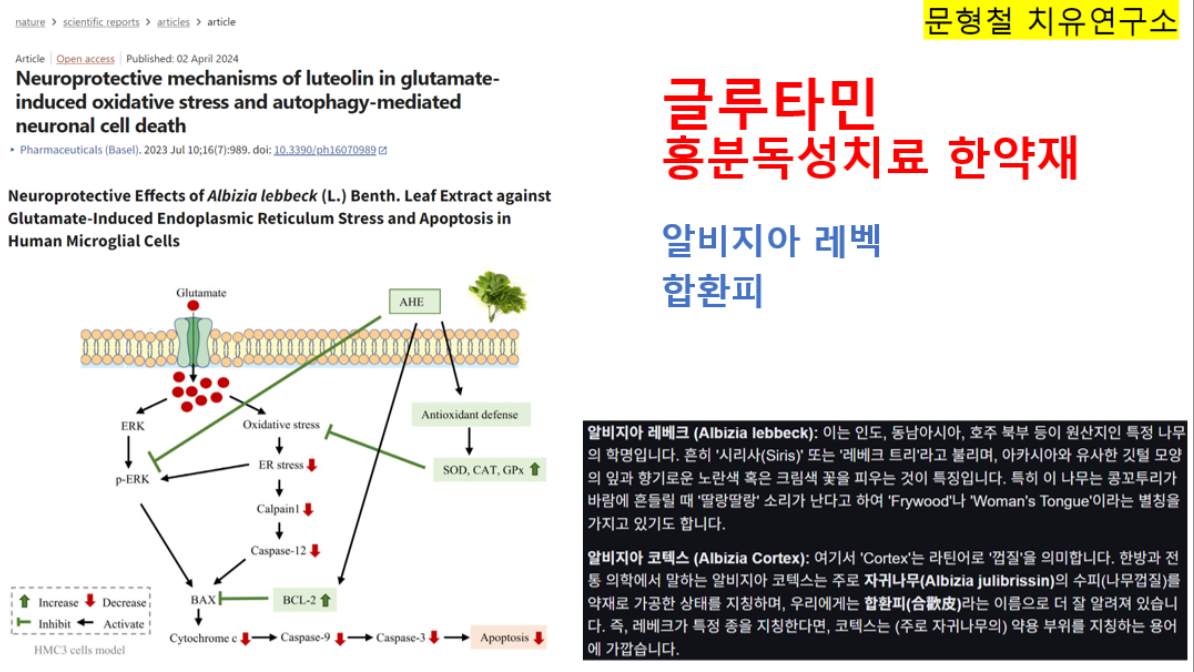
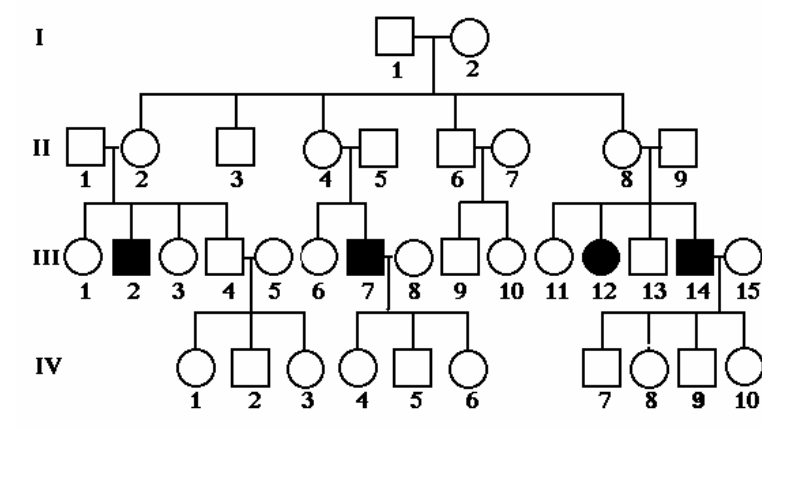
 - 발현 빈도는 약 2만~10만 명당 1명입니다. 인종과 민족 간 차이가 있습니다. - 근 긴장성 경직과 추체외로계 증후군을 특징으로 합니다. - SCA 1의 임상 증상은 보통 20~30대에 나타납니다. 이보다 더 빠르게 혹은 더 느리게 나타나기도 합니다. - 보행 장애, 불분명한 발음, 균형 잡기의 어려움, 근육 운동 조정의 어려움, 불수의적 행동(갑자기 경련성으로 움찔하거나, 무의식적인 움직임), 과대 반사 작용, 근력 저하, 연하장애 등이 나타납니다. - 10~20년의 기간을 두고 병이 진행되며, 결국에는 사망합니다. 보통 이른 나이(13세 이하)에 증상이 발현된 환자는 증상이 더 심하고 빠르게(16세 이전에 사망) 진행됩니다. ② SCA 2 - 발현 빈도는 약 2만~10만 명당 1명입니다. 인종과 민족 간의 차이가 있습니다. - 소뇌성 운동실조와 추체로 징후(pyramidal signs)를 특징으로 합니다. - 우성으로 유전되는 소뇌성 운동실조증 중 13% 정도가 SCA 2입니다. - 보통 40대에 증상이 나타납니다. 이보다 더 빠르게 혹은 더 느리게 나타나기도 합니다. - 보행 장애, 불분명한 발음, 눈의 이상 및 시각 장애(안구 진탕, 안구 부전마비 등), 균형 잡기의 어려움, 근육 운동 조정의 어려움, 불수의적 행동(갑자기 경련성으로 움찔하거나, 무의식적인 움직임), 반사작용 감소, 근력 저하 등이 나타납니다. - 10~15년의 기간을 두고 진행되며, 결국에는 사망합니다. 이른 나이(20세 이하)에 증상이 발현된 환자는 증상이 더 심하고 빠르게 진행됩니다. ③ SCA 3 - Machado-Joseph 병(MJD)이라고도 알려져 있습니다. - 주로 포르투갈 민족에서 많이 나타납니다. - 근 긴장성 경직, 파킨슨 증후군을 동반하는 소뇌성 운동실조증, 근긴장 이상과 말초신경병증을 특징으로 합니다. - 발병 평균연령은 40세 정도입니다. - 초기 증상은 보행 장애, 구음 장애, 돌출된 눈, 과다반사항진, 둔함(clumsiness), 시각 장애 등입니다. 증상이 진행됨에 따라 근육의 황폐, 얼굴과 사지의 경련이 나타납니다. 결국 환자는 휠체어에 의존하게 되고, 앉고 일어나는 행동에 어려움을 겪습니다. - 발병 후 15~20년이 지나면, 환자는 대부분 호흡기계의 합병증이나, 음식물 섭취의 어려움으로 인해 사망합니다. ④ SCA 6 - 느리게 진행되는 소뇌성 운동실조, 구음 장애, 안구진탕 등을 특징으로 합니다. - 발현 빈도는 여건에 따라 다양하게 나타납니다. 약 30만~50만 명당 1명입니다. 인종과 민족 간 차이가 있습니다. - 발병 연령은 19~71세 사이입니다. 발병 평균 연령은 43~52세입니다. - 표현촉진(Anticipation)은 나타나지 않습니다. - 초기 증상으로는 환자의 90% 정도가 불안정한 걸음(다리가 휘청거림), 비틀거림(stumbling), 불균형을 겪습니다. 환자의 10% 정도가 구음 장애를 겪습니다. - 증상은 서서히 진행됩니다. 결국 모든 환자는 보행운동실조, 상지의 조정 장애, 기도진전(intention tremor), 구음 장애를 보입니다. 약 50%가 복시증(diplopia)을 겪습니다. 수평주시안진(70~10%), 수직안진(65~83%)이 나타날 뿐만 아니라 사물을 고정시키거나 움직이는 사물과 관련된 시각에 장애가 나타납니다. - 연하곤란이나, 질식 등은 보편적으로 발생합니다. 반사이상항진, 족저의 신근반사(40~50%)이 발생합니다. 근긴장, 안검경련과 같은 대뇌 기저핵 증후(25%)가 나타납니다. 정신 상태는 온전하지만, 나이가 많은 환자의 10% 이상이 치매를 보입니다. 수명은 짧지 않습니다. ⑤ SCA 7 - 구음 장애와 연하 장애를 동반하는 진행성 소뇌 운동실조증과 중심 시력의 소실로 진행되어 실명에 이르는 망막 이영양증을 특징으로 합니다. - 발현 빈도는 10만 명당 1명 미만입니다. 인종과 민족 간 차이가 있습니다. - 평균 발병 연령은 10대 후반~20대 초반입니다. - 초기 증상으로 소뇌성 운동실조가 발생하기 전에 색각과 중심 시력의 이상이 항상 먼저 나타납니다. 망막의 퇴화는 진행성이며, 결국에는 실명에 이릅니다. - 소뇌성 운동실조 증상으로는 운동조절 이상, 길항운동반복불능증, 조정능력 저하가 선행되며, 이후 구음 장애, 연하 장애, 운동 근육의 조절 능력 소실로 진행됩니다. - 이른 나이에 발병할수록 질환이 진행하는 속도도 빠르고 증상이 심합니다. 늦게 발병하는 경우에는 느리게 진행되며 증상이 경미합니다. - 표현 촉진(Anticipation)이 나타납니다. - 50~60대에 첫 증상을 보이는 환자는 정상 수명을 유지합니다. |
유전성 소뇌실조증(hereditary cerebellar ataxia)은
유전적 원인으로 인해 소뇌의 퇴행성 변화가 발생하는 질환군으로,
임상적으로 매우 이질적입니다.
