또한 저항성 운동 후
LC-II 함량 감소는
운동 후 필수 아미노산 섭취에 따라 달라지는 것으로 나타났는데,
이는 저항성 운동으로 인한 자가포식 조절이
영양소 가용성에 따라 달라진다는 것을 시사합니다(Glynn 외. 2010).
그러나
아미노산 섭취와 함께
저항성 운동에 의한 자가포식의 명백한 조절은
운동 후 2시간 동안의 전반적인 단백질 합성이나
단백질 분해의 변화와 관련이 없었습니다(Glynn 외. 2010).
이는 자가포식을 매개로 한 아미노산 방출이
초기 회복 기간의 동화 작용/이해 작용 과정에
관여하지 않는다는 것을 나타냅니다.
저항성 운동 시합 후 초기 회복 기간 동안
인간 골격근에서 LC3-II 및 LC3-II/I 함량이 낮아진다는 보고는
최근 여러 연구에서 요약되었습니다(Dickinson 외. 2016; Francaux 외. 2016; Smiles 외. 2016).
이는 LC3-II/I가
자가포식 플럭스에 대한 유효한 마커라고 가정할 때
저항성 운동에서 회복하는 동안
자가포식이 비활성화된다는 것을 시사할 수 있습니다.
이와는 대조적으로,
저항 운동 후 다양한 시점에서
자가포식 마커의 변화가 없거나(Smiles 등. 2015) 증가(Ogborn 등. 2015)한다는 보고도 있습니다.
그러나 앞서 설명한 바와 같이
LC3-II 자체는
오토파지가 리소좀과 융합할 때 분해되는데,
이는 특정 경우 LC3-II 함량의 감소가 실제로
자가포식 플럭스 증가의 결과일 수 있음을 시사합니다(미즈시마 및 요시모리 2007; 루빈츠테인 외. 2009).
따라서 최근 연구에 따르면
저항성 운동으로 인한
샤프론 보조 선택적 자가포식의 활성화와
회복 후 24시간까지 인간 골격근에서
LC3 푼타 형성이 생쥐의 러닝머신 달리기 후 관찰된 것과 유사하게(Ulbricht et al. 2015)
나타난 것은 주목할 만한 결과입니다(Grumati et al.2011; He et al. 2012).
따라서
저항 운동이 자가포식 플럭스에 어떤 영향을 미치는지는 불확실합니다.
이를 해결하기 위해 사용할 수 있는 접근법은
지구력 운동에 반응하는 생쥐의 자가포식 플럭스를 측정하는 데
이전에 사용된 콜히친에 의한 리소좀 분해 차단입니다(Vainshtein 외. 2015b).
그러나 설치류의 저항 운동 모델은 인간과 비교할 수 없는데, 이는 '저항 운동'이 쥐 골격근의 LC3-II 수준에 영향을 미치지 않는다는 관찰에 의해 강조됩니다(오가사와라 외. 2016). 반면, PI3K 복합체의 구성 요소인 VPS34의 활성은 쥐 골격근의 고저항 전기 자극 수축에 따라 증가하는 것으로 나타났습니다(MacKenzie 등. 2009).
이는 저항성 운동이
인간 근육의 자가포식 플럭스에 미치는 영향을 직접 평가하기 위해서는
추가 연구가 필요하지만,
지구력 운동과 유사하게
저항성 운동에서 회복하는 동안
자가포식이 활성화된다는 것을 시사합니다.
MOLECULAR MEDIATORS OF EXERCISE-INDUCED AUTOPHAGY
AMPK
Cell culture studies have indicated that the 5′ AMP-activated protein kinase (AMPK) regulates autophagy through ULK1 Ser-317 and Ser-555 phosphorylation (Meley et al. 2006; Egan et al. 2011) during conditions of cellular energy demand. Accordingly, ULK1 Ser-317, and Ser-555 phosphorylation has been shown to increase in mouse muscle in response to a single exercise bout (Pagano et al. 2014). Furthermore, it has been shown that the catalytic AMPK α2 subunit is required for exercise-induced LC3 lipidation and ULK1 Ser-777 phosphorylation in mouse skeletal muscle (Liu et al. 2015). The link between AMPK and autophagy is also supported by the observation that BCL2 AAA mutated (Thr69Ala, Ser70Ala, and Ser84Ala) mice with impaired dissociation of the Beclin-1/BCL2 complex, causing inhibition of PI3K complex formation and blunted exercise-induced activation of autophagy, showed lower AMPK phosphorylation levels (He et al. 2012). On the other hand, inducible muscle-specific knockout (KO) of ATG7 resulted in impaired exercise-induced autophagy, but did not affect AMPK phosphorylation in mouse skeletal muscle (Lo et al. 2014). Moreover, an association between exercise-induced AMPK activation, ULK1 phosphorylation and modulation of autophagy is supported by results from some human studies (Moller et al. 2015; Schwalm et al. 2015). However, others have reported that there was no correlation between exercise-induced AMPK activation and LC3 lipidation in human muscle as well as no apparent effects on LC3 lipidation with pharmacological activation of AMPK in incubated mouse skeletal muscle or human myotubes (Fritzen et al. 2015). Still, AMPK seems to be persistently required for ULK1 Ser-555 phosphorylation (Egan et al. 2011; Fritzen et al. 2015). Taken together, evidence suggests that AMPK-dependent activation of autophagy can occur in skeletal muscle in response to acute exercise. Because AMPK is known as an intracellular energy sensor, this may suggest that exercise-induced autophagy activation occurs in response to lowered intracellular energy charge. However, a recent study showed that exercise-induced activation of autophagy was independent of continuous systemic glucose infusion during exercise (Moller et al. 2015), suggesting that activation of autophagy in skeletal muscle during exercise is not caused by insufficient substrate supply.
운동 유발 자가포식의 분자 매개체
AMPK
AMP-activated protein kinase
세포 배양 연구에 따르면
5′ AMP 활성화 단백질 키나아제(AMPK)는
세포 에너지 요구 조건에서
ULK1 Ser-317 및 Ser-555 인산화를 통해
자가포식을 조절합니다(Meley 외. 2006; Egan 외. 2011).
따라서,
한 번의 운동 시합에 대한 반응으로
마우스 근육에서 ULK1 Ser-317 및 Ser-555
인산화가 증가하는 것으로 나타났습니다(Pagano 등. 2014).
또한,
촉매 AMPK α2 서브유닛은
마우스 골격근에서 운동으로 유도된 LC3 지질화 및 ULK1 Ser-777
인산화에 필요한 것으로 나타났습니다(Liu et al. 2015).
AMPK와 자가포식 사이의 연관성은
Beclin-1/BCL2 복합체의 해리가 손상되어
PI3K 복합체 형성이 억제되고
운동으로 인한 자가포식 활성화가 둔화된 BCL2 AAA 돌연변이(Thr69Ala, Ser70Ala 및 Ser84Ala) 마우스가
낮은 AMPK 인산화 수준을 보인다는 관찰에서도 뒷받침됩니다(He et al. 2012).
