- nature
- molecular psychiatry
- expert review
- article
The molecular pathology of schizophrenia: an overview of existing knowledge and new directions for future research
- Expert Review
- Open access
- Published: 06 March 2023
The molecular pathology of schizophrenia: an overview of existing knowledge and new directions for future research
Molecular Psychiatry volume 28, pages1868–1889 (2023)Cite this article
Abstract
Despite enormous efforts employing various approaches, the molecular pathology in the schizophrenia brain remains elusive. On the other hand, the knowledge of the association between the disease risk and changes in the DNA sequences, in other words, our understanding of the genetic pathology of schizophrenia, has dramatically improved over the past two decades. As the consequence, now we can explain more than 20% of the liability to schizophrenia by considering all analyzable common genetic variants including those with weak or no statistically significant association. Also, a large-scale exome sequencing study identified single genes whose rare mutations substantially increase the risk for schizophrenia, of which six genes (SETD1A, CUL1, XPO7, GRIA3, GRIN2A, and RB1CC1) showed odds ratios larger than ten. Based on these findings together with the preceding discovery of copy number variants (CNVs) with similarly large effect sizes, multiple disease models with high etiological validity have been generated and analyzed. Studies of the brains of these models, as well as transcriptomic and epigenomic analyses of patient postmortem tissues, have provided new insights into the molecular pathology of schizophrenia. In this review, we overview the current knowledge acquired from these studies, their limitations, and directions for future research that may redefine schizophrenia based on biological alterations in the responsible organ rather than operationalized criteria.
다양한 접근법을 활용한 막대한 노력에도 불구하고,
정신분열증 뇌의 분자 병리학은 여전히 불분명하다.
반면, 질병 위험과 DNA 서열 변화 간의 연관성에 대한 지식,
즉 정신분열증의 유전적 병리학에 대한 이해는
지난 20년간 획기적으로 발전했다.
그 결과,
통계적으로 유의미한 연관성이 약하거나 없는 변이까지 포함해
분석 가능한 모든 일반적 유전적 변이를 고려함으로써
현재 정신분열증 발병 가능성의 20% 이상을 설명할 수 있게 되었다.
또한 대규모 엑솜 시퀀싱 연구를 통해
정신분열증 위험을 크게 증가시키는 희귀 돌연변이를 가진 단일 유전자들을 확인했으며,
그중 6개 유전자(SETD1A, CUL1, XPO7, GRIA3, GRIN2A, RB1CC1)는
10보다 큰 오즈비(odds ratio)를 나타냈다.
이러한 연구 결과와 유사한
큰 효과 크기를 가진 복제 수 변이(CNV)에 대한 선행 연구를 바탕으로,
높은 병인학적 타당성을 지닌 여러 질병 모델이 생성 및 분석되었습니다.
이러한 모델의 뇌 연구와 환자 사후 조직의 전사체 및 후성유전체 분석은 정신분열증의 분자 병리학에 대한 새로운 통찰력을 제공했습니다. 본 리뷰에서는 이러한 연구를 통해 얻은 현재의 지식, 그 한계점, 그리고 운영 기준이 아닌 관련 기관의 생물학적 변화에 기반하여 정신분열증을 재정의할 수 있는 향후 연구 방향을 개괄합니다.
Similar content being viewed by others

Rare coding variants in ten genes confer substantial risk for schizophrenia
Article 08 April 2022
Article 08 March 2021
The genetic architecture of schizophrenia: review of large-scale genetic studies
Article 12 July 2022
Introduction
The term pathology is defined as the study of the essential nature of disease [1]. In the pathology of physical diseases, abnormal changes in responsible organs or systems, such as invasion of malignant cells in cancers, and loss of nigrostriatal dopaminergic neurons in Parkinson’s disease, are examined typically by microscopes. On the other hand, a field of study of pathological cognition and behaviors often observed in patients with psychiatric disorders, psychopathology, does not analyze the brain, the organ presumably responsible for these disorders. This may not be unreasonable given that psychiatry is etymologically a study of the psyche, a Greek word ψυχή whose derived meaning includes invisible spirit or soul. Nevertheless, It has long been thought that mental illness is fundamentally a disease of the brain, as classically advocated by Wilhelm Griesinger, Emil Kraepelin, and others [2, 3]. Based on this concept, the brain pathology of schizophrenia, a common psychiatric disorder with a lifetime prevalence of ~1% [4], has been investigated in numerous studies. However, despite many efforts, no definite pathological changes, like senile plaques and neurofibrillary tangles in Alzheimer’s disease, were identified in their postmortem brains. Meanwhile, recent rapid advances in molecular biology and engineering have prompted the development of methods to analyze molecules such as nucleotides and proteins more comprehensively, more sensitively, with a higher cellular and spatial resolution, and quantitatively. Besides, multiple collaborative research frameworks have been established to ensure sample sizes sufficient to solve the critical problem of multiple testing and relevant statistical power. This has especially been true in comprehensive studies of variants in DNA, which encodes fundamental biological information for all organs, including the brain and can be analyzed by using easily accessible peripheral tissue samples, resulting in an accumulation of statistically robust findings. Given these, while we should refrain from being overly optimistic, perhaps now might be the time to bring together these technologies and resources to elucidate the molecular pathology of schizophrenia. In this review, we overview the findings from research aiming to decipher the molecular pathology of schizophrenia, emphasizing large-scale omics studies with substantial statistical power and analyses of etiologically valid disease models, and summarize the current achievements. Following that, the challenges and obstacles to be overcome and the future research directions will be discussed, along with an introduction of preliminary results from studies utilizing cutting-edge technologies that will surely facilitate the elucidation of the fundamental pathology of schizophrenia.
서론
병리학(pathology)이란
질병의 본질적 특성을 연구하는 학문으로 정의된다[1].
신체 질환의 병리학에서는
암의 악성 세포 침습이나 파킨슨병의 흑질-선조체 도파민 신경세포 소실과 같이
해당 장기나 시스템의 비정상적 변화를 주로 현미경으로 관찰한다.
반면,
정신질환 환자에서
흔히 관찰되는 병리적 인지 및 행동을 연구하는 분야인 정신병리학은
이러한 장애의 원인으로 추정되는 기관인 뇌를 분석하지 않는다.
정신의학이 어원적으로 '정신(psyche)'의 연구라는 점을 고려하면 이는 불합리하지 않을 수 있다. '정신(psyche)'은 그리스어 ψυχή에서 유래한 용어로, 그 파생된 의미에는 보이지 않는 영혼이나 정신이 포함된다. 그럼에도 불구하고, 정신 질환은 근본적으로 뇌의 질환이라는 관점이 오랫동안 유지되어 왔으며, 빌헬름 그라이징어(Wilhelm Griesinger), 에밀 크라펠린(Emil Kraepelin) 등이 이를 고전적으로 주창했다[2, 3].
이러한 개념에 기반하여,
평생 유병률이 약 1%인 흔한 정신 질환인 조현병(정신분열증)의 뇌 병리학은
수많은 연구에서 조사되어 왔다[4].
그러나 수많은 노력에도 불구하고,
알츠하이머병의 노인성 플라크나 신경섬유 엉킴과 같은
명확한 병리학적 변화는 사후 뇌에서 확인되지 않았다.
한편, 최근 분자생물학과 공학의 급속한 발전으로
뉴클레오티드나 단백질 같은 분자를 더 포괄적이고 민감하게,
세포 및 공간 해상도를 높여 정량적으로 분석하는 방법들이 개발되었다.
또한, 다중 검정과 관련 통계적 검정력이라는
중대한 문제를 해결하기에 충분한 표본 크기를 확보하기 위한
다중 협력 연구 체계가 구축되었다.
이는 특히 뇌를 포함한 모든 장기의 기초 생물학적 정보를 암호화하는
DNA 변이 종합 연구에서 두드러지며,
쉽게 접근 가능한 말초 조직 샘플을 활용하여 분석할 수 있어
통계적으로 견고한 연구 결과의 축적이 가능해졌다.
이러한 점을 고려할 때 지나친 낙관론은 자제해야 하지만,
아마도 지금이 이러한 기술과 자원을 통합하여
조현병의 분자 병리를 규명할 시기일 수 있습니다.
본 리뷰에서는
통계적 검정력이 충분한 대규모 오믹스 연구와 병인학적으로 타당한 질병 모델 분석을 중심으로
조현병 분자 병리 해독을 목표로 한 연구 결과를 개관하고
현재의 성과를 요약합니다.