주로 운동 실조(ataxia),
보행 불안정,
언어 장애(dysarthria),
안구 운동 이상(nystagmus) 등이 나타나며,
유전 양식에 따라 분류됩니다.
주요 분류는 다음과 같습니다:
1. 상염색체 우성 유전(Autosomal Dominant Cerebellar Ataxias, ADCAs)
가장 흔한 형태로,
전체 유전성 소뇌실조증의 약 50-70%를 차지합니다.
주로 척수소뇌실조증(Spinocerebellar Ataxias, SCAs)로 불리며,
50개 이상의 유형(SCA1~SCA50+)이 알려져 있습니다.
| SCA1 (Spinocerebellar Ataxia Type 1) 진행 과정 분류 및 요약 SCA1은 ATXN1 유전자의 CAG 반복 확장으로 인한 폴리글루타민 질환으로, 빠른 진행성을 보이는 상염색체 우성 유전성 소뇌실조증입니다. 주요 증상으로는 운동 실조, 피라미드 증상, 인지 저하가 있으며, 성별에 따라 인지 증상이 다를 수 있습니다. 최신 논문(2025년 Colucci F et al.)에 따르면, 운동 및 인지 증상이 진행되며, 성별이 인지 결과에 영향을 미칠 수 있으나, 구체적인 휠체어 사용 시기(보통 15년 내)나 생존 감소에 대한 정량 데이터는 부족합니다. 진행 속도는 빠르며, SARA 점수 상승이 급격할 수 있으나, 최신 연구에서 바이오마커 개발이 강조됩니다. SCA2 (Spinocerebellar Ataxia Type 2) 진행 과정 분류 및 요약 SCA2는 ATXN2 유전자의 CAG 반복으로 인한 형태로, 중간 진행성을 보이며 파킨슨 증상(느린 운동, 강직)이 동반될 수 있습니다. 발병 연령은 중년(median 60세, 범위 21-87세)으로, 초기 에피소딕 증상(약 50%) 후 천천히 진행하는 소뇌 증상(보행 실조)이 주를 이룹니다. 2025년 Pulst SM의 리뷰에서 초기 발견부터 치료 개발까지의 진척을 강조하며, 소아 케이스(19례 보고)에서 더 빠른 진행을 보입니다. SARA 점수나 휠체어 사용 시기(10-20년 내 가능), 생존 기간에 대한 정량 데이터는 제한적이지만, 바이오마커로 진행 추적이 필요합니다. Galecio-Castillo et al.(2025)에서 라틴아메리카 코호트에서 10% 빈도입니다. SCA3 (Spinocerebellar Ataxia Type 3, Machado-Joseph Disease) 진행 과정 분류 및 요약 SCA3는 ATXN3 유전자의 CAG 반복으로, 중간 진행성을 띠며 파킨슨 증상과 함께 다계통 침범(실조, 신경병증)이 특징입니다. 발병 연령 median 60세(범위 21-87세)로, 초기 에피소딕 증상(약 50%) 후 천천히 진행합니다. 2024년 Pellerin D et al.에서 천천히 진행하는 소뇌 증상을 강조하며, Camós-Carreras A et al.(2024)에서 망막 신경절 세포 손실이 진행과 관련됩니다. Paulino LA et al.(2025)에서 세계적으로 가장 흔한 ADCA로, 약물 개입으로 증상 관리 가능하나, 휠체어 사용(10-15년 내)이나 생존 감소에 대한 구체적 데이터 부족. Galecio-Castillo et al.(2025)에서 라틴아메리카 15% 빈도입니다. SCA6 (Spinocerebellar Ataxia Type 6) 진행 과정 분류 및 요약 SCA6는 CACNA1A 유전자의 CAG 반복으로 인한 순수 소뇌 증상(보행 실조, 진동안진, 발음 장애)이 주된 느린 진행성 형태입니다. 좋은 예후를 보이며, 비호모지고트보다 호모지고트에서 조기 발병 및 빠른 진행을 보입니다. 2025년 Kim JM et al.에서 호모지고트의 더 심한 진행과 시선반사 이상을 보고하며, Tarnutzer AA et al.(2024)에서 안구 운동 지표로 진행 평가를 제안합니다. SARA 진행 속도(연간 약 0.80점)는 느리며, 휠체어 사용이 늦거나 드물 수 있으나, 구체적 시간 데이터 부족. Koren TJ et al.(2025)에서 디스토니아/파킨슨 동반 가능성을 언급합니다. SCA7 (Spinocerebellar Ataxia Type 7) 진행 과정 분류 및 요약 SCA7은 ATXN7 유전자의 CAG 반복으로, 소뇌 증상 외에 망막 퇴행(황반 변성)으로 시력 상실이 동반되는 진행성 형태입니다. 2024년 멕시코 코호트 연구에서 진행성 소뇌 증상과 황반 변성을 강조하며, 자연 경과 관찰이 중요합니다. 또 다른 2024년 연구에서 릴루졸 치료(평균 4.8년) 중 SARA 점수로 진행 추적을 보고하나, 휠체어 사용 시기(10-20년 내)나 생존 기간에 대한 정량 데이터 부족. 시력 상실이 주요 합병증으로, 전문 치료의 도전성을 지적합니다. SCA27B (Spinocerebellar Ataxia Type 27B) 진행 과정 분류 및 요약 SCA27B는 FGF14 유전자의 GAA 반복 확장으로 인한 늦은 발병(5-7번째 10년, median 60세, 범위 21-87세) 느린 진행성 형태입니다. 주요 증상은 보행 실조(95%), 안구 증상(80%)으로, 알코올/활동으로 유발되는 조기 현기증/복시가 특징입니다. 2024-2025년 연구(Pellerin D et al.)에서 SARA 진행 속도(연간 0.23-0.40점, SCA6 0.80점보다 느림)를 확인하며, 휠체어 사용이 드물고, 에피소딕 증상(약 50%) 후 천천히 악화됩니다. 장기 시퀀싱 진단 발전과 자연 경과 연구 업데이트가 강조됩니다. |
- 반복 확장 돌연변이(Repeat Expansion Disorders): CAG 반복 확장으로 인한 폴리글루타민(polyQ) 단백질 축적 (e.g., SCA1(ATXN1), SCA2(ATXN2), SCA3(ATXN3, 가장 흔함), SCA6(CACNA1A), SCA7(ATXN7, 망막 퇴행 동반), SCA17(TBP)). 비코딩 반복 (e.g., SCA8(ATXN8OS), SCA10(ATXN10), SCA12(PPP2R2B), SCA31(BEAN1), SCA36(NOP56), SCA37(DAB1)).
- 점 돌연변이(Point Mutations): 미스센스나 결실 (e.g., SCA5(SPTBN2), SCA11(TTBK2), SCA13(KCNC3), SCA27A(FGF14 missense), SCA35(TGM6), SCA42(CACNA1G), SCA48(STUB1)).
- 기타: Dentatorubral-pallidoluysian atrophy (DRPLA, ATN1 CAG 반복), Episodic Ataxias (EA1-9, e.g., EA1(KCNA1), EA2(CACNA1A), 발작성 증상 중심).
- 최근 연구에서 SCA27B(FGF14의 GAA 반복 확장)는 성인 발병 유전성 아탁시아의 9-61%를 차지하는 빈번한 원인으로 밝혀졌습니다. 이는 유럽 코호트에서 SCA3(19%), SCA1(12%)와 비슷한 빈도입니다.
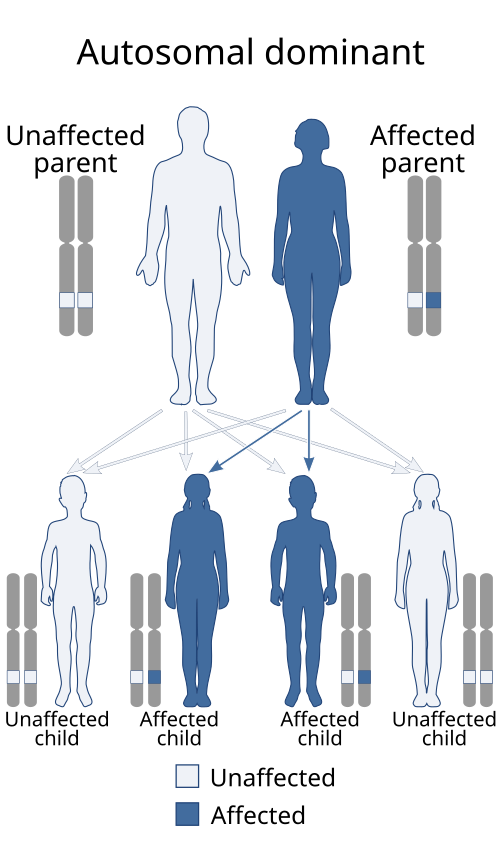
2. 상염색체 열성 유전(Autosomal Recessive Cerebellar Ataxias, ARCAs)
200개 이상의 유전자가 관여하며,
주로 어린 시절이나 청소년기 발병.
