beyond reason
어떤 사람에게는 좋은 음식이 어떤 사람에게는 독이 된다
근육검사
루게릭 병은 식이요법, orthomolecular medicine으로 완치가 가능한 질병이다 yes
루게릭 병의 주 원인은 글루텐이다. yes

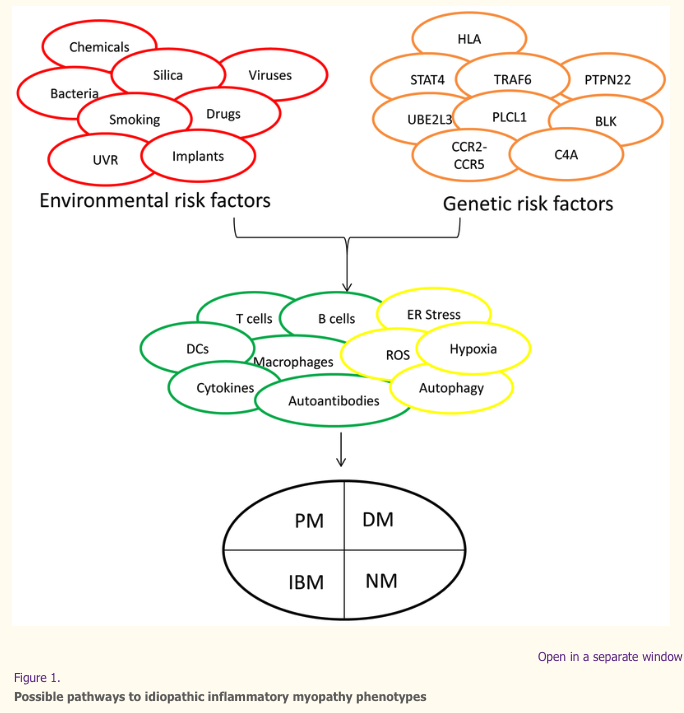
The idiopathic inflammatory myopathies (IIM) probably consist of multiple phenotypes, each of which might be defined by unique combinations of symptoms, signs, and laboratory abnormalities. The major IIM phenotypes—polymyositis (PM), dermatomyositis (DM), necrotizing myopathy (NM), and inclusion body myositis (IBM)—are shown in the black circle at the bottom. Each phenotype could result from different pathogenic mechanisms, as represented by green circles (showing immunologic processes) and yellow circles (showing non-immunologic processes) in the center of the figure, because of the interactions between genetic risk factors (orange circles) and environmental risk factors (red circles). Some combinations of genotypes and environmental exposures induce certain mechanisms and disease phenotypes, whereas other combinations might have no effect or could be protective. C4A, complement 4A; DCs, dendritic cells, ER, endoplasmic reticulum; HLA, human leukocyte antigen; ROS, reactive oxygen species; UVR, ultraviolet radiation.
Infectious and non-infectious agents evaluated in epidemiological studies suggest a wide array of risk and protective factors with different strengths of association with idiopathic inflammatory myopathy (IIM) phenotypes. CTD, connective tissue disease; OR, odds ratio; RR, relative risk. a. Infection in the year before IIM diagnosis. b. Infection in the year before IIM onset. c. Inflammatory lung disease at study inclusion.
Risk Factors and Disease Mechanisms in Myositis
Abstract
Autoimmune diseases develop as a result of chronic inflammation owing to interactions between genes and the environment. However, the mechanisms by which autoimmune diseases evolve remain poorly understood. Newly discovered risk factors and pathogenic processes in idiopathic inflammatory myopathy (IIM) phenotypes have illuminated innovative approaches for understanding these diseases. The HLA 8.1 ancestral haplotype is a key risk factor for major IIM phenotypes in white populations, and genetic risk variants for other autoimmune diseases have been identified as IIM risk factors.
Environmental risk factors are less studied but might include viruses, bacteria, ultraviolet radiation, smoking, occupational and perinatal exposures and a growing list of drugs, biologics, and dietary supplements. Disease mechanisms vary by phenotype, with evidence for shared innate and adaptive immune and metabolic pathways in some phenotypes but unique pathways in others. The heterogeneity and rarity of the IIMs make advancements in diagnosis and treatment cumbersome. Novel approaches, better-defined phenotypes, and international, multidisciplinary consensus have contributed to progress, and hopefully these methods can eventually enable therapeutic intervention before the onset or major progression of disease. In the future, preemptive strategies to IIM management might be possible.