반면,
ATG7의 유도성 근육 특이적 녹아웃(KO)은
운동으로 인한 자가포식 장애를 초래했지만,
마우스 골격근의 AMPK 인산화에는 영향을 미치지 않았습니다(Lo et al. 2014).
또한
운동으로 인한 AMPK 활성화,
ULK1 인산화 및 자가포식 조절 사이의 연관성은
일부 인간 연구 결과에 의해 뒷받침됩니다(Moller 외. 2015; Schwalm 외. 2015).
그러나
다른 연구자들은
인간 근육에서 운동으로 인한 AMPK 활성화와
LC3 지질화 사이에는 상관관계가 없으며,
배양된 마우스 골격근이나 인간 근육에서
AMPK의 약리학적인 활성화가 LC3 지질화에 미치는 영향도
뚜렷하지 않다고 보고했습니다(Fritzen 등. 2015).
그럼에도 불구하고
AMPK는
ULK1 Ser-555 인산화에 지속적으로 필요한 것으로 보입니다(Egan 외. 2011; Fritzen 외. 2015).
이러한 증거를 종합해 볼 때,
급성 운동에 대한 반응으로
골격근에서
AMPK 의존적인 자가포식 활성화가
일어날 수 있음을 시사합니다.
AMPK는
세포 내 에너지 센서로 알려져 있기 때문에
운동으로 인한 자가포식 활성화는
세포 내 에너지 전하 감소에 반응하여 발생한다는 것을 시사할 수 있습니다.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8919726/

그러나
최근 연구에 따르면
운동에 의한 자가포식 활성화는
운동 중 지속적인 전신 포도당 주입과 무관한 것으로 나타났는데(Moller 등. 2015),
이는 운동 중 골격근의 자가포식 활성화가 기질 공급 부족으로 인한 것이 아님을 시사합니다.
mTOR
The mammalian target of rapamycin (mTOR) has been identified as a major negative regulator of autophagy through an AMPK-opposing mechanism (Kim et al. 2011). During conditions of high substrate availability, a signaling pathway is activated involving the insulin-stimulated class I PI3K, which recruits Akt leading to activation of mTOR. Consequently, ULK1 is phosphorylated by mTOR at Ser-757, which, contrary to AMPK-mediated phosphorylation, inhibits formation of the ULK1 kinase complex preventing autophagosome maturation (Kim et al. 2011). AMPK activation inhibits mTOR signaling and, indeed, ULK1 Ser-757 phosphorylation has been shown to decrease in mouse skeletal muscle both during exercise (Pagano et al. 2014) and 10 hours into recovery (Halling et al. 2016) from a single treadmill running bout. This suggests that deactivation of mTOR signaling contributes to endurance exercise-induced autophagy. In addition, autophagy activation following high-resistance electrically stimulated contraction of rat skeletal muscle correlated with mTOR activity, while leucine treatment, but not amino acid starvation, of C2C12 cells also increased VPS34 activity (MacKenzie et al. 2009). Furthermore, inhibition of mTOR signaling attenuated protein synthesis rates up to 24 hours after a single resistance exercise bout in rat skeletal muscle (Ogasawara et al. 2016). Together, this suggests that it may be the increased amino acid turnover per se that regulates autophagy through mTOR signaling in response to resistance exercise potentially allowing further stimulation of protein synthesis at later timepoints.
mTOR
포유류의 라파마이신 표적(mTOR)은
AMPK에 대항하는 메커니즘을 통해
오토파지의 주요 음성 조절 인자로 확인되었습니다(Kim et al. 2011).
기질 가용성이 높은 조건에서는
인슐린 자극 클래스 I PI3K와 관련된 신호 경로가 활성화되고,
이 신호 경로가 Akt를 모집하여
mTOR를 활성화합니다.
결과적으로
ULK1은 mTOR에 의해 Ser-757에서 인산화되는데,
이는 AMPK 매개 인산화와는 반대로
자가포식체 성숙을 막는 ULK1 키나아제 복합체의 형성을 억제합니다(Kim et al. 2011).
AMPK 활성화는
mTOR 신호를 억제하며,
실제로 운동 중(Pagano 등. 2014)과
한 번의 러닝머신 달리기 시합 후 10시간 후(Halling 등. 2016) 마우스 골격근에서
ULK1 Ser-757 인산화가 감소하는 것으로 나타났습니다.
이는
mTOR 신호의 비활성화가
지구력 운동으로 인한 자가포식에 기여한다는 것을 시사합니다.
또한 쥐 골격근의 고저항 전기 자극에 따른 자가포식 활성화는 mTOR 활성과 상관관계가 있으며, C2C12 세포의 아미노산 결핍이 아닌 류신 처리도 VPS34 활성을 증가시켰습니다(MacKenzie et al. 2009). 또한, 쥐 골격근에서 한 번의 저항 운동 시합 후 최대 24시간까지 mTOR 신호의 억제가 단백질 합성 속도를 감소시켰습니다(오가사와라 외. 2016).
이는
저항성 운동에 대한 반응으로
mTOR 신호를 통해
오토파지를 조절하는 아미노산 회전율 자체가 증가하여
이후 시점에 단백질 합성을 더 자극할 수 있음을 시사합니다.
FOXO
Several stress-induced transcription factors have been proposed to regulate the expression of genes involved in autophagy (Pietrocola et al. 2013) of which the best-described example is the forkhead box O (FOXO) family of transcription factors. In particular, FOXO3 has been shown to play an important role in regulating core ATGs, including LC3 and BNIP3 in skeletal muscle (Mammucari et al. 2007; Zhao and Klionsky 2011). FOXOs are responsive to nutrient deprivation and oxidative stress through posttranslational modifications. This includes Akt (PKB)-mediated phosphorylation of FOXO3 on multiple residues leading to inhibition and retention of FOXO3 in the cytosol (Brunet et al. 1999). The observation that Akt 1 and 2 KO in mice increased BNIP3 mRNA in skeletal muscle (Reynolds et al. 2012) supports the relevance of Akt-mediated FOXO3 regulation in autophagy. Furthermore, the findings that the AMPK activator, AICAR, increased FOXO3 phosphorylation with concomitant transient relocalization of FOXO3 to the nuclei and increase in mRNA content of autophagy-related proteins in muscle cells (Sanchez et al. 2012) provide strong evidence that AMPK is involved in posttranslational regulation of FOXO3. In addition, acetylation of FOXO3 has been reported to induce translocation of FOXO3 to the cytosol followed by ubiquitination and degradation of FOXO3 in the proteasome, whereas sirtuin-mediated deacetylation and activation of FOXO3 has been suggested to promote autophagy (Bertaggia et al. 2012). Moreover, AMPK-induced modulation of sirtuin activity is thought to reduce FOXO3 acetylation. Taken together, multiple posttranslational modifications seem to contribute in the regulation of FOXO3 activity and localization. Interestingly, increased FOXO3 mRNA and protein content as well as stimulatory dephosphorylation of FOXO3 has been observed in human and mouse skeletal muscle following acute endurance exercise (Louis et al. 2007; Jamart et al. 2012a, 2013; Wang et al. 2015). This indicates that FOXO3 regulation, possibly mediated by Akt/AMPK, contributes to transcriptional regulation of autophagy components in response to exercise.