전 세계 유병률 3-6/100,000.

- 반복 확장: Friedreich's ataxia (FRDA, FXN의 GAA 반복, 가장 흔함, 1:40,000), CANVAS (RFC1의 AAGGG 반복, 감각 신경병증+전정 반사 소실).
- DNA 수복/유지: Ataxia-telangiectasia (AT, ATM), Ataxia with oculomotor apraxia (AOA1/APTX, AOA2/SETX).
- 효소/기질 대사: Cerebrotendinous xanthomatosis (CTX, CYP27A1), Adult polyglucosan body disease (APBD, GBE1), Vitamin E deficiency (AVED, TTPA).
- 미토콘드리아 관련: COQ8A ataxia (CoQ10 결핍), POLG-related disorders.
- 기타: ARSACS (SACS, 칼슘 신호 장애), Niemann-Pick type C (NPC1, 리소좀 저장 장애).
- 최근 WGS(whole-genome sequencing)로 RFC1 관련 질환이 FRDA 다음으로 흔한 ARCA로 확인되었으며, 25-50%의 진단율을 보입니다.
3. X-연관 유전(X-linked):
드물며, Fragile X-associated tremor/ataxia syndrome (FXTAS, FMR1 premutation, 남성에서 5번째 10년에 발병).
4. 미토콘드리아 유전(Mitochondrial):
POLG, OPA1 등, 모계 유전.
cf) https://m.blog.naver.com/sangmi001/222483894631



전체적으로 유전성 아탁시아의 33-50%에서 유전자 진단이 가능하며,
WES/WGS가 1차 검사로 사용됩니다.
최근 연구에서 반복 확장 분석과 WES 병용 시 진단율이 50%까지 상승합니다.
유전성 소뇌실조증의 예후
유전성 소뇌실조증은 대부분 진행성으로,
발병 후 10-20년 내에 보행 보조기나 휠체어가 필요해지며,
생존 기간은 유형에 따라 다릅니다.
구체적인 예후는 다음과 같습니다:
- 일반적 특징: 천천히 진행하나, 다계통 침범(신경병증, 피라미드 증상, 인지 장애, 심장/호흡 문제)으로 삶의 질 저하. SARA(Scale for the Assessment and Rating of Ataxia) 점수로 진행 추적 (e.g., ARCA에서 연간 0.6-1.3점 상승).
- ADCAs/SCAs: SCA1(빠른 진행, 15년 내 휠체어, 생존 감소), SCA2/SCA3(중간 진행, 파킨슨증상 동반), SCA6(느린 진행, 순수 소뇌 증상, 좋은 예후), SCA7(망막 문제로 시력 상실). SCA27B: 5-7번째 10년에 발병, 연간 SARA 0.23-0.40점 진행 (SCA6 0.80점보다 느림), 휠체어 사용 드물며, 증상(보행 실조 95%, 안구 증상 80%)이 천천히 악화. 조기 증상(현기증, 복시)은 알코올/활동으로 유발.
- ARCAs: FRDA(청소년 발병, 심근병증/당뇨 동반, 10년 내 휠체어, 생존 35-50년), AT(암/면역결핍 위험, 짧은 생존), CANVAS(늦은 발병, 느린 진행), CTX(조기 치료 시 안정화 가능). 반복 길이와 발병/중증도 상관성 약함.
- 기타: FXTAS(진행성 떨림/인지 저하), 미토콘드리아 형태(변동성 높음).
| FRDA (Friedreich's Ataxia)진행 과정 분류 및 요약 FRDA는 FXN 유전자의 GAA 반복 확장으로 인한 가장 흔한 상염색체 열성 소뇌실조증으로, 청소년기 발병(보통 10-16세, 늦은 발병은 25세 이후)이 특징입니다. 관련 증상으로는 심근비대증(심근병증)과 당뇨가 동반되며, 진행성으로 보행 및 사지 실조, 발음 장애, 깊은 감각 상실, Babinski 징후, pes cavus 등이 나타납니다. 치료 미실시 시 mFARS 점수 연간 2-2.2점 상승(성인 기준, 청소년기 더 빠름)으로, 발병 후 10-15년 내에 보행 보조기나 휠체어가 필요해집니다. 예후는 진행성 신경퇴행으로 생존 기간 평균 35-50년(약 38년)이며, 심장 합병증이 주요 사망 원인입니다. 반복 길이와 발병/중증도 상관성은 일반적으로 역상관(긴 반복=조기 발병/중증)이지만, 일부 코호트(예: 세르비아 30명)에서 발병 연령과 유의미한 상관 없음(p=0.552 for GAA1). AT (Ataxia-Telangiectasia) 진행 과정 분류 및 요약 AT는 ATM 유전자 돌연변이로 인한 희귀 질환(1/100,000)으로, 2세경 소뇌 실조로 시작해 3세경 안구피부 모세혈관확장증이 나타납니다. 진행은 심각하며, 보통 10-12세에 독립 보행 상실(휠체어 필요)로 이어집니다. 예후는 나쁘며, 중간 생존 기간 17-25년으로 성인기 초반 이후 생존 드물며, 사망 원인으로는 림프종 등 암 위험이 높고, 세포/체액 면역결핍으로 인한 반복 감염(부비동폐염, 림프구감소, IgA 결핍)이 주요합니다. 방사선에 의한 DNA 손상 민감성으로 암 발생 위험이 증가합니다. CANVAS (Cerebellar Ataxia with Neuropathy and Vestibular Areflexia Syndrome) 진행 과정 분류 및 요약 CANVAS는 RFC1 유전자의 AAGGG 반복 확장으로 인한 늦은 발병(성인 후반) 진행성 신경퇴행 질환으로, 소뇌 실조, 감각 신경병증, 양측 전정 반사 소실이 특징입니다.  초기 증상은 균형 장애와 현기증이며, 느린 진행으로 10년 후 약 50%가 지팡이 사용, 15년 후 25%가 휠체어 필요합니다. 예후는 천천히 악화되며, 운동 기능 저하가 주요이나 생명 위협은 적습니다. 확장 크기와 안구 운동 이상 등 중증도 상관성이 관찰됩니다. CTX (Cerebrotendinous Xanthomatosis) 진행 과정 분류 및 요약 CTX는 CYP27A1 유전자 돌연변이로 인한 희귀 지질 저장 장애로, 진행성 신경 증상(실조, 인지 저하), 건황색종, 백내장, 설사 등이 나타납니다. 치료 미실시 시 심각한 만성 진행으로 평생 관리가 필요하며, 진단 지연(평균 5.7년, 성인 11.6년)이 합병증 증가 원인입니다. CDCA(chenodeoxycholic acid) 치료 시 조기 시작(28세 이전)이 핵심으로, 28세 전 치료 시 100% 신경 안정화, 24세 전 시작 시 유의미한 예후 개선(중간 추적 8년); 늦은 치료라도 일부 이점 있으나 비가역적 손상 위험. 2025년 FDA 승인 약물(ctexli, chenodiol)이 삶의 질 개선 확인. 반복 길이와 발병/중증도 상관성 요약 유전성 소뇌실조증에서 반복 길이와 발병/중증도 상관성은 유형에 따라 다르지만, 약함으로 보입니다. FRDA에서 일반적으로 긴 GAA 반복이 조기 발병/중증과 상관되나, 세르비아 코호트 연구(2025)에서 발병 연령과 유의미한 상관 없음(p>0.5); 더 긴 GAA1은 Babinski 징후, GAA2는 진동 감각 장애와 연관. 다른 형태(ZFHX3, GAA-FGF14)에서는 긴 반복이 조기 발병/중증과 상관. 전체적으로 반복 길이가 예측 인자이나, 완전하지 않음 |
MRI: 소뇌 위축(vermian atrophy) 흔함. 병리: Purkinje 세포 손실.
최신 논문 보완
주어진 논문(PMC10819088)은 SCA27B의 분류(상염색체 우성, FGF14 GAA 반복 >250)와 예후(늦은 발병, 느린 진행, 4-아미노피리딘(4-AP)으로 증상 개선 가능)를 중점으로 다루며, 이는 최근 연구에서 성인 발병 유전성 아탁시아의 주요 원인으로 강조됩니다.
2024-2025 최신 리뷰에서 보완:
- 분류 업데이트: SCA27B의 빈도가 높아 (프랑스 캐나다인 61%), RFC1이 ARCA의 2위 원인. 새로운 유형 (e.g., SCA49/SAMD9L, SCA50/NPTX1) 발견, WGS로 미진단 케이스 33% 해결.