Introduction
The pathomechanisms of most immune-mediated diseases relate to chronic organ inflammation that can be caused by specific interactions between genetic and environmental risk factors. Immune activation in such diseases often involves both innate and adaptive mechanisms, as well as other non-immune mechanisms; however, the details and the interactions of different pathways are usually not clear. The rarity and heterogeneity of the idiopathic inflammatory myopathies (IIM), a group of systemic autoimmune diseases which include polymyositis, dermatomyositis, necrotizing myopathy, myositis that is found in conjunction with other autoimmune diseases, called overlap syndromes, and inclusion body myositis (IBM) (Box 1), have hampered our understanding of their risk factors and pathogenesis. Nonetheless, considerable progress has been made in this area over the last decade (Figure 1).

The idiopathic inflammatory myopathies (IIM) probably consist of multiple phenotypes, each of which might be defined by unique combinations of symptoms, signs, and laboratory abnormalities. The major IIM phenotypes—polymyositis (PM), dermatomyositis (DM), necrotizing myopathy (NM), and inclusion body myositis (IBM)—are shown in the black circle at the bottom. Each phenotype could result from different pathogenic mechanisms, as represented by green circles (showing immunologic processes) and yellow circles (showing non-immunologic processes) in the center of the figure, because of the interactions between genetic risk factors (orange circles) and environmental risk factors (red circles). Some combinations of genotypes and environmental exposures induce certain mechanisms and disease phenotypes, whereas other combinations might have no effect or could be protective. C4A, complement 4A; DCs, dendritic cells, ER, endoplasmic reticulum; HLA, human leukocyte antigen; ROS, reactive oxygen species; UVR, ultraviolet radiation.
In this Review, we describe the major advances from the past few years in our understanding of the genetic risk factors, associated environmental exposures, and immune mechanisms and non-immune-mediated mechanisms in IIM phenotypes. Given space limitations, we do not cover many historical features or the details and biologic differences among the phenotypes. The classification, outcome assessments, detailed autoantibody developments and therapies in the IIMs are reviewed in accompanying articles.
Genetic risk factors
Genetic epidemiology of IIM
Owing to the rarity of IIMs, there are few reports of familial occurrence1,2; hence, the heritability of IIMs is unknown. This caveat contrasts with other autoimmune disorders, such as rheumatoid arthritis or type I diabetes, which have reported heritabilities of 66%–88%3,4. In comparison with healthy individuals, higher prevalences of autoimmune disease, such as systemic lupus erythematosus, autoimmune thyroid disease, or type 1 diabetes, have been reported concurrently in patients with IIM, as well as in first-degree relatives of both adult and juvenile patients with IIM, on the basis of data from a relatively small number of patients5,6. Conversely, a nationwide study in Taiwan that investigated co-aggregation of autoimmune disease in families with systemic lupus erythematosus and systemic sclerosis, identified higher relative risk of IIM than that of the general population7,8, and a national study in China suggested an increased risk of IIM and certain other autoimmune diseases in relatives of patients with systemic sclerosis8. These findings of aggregation of autoimmune diseases within families of patients with IIM suggest that shared genetic and/or environmental factors might contribute to disease risk. However, large, well-powered, epidemiological studies are needed to robustly evaluate these data. Notably, in 2015, a systematic review demonstrated that the reported incidence and prevalence rates for IIMs have increased over time9. Although this finding might reflect a true increase in disease burden, this increase could also be due to a wider recognition or more accurate recording of disease than in the past.
HLA loci associated with IIM
Genome-wide single-nucleotide polymorphism (SNP) association studies in adult and juvenile individuals of European ancestry who have dermatomyositis or polymyositis identified the strongest disease associations within the MHC region on chromosome 610,11, one of the most complex regions of the genome, which has a high concentration of genes encoding proteins with immunological functions. The development of the so-called Immunochip (Illumina, USA), a cost-effective genotyping array that includes ~200,000 genetic variants associated with autoimmune diseases, combined with accurate imputation of HLA alleles and amino acids from SNP data12, greatly improved our ability to dissect HLA associations. The Myositis Genetics Consortium conducted the largest genetic study in IIM, which included 2,566 patients with IIM from 14 countries. The study demonstrated the strongest disease association with alleles of the 8.1 ancestral haplotype—HLA-DRB1*03:01 and HLA-B*08:01 in polymyositis and dermatomyositis, respectively—whilst conditional analysis revealed that multiple variants on this haplotype might contribute independently to disease risk13. In IBM, HLA-DRB1*03:01, HLA-DRB1*01:01, and HLA-DRB1*13:01 were independently associated with disease14; the latter two alleles are uniquely associated with IBM. Different risk factors are seen in other ethnic groups, including HLA-DRB1*08:03, which is associated with IIM in Japanese patients15 and HLA-DQA1*01:04 and HLA-DRB1*07, which are associated with dermatomyositis in Chinese populations16.