FOXO
자가포식에 관여하는 유전자의 발현을 조절하는
여러 스트레스 유발 전사인자가 제안되었으며(Pietrocola 등. 2013),
그 중 가장 잘 설명된 예는 포크헤드 박스 O(FOXO) 전사인자 계열입니다.
forkhead box O (FOXO) family of transcription factors
특히
FOXO3는
골격근에서 LC3 및 BNIP3를 포함한
핵심 ATG를 조절하는 데 중요한 역할을 하는 것으로 밝혀졌습니다(Mammucari et al. 2007; Zhao and Klionsky 2011).
FOXO는
번역 후 변형을 통해
영양소 결핍과 산화 스트레스에 반응합니다.
여기에는 세포질에서 FOXO3의 억제 및 유지로 이어지는 여러 잔기에서 Akt (PKB)를 매개로 한 FOXO3의 인산화가 포함됩니다(Brunet et al. 1999). 생쥐의 Akt 1 및 2 KO가 골격근에서 BNIP3 mRNA를 증가시킨다는 관찰(Reynolds et al. 2012)은 오토파지에서 Akt 매개 FOXO3 조절의 관련성을 뒷받침합니다.
또한,
AMPK 활성화제인
AICAR이 FOXO3의 핵으로의 일시적 재배치와
근육 세포에서 자가포식 관련 단백질의 mRNA 함량 증가와 함께
FOXO3 인산화를 증가시킨다는 결과(산체스 외. 2012)는
AMPK가 FOXO3의 번역 후 조절에 관여한다는 강력한 증거를 제시합니다.
또한,
FOXO3의 아세틸화는
FOXO3의 세포질로의 전위를 유도한 후 프로테아좀에서
FOXO3의 유비퀴틴화 및 분해를 유도하는 것으로 보고된 반면,
시르투인 매개 탈아세틸화 및 활성화는
FOXO3의 자가포식을 촉진한다고 제안되었습니다(Bertaggia et al. 2012).
또한
AMPK에 의한 시르투인 활성의 조절은
FOXO3 아세틸화를 감소시키는 것으로 생각됩니다.
종합하면, 여러 번역 후 변형이 FOXO3 활성 및 국소화에 기여하는 것으로 보입니다.
흥미롭게도
급성 지구력 운동 후 인간과 생쥐 골격근에서
FOXO3의 자극적 탈인산화뿐만 아니라
FOXO3 mRNA 및 단백질 함량 증가가 관찰되었습니다(Louis 외. 2007; Jamart 외. 2012a, 2013; Wang 외. 2015).
이는 아마도
Akt/AMPK에 의해 매개되는 FOXO3 조절이
운동에 대한 반응으로 자가포식 성분의 전사 조절에 기여한다는 것을 나타냅니다.
TFEB
The transcription factor EB (TFEB) has been shown to be a key factor in autophagy and lysosome function through transcriptional regulation of genes encoding autophagy and lysosome proteins (Settembre et al. 2011). TFEB localization and activity is regulated by mTORC1-mediated phosphorylation of multiple residues and dephosphorylation by calcineurin. In accordance, exhaustive running exercise in mice has been reported to induce a calcineurin-mediated dephosphorylation of TFEB in skeletal muscle with concomitant nuclear translocation of TFEB and overexpression of a calcineurin inhibitor in muscle by electroporation prevented these effects (Medina et al. 2015). A simultaneous exercise-induced reduction in mTOR-mediated phosphorylation and inhibition of TFEB has been suggested to contribute to ensuring that TFEB translocates to the nucleus in response to muscle contractions (Medina et al. 2015). Together, this shows the potential of TFEB to regulate exercise training induced long-term adaptations in the autophagy and lysosome pathways.
전사인자 EB(TFEB)는
오토파지 및 리소좀 단백질을 암호화하는
유전자의 전사 조절을 통해
오토파지 및 리소좀 기능의 핵심 인자로 밝혀졌습니다(세템브레 외. 2011).
TFEB의 국소화 및 활성은
mTORC1에 의해 매개되는 여러 잔기의 인산화와
칼시뉴린에 의한 탈인산화에 의해 조절됩니다.
따라서 생쥐에서 무리한 달리기 운동은 골격근에서 칼시뉴린 매개 TFEB의 탈인산화를 유도하여 TFEB의 핵 전위를 수반하는 것으로 보고되었으며, 전기 천공을 통한 근육 내 칼시뉴린 억제제의 과발현은 이러한 효과를 방지했습니다(Medina et al. 2015). 운동으로 인한 mTOR 매개 인산화 감소와 TFEB의 억제는 근육 수축에 반응하여 TFEB가 핵으로 전위되도록 하는 데 기여하는 것으로 제안되었습니다(Medina 외. 2015). 이는 운동 훈련으로 유도된 자가포식 및 리소좀 경로의 장기적 적응을 조절할 수 있는 TFEB의 잠재력을 함께 보여줍니다.
p53
The tumor suppressor p53 has also been shown to be involved in regulation of autophagy. However, whether p53 functions as an activator or suppressor of autophagy seems to be dependent on the context and subcellular localization, because cytosolic p53 has been shown to inhibit autophagy (Tasdemir et al. 2008), whereas p53 induces transcription of key stimulatory autophagy genes in the nucleus (Maiuri et al. 2009; Kenzelmann et al. 2013). Furthermore, the findings that the nuclear abundance of p53 is increased in human muscle (Tachtsis et al. 2016) and decreased in mouse muscle (Saleem and Hood 2013) during recovery from exercise underline the complexity of p53 regulation. Together, the possible role of p53 in the regulation of exercise-induced autophagy remains to be fully elucidated. However, the finding that p53 is dispensable for metabolic adaptations to exercise training in mice (Saleem et al. 2009) suggests that p53-mediated exercise-induced autophagy may not be required for the beneficial metabolic effects of exercise.
p53
종양 억제 인자인 p53도
자가포식 조절에 관여하는 것으로 나타났습니다.
그러나
세포질 p53은
자가포식을 억제하는 것으로 나타난 반면(Tasdemir 등. 2008),
p53은 핵에서 주요 자극성 자가포식 유전자의 전사를 유도하기 때문에(Maiuri 등. 2009; Kenzelmann 등. 2013),
p53이 자가포식의 활성화 또는 억제자로서 기능하는지는
맥락과 세포 내 국소화에 따라 달라지는 것으로 보입니다.