- 예후 및 치료 발전: 진행 속도 느림 (e.g., ARSACS/SPG7 0.6-0.9 SARA/년). FRDA에 omaveloxolone(FDA 승인, Nrf2 활성화로 진행 지연). ASO/RNAi/CRISPR로 유전자 치료 시도 (e.g., SCA3/ASO 임상, FRDA/AAV gene therapy). 4-AP, riluzole 등 증상 치료 효과 확인 (e.g., SCA27B, 다양한 SCA에서 SARA 개선). 재활 치료로 장기 효과 (e.g., 연간 집중 재활로 순수 소뇌 아탁시아 안정화).
소뇌실조증의 임상 분류 (Spinocerebellar Ataxia 중심) 주어진 논문(PMC6373366, "Spinocerebellar ataxia: an update", 2019년)은 주로 자배 우성 소뇌실조증(autosomal dominant cerebellar ataxias, ADCAs)인 spinocerebellar ataxias(SCAs)를 중점으로 다루며, Harding의 분류 체계를 기반으로 임상적으로 분류합니다. SCAs는 유전적으로 이질적이며, 보행 실조(gait ataxia), 안구 진탕(nystagmus), 발음 장애(dysarthria)를 핵심 증상으로 하며, 추가 증상(피라미드 징후, 추체외로 징후, 인지 장애 등)에 따라 분류됩니다. 논문은 Harding의 ADCA 분류를 여전히 유용하다고 강조하며, 다음과 같이 나눕니다:
또한, International Parkinson and Movement Disorders Task Force의 새로운 제안으로 '순수 또는 상대적으로 순수한 실조(pure or relatively pure ataxia)'와 '복합 실조(complex ataxia)'로 나누는 분류를 소개하며, 이는 Harding 분류와 중복됩니다. 진단 접근으로는 phenotype-first (임상 증상 우선) 방식을 권장합니다. 소뇌실조증의 예후 논문에 따르면, SCA의 예후는 subtype과 기저 기전에 따라 다양하며, 대부분 천천히 진행성입니다. 채널병증(channelopathies, e.g., CACNA1A, CACNA1G, KCND3, KCNC3 관련 SCA)은 가장 이른 발병 연령, 가장 긴 질병 지속 기간, 가장 느린 진행 속도, 가장 순수한 소뇌 증상을 보이는 좋은 예후를 가집니다. SCA2에서는 미토콘드리아 기능 장애와 산화 스트레스가 증상 전에 나타나며, 항산화제(예: coenzyme Q10)로 개선 가능성을 시사합니다. 유전 수정자(genetic modifiers, e.g., FAN1, PMS2의 SNPs)는 polyglutamine SCA의 발병 연령을 영향을 미칩니다. 비-polyglutamine 또는 신규 SCA에 대한 구체적 예후는 제한적이며, 일반적으로 진행 속도에 초점을 맞춥니다. 최신 논문 보완 (2024-2025) 최근 리뷰에서 SCA는 50개 이상의 subtypes로 확장되었으며, 돌연변이 유형에 따라 분류: 반복 확장(repeat expansions) - polyQ CAG 확장 (SCA1,2,3,6,7,17)과 비코딩 반복 (SCA8,10,12,31,36,37); 점 돌연변이(point mutations, 대부분 missense). 최근 발견된 SCA27B (FGF14의 GAA 반복 확장)는 후기 발병 late-onset ataxia로, 전사 간섭과 FGF14 기능 상실을 유발합니다. 분류는 반복 확장 vs. 비반복 돌연변이로 나뉘며, 분자 유전자 검사(진단 바이오마커)가 subtype 확인에 핵심입니다. 예후 측면에서 CAG 반복 길이는 polyQ SCA (SCA1,2,3,6)에서 발병 연령 변동의 44-75%를 설명하며, 긴 반복은 빠른 진행과 약한 상관성을 보입니다. SCA27B에서 반복 길이와 발병 연령 관계는 미확립. 바이오마커 발전: 혈중 neurofilament light chain (NfL)은 축삭 손상을 나타내며, ataxic/pre-ataxic 단계에서 상승; 높은 NfL은 SCA1에서 실조 전환 시간 단축, SCA2에서 소뇌 용적 감소 예측. MRI (pons, cerebellum, brainstem 용적)는 진행 예측에 민감하며, pre-ataxic SCA3에서 변화 감지. 예후 개선으로 치료 시도: trehalose (SCA3에서 SARA 점수 개선), 유전자 편집, RNA 간섭, antisense oligonucleotides 등 임상 시험 중; SCA3/MJD에 대한 증상 관리로 삶의 질 향상, 최근 SCA 치료제 troriluzole이 진행 50-70% 지연 효과 보이나 FDA 완전 응답 서한 발급. 전체적으로 예후는 젊은 발병, 짧은 질병 기간, 낮은 SARA 점수가 더 나은 반응과 관련. |
<!--br {mso-data-placement:same-cell;}-->
연관 임상 증상유전 subtypes
| 말초 신경병증 | SCA1, 2, 3, 4, 18, 25, 38, 43, 46 |
| 피라미드 징후 | SCA1, 3, 7, 8, 10, 14, 15, 17, 35, 40, 43 |
| 근긴장 이상(dystonia) | SCA3, 14, 17, 20, 35 |
| 근간대성 근경련(myoclonus) | SCA14 |
| 파킨슨증상 | SCA2, 3, 10, 14, 17, 19/22, 21 |
| 떨림(tremor) | SCA12, 15, 27 |
| 무도증(chorea) | SCA17, 27, DRPLA |
| 인지 장애 | SCA2, 8, 13, 17, 19/22, 21, 36, 44, DRPLA |
| 정신 증상 | SCA2, 17 |
| 안근 마비(ophthalmoplegia) | SCA2, 3, 28, 40 |
| 시각 장애 | SCA7 |
| 안면/설근 섬유속수축(fasciculation) | SCA36 |
| 어류피부증(ichthyosiform plaques) | SCA34 |
| 발작 | SCA10, 19/22, ATN1 |
| 기면증(narcolepsy) | DNMT1 |
| 청력 상실 | SCA31, 36, DNMT1 |
J Neurol
. 2018 Oct 3;266(2):533–544. doi: 10.1007/s00415-018-9076-4
Spinocerebellar ataxia: an update
Roisin Sullivan 1,✉, Wai Yan Yau 1, Emer O’Connor 1, Henry Houlden 1
- Author information
- Article notes
- Copyright and License information
PMCID: PMC6373366 PMID: 30284037
Abstract
Spinocerebellar ataxia (SCA) is a heterogeneous group of neurodegenerative ataxic disorders with autosomal dominant inheritance. We aim to provide an update on the recent clinical and scientific progresses in SCA where numerous novel genes have been identified with next-generation sequencing techniques. The main disease mechanisms of these SCAs include toxic RNA gain-of-function, mitochondrial dysfunction, channelopathies, autophagy and transcription dysregulation. Recent studies have also demonstrated the importance of DNA repair pathways in modifying SCA with CAG expansions. In addition, we summarise the latest technological advances in detecting known and novel repeat expansion in SCA. Finally, we discuss the roles of antisense oligonucleotides and RNA-based therapy as potential treatments.
Electronic supplementary material
The online version of this article (10.1007/s00415-018-9076-4) contains supplementary material, which is available to authorized users.