Specific amino acid associations in the HLA region, such as position 57 of HLA-DQB1, position 77 of HLA-DRB1*03:01, and positions 26 and 11 of HLA-DRB1*03:01, differentiate dermatomyositis, polymyositis, and IBM, respectively13,14, and suggest different predominating pathophysiology in different clinical subgroups. Conversely, although HLA allele associations differ in non-white populations, the amino acid associations might be consistent across different populations. Amino acid sequence variations might alter the structure of the HLA molecule’s peptide-binding groove and thereby increase disease susceptibility by influencing antigen repertoires and the affinity of peptides presented to the immune system. If so, computational modelling of predicted antigen-HLA binding to identify the immunogenic peptides could help to determine disease mechanisms.
The finding that different HLA alleles have been associated with various myositis-specific autoantibody (MSA)-defined subgroups17 agrees with the finding that many MSAs are mutually exclusive. In general, HLA risk alleles are more strongly associated with MSA-defined subgroups than clinically-defined subgroups (which are less homogenous than the MSA-defined subgroups) despite smaller sample sizes in the MSA groups. For example, the presence of anti-histidyl-tRNA synthetase (anti-Jo1) autoantibodies in IIM is strongly associated with multiple alleles of the 8.1 ancestral haplotype, including HLA-B*08:01, DQB1*02:01, and DRB1*03:01, particularly when multiple alleles are considered together as a haplotype11. The presence of other autoantibodies, including autoantibodies to Mi-2 (also known as chromodomain-helicase-DNA-binding proteins), SUMO-activating enzyme (SAE), melanoma differentiation-associated gene 5 (MDA5), signal recognition particle (SRP), transcription intermediary factor 1 (TIF1) and anti-PL-7, have been associated with specific HLA alleles17,18. Although most HLA associations are the same in adult and juvenile IIM phenotypes, HLA associations can occasionally differ between adult and juvenile patients, as is the case with anti-3-hydroxy-3-methylglutaryl-coenzyme A reductase (HMGCR) autoantibodies, with associations of HLA-DRB1*11:01 and HLA-DRB1*07:01 for adult and juvenile patients with IIM, respectively19,20.
Non-HLA loci associated with IIM
Several non-HLA loci have been associated with IIM (Table 1), including PTPN2213,21; this locus was associated with polymyositis (reaching genome-wide significance, defined as p<5×10−8), but not with adult or juvenile dermatomyositis13, again suggesting that different clinical subgroups have different pathophysiologies. Other genes, including STAT4, TRAF6, and UBE2L3 in IIM13,22; PLCL1 and BLK in dermatomyositis10,23; and CCR5 in IBM14, have been associated with disease, reaching a suggestive level of significance (defined as p<2.25×10−5). Complement 4A (C4A) deficiency, owing to copy number variation, has been linked to an increased risk of developing juvenile dermatomyositis, although the strongest risk factor identified in the study was attributable to the presence of HLA-DRB1*03:01 along with C4A deficiency24. In IBM, sequencing of candidate genes involved in related neuromuscular or neurodegenerative diseases25,26 and whole-exome sequencing of proteins overrepresented in the skeletal muscle rimmed vacuoles—defined as a space within the cytoplasm of a muscle cell with a purplish staining rim on trichrome staining—of patients with IBM27 identified rare variants in VCP, SQSTM1, and FYCO1associated with disease, suggesting impaired autophagy as a mechanism of IBM pathogenesis.
Table 1.