또한,
운동 후 회복 중에
p53의 핵 농도가 인간 근육에서는 증가하고(Tachtsis 등. 2016),
마우스 근육에서는 감소한다는 연구 결과(Saleem and Hood 2013)는
p53 조절의 복잡성을 강조합니다.
운동으로 인한 자가포식 조절에서
p53의 가능한 역할은 아직 완전히 규명되지 않았습니다.
그러나 생쥐의 운동 훈련에 대한 대사 적응에 p53이 필수적이지 않다는 연구 결과(Saleem 외. 2009)는 운동의 유익한 대사 효과에 p53 매개 운동 유도 자가포식이 필요하지 않을 수 있음을 시사합니다.
PGC-1α
The exercise responsive transcriptional co-activator peroxisome proliferator-activated receptor γ coactivator (PGC)-1α has been identified as a key regulator of mitochondrial biogenesis (Wu et al. 1999; Pilegaard et al. 2003; Lin et al. 2005). Moreover, several recent studies have suggested that PGC-1α plays a role in exercise-induced autophagy. Hence, the finding that muscle-specific PGC-1α overexpression (MCK) was associated with increased BNIP3 protein content and increased basal autophagy (Lira et al. 2013) indicates that PGC-1α contributes to the regulation of autophagy in resting skeletal muscle. This was accompanied by results showing that autophagy levels are higher in oxidative than glycolytic muscles (Lira et al. 2013). The finding that exercise increased LC3-II protein content during recovery from a single treadmill running bout in PGC-1α MCK mice, but not in wild-type (WT) mice, suggests that elevated levels of PGC-1α also contributes in inducing autophagy in response to acute exercise. This was supported by a blunted treadmill exercise-induced increase in skeletal muscle LC3-II protein in muscle-specific PGC-1α KO mice (Halling et al. 2016). In addition, an attenuated treadmill exercise-induced increase in mitochondrial LC3-II flux in skeletal muscle from whole-body PGC-1α KO mice determined by monitoring LC3-II accumulation after treatment with the autophagy inhibitor colchicine further underlines the impact of PGC-1α in exercise-induced autophagy (Vainshtein et al. 2015b). Together, this may reflect that the metabolic profile of PGC-1α overexpression and KO mice affects the exercise-induced autophagy response. Thus, PGC-1α may influence autophagy indirectly through regulation of muscle oxidative capacity. However, it is also possible that PGC-1α affects exercise-induced autophagy through transcriptional regulation of autophagy proteins. Hence, the observations that PGC-1α KO mice had lower TFEB protein in skeletal muscle than WT mice (Vainshtein et al. 2015a) and did not increase the mRNA content of LC3, p62, and Niemann–Pick C1 in response to treadmill running (Vainshtein et al. 2015b) indicate that PGC-1α also plays a role in the endurance-exercise-induced adaptive gene responses of autophagy proteins in skeletal muscle. Taken together, this suggests that PGC-1α influences exercise training-mediated adaptations in key autophagy markers resulting in increased capacity of the autophagy machinery with exercise training.
PGC-1α
운동 반응성 전사 공동 활성화제인
퍼옥시좀 증식인자 활성화 수용체 γ 공동 활성화제(PGC)-1α는
미토콘드리아 생물 생성의 핵심 조절 인자로 확인되었습니다(Wu 등, 1999; Pilegaard 등, 2003; Lin 등, 2005).
또한,
최근 여러 연구에 따르면
PGC-1α는 운동으로 인한 자가포식에 중요한 역할을 한다고 합니다.
따라서
근육 특이적 PGC-1α 과발현(MCK)이
BNIP3 단백질 함량 증가 및 기저 자가포식 증가와 관련이 있다는 발견(Lira 등. 2013)은
PGC-1α가 휴식 중인 골격근의 자가포식 조절에 기여한다는 것을 시사합니다.
이는 당분해 근육보다 산화 근육에서 자가포식 수치가 더 높다는 결과와 함께 나타났습니다(Lira 외. 2013). PGC-1α MCK 마우스에서 러닝머신 달리기 한 시합에서 회복하는 동안 운동이 LC3-II 단백질 함량을 증가시켰지만 야생형(WT) 마우스에서는 그렇지 않았다는 발견은 PGC-1α의 높은 수준이 급성 운동에 대한 반응으로 자가포식을 유도하는 데 기여한다는 것을 시사합니다. 이는 트레드밀 운동으로 인한 근육 특이적 PGC-1α KO 마우스에서 골격근 LC3-II 단백질의 증가가 둔화됨으로써 뒷받침되었습니다(Halling 외. 2016). 또한, 자가포식 억제제 콜히친으로 처리한 후 LC3-II 축적을 모니터링하여 확인한 전신 PGC-1α KO 마우스의 골격근에서 트레드밀 운동으로 인한 미토콘드리아 LC3-II 플럭스의 약화된 증가는 운동으로 인한 자가포식에서 PGC-1α의 영향을 더욱 강조합니다(Vainshtein et al. 2015b). 이는 PGC-1α 과발현 및 KO 마우스의 대사 프로필이 운동으로 유도된 자가포식 반응에 영향을 미친다는 것을 반영할 수 있습니다. 따라서 PGC-1α는 근육 산화 능력 조절을 통해 간접적으로 오토파지에 영향을 미칠 수 있습니다. 그러나 PGC-1α가 오토파지 단백질의 전사 조절을 통해 운동 유발 오토파지에 영향을 미칠 가능성도 있습니다. 따라서 PGC-1α KO 마우스가 WT 마우스보다 골격근에서 TFEB 단백질이 낮고 (Vainshtein et al. 2015a) 트레드밀 달리기에 대한 반응으로 LC3, p62 및 Niemann-Pick C1의 mRNA 함량이 증가하지 않는다는 관찰 (Vainshteinetal. 2015b)은 PGC-1α가 골격근에서 자가포식 단백질의 지구력-운동 유발 적응 유전자 반응에도 역할을한다는 것을 시사합니다. 종합하면, 이는 PGC-1α가 운동 훈련에 따른 주요 자가포식 마커의 운동 훈련 매개 적응에 영향을 미쳐 운동 훈련을 통해 자가포식 기계의 용량을 증가시킨다는 것을 시사합니다.