Keywords: Spinocerebellar ataxia, Molecular diagnosis, Next-generation sequencing
Introduction
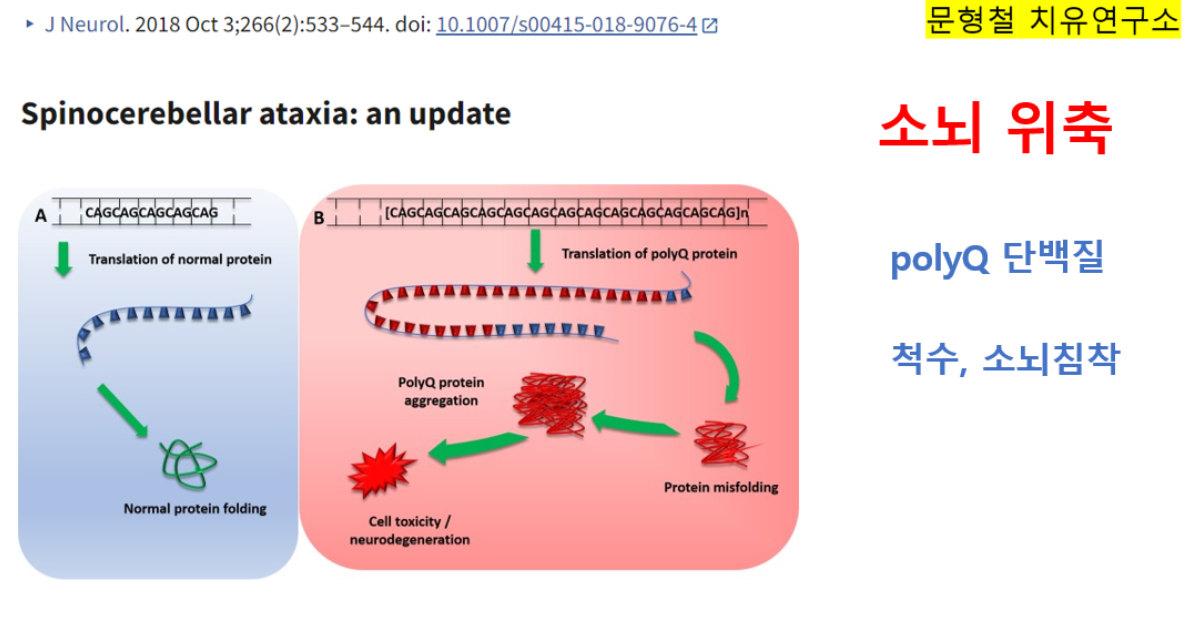
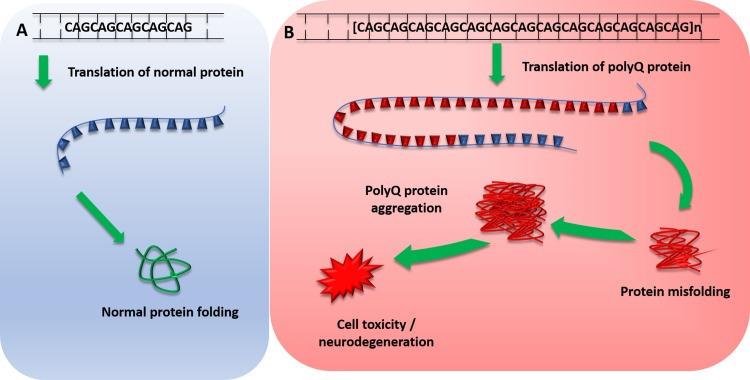
The Spinocerebellar ataxias (SCA) are a subset of hereditary cerebellar ataxias that are autosomal dominantly transmitted. They are progressive neurodegenerative diseases that share the clinical features of ataxia, which arise from the progressive degeneration of the cerebellum but can also affect other connected regions, including the brain stem. They are a highly heterogenous group of disorders with a complex genotype–phenotype spectrum; many SCAs are caused by CAG nucleotide repeat expansions that encode polyglutamine, and therefore, involve the toxic polyglutamine protein (polyQ) (Fig. 1) [1]. Recent advances in next-generation sequencing have identified new genes implicated in SCAs providing insights into disease transmission and pathogenesis. Here, we discuss updates in epidemiology, clinical features, molecular mechanisms and their potential implications in the future.
Fig. 1.
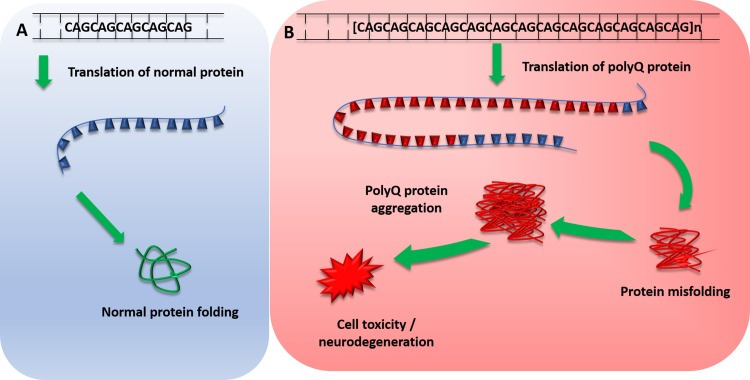
Mechanism of polyglutamine protein expansion repeats. a Normal translation of polyglutamine repeat within normal repeat range, producing normal protein transcript and protein folding. b Pathogenic polyglutamine expansion repeat length leads to translation of expanded abnormal PolyQ repeat, which leads to protein misfolding. Misfolded polyQ proteins form aggregates which lead to various cellular process dysfunctions, leading to cell toxicity and degeneration. PolyQ polyglutamine proteins
What is new in the epidemiology of SCA and its subtypes?
A recent systemic review shows that the global prevalence of SCA is 3 in 100,000 [2], however, a wide regional variation exists. SCA3 is commonest subtype around the globe [3–5], SCA2 is more prevalent in Cuba than SCA3 whilst SCA7 is the most frequent subtype in Venezuela due to strong founder’s effect [6, 7]. SCA6 is one of the most common ADCA in the North of England, with a global prevalence of 5.2/100,000 [8]. There are various mutations described in SCA, although repeat expansions still account for almost half of SCA diagnosis in European cohort. In 412 undiagnosed autosomal dominant cerebellar ataxia (ADCA) without known repeat expansion, 59 individuals (14.3%) were found to harbor pathogenic variants [9]. Thirty five of these variants (8.5%) belong to channel genes. In contrast, conventional mutations in channel genes are rare in Han Chinese cohort [10]. In another cohort of 194 individuals with undiagnosed ADCA, SCA14 accounts for 6.7% of the studied population [11]. Other similar studies in Germany, United Kingdom, France, United States, Japan and Taiwan confirm the relative rarity of SCA 8, 23, 35, 36 and 42. They are each responsible for less than 1% of undiagnosed ADCA [12–18] although the advance in diversity genetics will further reveal the frequency of these genes in other populations.
Make sense of SCA clinical features
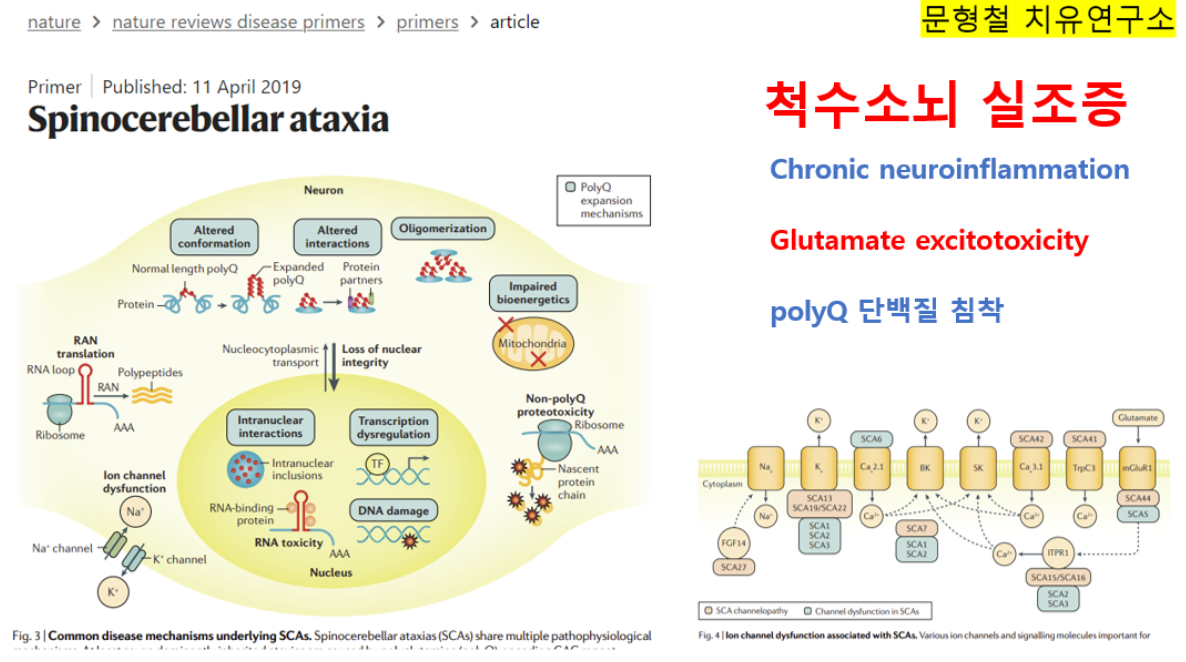
The core triad of symptoms of SCAs include gait ataxia and incoordination, nystagmus/visual problems and dysarthria. Patients can present with additional features such as pyramidal, extrapyramidal signs, ophthalmoplegia and cognitive impairment in specific SCAs. Harding’s classification of ADCA in 1982 is still useful in the clinical setting (Fig. 2) [19]. ADCA type 1 describes cerebellar ataxia with variable additional signs. This list is ever expanding and includes SCA1–4, 8, 10, 12–14, 15, 17–22, 25, 27, 28, 31, 32, 34–37, 38, 42–44, 46, 47, ataxia with DNMT1 and DRPLA [20–23]. ADCA type 2 describes cerebellar ataxia with pigmentary macular degeneration and consists of only SCA 7 [20]. ADCA type 3 refers to ‘pure’ cerebellar ataxia, which includes SCA 5, 6, 11, 23, 26, 30, 37, 41 and 45 [20, 24]. Several SCAs have characteristic clinical features in addition to cerebellar ataxia and helps distinguish them from other subtypes. For instance, SCA 12, 15 and 27 have upper limb postural tremor [25–27]; SCA 14 may have myoclonus and task-specific dystonia [28]; and a subset of SCA 36 have facio-lingual fasciculation with sensorineural hearing loss [29]. Table 1 shows a non-exhaustive list of distinctive clinical signs that feature prominently with cerebellar ataxia, adapted from a recent systematic review [30]. International Parkinson and Movement disorders Task Force recently proposes a new classification of SCA, dividing them into pure or relatively pure ataxia and complex ataxia, which overlaps with above mentioned ADCA classification [31]. A phenotype-first approach remains pertinent in molecular diagnosis of rare genetic disorders (Fig. 3) [32]. Clinicians should also consider genetic testing for primary episodic ataxias (EA), especially with history of episodic attacks of imbalance, dysarthria, vertigo and/or diplopia lasting hours–days. EAs are autosomal dominant channelopathies and they mostly manifest before age 20 years [33]. They can be associated with other paroxysmal neurological disorders such as migraines, epilepsy and dystonia. Patients with EA type 1 also have interictal myokymia. However, progressive cerebellar ataxia may also occur in a proportion of patients with non-expansion mutations in KCNA1 and CACNA1A, especially later in the disease course [34, 35]. Unfortunately, the utility of clinical–genetic classification in SCA is limited by high level of phenotype–genotype overlap.