이어서 극복해야 할 과제와 장애물, 향후 연구 방향을 논의하고,
정신분열증의 근본적 병리 규명에 확실히 기여할
첨단 기술을 활용한 연구의 예비 결과를 소개한다.
The genetic pathology of schizophrenia
Almost unquestionably, the brain is the organ primarily responsible for the pathogenesis of schizophrenia. However, the human brain is covered by thick cranial bones, and therefore it is almost impossible to access and analyze living human brain tissues at the molecular level in a non-invasive manner, nor is it easy to collect postmortem brains on a large scale. On the other hand, the sequences of DNA, the molecule encoding fundamental biological information of all tissues, can be analyzed without accessing the brain, since its sequences are in principle identical in every cell and invariant throughout life, with few exceptions. Based on this relative ease in obtaining samples as well as the high heritability of schizophrenia, reported to be 60–80% in epidemiological studies [5,6,7,8], large-scale genetic studies, analyzing samples from more than 100,000 individuals these days, have been conducted. Reflecting this large number, among various research on the molecular pathology of schizophrenia, statistically robust findings have been particularly accumulated from human genetics studies analyzing variations of the sequences of DNA molecules. For this reason, we begin with an overview of our knowledge of the genetic pathology of schizophrenia.
정신분열증의 유전적 병리학
의심의 여지없이
뇌는 정신분열증 발병의 주요 책임 기관이다.
그러나
인간의 뇌는 두꺼운 두개골 뼈로 덮여 있어,
비침습적 방식으로 살아있는 인간 뇌 조직을 분자 수준에서 접근 및 분석하는 것은 거의 불가능하며,
사후 뇌를 대규모로 수집하는 것도 쉽지 않다.
반면,
모든 조직의 근본적인 생물학적 정보를 암호화하는 분자인
DNA의 염기서열은 뇌에 접근하지 않고도 분석할 수 있습니다.
왜냐하면
그 염기서열은 원칙적으로 모든 세포에서 동일하며,
극히 일부 예외를 제외하고는 일생 동안 불변하기 때문입니다.
이러한 상대적인 시료 확보 용이성과 더불어,
역학 연구에서 60~80%로 보고된 정신분열증의 높은 유전율[5,6, 7,8]에 따르면,
최근에는 10만 명 이상의 개인 샘플을 분석하는 대규모 유전 연구가 수행되고 있다.
이러한 대규모 연구를 반영하여, 정신분열증의 분자 병리학에 관한 다양한 연구 중에서도 DNA 분자 서열의 변이를 분석하는 인간 유전학 연구에서 통계적으로 견고한 결과가 특히 많이 축적되었다. 이러한 이유로, 우리는 정신분열증의 유전적 병리학에 대한 우리의 지식을 개관하는 것으로 시작한다.
Robust findings from large-scale analyses of common and rare variants
Although schizophrenia is a highly heritable disorder, no single variant explaining a large portion of the overall heritability has been identified. Therefore, comprehensive studies of various allele frequencies and effect sizes of genetic variants contributing to its risk are warranted. In a simplified view, there are two major types of genetic studies on different frequencies and effect sizes of variants, that is, genome-wide association studies (GWAS) of common single nucleotide polymorphisms (SNPs) with small effect sizes (typically odds ratio [OR] < 1.2) and sequencing studies of rare variants potentially with large effect sizes (sometimes OR > 10). The scales of these two types of studies have consistently grown [9,10,11,12,13,14,15,16,17,18,19], and the results of the two largest studies to date, each looking into common SNPs [20] or rare variants [21], have recently been published after peer review.
일반적 및 희귀 변이체에 대한 대규모 분석의 확고한 결과
정신분열증은 높은 유전성을 지닌 질환이지만,
전체 유전성의 상당 부분을 설명하는 단일 변이체는 확인되지 않았다.
60-80% 유전율
따라서
위험에 기여하는 유전적 변이체의 다양한 대립유전자 빈도와 효과 크기에 대한
포괄적 연구가 필요하다.
간단히 말해,
변이의 빈도와 효과 크기에 대한 유전 연구는
크게 두 가지 유형으로 나뉜다.
즉, 효과 크기가 작은(일반적으로 오즈비[OR] < 1.2)
흔한 단일염기다형성(SNP)에 대한 전장유전체연관분석(GWAS)과
효과 크기가 클 가능성이 있는(때로는 OR > 10) 희귀 변이에 대한 염기서열 분석 연구이다.
genome-wide association studies (GWAS) of
common single nucleotide polymorphisms (SNPs)
| 'Schizophrenia'라는 용어는 그리스어에서 유래. 'schizo-'는 '분열되다' 또는 '나뉘다'라는 뜻이고, 'phren'은 '정신' 또는 '마음'을 의미. '조현병'이라는 병명은 '현악기의 줄을 고르다'라는 뜻의 '조현(調絃)'. 이는 조현병 환자의 상태가 마치 조율되지 않은 현악기처럼 혼란스러운 상태를 의미 GWAS는 우리 몸의 모든 유전 정보를 조사해서, 어떤 유전자 변이들이 특정 질병과 관련이 있는지 찾는 것 . 마치 형사들이 사건 현장에서 단서를 찾는 것처럼. 이때 주로 살펴보는 단서가 커먼 싱글 뉴클레오타이드 폴리모피즘, common single nucleotide polymorphism 줄여서 SNP라고 불림. 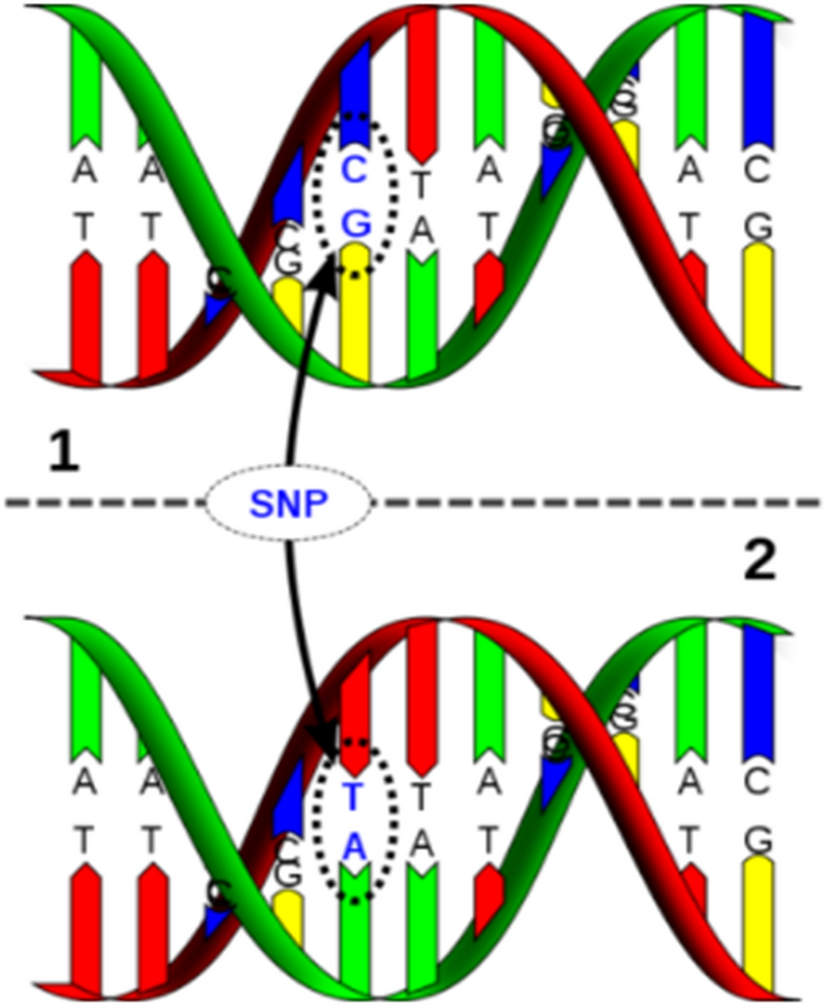 Polymorphism'은 한국어로 '다형성' 이는 하나의 유전자 정보가 여러 형태로 나타날 수 있는 것을 의미. 예를 들어, 머리카락 색깔이 검은색, 갈색 등으로 다양하게 나타나는 것처럼 유전체 전반의 단일 염기 변이 정보를 이용한 대규모 연관 분석인 GWAS는, 질병과 관련된 유전적 변이를 발굴하는 데 핵심적인 역할. 특히, 유전체 전반에 걸쳐 빈번하게 나타나는 SNP는, 질병 감수성과 연관된 유전적 변이 식별의 중요한 마커로 활용. GWAS는 통계적 기법을 통해 SNP와 표현형 사이의 연관성을 탐색하며, 이를 통해 질병 관련 유전자를 찾아낼 수 있음. 하지만 이러한 연구는 대규모 데이터를 필요로 하며, 결과 해석 시에는 인종이나 민족 간의 차이 등을 고려해야 |
In the newest GWAS of common SNPs by the Psychiatric Genomics Consortium (PGC) analyzing 76,755 schizophrenia cases and 243,649 control individuals [20], 287 distinct loci with genome-wide significant association (P < 5 × 10−8) were identified. The SNP with the largest effect size was the rs140365013 variant on chromosome 6 near the major histocompatibility complex region, with an OR of 1.23, confirming that individual SNPs do not greatly increase the disease risk. On the other hand, the proportion of the variance in schizophrenia liability explained by all measured SNPs, including numerous variants that did not show statistically significant association with schizophrenia, was reported to be 24%. This proportion is much larger than that calculated solely from loci associated with genome-wide significance. Therefore, a significant part of the heritability is attributable to common SNPs that individually show weak associations and effects, and it can be said that schizophrenia is a highly polygenic disorder. Since each of the 287 associated loci often contains multiple genes (specifically, of the 287 loci, 206 and 108 contain ≥2 and ≥5 genes, respectively), it is essential to identify functionally important causal genes and variants in order to understand the molecular pathology from the genetic pathology. To this end, gene prioritization was performed in this GWAS by PGC using various approaches such as fine-mapping of credible sets of causal SNPs by an adaptation of a Bayesian inference algorithm [22] and Mendelian randomization to identify SNPs whose causal effects are likely to be mediated through regulation of gene expression [23]. As a result, a total of 120 prioritized genes were nominated, of which 70 and 55 received support from the fine-mapping and Mendelian randomization analyses, respectively. There were five genes supported by both lines of evidence (CUL9, FURIN, LINC00320, SNAP91, and ZNF823). In addition, two other prioritized genes (GRIN2A, and SP4) are supported by statistical evidence significant after conservative multiple testing correction in the rare variant study described below (Table 1a, b).