AUTOPHAGY-DEPENDENT BENEFICIAL EFFECTS OF EXERCISE
Exercise Performance and Oxidative Capacity
The possibility that exercise-induced autophagy influences exercise performance is supported by the finding that BCL2 AAA mutated mice that are incapable of exercise-induced BCL2–Beclin-1 disassociation and autophagy activation have lower maximal exercise capacity than control mice (He et al. 2012). Of notice is that the lower maximal exercise capacity was not because of differences in basal muscle properties, but the inability to induce autophagy during running. Maximal running distance was also shown to be lower in ATG6 (Beclin-1)+/− mice with reduced ATG6 protein content in skeletal muscle, but normal basal autophagy (He et al. 2012), although others did not observe changes in time to exhaustion during treadmill running in ATG6+/− mice (Lira et al. 2013). This suggests that the impact of the level of ATG6 on exercise performance may depend on exercise type, intensity, and/or duration. In accordance, exercise performance during regular treadmill running was not impaired in inducible skeletal-muscle-specific ATG7 KO mice, despite a blunted exercise-induced autophagy response (Lo et al. 2014), supporting that acute autophagy activation is not universally required for sustaining muscle contractions. However, the inducible deletion of ATG7 in skeletal muscle reduced performance and caused profound mitochondrial membrane depolarization in skeletal muscle during downhill running (eccentric muscle contractions) in female mice (Lo et al. 2014), suggesting that autophagy influences exercise performance during more damaging muscle contractions (Fig. 2).
운동의 자가포식 의존적 유익한 효과
운동 수행 능력과 항산화 능력
운동으로 유도된 자가포식이
운동 수행 능력에 영향을 미칠 수 있다는 가능성은
운동으로 유도된 BCL2-Beclin-1 분리 및
자가포식 활성화가 불가능한 BCL2 AAA 돌연변이 마우스가
대조군 마우스보다 최대 운동 능력이 낮다는 발견으로 뒷받침됩니다(He et al. 2012).
주목할 점은 최
대 운동 능력이 낮은 것은
기초 근육 특성의 차이 때문이 아니라
달리기 중 자가포식을 유도할 수 없기 때문이라는 점입니다.
최대 달리기 거리는
골격근의 ATG6 단백질 함량은 감소했지만
기저 자가포식은 정상인 ATG6 (Beclin-1)+/- 마우스에서도 더 낮은 것으로 나타났지만(He et al. 2012),
다른 연구에서는 ATG6+/- 마우스에서 러닝머신 달리기 중 탈진까지의 시간 변화가 관찰되지 않았습니다(Lira et al. 2013).
이는 ATG6 수준이 운동 수행 능력에 미치는 영향이
운동 유형, 강도 및/또는 지속 시간에 따라
달라질 수 있음을 시사합니다.
이에 따라,
운동으로 인한 자가포식 반응이 둔화되었음에도 불구하고
유도성 골격근 특이적 ATG7 KO 마우스에서는
규칙적인 러닝머신 달리기 동안 운동 수행 능력이 손상되지 않았으며(Lo et al. 2014),
이는 급성 자가포식 활성화가 근육 수축을 지속하는 데
보편적으로 필요하지 않다는 것을 뒷받침합니다.
그러나
골격근에서 ATG7의 유도성 결실은
암컷 마우스의 내리막 달리기 (편심 근육 수축) 동안
골격근에서 성능을 감소시키고
심각한 미토콘드리아 막 탈분극을 유발하여 (Lo 외 2014),
자가 포식이 더 손상된 근육 수축 동안 운동 성능에 영향을 미친다는 것을 시사합니다 (그림 2).
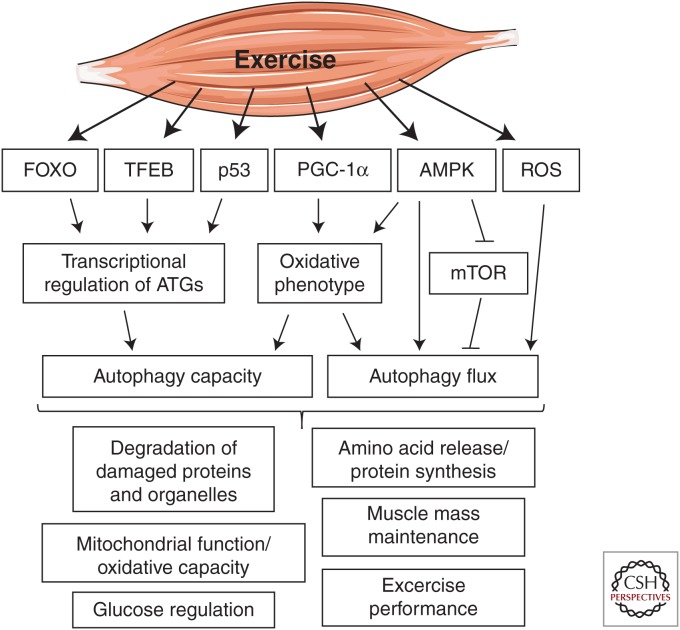
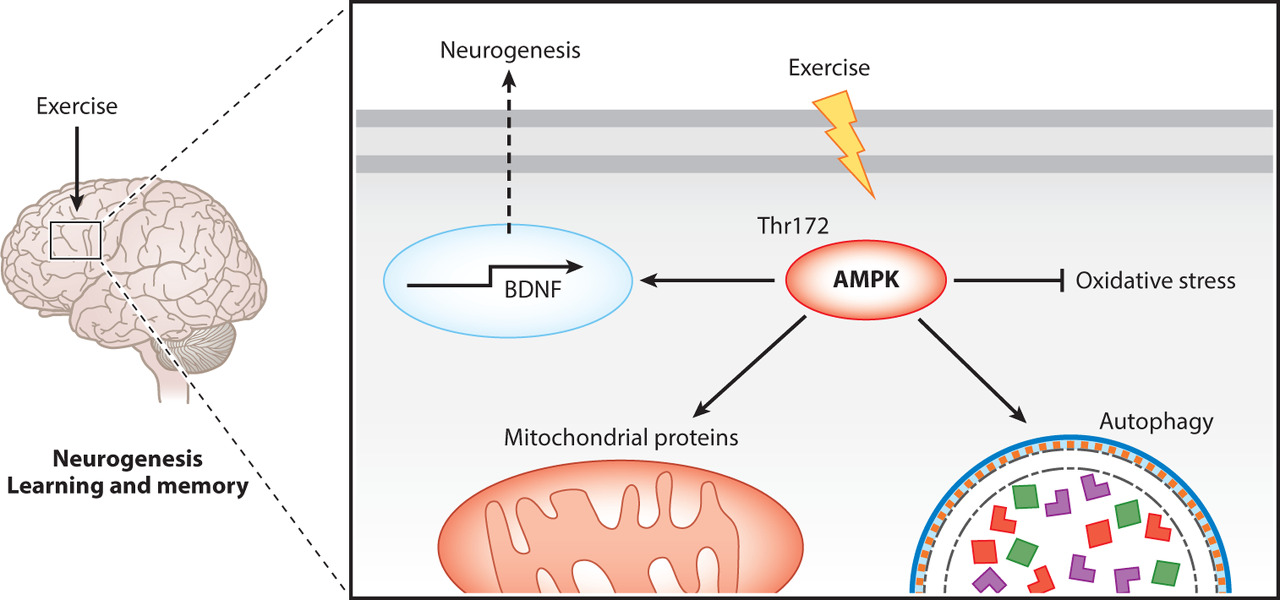
Overview of molecular mediators of exercise-induced autophagy and the autophagy-dependent beneficial effects of exercise. Exercise-induced activation of autophagy in skeletal muscle has been shown to be influenced by transcriptional regulation of autophagy-related genes (ATGs) mediated by forkhead box O (FOXO) family of transcription factors, transcription factor EB (TFEB), p53, and peroxisome proliferator-activated receptor γ coactivator 1α (PGC-1α) to increase the autophagic capacity. The oxidative phenotype of skeletal muscle is linked to autophagy capacity/flux and is positively regulated by the exercise responsive 5′ AMP-activated protein kinase (AMPK) and PGC-1α. AMPK and mammalian target of rapamycin (mTOR) act as an activator and suppressor of exercise-induced autophagy, respectively. Exercise-induced generation of reactive oxygen species (ROS) seems to serve as a signal for autophagy activation. Autophagy-dependent beneficial effects of exercise include degradation of oxidatively damaged proteins and organelles, improved mitochondrial oxidative capacity, improved glucose regulation, protein synthesis, preservation of muscle strength and mass, and improved endurance exercise performance.