Representative genetic loci associated with IIMs
| IIM phenotype | Gene or allele (associated SNP or amino acid position) | p value | OR (95% CI) | Study type | Putative function | Associations with other autoimmune disease | Reference |
|---|---|---|---|---|---|---|---|
| IIM | HLA-DRB1*03:01† | 2.58×10−135 | 1.88 (1.68–2.11) | Immunochip & HLA imputation | Regulation of immune system | Type I diabetes, SLE, Crohn’s disease | 13 |
| HLA-B*08:01† | 3.23×10−14 | 1.58 (1.41–1.78) | Immunochip & HLA imputation | Regulation of immune system | Unknown | 13 | |
| STAT4(rs4853540) | 1.57×10−6 | 0.83 (0.77–0.89) | Immunochip | T cell differentiation | Celiac disease, Crohn’s disease, IBD | 13 | |
| TRAF6(rs570676) | 9.42×10−6 | 0.87 (0.82–0.92) | Immunochip | TNF receptor signalling | RA | 13 | |
| UBE2L3(rs5754467) | 4.67×10−7 | 1.21 (1.12–1.30) | Immunochip | NF-κB signalling | Celiac disease, psoriasis SLE | 13 | |
| IIM (Japanese population) | HLA-DRB1*08:03 | 0.02 | 1.9 (1.1–3.2) | Candidate gene | Regulation of immune system | Allergic rhinitis | 15 |
| Polymyositis | HLA-DRB1*03:01† | 6.11×10−80 | 1.99 (1.67–2.36) | Immunochip & HLA imputation | Regulation of immune system | Type I diabetes, SLE, Crohn’s disease | 13 |
| HLA-B*08:01† | 4.17×10−9 | 1.71 (1.43–2.05) | Immunochip & HLA imputation | Regulation of immune system | Unknown | 13 | |
| HLA-DRB1(asparagine 77) | 1.65×10−80 | 2.93 (2.53–3.17) | Immunochip & HLA imputation | Regulation of immune system | Type I diabetes, autoimmune thyroid disease | 13 | |
| PTPN22(rs2476601) | 7.90 × 10−11 | 1.58 (1.38–1.81) | Immunochip | T cell signalling | RA, SLE, type I diabetes, autoimmune thyroid disease | 13 | |
| Adult dermatomyositis | HLA-B*08:01 | 2.46×10−42 | 1.90 (1.66–2.17) | Immunochip & HLA imputation | Regulation of immune system | Unknown | 13 |
| HLA-DQB1(alanine 57) | 1.29×10−12 | 1.62 (1.44–1.83) | Immunochip & HLA imputation | Regulation of immune system | Type I diabetes | 13 | |
| Adult dermatomyositis (Chinese population) | HLA-DQA1*01:04 | 0.01 | 2.58 (1.18–5.64) | Candidate gene | Regulation of immune system | Unknown | 16 |
| HLA-DRB1*07 | 0.01 | 2.26 (1.12–4.59) | Candidate gene | Regulation of immune system | Unknown | 16 | |
| Juvenile dermatomyositis | HLA-DRB1*03:01 | 7.91×10-14 | 1.90 (1.61–2.22) | Immunochip & HLA imputation | Regulation of immune system | Type I diabetes, SLE, Crohn’s disease | 13 |
| C4Adeficiency | 8.2×10−6 | 3.00 (1.87–4.79) | Candidate gene | Classical complement pathway | SLE, Bechet syndrome | 24 | |
| Juvenile and adult dermatomyositis | PLCL1(rs7572733) | 6.18×10−6 | 0.80 (0.72–0.88) | GWAS & candidate gene | Inositol phospholipid signalling | SLE | 10 |
| BLK(rs2736340) | 0.000065 | 1.25 (1.12–1.40) | GWAS | B cell signalling and development | RA, SLE | 10 | |
| IBM | HLA-DRB1*03:01† | 5.77×10−34 | 7.97 (5.88–10.95) | Immunochip & HLA imputation | Regulation of immune system | Type I diabetes, SLE, Crohn’s disease | 14 |
| DRB1*01:01† | 1.56×10−16 | 4.64 (3.33–6.49) | Immunochip & HLA imputation | Regulation of immune system | RA | 14 | |
| DRB1*13:01† | 3.28×10−8 | 3.19 (2.14–4.72) | Immunochip & HLA imputation | Regulation of immune system | Primary sclerosing cholangitis | 14 | |
| HLA-DRB1(tyrosine 26) | 1.19×10−16 | 3.83 (2.80–5.29) | Immunochip & HLA imputation | Regulation of immune system | SLE | 14 | |
| CCR2–CCR5region | 1.93×10−6 | 0.42 (0.29–0.60) | Immunochip | Binds pro-inflammatory chemokines | JIA, ulcerative colitis, celiac disease | 14 | |
| VCP‡ | N/A | N/A | Targeted sequencing | Protein homeostasis | IBMPFD§, ALS§ | 26,27 | |
| SQSTM1‡ | N/A | N/A | Targeted sequencing | NF-κB signalling | ALS§ | 26,27 | |
| FYCO1 | 0.003 | N/A | Whole-exome sequencing | Autophagic adaptor protein | Unknown | 26,27 | |
| Anti-Jo1 positive IIM | AH8.1 alleles^ (HLA-B*08:01, HLA-DQB1*02:01 and HLA-DRB1*03:01) | 1.20×10−82 | N/A | GWAS & HLA imputation | Regulation of immune system | Type I diabetes, SLE, Crohn’s disease | 11 |
| Anti-HMGCR positive adult myopathy | HLA-DRB1*11:01 | 1.2×10−6 | 10.4 (3.6–31.4) | Candidate gene | Regulation of immune system | JIA | 19,20 |
| Anti-HMGCR positive juvenile myositis | HLA-DRB1*07:01 | 0.01 | N/A | Candidate gene | Regulation of immune system | Pancreatitis | 19,20 |
95% CI, 95% confidence interval; AH8.1, 8.1 ancestral haplotype; ALS, amyotrophic lateral sclerosis; GWAS, genome-wide association study; HMGCR, anti-3-hydroxy-3-methylglutaryl-coenzyme A reductase; IBD, inflammatory bowel disease; IBM, inclusion body myositis; IBMPFD, IBM with Paget’s disease of bone and frontotemporal dementia; IIM, idiopathic inflammatory myopathy; JIA, juvenile idiopathic arthritis; N/A, not applicable; NF-κB, nuclear factor κB; OR, odds ratio; RA, rheumatoid arthritis; SLE, systemic lupus erythematosus.