이러한 두 유형의 연구 규모는 지속적으로 확대되어 왔으며[9,10,11,12,13,14,15,16,17,18,19],
현재까지 수행된 가장 대규모 연구 두 건(각각 흔한 SNP[20]
또는 희귀 변이[21]를 대상으로 함)의 결과가 최근 동료 평가를 거쳐 발표되었다.
https://pmc.ncbi.nlm.nih.gov/articles/PMC9392466/

https://pmc.ncbi.nlm.nih.gov/articles/PMC9805802/


정신병리 유전체학 컨소시엄(PGC)이 수행한
최신 일반적 SNP GWAS 연구[20]에서는
76,755명의 정신분열증 환자와 243,649명의 대조군을 분석하여
유전체 수준 유의미한 연관성(P値 < 5×10−8)을 보이는
287개의 고유한 유전좌위를 확인했습니다.
가장 큰 효과 크기를 보인 SNP는
주요 조직적합성 복합체(MHC) 영역 근처 6번 염색체상의 rs140365013 변이체로,
오즈비(OR) 1.23을 나타내며
개별 SNP가 질병 위험을 크게 증가시키지 않음을 확인했습니다.
반면, 정신분열증 발병 가능성의 변동성 중
통계적으로 유의미한 연관성을 보이지 않은 수많은 변이체를 포함한
모든 측정된 SNP가 설명하는 비율은 24%로 보고되었다.
이 비율은
전장 유전체 유의성(GWAS)과 연관된 유전자좌만으로 계산된 것보다 훨씬 크다.
따라서
유전율의 상당 부분은 개별적으로 약한 연관성과 효과를 보이는 흔한 단일핵산다형성(SNP)에 기인하며,
정신분열증은 고도로 다유전적 장애라고 할 수 있다.
287개 연관 유전자좌 각각은
종종 다수의 유전자를 포함하므로(구체적으로 287개 유전자좌 중 206개와 108개가 각각 ≥2개 및 ≥5개 유전자를 포함함),
유전병리학으로부터 분자병리학을 이해하기 위해서는
기능적으로 중요한 원인 유전자와 변이를 식별하는 것이 필수적이다.
이를 위해 본 GWAS에서는
PGC를 통해 다양한 접근법(예: 베이지안 추론 알고리즘의 변형 적용을 통한 신뢰할 수 있는 인과적 SNP 집합의 정밀 매핑[22], 유전자 발현 조절을 통해 인과적 효과가 매개될 가능성이 높은 SNP를 식별하기 위한 멘델식 무작위화[23])을 활용하여 유전자 우선순위화를 수행했습니다. 그 결과 총 120개의 우선순위 유전자가 선정되었으며, 이 중 70개는 정밀 매핑 분석에서, 55개는 멘델식 무작위화 분석에서 각각 지지받았다. 두 가지 증거 모두에서 지지받은 유전자는 다섯 개였다(CUL9, FURIN, LINC00320, SNAP91, ZNF823). 추가로, 다른 두 우선순위 유전자(GRIN2A 및 SP4)는 아래에서 설명하는 희귀 변이 연구에서 보수적인 다중 검정 보정 후에도 통계적으로 유의미한 증거를 받았습니다(표 1a, b).
Table 1 Prioritized schizophrenia genes from common and rare variant studies.
In a companion study of rare variants by the Schizophrenia Exome Meta-Analysis (SCHEMA) Consortium, rare single nucleotide variants (SNVs, any frequency of single nucleotide substitutions including both SNPs and rare variants) and short insertion/deletions (indels) in protein-coding regions were systematically analyzed by using exome (all protein-coding exonic regions) sequencing data of 24,248 schizophrenia cases, 97,322 controls, and 3402 trios consisting of schizophrenia probands and their unaffected parents. By evaluating the burden of rare loss-of-function (LOF: nonsense, splice site, and frameshift indel) variants (also known as protein-truncating variants: PTVs) and damaging missense variants (defined by the missense badness, PolyPhen-2, and constraint [MPC] score), including those arisen de novo in the probands, this study identified ten genes (SETD1A, CUL1, XPO7, TRIO, CACNA1G, SP4, GRIA3, GRIN2A, HERC1, and RB1CC1; Table 1b) surpassing the exome-wide significance threshold (defined as 2.14 × 10−6 based on the number of protein-coding genes analyzed) and 22 additional genes at a false discovery rate (FDR) < 0.05. Considering their effect sizes, six (SETD1A, CUL1, XPO7, GRIA3, GRIN2A, and RB1CC1) out of the ten exome-wide significant genes were enriched for rare PTV and damaging missense (MPC > 3) variants with OR > 10, indicating that rare deleterious variants of these genes confer the schizophrenia risk with large effects as well as robust statistical significance. Another notable thing is that five (SETD1A, TRIO, CACNA1G, GRIA3, and GRIN2A) of the ten genes are also implicated in other neurodevelopmental disorders, such as intellectual disability and epilepsy, as registered on the Online Mendelian Inheritance in Man (OMIM) database [24]. Therefore, on the one hand, it is possible that these cases with deleterious variants in known neurodevelopmental disorder genes might be individuals who should have been molecularly diagnosed as patients of highly heritable neurodevelopmental diseases, while on the other hand, another possibility is that the carriers of these variants did not exhibit developmental and physical symptoms sufficient to be diagnosed with a neurodevelopmental disease due to some modifying factors and were operationally diagnosed with schizophrenia. If the former is true, this suggests that genetic testing may detect overlooked patients with highly heritable neurodevelopmental diseases and provide clues for better intervention. If the latter is the case, it implies that some modifying factors influence the severity of the symptoms. Indeed, there is accumulating evidence supporting the existence of modifying factors, such as common and rare variants other than the diagnostic mutation, in patients with neurodevelopmental diseases [25,26,27,28]. A more detailed analysis of such modifying factors may pave the way toward the development of new treatment and prevention strategies.