Several studies suggest that autophagy is essential for cellular adaptations to exercise training. A potential link between exercise-induced autophagy and exercise training-mediated adaptations was examined using ATG6+/− mice. Exercise training did not improve endurance exercise performance and did not increase the protein content of the oxidative markers cytochrome c and COXIV, or LC3 and BNIP3 protein in skeletal muscle of ATG6+/− mice (Lira et al. 2013). This suggests that exercise-induced autophagy is required for exercise training-mediated adaptations in skeletal muscle oxidative capacity and exercise performance (Fig. 2). This is supported by the finding that there was a positive correlation between LC3-II content and the conversion of glycolytic type IIX muscle fibers toward the more oxidative type IIA fibers in response to endurance exercise training in rats (Tam et al. 2015).
운동으로 유도된
자가포식의 분자 매개체와
운동의 자가포식 의존적 유익한 효과에 대한 개요.
운동에 의한 골격근의 자가포식 활성화는
포크헤드 박스 O(FOXO) 전사인자 계열,
전사인자 EB(TFEB),
p53,
퍼옥시좀 증식인자 활성화 수용체 γ 보조인자 1α(PGC-1α)를 매개로 하는
자가포식 관련 유전자(ATG)의 전사 조절에 영향을 받아
자가포식 능력을 증가시키는 것으로 나타났습니다.
골격근의 산화적 표현형은
자가포식 능력/유량과 관련이 있으며
운동 반응성 5′ AMP 활성화 단백질 키나아제(AMPK)와
PGC-1α에 의해 긍정적으로 조절됩니다.
AMPK와 포유류 라파마이신 타겟(mTOR)은
각각 운동으로 인한 자가포식의
활성화제와 억제제 역할을 합니다.
운동으로 인한 활성 산소종(ROS)의 생성은
오토파지 활성화의 신호로
작용하는 것으로 보입니다.
운동의 자가포식에 의존하는 유익한 효과로는
산화적으로 손상된 단백질과
세포 소기관의 분해,
미토콘드리아 산화 능력 향상,
포도당 조절 개선,
단백질 합성,
근력 및 질량 보존,
지구력 운동 능력 향상 등이 있습니다.
여러 연구에 따르면
자가포식이 운동 훈련에 대한
세포의 적응에 필수적이라고 합니다.
ATG6+/- 마우스를 사용하여 운동으로 인한 자가포식과 운동 훈련 매개 적응 사이의 잠재적 연관성을 조사했습니다. 운동 훈련은 지구력 운동 능력을 향상시키지 못했고, ATG6+/- 마우스의 골격근에서 산화 마커인 사이토크롬 c와 COXIV 또는 LC3와 BNIP3 단백질의 단백질 함량을 증가시키지 않았습니다(Lira et al. 2013). 이는 운동으로 인한 자가포식이 골격근 산화 능력과 운동 수행 능력의 운동 훈련 매개 적응에 필요하다는 것을 시사합니다(그림 2). 이는 쥐의 지구력 운동 훈련에 대한 반응으로 LC3-II 함량과 당분해성 IIX형 근육 섬유가 더 산화적인 IIA형 섬유로 전환되는 것 사이에 양의 상관관계가 있다는 발견에 의해 뒷받침됩니다(Tam et al. 2015).
Muscle Mass
Given the reported effects of resistance exercise on regulation of autophagy, it can be speculated that autophagy-induced amino acid replenishment contributes to the hypertrophic response to resistance exercise training. In addition, the ability of resistance exercise training to preserve muscle mass during aging has been associated with regulation of autophagy (Luo et al. 2013). Thus, aged rats subjected to 9 weeks of weight-bearing exercise had higher muscle cross-sectional area and strength as well as higher cathepsin L activity, Beclin-1, ATG5 and ATG7 protein content, whereas LC3-II and p62 protein content was lower than in aged untrained rats, altogether indicating increased basal autophagic flux with resistance exercise training (Luo et al. 2013). Furthermore, mice lacking collagen-type VI α (COL6A KO) with impaired autophagy have been shown to display a blunted exercise-induced increase in LC3II/LC3I with concomitant accumulation of dysfunctional mitochondria, altered mitochondrial network (based on SDH staining), muscle fiber degeneration, and decreased muscle strength (Grumati et al. 2010, 2011). These observations showing that an exercise bout elicited extensive muscle damage in COL6A KO mice, but not WT mice, indicate that exercise-induced autophagy is an important myoprotective process. Accordingly, 3 months of exercise training, increasing SDH staining in WT skeletal muscle, reduced skeletal muscle LC3-II/I and exacerbated the dystrophic phenotype with extensive muscle degradation in COL6A KO mice (Grumati et al. 2011). Taken together, this suggests that exercise-induced autophagy is crucial for maintaining muscle mass, ultrastructure, and function (Fig. 2).
Glucose Regulation
A few studies have examined the role of exercise-mediated autophagy on metabolic adjustments in response to exercise. The observation that plasma lactate, glucose, and free fatty acid concentrations after exercise were unaffected by inducible muscle-specific KO of ATG7 with impaired exercise-induced autophagy (Lo et al. 2014) suggests that autophagy is not required for exercise-induced adjustments in circulating substrates and metabolites. In contrast, pharmacological inhibition of autophagy reduced insulin-stimulated glucose uptake in C2C12 myotubes (Liu et al. 2015), supporting a role of autophagy in glucose regulation. In accordance, the impaired exercise-induced activation of autophagy in BCL AAA and ATG6+/− mice was associated with reduced GLUT4 translocation to the sarcolemma in response to exercise and lower radiolabeled glucose uptake in the isolated soleus muscle. This suggests an association between exercise-regulated autophagy and glucose uptake in skeletal muscle. BCL2 AAA mutated mice also showed impaired exercise training–mediated protection against high fat diet–induced glucose intolerance (He et al. 2012), further supporting that exercise-induced activation of autophagy is required for the beneficial effects of exercise on glucose metabolism (Fig. 2).