Fig. 2.
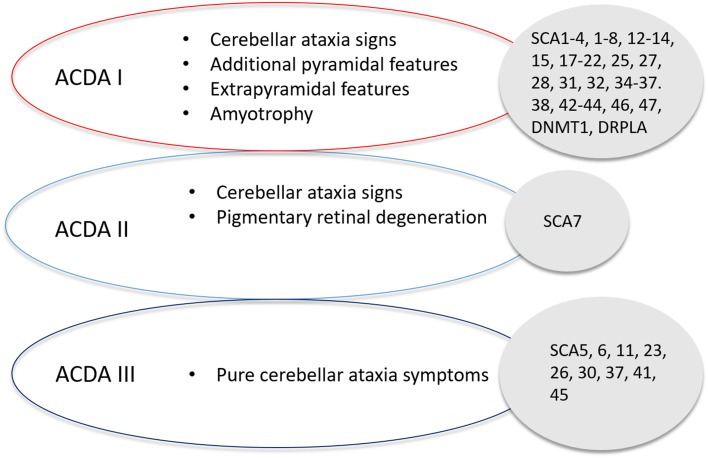
Harding’s classification of Spinocerebellar Ataxia, detailing the classification of SCA based on symptom presentation and the associated SCAs with that classification
Table 1.
SCA subtypes with associated clinical signs that feature prominently with cerebellar ataxia
Associated clinical featuresGenetic subtypes
| Peripheral neuropathy | 1, 2, 3, 4, 18, 25, 38, 43, 46 |
| Pyramidal signs | 1, 3, 7, 8, 10, 14, 15, 17, 35, 40, 43 |
| Dystonia | 3, 14, 17, 20, 35 |
| Myoclonus | 14 |
| Parkinsonism | 2, 3, 10, 14, 17, 19/22, 21 |
| Tremor | 12, 15, 27 |
| Chorea | 17, 27, DRPLA |
| Cognitive impairment | 2, 8, 13, 17, 19/22, 21, 36, 44, DRPLA |
| Psychiatric symptoms | 2, 17 |
| Ophthalmoplegia | 2, 3, 28, 40 |
| Visual impairment | 7 |
| Face/tongue fasciculation | 36 |
| Ichthyosiform plaques | 34 |
| Seizures | 10, 19/22, ATN1 |
| Narcolepsy | DNMT1 |
| Hearing loss | 31, 36, DNMT1 |
ATN1 atrophin 1, mutation responsible for dentatorubral–pallidoluysian atrophy, DNA methyltransferase 1, mutation responsible for ADCA-deafness and narcolepsy
Fig. 3.
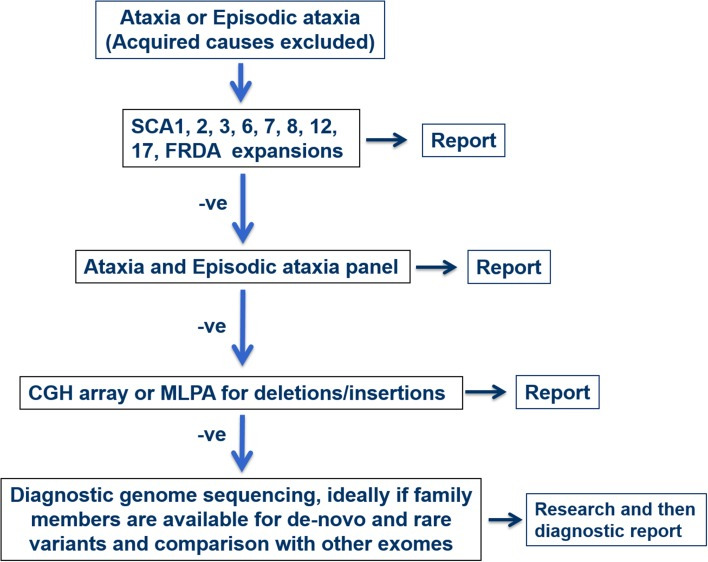
Flowchart of diagnosis pathway based on either positive or negative result of each diagnostic test. −ve—negative
Advances in molecular diagnoses and disease mechanisms
Advances in next-generation sequencing (NGS) have facilitated further insights into the molecular causes of SCA. When NGS becomes translated to clinical practise, it has the potential to increase the success of molecular diagnosis for patients currently undiagnosed by standard genetic testing. Recent successes include conventional mutations in CCDC88C, TRPC3, CACNA1G, MME, GRM1, FAT2, PLD3 and PUM1 [23, 24, 36–40] since the last update in this journal in 2015 (see Table 2) [41]. Exome sequencing has an average diagnostic rate of 36% compared to target resequencing panel of 17% [42]. Exome sequencing identifies less classical phenotype–genotype correlations and detects new mutations in known cerebellar genes [43]. We outline the roles of toxic RNA gain-of-function, mitochondrial defects, channelopathy, autophagy and transcriptional dysregulation in pathogenesis of SCA.
Table 2.
Summary of major clinical characteristics of novel SCA genes described since 2015
Gene/locusMutationNumber of pedigreesClinical featuresPathogenic mechanisms
| CCDC88C (SCA 40) [36] | Missense c.1391G>A (pR464H) | 4 Probands from 1 family | Cerebellar ataxia, hyperreflexia | JNK pathway hyperphosphorylation induced cellular apoptosis |
| TRPC3 (SCA 41) [37] | Missense c.2285G>A (pR762H) | 1 Proband from 1 family | Cerebellar ataxia | Toxic gain-of-function, channelopathy |
| CACNA1G (SCA 42) [38, 57] | Missense c.5144G>A (pR1715H) | 30 Probands from 5 families | Cerebellar ataxia | Haplo-insufficiency of T-type calcium channel |
| MME (SCA 43) [39] | Missense c.428G>A (p.C143Y) | 7 Probands from 1 family | Cerebellar ataxia with peripheral neuropathy | Haplo-insufficiency of neprilysin, a zinc-dependent metalloproteinase |
| GRM1 (SCA 44) [40] | Missense c.2375A>G (p.Y792C) c.785A>G (p.Y262C) | 7 Probands from 2 families | Cerebellar ataxia with pyramidal sign | Toxic gain-of-function metabotropic glutamate receptor 1 |
| FAT2 (SCA 45) [24] | Missense c.10946G>A (p.R3649Q) c.10758G>C (p.K3586N) | 6 Probands from 1 family | Cerebellar ataxia | ?affect cell adhesion |
| PLD3 (SCA 46) [24] | Missense c.923T>C (L308P) | 11 Probands from 1 family | Cerebellar ataxia with peripheral neuropathy | Haplo-insufficiency of phospholipase D activity |
| PUM1 (SCA 47) [23] | Missense g.31414862 T>A (p.T1035S) | 9 Probands from 1 family | Cerebellar ataxia | Haplo-insufficiency of PUM1 |
CCDC88C coiled-coil domain containing 88C, JNK c-Jun N-terminal kinase, TRPC3 transient receptor potential cation channel subfamily C member 3, CACNA1G voltage sensor S4 segment of domain IV in Cav3.1T-type calcium channel protein MME neprilysin, GRM1 glutamate metabotropic receptor 1, FAT2 FAT atypical cadherin 2, PLD3 phospholipase D3, PUM1 RNA-binding protein Pumilio1
Toxic RNA gain-of-function
Non-coding repeat expansions are implicated in SCA subtypes that include SCA 8, 10, 12, 31, 36 and 37. The hallmarks are transcribed nuclear accumulations of repeat RNA-binding proteins that can cause RNA toxicity and lead to disease pathogenesis (see Fig. 4) [44]. An intronic ATTCT pentanucleotide repeat expansion in ATXN10 has been found to cause SCA10, with pathogenicity in the range of 800–4500 repeats [45]. Cytoplasmic and nuclear foci form in SCA10 cells and SCA10 transgenic mice brain from degradation-resistant, aggregated AUUCU RNA, which is formed from the splicing out of intron 9 from ATXN10 pre-mRNA. The expanded AUUCU RNA binds to heterogeneous nuclear ribonucleoprotein K (hnRNPK), a spicing factor, causing its sequestration and loss of function. Ultimately this leads to the mitochondrial accumulation of protein kinase C δ (PKCδ) and apoptosis of SCA10 cells [46]. An intronic GGCCTG hexanucleotide repeat expansion was found in the gene NOP56 using genome-wide linkage, responsible for SCA 36. Patients’ lymphoblastoid cells contain RNA foci, and transcription of MIR1292, a neighbouring miMRA is reduced. These implicate a toxic RNA gain-of-function pathological mechanism in SCA 36 [29]. A toxic gain of function effect was found to be implicated in SCA8 pathogenesis, with the CTG CAG repeat expansion, which is bidirectionally expressed, causing [CUG]n transcript accumulation of ribonuclear inclusions that localise with the RNA binding protein Mbnl1. The downstream effects and alternative splicing contribute to the movement disorder phenotype in a SCA8 mice model [47]. Bidirectional expression of sense [CUG]n and antisense [CAG]n has been found in other disorders such as at the DM1 locus [48] and in HDL2, which also forms ribonuclease inclusions [49]. Most recently an unstable intronic ATTTC repeat has been identified as the pathogenic cause of SCA37 in two Spanish cohorts, dysregulating reelin adaptor protein disabled-1 coding DAB1 expression, leading to alternative splicing, an RNA switch and an upregulation of reelin-DAB1 signalling in the SCA37 cerebellum [50].