Besides common SNPs analyzed in GWAS and rare SNVs/indels analyzed in exome sequencing studies, another important class of genetic variation of which several are known to be robustly associated with schizophrenia is copy number variants (CNVs) [29,30,31,32]. Among important works on CNVs, a large genome-wide study by the CNV Working Groups of PGC analyzing 21,094 schizophrenia cases and 20,227 controls [31] identified copy number losses at six loci (1q21.1, 2p16.3 involving NRXN1, 3q29, 15q13.3, distal 16p11.2, and 22q11.2) and gains at two loci (7q11.23 and proximal 16p11.2) that are significantly (P < 1.33 × 10−4 for losses and 4.33 × 10−5 for gains) associated with schizophrenia after multiple testing corrections (Table 1c). All of these are rare in controls and contribute to the risk for schizophrenia with ORs ranging from 3.8 to infinity. Also, like the known neurodevelopmental disorder genes identified in the SCHEMA study described above, all, or nearly all of these schizophrenia-associated CNVs are phenotypically pleiotropic and often more strongly associated with disorders other than schizophrenia, such as ASD and intellectual disability. Therefore, complex phenotype-genotype relationships should be considered when predicting disease risks from the information on CNVs and generating and analyzing CNV-based animal and cellular models.
Aggregating the findings from studies of common SNPs and rare SNVs/indels and CNVs, we can now explain a substantial part of the schizophrenia heritability (mainly by common SNPs) and have produced a list consisting of six genes and six CNVs associated with schizophrenia with observed ORs larger than ten. Also, there is a convergence of the results of studies of common and rare variants. At the level of individual genes, GRIN2A and SP4 are included in the list of 120 genes prioritized in the PGC GWAS and showed exome-wide significant enrichment of rare deleterious variants in the SCHEMA study, as described above. At the level of overall enrichment patterns, the sets of genes implicated in the SCHEMA study and studies of rare coding variants in other neurodevelopmental disorders (e.g., ASD and intellectual disability) are shown to be significantly enriched for common variant associations in the PGC GWAS.
We provide a compiled list of genes and CNVs identified through large-scale studies, which underlie the genetic pathology of schizophrenia, in Table 1, together with the landscape of their population frequencies and effect sizes in Fig. 1. Meanwhile, as shown in Table 1 and Fig. 1, it should be noted that there are wide ranges of confidence intervals for ORs for rare risk variants. Also, there is a possibility of the so-called winner’s curse [33] in genetic studies. To more accurately estimate their effect sizes and the robustness of the association, further larger studies are always warranted.
Fig. 1: Allelic spectrum of schizophrenia-associated variants.
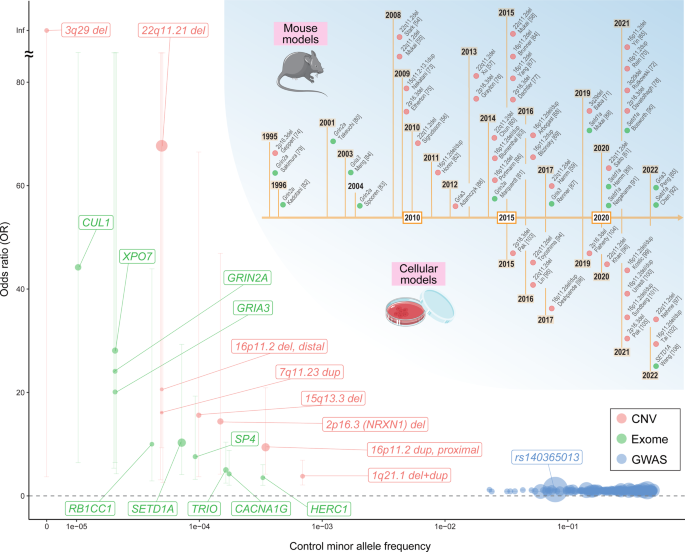
A plot of minor allele frequencies in controls (x-axis) and ORs (y-axis) for schizophrenia-associated genes/variants with robust statistical evidence, that is, the index SNPs of 287 genome-wide significant loci identified by the PGC GWAS (blue, ref. [20]), ten exome-wide significant genes in the SCHEMA exome sequencing study (green, ref. [21]) and the study-wide significant eight CNVs in the PGC CNV Working Groups study (red, ref. [31]). Genes and variants with OR > 1.2 were labeled. The sizes of points are proportional to the −log10 P values for the association. The error bars indicate 95% confidence intervals of the ORs. The upper right chronology summarizes the representative studies of etiologically valid mouse and cellular models of schizophrenia shown in Tables 2 and 3.
Functional convergence of the findings in genetic studies
As described above, genetic studies have identified a number of statistically robust new genes and variants; however, these in themselves only show “association” with the phenotype. Thus, further analysis is needed to translate genetic findings into the knowledge of the molecular brain pathology of schizophrenia. This can be facilitated, for example, by testing if specific molecular and biological pathways are enriched among the associated genes.
More specifically, gene ontology (GO) enrichment analyses were performed in both the PGC GWAS and the SCHEMA study by utilizing MAGMA [34] and DNENRICH [16] that appropriately control for confounding factors such as gene sizes and linkage disequilibrium. We, therefore, examined how the results of these two studies converge at the levels of molecular function. In the PGC GWAS and the SCHEMA study, the results of GO enrichment analysis for 7315 and 1491 terms are available, respectively. Of these, 1431 GO terms were commonly analyzed and 111 of them showed significant enrichment at uncorrected P < 0.05 in both of these two studies (Fig. 2a). The statistical significance (−log10 P value) for each term in the PGC GWAS and the SCHEMA study showed a highly significant correlation (Fig. 2b, Pearson’s correlation coefficient = 0.39, P = 6.67 × 10−54). This result indicates that there is a convergence of molecular and biological pathways implicated by common SNPs and rare deleterious variants. Specifically, four GO terms, all related to voltage-gated channels and synaptic transmissions, were significant after Bonferroni correction in both the PGC GWAS and the SCHEMA study (Fig. 2b). When the 32 GO terms with Bonferroni-corrected P < 0.05 in either dataset (respectively 25 and 11 terms in the PGC GWAS and the SCHEMA study, of which four are common as above) were visualized as networks by connecting them based on the similarity of the contained genes (Fig. 2c), we observed the formation of three clusters, each related to channel or transporter activities; neuronal components (synapse, axon, and dendrite); chromatin or histone organization. Though the cluster of chromatin or histone organization was only supported by SCHEMA, the former two clusters (“channel or transporter activities” and “neuronal components”) showed convergent enrichment in both studies. Taken together, these can be considered molecular pathways whose involvement in schizophrenia pathogenesis is supported by both common and rare variant studies.
Fig. 2: Functional convergence of the findings from studies of common and rare variants.