ROS Protection
The findings that COL6A KO mice had impaired exercise-induced activation of autophagy and accumulated damaged mitochondria (Grumati et al. 2011) suggest that exercise-induced autophagy serves to remove dysfunctional organelles. In accordance, skeletal muscle protein carbonylation (used as marker of oxidative damage) was shown to be reduced in the recovery period from a single exercise bout coinciding with an increase in LC3-II (Halling et al. 2016). This indicates that exercise-induced autophagy plays a role in removing oxidized proteins to prevent the accumulation of dysfunctional proteins. Furthermore, muscle-specific ATG7 KO mice have been shown to exhibit higher ROS accumulation and mitochondrial membrane depolarization in skeletal muscle than WT mice after eccentric contractions (Lo et al. 2014). This supports that the level of reactive oxygen species may play a role in the regulation of autophagy (Fig. 2). It should be noted that antioxidant treatment did not prevent exercise-induced activation of mitophagy and the ability of exercise to restore the mitochondrial membrane potential (Lo et al. 2014), showing that additional factors must also be involved. However, N-acetylcysteine (NAC) antioxidant treatment reduced basal mitophagy in mice (Lo et al. 2014; Qi et al. 2014). This may suggest a mechanism explaining the observation that antioxidant supplementation can have adverse effects on exercise performance as well as metabolic adaptations to exercise training (Gomez-Cabrera et al. 2008; Ristow et al. 2009; Gliemann et al. 2013; Olesen et al. 2014).
ROS 보호
운동으로 인한 자가포식 활성화가 손상되고
손상된 미토콘드리아가 축적되었다는 연구 결과(Grumati 등. 2011)는
운동으로 인한 자가포식이 기능 장애를 일으키는
세포 소기관을 제거하는 역할을 한다는 것을 시사합니다.
이에 따라 골격근 단백질 탄화(산화적 손상의 마커로 사용)는 LC3-II의 증가와 동시에 단일 운동 시합에서 회복 기간에 감소하는 것으로 나타났습니다(Halling et al. 2016).
이는
운동으로 인한
자가포식이
산화 단백질을 제거하여
기능 장애 단백질의 축적을 방지하는 역할을 한다는 것을 나타냅니다.
또한, 근육 특이적 ATG7 KO 마우스는 편심 수축 후 WT 마우스보다 골격근에서 더 높은 ROS 축적과 미토콘드리아 막 탈분극을 보이는 것으로 나타났습니다(Lo et al. 2014).
이는 활성 산소 종의 수준이
자가포식 조절에 중요한 역할을 할 수 있음을 뒷받침합니다(그림 2).
항산화 치료가
운동으로 인한 미토파지의 활성화와
운동이 미토콘드리아 막 전위를 회복시키는 능력을
막지 못한다는 점에 주목해야 합니다(Lo et al. 2014),
이는 추가적인 요인이 관여해야 함을 보여줍니다.
그러나
N-아세틸시스테인(NAC) 항산화제 처리는
생쥐의 기초 미토파지를 감소시켰습니다(Lo 외. 2014; Qi 외. 2014).
이는 항산화 보충제가
운동 수행 능력과 운동 훈련에 대한 대사 적응에 부정적인 영향을 미칠 수 있다는 관찰을 설명하는 메커니즘을 제시할 수 있습니다(Gomez-Cabrera 외. 2008; Ristow 외. 2009; Gliemann 외. 2013; Olesen 외. 2014).
Disease States
The impact of exercise-induced autophagy has also been studied through inhibition of autophagy by treatment with the lysosomal inhibitor chloroquine 5 days/week for 16 weeks resulting in sporadic inclusion body myositis, a condition causing muscle weakness and wasting (Kwon et al. 2015). Resistance exercise training was shown to improve muscle strength in chloroquine-treated rats and to prevent chloroquine-induced increases in ATG6 (Beclin-1) and p62 (Kwon et al. 2015). This suggests that resistance exercise training modulates autophagy in atrophying skeletal muscle with potential protective effects on muscle function. Although exercise-induced autophagy normally seems to exert beneficial effects, uncontrolled enhancement of autophagy may lead to muscle wasting and oxidative stress. Hence, treating rats with the autophagy-stimulating antitumor agent doxorubicin was shown to increase LC3-II/I, oxidative stress, and muscle degradation. However, exercise training prevented doxorubicin-induced apoptosis, and elevated the mRNA and protein content of autophagy genes in skeletal muscle (Smuder et al. 2011). This suggests that, in addition to the bona fide autophagy-dependent beneficial effects of exercise, exercise training can contribute in adjusting the level of autophagy in disease states, which may serve to reduce the disease burden.
질병 상태
운동으로 인한 자가포식의 영향은
리소좀 억제제인 클로로퀸을
주 5일씩 16주 동안 투여하여
자가포식을 억제함으로써
근육 약화 및 소모를 유발하는 산발성 봉입체 근염을 유발하여 연구되었습니다(권 외. 2015).
저항성 운동 훈련은 클로로퀸으로 치료받은 쥐의 근력을 개선하고 클로로퀸으로 인한 ATG6(Beclin-1) 및 p62의 증가를 예방하는 것으로 나타났습니다(권 외. 2015). 이는 저항성 운동 훈련이 위축된 골격근에서 자가포식을 조절하여 근육 기능에 잠재적인 보호 효과를 준다는 것을 시사합니다. 운동으로 유도된 자가포식은 일반적으로 유익한 효과를 발휘하는 것으로 보이지만, 자가포식의 통제되지 않은 강화는 근육 소모와 산화 스트레스로 이어질 수 있습니다. 따라서 쥐에게 자가포식 자극 항암제인 독소루비신을 투여한 결과 LC3-II/I, 산화 스트레스 및 근육 분해가 증가하는 것으로 나타났습니다. 그러나 운동 훈련은 독소루비신에 의한 세포 사멸을 예방하고 골격근에서 자가포식 유전자의 mRNA 및 단백질 함량을 증가시켰습니다(Smuder 외. 2011). 이는 운동의 진정한 자가포식 의존적 유익한 효과 외에도 운동 훈련이 질병 상태의 자가포식 수준을 조절하여 질병 부담을 줄이는 데 기여할 수 있음을 시사합니다.
결론
수많은 연구에서
운동에 반응하여 골격근에서
자가포식이 활성화된다는 증거가 제시되었습니다.