In contrast to the more common immune-mediated diseases, where extensive meta-analyses have been conducted, a relatively small number of genetic risk variants has been identified for IIM. This small number likely reflects the sample sizes of patients with IIM, as well as the marked heterogeneity of these complex diseases, and highlights the importance of collaborative endeavours. Most large genetic studies of IIMs have focused on populations of European ancestry. Further large-scale studies are required to establish whether variants, pathways, and gene–environment interactions are shared across different ethnic groups.
Pathways implicated in IIM pathogenesis
By identifying the genes associated with IIM, studies can focus on the molecular pathways involved and thereby improve our understanding of IIM pathogenesis. The strong association between IIM subsets and HLA-DR and HLA-DQ genes supports a role for the adaptive immune system in the pathogenesis of IIM, as a key role of HLA class molecules is to present antigens to T cells. The roles of other associated variants have been investigated by functional annotation, for example, analysing the effects of coding variants on the translation of the encoded protein and/or regulatory effects on gene expression through expression quantitative trait loci (eQTL) analysis. Identification of eQTLs might also help to identify functionally relevant cell types through immune cell–specific eQTLs that affect spatial and temporal gene expression, the cellular response to stimulation, and/or the magnitude of the response. Associated genes (Table 1) implicate both the innate and adaptive immune responses through, for example, the roles of PTPN22 and STAT4 in the T cell receptor pathway, or BLK, UBE2L3, and TRAF6 in B cells and the nuclear factor κB (NF-κB) signalling pathway. In IBM, specific genes implicate both inflammatory and degenerative changes, including mitochondrial abnormalities, in disease pathogenesis.
Despite the small contribution of identified genetic variants to clinical phenotypes, drugs targeting the pathways affected by genetic variations might be disproportionately effective. In IIM, the application of drugs repurposed from other diseases will probably become more important as our understanding of disease mechanisms evolve. Refinements in defining the genetic factors that drive different phenotypes will be important in clinical decision-making for early and effective diagnosis, classification, and therapeutic management, by targeting therapy to patients most likely to respond.
Missing heritability
The pathogenesis of IIM cannot be explained solely by genetic risk factors. Many of the variants identified have a relatively small effect on disease risk individually, and only 5.5%–16% of the phenotypic variance can be explained by genetic variants identified from Immunochip studies28. Following accepted models of genetic architecture, rare variants, including single nucleotide and copy number variants, are probably involved in rare diseases such as the IIMs but have not yet been extensively investigated. This postulate is illustrated by Mendelian forms of monogenic juvenile-onset disorders that share clinical and immunologic features with juvenile dermatomyositis, such as chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature (CANDLE) syndrome29.
Environmental risk factors
Although a number of genes have been associated with the IIMs, the physiologic effect of these genes might depend on their activation or modification by environmental factors. Compared with genetic studies, however, fewer investigations have explored the role of environmental exposures in the development of IIM. Thus, the field is decades behind other areas, such as oncology, which has identified modifiable risk and protective factors for different cancers; by contrast, in many cases, only a single or small number of studies have identified specific environmental exposures associated with particular IIM phenotypes.