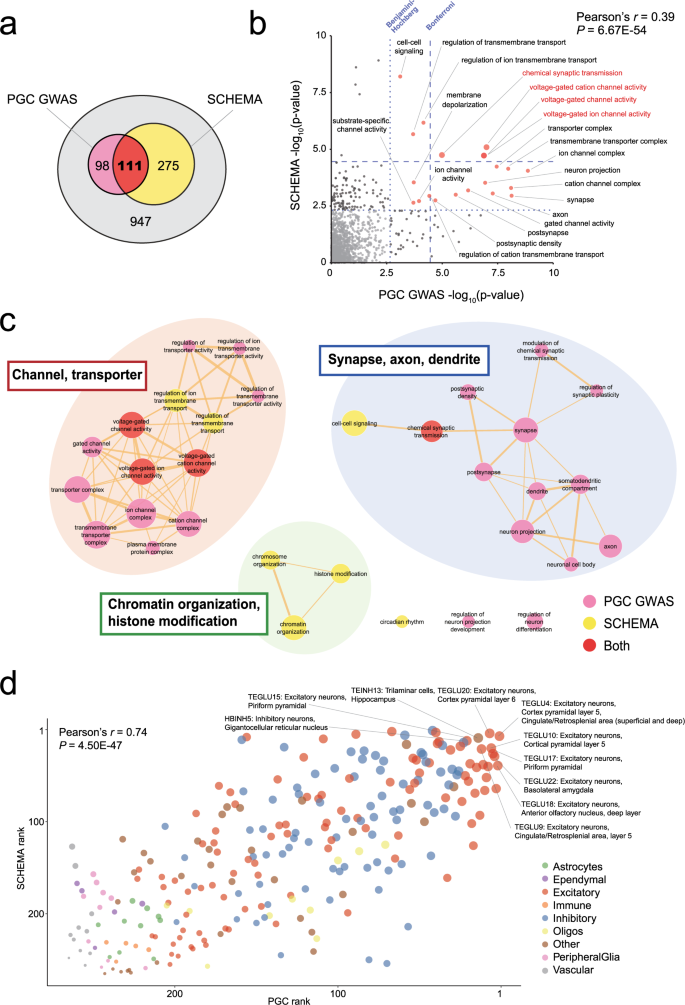
a A Venn diagram showing the overlap of GO terms that showed enrichment (uncorrected P < 0.05) in the PGC GWAS and the SCHEMA exome sequencing study. b A plot of the enrichment of 1431 GO terms commonly analyzed in the PGC GWAS and the SCHEMA study. The x- and y-axes indicate −log10 uncorrected P values in the PGC GWAS and the SCHEMA study, respectively. The blue dotted lines indicate Bonferroni- or Benjamini-Hochberg-corrected significance thresholds. GO terms with Benjamini-Hochberg-corrected P < 0.05 in both studies are indicated by labeled red dots, of which four terms with Bonferroni-corrected P < 0.05 in both studies (voltage-gated cation channel activity, voltage-gated channel activity, voltage-gated ion channel activity, and chemical synaptic transmission) are labeled in red. The correlation between the two studies (Pearson’s r = 0.39. P = 6.67 × 10−54) is shown in the upper right. c Network visualization of the GO terms enriched in the PGC GWAS and the SCHEMA study. Significant GO terms after the Bonferroni correction in either or both studies are displayed. Nodes of significant GO terms are color-coded as follows: pink, the PGC GWAS; yellow, the SCHEMA study; red, both studies. The sizes of nodes are proportional to –log10 meta-analysis P values calculated by combining uncorrected P values in the PGC GWAS and the SCHEMA study using Fisher’s method. Nodes are connected when the similarity score ≥0.4, and the edge width is proportional to the similarity score. We observed three major clusters of GO terms, each related to channel or transporter activities; neuronal components (synapse, axon, and dendrite); chromatin or histone organization. d A plot of the results of cell type enrichment analyses in the PGC GWAS and the SCHEMA study. In both studies, the data of 265 cell types defined in the Zeisel et al. study [36] were used. The x- and y-axes indicate enrichment ranks in the PGC GWAS and the SCHEMA study, respectively. The top ten cell types for which the sum of the ranks in the PGC GWAS and the SCHEMA study is the smallest are labeled with the cell cluster ID, major cell type, and likely location. Each circle indicates each cell type, which is color-coded as indicated in the lower right. The sizes of the circles are proportional to the enrichment ranks. The correlation between the two studies (Pearson’s r = 0.74. P = 4.50 × 10−47) is shown in the upper left.
In addition to molecular pathways, both the PGC GWAS and the SCHEMA study performed cell-type enrichment analysis utilizing data from single-cell RNA sequencing, which is rapidly developing in recent years. For this analysis, both studies employed the statistical method described by Skene et al. [35] and used the data of 265 cell types defined in a single-cell RNA sequencing study of the mouse nervous system by Zeisel et al. [36]. When we plotted the enrichment ranks for the 265 cell types in the PGC GWAS and the SCHEMA study, which are detailed in Supplementary Fig. 3 and Supplementary Table 10 of the corresponding papers, respectively, we again found that there is a highly significant correlation (Fig. 2d, Pearson’s correlation coefficient = 0.74, P = 4.50 × 10−47) (note that we used the enrichment ranks because exact statistics were not available in Supplementary Fig. 3 of the PGC GWAS). Overall, enrichment was particularly strong in excitatory neurons, followed by inhibitory neurons, and less pronounced in other cell types such as glial, vascular, and immune cells. Among the top ten highest-ranked cell types, eight were excitatory neurons, of which five, including the top two (“TEGLU4: Excitatory neurons, Cortex pyramidal layer 5, Cingulate/Retrosplenial area (superficial and deep)” [1st in PGC GWAS and 7th in SCHEMA) and “TEGLU20: Excitatory neurons, Cortex pyramidal layer 6” [7th in PGC GWAS and 4th in SCHEMA]), were annotated as deep layer excitatory neurons. These results represent another form of functional convergence of the findings from genetic studies of common and rare variants, and provide insight into the cell types likely playing a critical role in the molecular pathology of schizophrenia.
Other types of variants potentially explain still-missing heritability
Although large-scale genetic studies and refinements in statistical methods have elucidated a substantial part of the genetic architecture of schizophrenia, there remains a large gap between the overall heritability reported in epidemiological studies (60–80%) [5,6,7,8] and that explained by common SNPs (24% in ref. [20]) or rare gene-disruptive rare SNVs, indels, and CNVs (<10% according to refs. [37, 38]). Provisional evidence suggests other types of variants that are not captured by GWAS or exome sequencing are likely to be involved, including rare non-coding variants [39] and tandem repeat variants [40] identified through whole genome sequencing. Also, there is preliminary evidence suggesting the role of postzygotic somatic variants [31, 41,42,43,44], while these are not transmitted and do not contribute to heritability. More detailed information on results from these pioneering but preliminary studies can be found in Supplementary Note. To more accurately estimate the contributions from these under-studied types of variants, further large-scale studies are mandatory.
Transcriptomic and epigenomic pathology in schizophrenia
GO enrichment analysis of rare coding variants identified regulation of transcription and chromatin organization as one of the molecular pathways implicated in schizophrenia (Fig. 2c). Also, multiple single genes with large effect sizes, such as SP4 encoding the transcription factor Sp4 and SETD1A encoding a histone methyltransferase, are core components of transcriptional and epigenetic regulation. In line with these findings, studies of transcriptomic and epigenomic pathology of schizophrenia using patient-derived tissues have been conducted at scale.
One of the largest studies of transcriptomic brain pathology in schizophrenia was conducted by the PsychENCODE consortium [45]. In this study, gene- and transcript isoform-level differential expression was comprehensively analyzed by performing RNA-sequencing (RNA-seq) of bulk postmortem cerebral cortex tissues from 559 schizophrenia cases and 936 control individuals, together with 51 ASD and 222 bipolar disorder brains. They identified that the expression of 4821 genes and 3803 isoforms significantly differed between schizophrenia and controls (FDR < 0.05). Genes related to “inflammatory response” and “receptor activity” were enriched in the significantly upregulated and downregulated genes/isoforms, respectively. The enrichment of the genes related to receptor activities is consistent with the results summarized in the previous section. Schizophrenia heritability was enriched among genes and transcripts dysregulated in schizophrenia brains, especially in down-regulated transcript isoforms.
Regarding epigenomic brain pathology, a recent large-scale study analyzed two major histone modifications, histone 3 lysine 27 acetylations (H3K27ac) and histone 3 lysine 4 trimethylations (H3K4me3) [46], in postmortem prefrontal cortical samples (sorted neurons or bulk tissues) from 303 schizophrenia cases and 388 controls along with 48 bipolar disorder brains by chromatin immunoprecipitation sequencing (ChIP-seq) [47]. In the analysis of differential H3K4me3/H3K27ac peaks, 6219 differential H3K27ac peaks (FDR < 0.05) between schizophrenia and controls were identified though there were no differential H3K4me3 peaks. Of these, schizophrenia heritability based on GWAS [20] was enriched in the H3K27ac peaks hyper-acetylated in schizophrenia. Subsequently, this study mapped cis-regulatory domains (CRDs), which often overlap with topologically associating domains defined by the analysis of 3D chromosomal conformations but constitute smaller regulatory units of 104–106 bp [48, 49], in the brain using the information of inter-individual correlations between histone peaks. In an analysis integrating information on CRDs and differential H3K27ac peaks, it was shown that schizophrenia heritability is strongly enriched at differential H3K27ac peaks in CRDs hyper-acetylated in schizophrenia, suggesting that dysregulated H3K27ac peaks within dysregulated CRDs particularly are associated with the genetic schizophrenia risk.