Fig. 4.
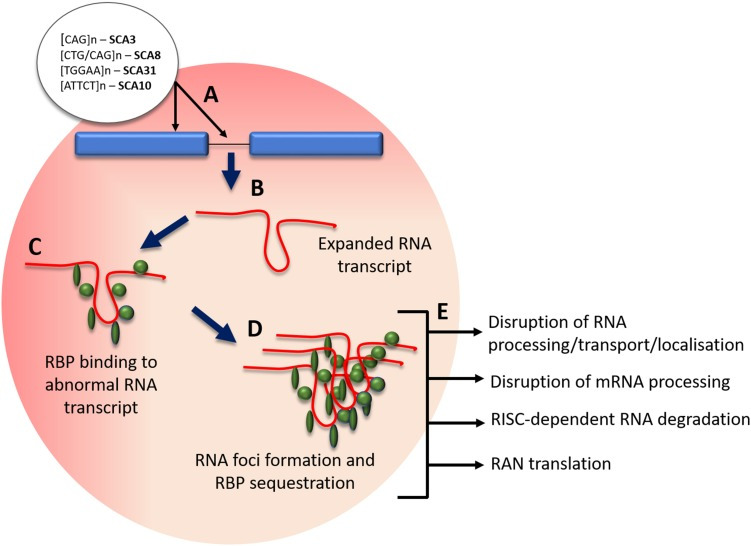
Mechanism of RNA foci formation and effects. a Pathogenic SCA intronic and exonic expansion repeats; b transcription of expanded repeat into expanded mRNA/pre-RNA; c binding of regulatory binding proteins (RBP) to abnormal mRNA transcript; d RBP protein sequestration and abnormal transcript aggregation; e effects of RBP sequestration on cellular processes. RBP regulatory binding proteins, RAN repeat-associated non-ATG
Mitochondrial dysfunction
Recently advances have been made in the understanding of mitochondrial dysfunction and ataxia, with several genes related to mitochondrial function identified. These include mutations in OPA1, a mitochondrial dynamin-like GTPase and POLG [9], coding for the catalytic subunit of mitochondrial DNA polymerase gamma, as well as MTATP6 [22]; both of which involve mitochondrial dysfunction. Functional mitochondria are required for all cell processes including cell proliferation, differentiation, apoptotic cell death and are a crucial part of signalling cascades [51]. Findings have suggested that mitochondrial dysfunction and reactive oxygen species (ROS) may be implicated in SCA2 pathogenesis, with an increase in mitochondrial oxidative stress in SCA2 patient fibroblasts, as well as changes in mitochondrial respiratory chain (MRC) enzymes and in mitochondrial morphology. Importantly these effects were increased in a SCA2 fibroblast line with an expanded CAG repeat from a patient yet to exhibit clinical symptoms, suggesting that mitochondrial dysfunction may precede disease onset. Furthermore, the antioxidant coenzyme Q10 (CoQ10) was found to improve oxidative stress in fibroblasts [52]. Oxidative damage is a frequent feature of neurodegenerative conditions, including Alzheimer’s disease and Parkinson’s disease, due to the brains high oxygen utilisation and high content of oxidisable polyunsaturated fatty acids [53]. The polymorphic locus A100398G has been implicated in early age of onset in Cuban SCA2 patients [54] and has recently been described in a more severe cognitive phenotype in a 42 SCA2 cohort, corroborating with prior findings in the Cuban cohort [55].
Channelopathies
Mutations coding for ion channel subunits or regulatory proteins, channelopathies, are frequently involved in the pathology of SCAs. Inherited channelopathies can alter ion channel function by mechanisms including inhibiting the ion movement through an open channel pore and altering ion channel gating through changes to channel opening processes or inactivation processes [56]. A recent study using amplicon-based panel sequencing for 65 genes on 412 index patients found that channelopathies (with mutations in CACNA1A, CACNA1G, KCND3 and KCNC3 in particular) had the earliest age of onset, longest disease duration and slowest disease progression, as well as purest cerebellar presentation. Interestingly they found a prominent implication of SPG7 and CACNA1A in ataxic point mutations [9].
In fact, several newly described SCA gene mutations (GRM1, CACNA1G, TRPC3) are implicated on physiological functions of the cellular channels [37, 38, 40, 57]. Transient receptor potential C3 (TRPC3) mutation occurs in a highly conserved region of the nxxxxonselective cation channel and is proposed to affect regulation of channel gating [37]. It has only been described in a sporadic case of adult-onset cerebellar ataxia. Calcium voltage-gated channel subunit alpha 1 G (CACNA1G) mutation leads to altered physiological property in the low voltage-gated calcium channel and causes a relatively pure cerebellar ataxia [38, 57]. CACNA1G channels are most prominently expressed in Purkinje cells and deep cerebellar nuclei. Electrophysiological study demonstrates that the mutation shifts the current–voltage and the steady-state activation curves of the mutant transfected cells positively [57]. In silico this causes decreased neuronal excitability. Glutamate metabotropic receptor 1 (GRM1) gain-of-function mutations result in excessive glutamate receptor signalling and intracellular calcium level [40]. The authors hypothesised excitotoxicity in cerebellar Purkinje cells as the cause of ADCA in two families.
Autophagy
Autophagy is one of the main pathways for degradation of misfolded proteins; the other is the ubiquitin–proteasome system. Autophagy has been linked to neurodegeneration [58]. Both of SCA 3 fibroblast and SCA 7 mouth model demonstrate impaired autophagy [59, 60]. A mammalian target of rapamycin (mTOR) inhibitor that upregulates autophagy clears ataxin-3 and aggregates in brain in a SCA 3 mouse model. This appears to slow motor deterioration [61]. Until recently, it remains unclear whether the misfolded protein accumulation causes the dysfunction of autophagy or whether impaired autophagy leads to accumulation of misfolded protein. Ashkenazi et al. demonstrate that wide-type ataxin-3 polyglutamine repeat interacts with beclin 1, a key initiator of autophagy [62]. This interaction permits the deubiquitinase activity of ataxin-3 to protect beclin 1 from proteasome-mediated degradation. Thus, it allows autophagy to occur. Long polyglutamine expansion in mutant ataxin-3 competes with wide type ataxin-3 interaction with beclin 1 and leads to impaired starvation-induced autophagy. This highlights the direct role abnormally repeat expansion plays in neurodegeneration of SCA other than protein aggregation.
Transcriptional dysregulation
The molecular mechanisms of number SCAs (SCA 1, 2, 3, 7, 17) involve interference with transcription through different mechanisms [63]. These include protein–DNA interactions, acetylation, phosphorylation and RNA interference. The mutant protein Ataxin-1, involved in SCA1 pathogenesis, is a transcription activator, whilst the polyglutamine expansion of SCA 17 occurs within the TATA box-binding protein (TBP), an essential transcription factor [64, 65]. A recently described mutation in RNA-binding protein Pumilio (PUM1) results in ADCA, coined SCA 47 [23]. Experiments in patients’ fibroblasts demonstrated that PUM1 protein acts as atranscription repressor. Reduced expression of PUM1 suppresses dendritic arborization. Interestingly, the phenotypic severity of PUM1 mutation varies with the degree which the missense mutations/deletion reduce PUM1 protein levels. Around 50% reduction of PUM1 protein leads to a severe syndromic development delay whilst 25% reduction produces adult-onset cerebellar ataxia.