Multiple lines of evidence suggest that autoimmune diseases have an environmental component: the concordance rates for autoimmune diseases in monozygotic twins is much less than 50%30,31; there are strong temporal associations between certain exposures (infectious agents and drugs in particular) and the subsequent development of some autoimmune diseases32–34; in some individuals, disease improves after removing a suspected environmental agent (dechallenge) and/or worsens or reoccurs after re-exposure to the suspected agent (rechallenge)34–36; the incidence of many autoimmune diseases has increased over time37,38; there are seasonal and geographic variations in disease onset and in birth dates of individuals who have developed an autoimmune disease34,39–42; data from relevant animal models have demonstrated the plausibility of multiple environmental agents potentially triggering autoimmune disease43–45; the major genetic risk factors for autoimmunity are polymorphic genes that regulate responses to environmental agents46,47; variations in the human immune system are largely driven by nonheritable influences48; and associations between specific exposures and autoimmune diseases have been documented in large epidemiologic studies49,50.
In addition to their role as possible initiators of autoimmunity, environmental factors might alter the rate of progression to clinical disease, the specific manifestations of disease expression, and/or the course of illness34,51. After certain disease-initiating exposures, decades can pass before autoimmune disease manifests, and, as for many cancers, there can be a progression of stages from autoimmunity to preclinical disease with immune alterations to classifiable autoimmune disease52. A panel of international investigators convened by the U.S. National Institute of Environmental Health Sciences in 2012 defined a number of well-supported associations between environmental exposures and autoimmune diseases50 and proposed criteria to define environmentally associated autoimmune diseases in clinical care and epidemiologic settings36. Thus, there are many possible ways in which environmental agents might influence individuals over the lifetime to result in perturbations that could eventually result in the autoimmune phenotypes we recognize today. However, it is critical to develop consensus on how to best recognize and diagnose environmentally associated autoimmunity.
Specific environmental agents of concern
As with other autoimmune diseases, many environmental studies in IIM have been based on animal models, case reports, and/or case series, and which suggest environmental risk factors that might vary in IIM phenotypes53,54. Such studies have investigated possible disease associations of selected IIM phenotypes with many viral, bacterial, and parasitic infections55; foods and dietary supplements56; collagen and silicone implants57,58; dozens of biologic agents and chemicals prescribed as drugs46; seasonal variations10; birth date assocations42; exposure to ultraviolet light41,59,60; and occupational exposures to dust, gases, or fumes61,62.
A number of specific infectious agents are implicated in IIM pathogenesis on the basis of reported occurrences of suspected infection-induced disease or biologic plausibility from animal models52. Examples include hepatitis B virus in polymyositis63 and dermatomyositis64; hepatitis C virus in IBM65; retroviruses, particularly human immunodeficiency virus (HIV) and human T-lymphotropic virus-1 (HTLV-1) in polymyositis66, dermatomyositis67, and IBM68,69; Toxoplasma spp. and Borrelia spp. in polymyositis and dermatomyositis70; and influenza, picornaviruses, and echovirus in polymyositis, dermatomyositis, and juvenile dermatomyositis55. Case reports have documented associations between IIM development and medications, including D-penicillamine in polymyositis and dermatomyositis71; therapeutic cytokines, especially interferons72 and anti-TNF agents in dermatomyositis73; and statins in polymyositis, dermatomyositis, necrotizing myopathy, and IIM with anti-HMGCR autoantibodies74–76. On the basis of case series and animal models, vaccines (especially those containing aluminum hydroxide) have been hypothesized to be triggers of polymyositis, dermatomyositis, and focal forms of myositis that affect only a selected extremity or part of the body77.
Because of the limitations of animal models and case reports, and despite the rarity and heterogeneity of the IIM, investigators have also assessed environmental risk factors by using epidemiologic approaches. The environmental associations already discussed have in some cases, but not in others, been supported by the few epidemiologic studies reported to date, which often addressed only a single exposure and have in many cases not been replicated. Those studies have sometimes had conflicting results, probably due to differing approaches and study populations. The epidemiologic studies to date reveal a number of preliminary environmental associations (Figure 2): the risk of IIM appears to be increased after any infection and after gastrointestinal or respiratory tract infections or lung inflammation78; the risk of polymyositis and dermatomyositis is decreased after upper respiratory tract infections79; the risk of polymyositis or dermatomyositis is increased after excess physical exertion79; there is no association with vaccines in polymyositis or dermatomyositis79; smoking is a risk factor for anti-Jo-1 autoantibody-positive IIM (potentially interacting with HLA-DRB1*03 to increase this risk, an effect similar to the HLA–smoking interaction seen in rheumatoid arthritis) and for myositis overlap syndromes80; bovine collagen implants are associated with dermatomyositis57; and group A Streptococcus infections are associated with juvenile dermatomyositis81. Perinatal factors in mothers, including air pollution, smoking, and occupational exposure to dust and/or solvent, have also been proposed as important risk factors for juvenile dermatomyositis in a small study82.