Besides histone modifications, DNA methylation is another major epigenetic modifier with regulatory functions. Several studies have explored genome-wide DNA methylation status in postmortem schizophrenia brains. Among them, a study of microarray-based analysis of DNA methylation at ~450,000 loci in postmortem dorsolateral PFC (DLPFC) tissues from 526 individuals was reported [50]. In this study, a total of 2104 sites differentially methylated between quality-controlled 108 schizophrenia cases and 136 controls (Bonferroni-corrected P < 0.05), of which 97.1% were hypomethylated in schizophrenia, were identified. A GO enrichment analysis of genes in or near the differentially methylated sites showed an overrepresentation of genes related to embryo development, cell fate commitment, and nervous system differentiation. Also, modest enrichment of schizophrenia-associated loci among the differentially methylated sites (1.9% among differentially methylated sites vs. 1.3% among others, P = 0.004, Chi-square test) was observed. On the other hand, in a study of postmortem brain samples overlapping with the above-described microarray-based study (70 and 95 schizophrenia DLPFC and hippocampus, and 77 and 102 control DLPFC and hippocampus) using whole-genome bisulfite sequencing, a technique that can detect DNA methylation at the single base resolution, much smaller numbers of differentially methylated sites, none in DLPFC and 70 in the hippocampus, were identified despite less stringent multiple testing corrections (FDR < 0.05) [51]. This discrepancy could be explained by the difference in sample sizes as well as the sensitivity to detect differentially methylated sites and the number of hypotheses tested, as a larger number of sites are covered by whole-genome bisulfite sequencing.
In addition to the studies using postmortem brain tissues, there are large-scale studies of peripheral samples aiming to identify disease biomarkers. In a study by Aberg et al. [52], analyzing genome-wide DNA methylation profiles in blood samples from 759 schizophrenia cases and 738 controls by methyl-CpG–binding domain protein-enriched genome sequencing, 25 and 139 sites associated with the diagnosis at Bonferroni-corrected P < 0.05 and FDR < 0.01, respectively, were identified. The most significant association was observed in the region involving FAM63B, a part of networks regulated by microRNAs associated with neuronal differentiation and dopaminergic gene expression. This association was replicated in an independent cohort of >1000 individuals. The observed effect sizes for three associated methylation sites at FAM63B were moderate (Cohen’s d = 0.42–0.45). In a recent meta-analysis of blood DNA methylation profiles from 4483 participants from seven cohorts, including 1681 schizophrenia cases and 1583 controls, by Hannon et al. [53], 1013 differentially methylated loci with methylome-wide significance (P < 9 × 10−8), which were annotated to 692 genes, were identified. Among 158 schizophrenia-associated loci identified by GWAS [10], overall differential DNA methylation was observed at 21 loci after correcting for multiple testing, supporting co-localization of signals from GWAS and epigenome-wide association study. On the other hand, an integrative analysis of DNA methylation and genetic variants exploring the causal relationships was not performed in their study. Further studies of the interaction between genetic and epigenetic factors that are expected to provide additional insights into the molecular mechanisms underlying co-localization are warranted.
Overall, while the significant overlap between differentially expressed or modified genes and the genetic risk of schizophrenia has been reported in some studies, transcriptomic or epigenomic alterations of single genes that can biologically define the general population of schizophrenia or serve as a high-sensitivity and specificity biomarker have not been discovered, or perhaps there is no such universal molecular marker. Therefore, further studies are necessary to identify conclusive transcriptomic and epigenomic pathology in schizophrenia.
Studies of etiologically valid mouse and cellular models of schizophrenia
As summarized in Table 1 and Fig. 1, recent large-scale genetic studies have identified specific genes, SNVs/indels, and CNVs that confer a substantial risk of schizophrenia. Based on this, rodents or cells carrying the alleles equivalent to the above-described risk variants identified in humans have been generated and analyzed. In this section, we overview studies of such etiologically valid, i.e., having the same causal conditions as in human patients, models of schizophrenia. (Tables 2 and 3), which have provided various insights into the connection between genetic pathology and pathological changes at the levels of molecules (e.g., transcripts and proteins), cells, neural circuits, whole tissues, or individuals’ behaviors.
Table 2 Etiologically valid mouse models of schizophrenia.
Table 3 Etiologically valid cellular models of schizophrenia.
Mouse models
We systematically surveyed studies of mice with mutant alleles orthologous to the variants listed in Table 1 with an observed OR greater than ten. We found that there are studies on the following variants: 22q11.2 deletion, 16p11.2 deletion/duplication, 3q29 deletion, 15q11.2–13.1 duplication, 2p16.3 (NRXN1) deletion, GRIN2A LOF/PTV, GRIA3 LOF/PTV, and SETD1A LOF/PTV (Table 2) [54,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72,73,74,75,76,77,78,79,80,81,82,83,84,85,86,87,88,89,90,91,92]. While there are studies of mice with mutations in other genes, such as RB1CC1 (also known as FIP200), the introduced alleles were not equivalent to ones in human patients, and/or the mice were not analyzed in the context of neuroscience, and thereby not highlighted in this review.
Regarding the molecular phenotypes mainly analyzed by transcriptomic profiling, commonly dysregulated pathways include neural transmission and regulation of transcription [54, 63, 70, 88, 91, 92], in agreement with the findings from human genetics and genomics studies. Also, analyses utilizing results of large-scale human genetics studies have reported enrichment of genetic risk for schizophrenia in genes differentially expressed in the models or molecular targets of the genetically modified genes [88, 91]. Meanwhile, mutant mice originally created not as a schizophrenia model but to elucidate gene function in the central nervous system, such as knockout mice for GRIN2A or GRIA3 encoding a glutamate receptor subunit, have not been subjected to omics analysis. Molecular profiling of these etiologically valid models may provide further convergent insights into the brain pathology of schizophrenia.
Morphological analysis of neuronal cells in these models has reported reduced axonal and dendritic complexity and abnormal spine morphology in Setd1a heterozygous knockout mice and several mouse models with CNVs [54, 55, 57, 58, 88, 91]. Common electrophysiological phenotypes include altered synaptic transmissions, such as diminished excitability indicated by reduced excitatory postsynaptic currents [55, 58, 60, 75, 79, 82, 85, 88, 91] or deficits in long-term potentiation [68, 79], though some mice showed increased excitability or altered activities of other neuronal subtypes such as GABAergic neurons [70, 84].
Behavioral alterations common in these mice include deficits in sociality, cognitive performance, and prepulse inhibition [54, 61, 66,67,68, 70,71,72,73, 75,76,77, 79, 88, 90,91,92]. These phenotypes are generally consistent with those in human schizophrenia patients [93], though there would be biases that these phenotypes are more likely to be investigated and reported. Also, in some cases, there were inconsistent results even among models with mutations in the same gene. This could be explained by differences in the method of introduction of the mutations (e.g., CRISPR/Cas9 or gene targeting), genetic backgrounds (e.g., C57BL/6J and C57BL/6N), and other factors. In addition, the acquisition of more definitive results will be facilitated by strict standardization of analysis protocols and ensuring a sufficient sample size, as have been done in human genetics studies.
Cellular models
Recent technological advances have enabled the reproduction of pathological conditions in vitro by creating patient-derived or mutation-carrying induced pluripotent stem (iPS) cells and then differentiating them into central nervous system cells or miniature brains. Studies of etiologically valid cellular models of schizophrenia produced with this technology, including those with 22q11.2 deletion, 16p11.2 deletion/duplication, SETD1A LOF/PTV, and NRXN LOF/PTV, have been conducted and reported [94,95,96,97,98,99,100,101,102,103,104,105,106] (Table 3).
In line with the findings in etiologically valid mouse models, molecular profiling of these cellular models generally supports dysregulation of genes related to neural transmission, especially synaptic genes [95,96,97, 100, 101, 106], transcriptional regulators including microRNAs [94, 104], and schizophrenia-associated genes discovered by human genetics studies [96, 97, 104]. Also, morphological alteration of soma and dendrite were common except for iPS cell-derived neurons with NRXN1 deletion [94, 98, 101, 102, 106]. Abnormal neural activities are identified in multiple patient-derived or genetically engineered iPS cell-derived neurons, however, the directions of the abnormalities are sometimes inconsistent across models manipulating different genes [97, 101,102,103,104,105,106]. Though it may be due to artifacts depending on the differences in the experimental designs, another possibility is that imbalanced excitatory/inhibitory activities themselves [107], regardless of the direction of abnormality, are important in schizophrenia pathology. It is also worth noting that multiple studies have investigated interventions to improve abnormal phenotypes observed in cellular models [96, 100]. Overall, the iPS cell technology is a powerful tool to analyze the molecular pathology of schizophrenia using human samples, and further studies are warranted.
Considerations on the model validity
In the previous sections, we have defined etiologically valid models using the criteria that the modified gene showed a significant association with schizophrenia after stringent multiple testing correction and that the ORs observed in studies that found the association was greater than ten. However, we would like to explicitly state that there is still uncertainty regarding the validity of these models.