Genetic modifiers
The clinical diversity of the hereditary cerebellar ataxias in terms of age of onset, progression and severity of disease, strongly suggests the presence of modifying factors. We are beginning to dissect the complex mechanisms of genetic modifiers through the delineation of molecular pathways in the era of high-throughput sequencing. Common genetic variants, with a significant effect size, may act as genetic modifiers in rare mendelian conditions such as SCAs. A Genome-Wide Association Studies (GWAS) on Huntington Disease (HD), a (CAG)n expansion disorder, identified three significant loci with enrichment in DNA repair networks [66]. Subsequently, genotyping of these single nucleotide polymorphisms (SNPs) in 1462 subjects with CAG repeat SCAs and HD showed a significant association between DNA repair genes and the age at onset of SCAs and HD, with SNPs in FAN1 and PMS2 reaching the lowest p values [67]. FAN1 is a repair nuclease that is recruited to sites of crosslink damage and PMS2 endonuclease is a mismatch repair (MMR) protein [68, 69]. In another association study of 137 parent–child transmissions in SCA 3, a variant ERCC6 (Cockayne syndrome protein CSB) is associated with an expansion bias of (CAG)n [70]. Tight DNA repair regulation is an integral process to maintain integrity of expansions in replication and translation [71]. Dysregulation of DNA repair genes is postulated to result in somatic expansions in non-expanding cells of SCAs with trinucleotide repeat expansions and subsequent disease progression. An International GWAS of repeat expansion ataxia will be extremely valuable to provide further insights into the genetic factors influencing the clinical characteristics of these disorders.
Advances in diagnosis of repeat expansion disorders
The vast heterogeneity of SCA highlights the effectiveness of whole-exome and -genome sequencing (WES/WGS) as a diagnostic tool. Multiple reads are required for SCAs with conventional disease-causing mutations including single-nucleotide polymorphisms (SNPs), deletions and insertions, to span the full length of the nonreference allele [72]. Currently, high-throughput sequencing technologies are limited to read lengths of approximately 150 base pairs (bp), however, pathogenic expansion repeats can span to thousands of bp in size, therefore, being unidentifiable by short-read sequencing technologies.
Repeat primed-PCR and subsequent fragment analysis is widely used to detect repeat expansions, such as the repeat expansion in the C9orf72 locus implicated in both Amyotrophic Lateral Sclerosis (ALS) and Frontotemporal Dementia (FTD). However, these approaches have a frequent risk of misinterpretation due to false positives and negatives because of indels in the repeat flanking regions and variability in diagnostic laboratory protocols [73]. In addition, repeat length size cannot be estimated by these techniques. The current gold standard for estimating repeat length is Southern blotting which requires substantial amounts of DNA (approximately 10 µg) and is vulnerable to somatic heterogeneity, reducing the precision of size estimation [74]. A novel method named tethering PCR, has recently been proposed by Cagnoli et al. to identify pathogenic expansions and estimate repeat size in SCA1, 2, 3, 6 and 7, as well as recognise large alleles and repeat interruptions, negating the need to perform a secondary test (i.e. Southern blot) [75].
Long-read sequencing technologies such as the Oxford Nanopore sequencing and PacBio single-molecule real-time (SMRT) facilitate the sequencing of more than 10,000 bp DNA sequence lengths. Nanopore sequencing uses a protein nanopore covalently attached to an adaptor molecule to identify unlabeled nucleoside 5′ monophosphate molecules. It has an average accuracy of 99.8%. It can function without exonuclease and has a high accuracy of not registering the same nucleotide twice due to translocation through the nanopore [76]. By contrast, SMRT sequencing uses real-time imaging of fluorescently tagged nucleotides during DNA synthesis along template models, using DNA polymerase as a reaction driver [77]. PacBio reads have around a 15% average higher error rate [78], however, Oxford Nanopore platforms also have limitations with their MiniION sequencer having a reported estimated base-calling error rate of 38.2% [79].
Due to the popularity of WGS, developing a method for pathogenic repeat expansions has been of interest. Recently a software package called ExpansionHunter has been developed by Illumina that can determine the size of repeats of varying size, including very large pathogenic expansions much longer than the read length, using PCR-free WGS data [80]. They developed an algorithm able to identify reads in several different conditions; reads that span the full length, reads that fully contain the repeat [‘in-repeat’ reads (IRR)] and repeats that include the repeat and flanking sequence on one side of the repeat. The software correctly classified all C9orf72 expanded samples in an ALS cohort as either expanded, possibly expanded or wild-type as well as 8 other pathogenic repeat expansions, including samples from SCA1 and SCA3 cohorts. The tool facilitates the screening of repeat expansions using a single run of WGS, with PCR-free WGS data. Development is still in progress as currently the software requires an STR to be specified by reference coordinate and repeat motif. However, future association studies and available WGS-data will provide a genome-wide STR database, which will greatly enhance the utility of ExpansionHunter.
Future direction and potential treatments
Advancements in the understanding of pathophysiologic mechanisms facilitate the potential to find new therapeutic targets. Current treatment pipelines involve the use of pharmacological molecules to target affected downstream pathways, as well as genetic therapies to decrease toxic polyQ gene products. The former could benefit a larger proportion of patients if the targeted pathway is implicated in several neurodegenerative diseases, however, if a mutant protein affects multiple cellular processes it could prove difficult to target a vital pathway to provide effective treatment [81].
Antisense oligonucleotides
Antisense oligonucleotides (ASOs) are small single-stranded sequences of DNA that have the gene-targeting effect of reducing levels of toxic protein or making non-toxic modifications which are valuable tools for neurodegenerative diseases [82]. Successful uptake of ASOs facilitates their gene-modulating effects and relies on effective delivery. Mechanisms include lysosomal or endosomal compartmental internalisation, association with high- and low-binding plasma proteins and cellular trafficking.
ASOs decrease the expression of the target protein using Watson–Crick hybridization to bind to complementary mRNA transcripts and recruit RNase H enzymes [83]. ASO-mediated exon skipping has been successfully applied to SCA3 fibroblasts, removing the central 88 amino acid region of the ataxin-3 protein in one study to halt production of potentially toxic cleavage fragments; however, protein-modification effect was at a low-level [84, 85]. ASO therapy targeting SCA3 mice models has also been shown to reduce disease protein levels by greater than 50% in the cerebellum, diencephalon, cervical spinal cord and forebrain, presenting with no signs of microgliosis and astrogliosis, indicating its potential for a well-tolerated preventative therapy despite no observable reduction in motor phenotype [86, 87]. ASO therapy targeting ATXN2 has shown slowed progression of the motor phenotype and improved survival in both SCA2 [81] and ALS transgenic mouse model TDP-43 [88].
RNA based therapy
Therapeutics based on RNA interface (RNAi) harness the cellular mechanism of gene expression silencing to reduce the expression of pathological proteins. Synthetic small interfering RNA (siRNAs) and short hairpin RNA (shRNAs) control the RNAi process of target mRNA enzymatic cleavage in a predictable and consistent action [89]. RNAi has been successfully used to reduce mutant ataxin-7 in SCA7 mouse models in a nonallele- [90] and allele-specific manner [91]. ATXN3 has also been suppressed in Machado–Joseph disease (MJD) mouse models, with reports of improved motor symptoms and neuropathology [92, 93].
Stem cell based therapy
Some stem cell-based therapies have been performed on several cerebellar mutant mice, such as SCA1 mouse models [94–98], with reports of normalized motor deficits and reduced Purkinje cell loss and SCA2 models which showed delayed onset of motor function deterioration [99]. Phase I and II clinical trials, one of which involved intravenously infused human umbilical cord mesenchymal stem cell (UC-MSC) into SCA1, 2 or 3 cohorts (n = 16) reported no transplantation side effects and an improved International Cooperative Ataxia Rating Scale (ICARS) and Berg Balance Scale (BBS) score after 6 months post-transplantation [100]. Another trial on 14 SCA patients using intrathecal injection of UC-MSC reported significant ICARS and Activity of Daily Living Scale (ADL) scores, which decreased after 1 month of treatment, although 8 patients remained stable for 6–9 months post-transplantation [101]. These suggest a potential proof of principle for stem cell therapy as a therapeutic intervention, however, assessment of efficacy and safety requires further clinical trials.
Conclusions
Tremendous scientific progress has occurred in the understanding of spinocerebellar ataxia. Next-generation sequencing has helped improve the diagnostic accuracy of SCAs and discover new disease mechanisms. New technologies such as nanopore and ExpansionHunter may help improve diagnosis of known and new SCAs with repeat expansions in future. As outlined, evidence suggests that genes in DNA repair pathways appear to play a modifying role. An international GWAS of repeat expansion ataxia would be worthwhile to pursue these potential therapeutic targets. Meanwhile, emerging therapies for neurogenetic diseases such as ASOs also provide physicians and patients of SCA hopes of effective treatments in the near future.