Infectious and non-infectious agents associated with IIM phenotypes from epidemiologic studies suggest a wide array of risk factors with different strengths of association. 95% CI, 95% confidence interval; CTD, connective tissue disease; DM, dermatomyositis; GI, gastrointestinal; HLA, human leukocyte antigen; IIM, idiopathic inflammatory myopathies; JDM, juvenile dermatomyositis; JPM, juvenile polymyositis; OR, odds ratio; PM, polymyositis; RR, relative risk; URI, upper respiratory infection.
The compelling data that reveal a link between the environment and autoimmunity, as well as the remarkable increases in the incidence and prevalence of many autoimmune diseases for unknown reasons37,83–85, underscore a critical need for both exploratory and confirmatory environmental investigations in this understudied field. Identifying factors that protect against disease is also important to decrease the prevalence of autoimmune disease. Much more work is needed in these areas. Enumerating genetic and environmental risk and protective factors in carefully defined disease phenotypes is an important first step for discovering gene–environment interactions that could lead to preventive strategies34,46.
Immune-mediated disease mechanisms
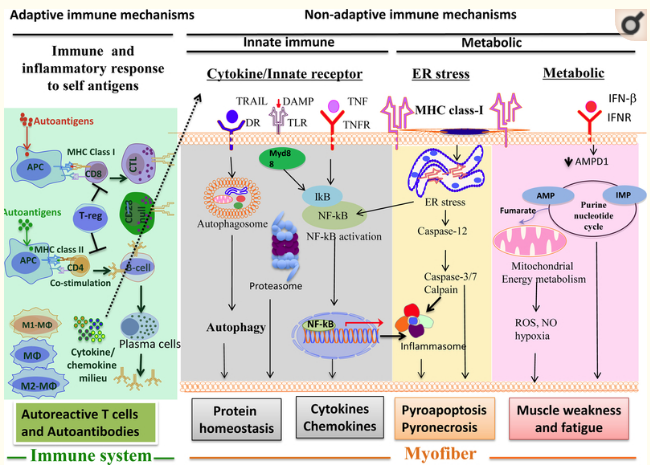
Preliminary data suggest that many adaptive and innate immune mechanisms, as well as non-immune mechanisms, are involved in the development of the IIMs (Figure 3).

Infectious and non-infectious agents evaluated in epidemiological studies suggest a wide array of risk and protective factors with different strengths of association with idiopathic inflammatory myopathy (IIM) phenotypes. CTD, connective tissue disease; OR, odds ratio; RR, relative risk. a. Infection in the year before IIM diagnosis. b. Infection in the year before IIM onset. c. Inflammatory lung disease at study inclusion.
T cells in IIM
Several studies have demonstrated the presence of T cells and B cells, especially invasive, granzyme B and perforin-expressing cytotoxic CD8+ effector T cells, in the muscles of patients with polymyositis or IBM86,87. The invasion of CD8+ effector cells into non-necrotic muscle fibres is considered a characteristic histological feature of polymyositis and IBM. The number of invading CD8+ and CD57+ T cells in the muscle correlates with the size of large granular lymphocyte populations in the blood in patients with IBM, and the autoimmune T cell expansion is proposed to evolve into a neoplastic-like or overtly neoplastic disorder of T cell aggressiveness88.
In the 1990s, investigations demonstrated the presence of clonal γδ T cells in the muscle of patients with polymyositis, and in 2012, investigators showed that these γδ T cells recognize aminoacyl-tRNA synthetases (an MSA target), suggesting a potential link between γδ T cells and autoantibody responses in autoimmune myositis89–91. Patients with γδ T cells in their muscle tissue seem to be more responsive to steroids compared to those without these cells in muscle89,92. Muscles affected by myositis also contain some unique subsets of highly cytotoxic, apoptosis-resistant, pro-inflammatory T cells, such as CD28null T cells (CD4+CD28null and CD8+CD28null)93–95. Persistence of CD244+ (CD28null) T cells in muscle tissue following immunosuppressive therapy is associated with poor outcome in patients with IIM95.