First, it should be noted that in general, there are wide ranges of confidence intervals for ORs for rare risk variants (Fig. 1 and Table 1). Indeed, another large-scale study (20,403 cases and 26,628 controls) analyzing the association of schizophrenia and CNVs implicated in neurodevelopmental disorders reported no statistically significant associations of 16p11.2 deletion and 15q11.2–13.1 duplication with schizophrenia and association of 2p16.3 (NRXN1) deletion with OR smaller than ten [30, 32]. Therefore, there is the possibility that ORs are overestimated in the existing data. Second, as mentioned in the section on genetic pathology, many of the genes and variants highlighted here, especially almost all CNVs, are associated not only with schizophrenia but also with other neurodevelopmental disorders. Given this, the mice and cells harboring such mutations should not be considered as specifically modeling schizophrenia. Third, particularly in the case of CNV-based animal models, evaluation of the model validity and the interpretation of the phenotypes require caution because CNVs usually contain multiple genes and non-coding regions whose structure is sometimes not well conserved between rodents and humans. Lastly, we would like to emphasize that a number of valuable findings interpreted as being relevant to the molecular pathology of schizophrenia have also been obtained from analyses of models in which genes not strongly supported by currently available evidence from human genetics studies were manipulated. For example, given the mechanisms of action of known antipsychotics, it is quite obvious that dysregulation of the monoaminergic system is involved in the pathophysiology of schizophrenia [108, 109], and therefore various genes in this pathway have been intensively investigated. Although these include genes that do not have strong genetic support, unlike DRD2 identified in GWAS [9, 20] and others, they form a foundation not only for the study of schizophrenia patients but also for the study of animal and cellular models with genetic etiological validity [60, 110,111,112,113,114]. Besides, as evidenced by the fact that heritability is better explained by considering numerous SNPs, not only those in genome-wide significant loci, it is clear that there are true schizophrenia risk genes among those that did not reach the stringent significance threshold with the current sample sizes. This indicates that further identification of robust risk genes in larger studies will certainly increase the value of existing research using animal and cellular models with modifications of such genes. Meanwhile, it is also true that there are mice that have been interpreted as models of schizophrenia based on their face validity, despite the lack of etiological validity in light of currently available knowledge. Moreover, sometimes models are interpreted as meeting face validity based on phenotypes in homozygous mutants, even though heterozygous variants are associated with schizophrenia in humans. Therefore, caution should be exercised in discussing the validity of a model solely on the strength of its face validity, regardless of the robustness of the association between the manipulated gene and schizophrenia risk.
Current achievements, limitations, and new directions for future research
Based on the above-described existing knowledge of the genetic and molecular pathology of schizophrenia as well as the emerging insights into their links to other scales of pathologies from studies of etiologically valid models, we summarize the current achievements, limitations, and new directions for future research as follows.
Regarding the genetic pathology, significant advances, such as the elucidation of the highly polygenic nature of schizophrenia, the explanation of more than 20% of disease liability by measurable genetic variants, and the identification of specific genes and CNVs associated with schizophrenia with large effect sizes, have been achieved. While a substantial part of the heritability is still unexplained and the number of disease-responsible genes identified so far is not as large as in ASD, where similarly large studies have been conducted, it is certainly expected that by simply increasing the sample size and investigating under-studied types of variants, such as rare non-coding mutations, we can better explain the genetic liability to schizophrenia and identify additional responsible genes. Indeed, a more recent target sequencing study of candidate schizophrenia-associated genes in 11,580 cases and 10,555 controls, followed by a meta-analysis with the SCHEMA dataset identified two novel exome-wide significant genes, AKAP11 and SRRM2 [115], confirming the importance of increasing sample sizes. In addition to the promotion of basic genetic research, the application of genetic information to clinical psychiatry is a subject of active discussion. As an example, there are attempts to utilize polygenic risk scores (PRS) based on the profiles of common SNPs to predict clinical courses [116, 117], though further studies are needed. Also, the identification of patients with Mendelian genetic disease among patients clinically diagnosed with schizophrenia based on operationalized criteria and the optimization of their treatment based on genetic diagnosis is expected to be implemented in the near future.
Compared to the knowledge of genetic pathology, our understanding of the molecular pathology of schizophrenia, such as transcriptomic and epigenomic alterations, is insufficient and no convincing single molecular markers that can biologically define the schizophrenic brain have been identified. Nevertheless, collectively interpreting the results of large-scale omics analyses of human postmortem brains and studies of models with high etiological validity, one might be able to argue that small transcriptional and/or epigenetic alterations of many schizophrenia-associated genes and genes involved in neuronal processes such as the formation and regulation of synapses would be the underlying molecular brain pathology of schizophrenia. To obtain a clearer picture, the following directions would be considered.
First, as summarized in Tables 2 and 3, multiple etiologically valid models of schizophrenia have been generated and analyzed. One of the next important steps will be to elucidate the alterations that are commonly observed across them, and studies seeking this goal should be facilitated by investigating two or more models in the controlled same experimental settings. This is because the results of studies of disease models are often confounded by subtle differences in experimental design and conditions, such as apparatus, experimenter, mouse strain and genetic background, and others. However, to our knowledge, there have been no publications reporting the results of the analysis of multiple schizophrenia models with high etiological validity listed in Tables 2 and 3 under the same conditions. By conducting such studies of multiple models, which have already been done for ASD [118,119,120,121], it is expected that we can obtain convergent insight into the pathological changes in schizophrenia.
Second, while it must be recognized that collecting human postmortem brains on a large scale requires a great endeavor, there is an open question of whether the sample sizes in studies to date are sufficient. Evaluating the inter-study reproducibility, which may help answer this question, it is true that the result of the analysis of genes differentially expressed between schizophrenia cases and controls in the aforementioned PsychEncode study [45] is well correlated with that of a preceding study by the CommonMind Consortium (CMC) [122] (correlation coefficient between the two studies for 687 genes with FDR < 0.05 in the CMC study = 0.799). On the other hand, of the 23 genes that showed statistical significance after Bonferroni correction in the CMC study (uncorrected P < 3.04 × 10−6, 0.05 divided by the number of genes analyzed, 16,423), only nine genes surpassed the same significance threshold in the PsychENCODE study. This number is much more than random expectation; however, this contrasts with the observation in GWAS that 116 out of the 128 loci genome-wide significantly (P < 5 × 10−8) associated with schizophrenia in the previous PGC GWAS in 2014 were replicated with the genome-wide significant association in the same local regions in 2022 GWAS (and supporting evidence was obtained for 11 of the remaining 12 loci in an extended analysis). Given these, it is warranted to further increase the sample sizes in postmortem brain studies of schizophrenia in order to obtain robust and reproducible results, as has been done in GWAS throughout its history. The same would be true for human brain imaging studies that explore structural and functional alterations associated with phenotypes in a brain-wide manner [123].
Third, as is true in any field of science, often important unresolved problems, such as the mystery of the molecular brain pathology of schizophrenia, are addressed by the utilization of new technologies. Considering the major limitations of current research on molecular pathology using patient or model brains, while the etiological validity of the model is undoubtedly important, in this context, there is an inherent concern that the molecular pathology in human schizophrenia patients may not be adequately reproduced in rodent or cellular models, even when the exact same variants that are pathogenic in humans are introduced. Perhaps this problem would be addressed by studying species closer to humans, specifically non-human primates. Owing to recent innovations in genome editing technology, genetically engineered non-human primates carrying mutations that are pathogenic in humans, such as cynomolgus monkeys with mutations in MECP2 [124], the gene responsible for Rett syndrome, or SHANK3 [125], an ASD gene in the Phelan–McDermid syndrome critical region, and common marmosets with a mutation in PSEN1 [126] causal for familial Alzheimer’s disease, have been created and analyzed. While research using primates is much more time- and cost-consuming than studies of mice, non-human primate models of schizophrenia will be an excellent resource to fill the gap between humans and rodents. Another major limitation is that while we can comprehensively analyze transcriptomic and epigenomic profiles in postmortem brain tissues, such analysis in the brain of living patients can not be performed. On the other hand, recent technological advances have allowed us to quantify some key molecules involved in the regulation of synapses and histone modifications, such as the synaptic vesicle glycoprotein 2A [127], AMPA-type glutamate receptors [128], and histone deacetylases (HDACs) [129] in the living human brain. By expanding the repertoire of measurable molecules and scaling up studies, we will better understand molecular changes in the brain of living schizophrenic patients.