운동에 의한 자가포식 조절에는
자가포식 플럭스의 증가와
중요한 자가포식 유전자의 전사 활성화가 모두 포함되며,
그 결과 자가포식 능력이 향상될 수 있습니다.
또한
운동으로 유도된 자가포식은 운
동의 많은 유익한 효과를 매개하는 데
관여하는 것으로 보입니다.
그러나
자가포식의 급성 활성화에 관여하는 분자 메커니즘과
그에 따른 효과에 대해서는 아직 더 많은 연구가 진행 중입니다. 특히, 인간의 운동 수행 능력과 대사 적응에 대한 자가포식의 잠재적 영향은 명확하지 않습니다.
CONCLUDING REMARKS
Numerous studies have provided evidence that autophagy is activated in skeletal muscle in response to exercise. The exercise-induced regulation of autophagy includes both increased autophagy flux as well as transcriptional activation of important autophagy genes potentially resulting in enhanced autophagy capacity. Furthermore, exercise-induced autophagy seems to be involved in mediating many of the beneficial effects of exercise. However, much more remains to be investigated regarding both the molecular mechanisms involved in acute activation of autophagy and the resulting effects. In particular, the potential impact of autophagy on exercise performance and metabolic adaptations in humans is not clear.
Footnotes
Editors: Juleen R. Zierath, Michael J. Joyner, and John A. Hawley
Additional Perspectives on The Biology of Exercise available at www.perspectivesinmedicine.org
REFERENCES
- Bertaggia E, Coletto L, Sandri M. 2012. Posttranslational modifications control FoxO3 activity during denervation. Am J Physiol Cell Physiol 302: C587–C596. [PubMed] [Google Scholar]
- Biolo G, Maggi SP, Williams BD, Tipton LD, Wolfe RR. 1995. Increased rates of muscle protein turnover and amino acid transport after resistance exercise in humans. Am J Physiol 268: E514–E520. [PubMed] [Google Scholar]
- Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Anderson MJ, Arden KC, Blenis J, Greenberg ME. 1999. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 96: 857–868. [PubMed] [Google Scholar]
- Chesley A, MacDougall JD, Tarnopolsky MA, Atkinson SA, Smith K. 1992. Changes in human muscle protein synthesis after resistance exercise. J Appl Physiol (1985) 73: 1383–1388. [PubMed] [Google Scholar]
- Dickinson JM, Reidy PT, Gundermann DM, Borack MS, Walker DK, D’Lugos AC, Volpi E, Rasmussen BB. 2016. The impact of post exercise essential amino acid ingestion on the ubiquitin proteasome and autophagosomal-lysosomal systems in skeletal muscle of older men. J Appl Physiol (1985) 10.1152/japplphysiol.00632.2016. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Egan DF, Shackelford DB, Mihaylova MM, Gelino S, Kohnz RA, Mair W, Vasquez DS, Joshi A, Gwinn DM, Taylor R, et al. 2011. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 331: 456–461. [PMC free article] [PubMed] [Google Scholar]
- Francaux M, Demeulder B, Naslain D, Fortin R, Lutz O, Caty G, Deldicque L. 2016. Aging reduces the activation of the mTORC1 pathway after resistance exercise and protein intake in human skeletal muscle: Potential role of REDD1 and impaired anabolic sensitivity. Nutrients. 8: E47. [PMC free article] [PubMed] [Google Scholar]
- Fritzen AM, Madsen AB, Kleinert M, Treebak JT, Lundsgaard AM, Jensen TE, Richter EA, Wojtaszewski J, Kiens B, Frosig C. 2015. Regulation of autophagy in human skeletal muscle—Effects of exercise, exercise training and insulin stimulation. J Physiol 594: 745–761. [PMC free article] [PubMed] [Google Scholar]
- Fry CS, Drummond MJ, Glynn EL, Dickinson JM, Gundermann DM, Timmerman KL, Walker DK, Volpi E, Rasmussen BB. 2013. Skeletal muscle autophagy and protein breakdown following resistance exercise are similar in younger and older adults. J Gerontol A Biol Sci Med Sci 68: 599–607. [PMC free article] [PubMed] [Google Scholar]
- Gliemann L, Schmidt JF, Olesen J, Bienso RS, Peronard SL, Grandjean SU, Mortensen SP, Nyberg M, Bangsbo J, Pilegaard, et al. 2013. Resveratrol blunts the positive effects of exercise training on cardiovascular health in aged men. J Physiol 591: 5047–5059. [PMC free article] [PubMed] [Google Scholar]
- Glynn EL, Fry CS, Drummond MJ, Dreyer HC, Dhanani S, Volpi E, Rasmussen BB. 2010. Muscle protein breakdown has a minor role in the protein anabolic response to essential amino acid and carbohydrate intake following resistance exercise. Am J Physiol Regul Integr Comp Physiol 299: R533–R540. [PMC free article] [PubMed] [Google Scholar]
- Gollnick PD, Armstrong RB, Saubert CW, Piehl K, Saltin B. 1972. Enzyme activity and fiber composition in skeletal muscle of untrained and trained men. J Appl Physiol 33: 312–319. [PubMed] [Google Scholar]
- Gomez-Cabrera MC, Domenech E, Romagnoli M, Arduini A, Borras C, Pallardo FV, Sastre J, Vina J. 2008. Oral administration of vitamin C decreases muscle mitochondrial biogenesis and hampers training-induced adaptations in endurance performance. Am J Clin Nutr 87: 142–149. [PubMed] [Google Scholar]
- Grumati P, Coletto L, Sabatelli P, Cescon M, Angelin A, Bertaggia E, Blaauw B, Urciuolo A, Tiepolo T, Merlini L, et al. 2010. Autophagy is defective in collagen VI muscular dystrophies, and its reactivation rescues myofiber degeneration. Nat Med 16: 1313–1320. [PubMed] [Google Scholar]
- Grumati P, Coletto L, Schiavinato A, Castagnaro S, Bertaggia E, Sandri M, Bonaldo P. 2011. Physical exercise stimulates autophagy in normal skeletal muscles but is detrimental for collagen VI-deficient muscles. Autophagy 7: 1415–1423. [PMC free article] [PubMed] [Google Scholar]
- Halling JF, Ringholm S, Nielsen MM, Overby P, Pilegaard H. 2016. PGC-1α promotes exercise-induced autophagy in mouse skeletal muscle. Physiol Rep 4: e12698. [PMC free article] [PubMed] [Google Scholar]
- He C, Bassik MC, Moresi V, Sun K, Wei Y, Zou Z, An Z, Loh J, Fisher J, Sun Q, et al. 2012. Exercise-induced BCL2-regulated autophagy is required for muscle glucose homeostasis. Nature 481: 511–515. [PMC free article] [PubMed] [Google Scholar]