Besides them, one of the most prominent new technologies that have become prevalent over the last decade is single-cell sequencing techniques, whose usefulness was mentioned in the above section describing the convergent results of cell type enrichment analysis in the PGC GWAS and SCHEMA study. Single-cell analyses are particularly powerful in studies of organs where different cell types are intermingled, such as the brain. By performing cell type-resolved analysis using single-cell technology, it may be possible to more clearly capture molecular pathology that was obscured in bulk tissues. At the end of this section, we highlight pioneering single-cell (nucleus) RNA sequencing studies of postmortem schizophrenia brains, while some of them have been posted to preprint servers and have not been published after peer review.
Single-nucleus RNA sequencing studies of postmortem schizophrenia brains
Technically, analysis of single “cells”, including cytoplasm, cannot be currently performed in studies of frozen postmortem brain tissues; therefore, single-“nucleus” RNA sequencing (snRNA-seq) studies of human schizophrenia brains have been conducted.
To our knowledge, there are four publications on snRNA-seq of postmortem schizophrenia brains, including two preprints that have not yet been peer-reviewed. Among them, the largest study by Ruzicka et al. analyzed 266,431 nuclei from 24 schizophrenia patients and 293,589 nuclei from 24 controls using frontopolar cortex (Brodmann area 10) samples [130]. In this study, 20 cell types/states were annotated based on their transcriptional profiles, and it was shown that the majority of genes differentially expressed in schizophrenia occurred in the neuronal population. The cell types with the strongest enrichment of schizophrenia-associated genes identified by GWAS among differentially expressed genes include cortico-cortical projection neurons in the deep layers V/VI, parvalbumin-positive basket interneurons, and excitatory neurons of a novel cell state enriched in the supragranular layers II/III. This novel type of supragranular excitatory neurons was more abundant in schizophrenia than in controls, but was preferentially found in schizophrenia individuals with less “transcriptional pathology score”, defined by overall schizophrenia-associated transcriptional dysregulation in each individual. Based on this observation, the authors speculated that this population of excitatory neurons, named Ex-SZTR, might be associated with “schizophrenia transcriptional resilience”. While further scrutinization through peer reviews is needed, this finding may contribute to conceptual advances in the understanding of the molecular/cellular pathology of schizophrenia.
The observation that the majority of differentially expressed genes are found in neuronal populations was also reported in another study by Reiner et al., where 127,930 and 145,120 nuclei from DLPFC of 12 schizophrenia and 14 control individuals were analyzed by snRNA-seq, respectively [131]. In their study, ~96% of differentially expressed genes were observed in neuronal cell types, including excitatory neurons across layers II-V and parvalbumin-positive interneurons.
In a study by Batiuk et al., not only snRNA-seq of sorted neurons from DLPFC of 9 schizophrenia patients and 14 controls (81,817 and 127,236 nuclei, respectively) but also follow-up immunohistochemistry, single-molecule fluorescence in situ hybridization, and spatial transcriptomics analyses in an extended cohort were performed [132]. Results of these analyses convergently suggested that transcriptional dysregulation and altered cellular composition within the upper cortical layer, involving both GABAergic interneurons and principal projection (excitatory in the cortex) neurons, might be a core substrate associated with the brain pathology of schizophrenia.
These results would collectively support that schizophrenia is primarily a disease of neuronal cells. On the other hand, a unique study focusing on cells constituting the blood-brain barrier (BBB) based on the neurovascular hypothesis of schizophrenia was conducted by Puvogel et al. [133]. In their study, a total of 178,009 nuclei (NEUN and OLIG2 negative nuclei enriched for BBB cells and NEUN positive and OLIG2 negative nuclei enriched for neuronal cells) from postmortem midbrain tissues of 15 schizophrenia patients and 14 controls were analyzed by snRNA-seq. The results showed that there was no significant difference in the relative proportions of the major BBB cell types between schizophrenia and controls. A limited number of genes were differentially expressed in schizophrenia (14 genes with log2 fold change > 0.3 and FDR < 0.05). These differentially expressed genes were restricted to ependymal cells and pericytes, suggesting that BBB cells are not broadly affected in schizophrenia.
Overall, many of the findings in these studies are detectable only when cell type-resolved analysis is performed, demonstrating the value of snRNA-seq. Nevertheless, some of the above-described results should be considered preliminary because half of the four studies highlighted here have not been peer-reviewed yet. Also, the numbers of individuals analyzed in these studies are not large, while the numbers of nuclei examined were huge. Therefore, it is necessary to consider whether the sample size is sufficient.
Perspectives: a decade after the best of times, the worst of times for psychiatric disease
In 2012, Karayiorgou et al. on behalf of the Genetic and Neural Complexity in Psychiatry 2011 Working Group described the situation at that time as “the best of times, the worst of times for psychiatric disease” [134]. This was because, on the one hand, the development and deployment of long-awaited new DNA sequencing technology (i.e., next-generation sequencing) made it possible to conduct genome-wide exploration of highly penetrant rare variants on a population scale (the best of times), while on the other hand, many pharmaceutical companies withdrew from the research and development of novel therapeutics due to their low success rates (the worst of the times). A decade later, as predicted, several robust risk genes with large effect sizes have been identified for schizophrenia, and the first results of pioneering studies using animal and cellular models created on the basis of the discovery of these genes are beginning to be harvested [88,89,90,91,92, 106]. Overall, it can be said that we have achieved the expected outcomes over the past ten years. Also, a number of powerful new technologies have been developed and implemented during this period. The important thing is to continue this progress, and such effort will reverse the retreat from research and development by pharmaceutical companies and other investors, which was recognized a decade ago and persists today.
In this context, it would be meaningful to provide a clearer picture of how the field of schizophrenia genetics and biology will further develop. In our view, the overarching challenge for the next decade will be how we translate the findings in basic genetic and biological research into clinical psychiatry. The first part of the path to resolving this problem has been clarified by the results of studies to date. Aggregating the existing knowledge, we are able to identify diagnostic genomic variants (e.g., Pathogenic or Likely Pathogenic variants in the American College of Medical Genetics and Genomics [ACMG] guidelines [135]) in 1–6% of schizophrenia patients by comprehensively analyzing rare variants [136,137,138,139], and to extract a small proportion of the population with high genetic risk (e.g., OR > 5) utilizing the overall profiles of common variants (i.e., PRS [140, 141]). On the other hand, to our knowledge, there are no genetic tests for schizophrenia approved by the government and covered by health insurance. Among several reasons for this situation, the primary one is that the clinical benefits gained from genetic testing are far less than the cost and potential side effects. More specifically, there are two major factors limiting the benefits: the performance of risk prediction from genetic information is insufficient, and the results of genetic testing rarely lead to changes in clinical actions. To improve the performance of genetic risk prediction, as described above, it is indispensable to expand the sample size and investigate various types of variants, which include not only common SNPs, rare coding SNVs, and CNVs but also non-coding rare variants, repeat element variants, complex structural variants, somatic variants, and others, with sufficient statistical power. In particular, the variants that are not common but not extremely rare, which can fill the blank region in Fig. 1, will be a major target in future research. Also, it is crucial to conduct sufficiently large studies in diverse ethnic populations. The importance of such studies is evident from the observation that the performance of PRS is greatly reduced when the ethnicity of the individuals being scored is different from that of the data used to construct the prediction model [142, 143]. Regarding the improvement of clinical actionability, it is expected that the generation and investigation of multiple etiologically valid schizophrenia models, as featured in this review, will play an important role. The realization of precision medicine, such as gene therapy, for specific genetic diseases frequently comorbid with schizophrenia (e.g., 22q11.2 deletion or SETD1A haploinsufficiency syndrome) leveraging the observations in studies of these models might be the achievable goal within the next decade. And beyond that, by integrating the results of human genetics and model studies as well as other areas of research, such as human functional imaging and brain circuity, it should be aimed to define biologically homogeneous schizophrenia subgroups and identify the optimal treatment and prevention for them.
The World Health Organization estimates that by 2030 mental disorders will be the leading cause of disease burden globally [144], of which a significant part should be accounted for by schizophrenia due to its chronic and often treatment-resistant nature. Studies toward the elucidation of the molecular pathology of schizophrenia, which forms the foundation for essential therapeutics, are of great social value. Therefore, continuous investments from academia, government, industry, and citizens, along with appropriate ethical, legal, and social considerations, are warranted.
References