The role of regulatory T (Treg) cells (defined by FoxP3 expression) and the effect of these cells on inflamed muscle were investigated in adult and juvenile forms of myositis96,97. It was proposed that FoxP3 Treg cells counterbalance muscle inflammation in myositis. The presence of significant inflammation in the muscle of juvenile dermatomyositis patients despite a high proportion of Treg cells in the milieu suggests that Treg cell function is impaired. In a muscular dystrophy model, Treg cells derived from the muscle express growth factors, such as amphiregulin, that act directly on muscle satellite cells in vitro and improve muscle repair in vivo98.
B cells in IIM
Despite initial reports of the predominance of B cells in the perivascular regions on muscle biopsy in dermatomyositis76, subsequent studies have demonstrated the presence of B cells, plasma cells, and immunoglobulin transcripts in the muscle of patients with polymyositis or IBM, indicating a humoral component in those disorders99–101. Autoantibodies are present in more than half of all patients with IIM. The presence of MSAs, such as anti-Jo1, anti-Mi2, anti-cytosolic 5’-nucleotidase 1A (cN1A), or anti-HMGCR autoantibodies, are well described in patients with polymyositis, dermatomyositis, IBM, or necrotizing myopathy, respectively102. In addition to the prior work confirming many genetic and clinical associations with the classic myositis autoantibodies, studies have highlighted unique associations of newly identified autoantibodies with additional clinical phenotypes. For example, autoantibodies targeting melanoma differentiation-associated protein 5 (MDA5) are associated with mucocutaneous lesions and severe lung disease in patients with dermatomyositis; autoantibodies that bind to nuclear matrix protein 2 (NXP2) are associated with joint contractures and calcinosis in juvenile dermatomyositis; and autoantibodies to SAE, transcription intermediary factor 1γ (TIF1γ), and transcription intermediary factor 1α (TIF1α) are associated with malignancy in dermatomyositis103.
Although the pathological roles of autoantibodies in IIM are unclear, their associations with distinct genotypes, clinical phenotypes, and interferon patterns are strong. For example, the spectrum of clinical phenotypes in antisynthetase syndrome, a group of IIM defined by the presence of autoantibodies to aminoacyl-tRNA-synthetases that are involved in protein synthesis, includes interstitial lung disease, arthritis, Raynaud phenomenon, and mechanic’s hands. Levels of anti-Jo1 autoantibodies are also strongly associated with clinical outcomes, suggesting that anti-Jo1 autoantibody levels might be a good biomarker for disease activity104. Furthermore, animal and in vitro studies demonstrate that the Jo1 autoantigen, histidyl-tRNA synthetase, might also play a role in inducing myositis105 or function as a chemokine106.
The role of autoantibodies in causing muscle damage and dysfunction is controversial because most of the autoantigens are intracellular and thus not easily accessible to circulating autoantibodies; however, additional work focusing on their role is warranted given their importance as biomarkers of unique genetic, clinical, therapeutic, and outcome phenotypes. Although cause-and-effect relationships between autoantibodies and disease phenotype and activity currently are not clear, the strong autoantibody associations with clinical phenotypes and disease severity are immensely useful for the diagnosis and prognosis of IIM.
Innate immune cells
Antigen processing and presentation by professional antigen-presenting cells, such as dendritic cells, are critical to initiating the adaptive immune response, and the muscle microenvironment of IIM is enriched with both myeloid and plasmacytoid dendritic cells107–111. The relative proportion of lymphocytes and macrophages in skeletal muscle seems to vary in different clinical entities; for example, in anti-HMGCR myopathy, M2 (alternatively activated) macrophages predominate over CD4+ and CD8+ T cells and CD123+ plasmacytoid dendritic cells112. A predominance of M2 macrophages in skeletal muscle is also seen in forms of inflammatory myopathy with abundant macrophages113. The presence of M2 macrophages is consistent with the known role of macrophages in skeletal muscle injury and repair114: M1 (classically activated) macrophages infiltrate the muscle in the early stages to help clear necrotic debris, whereas M2 macrophages arrive later to sustain tissue healing115.
Inflammatory mediators
The high serum levels of creatine kinase and other molecules that are released from skeletal muscle cells in patients with IIM could represent danger-associated molecular patterns, which sometimes serve as endogenous Toll-like receptor ligands. Skeletal muscle, as well as muscle-infiltrating cells, express abundant innate immune receptors, including Toll-like receptors116–118. Activation of innate immune receptors can lead to activation of NF-κB signalling and pro-inflammatory cytokine and chemokine secretion, which, in turn, further recruit immune cells into a milieu that is already ripe for antigen processing and presentation by dendritic cells. These immune cells and cytokines can then further activate T helper 1 (TH1), T helper 17 (TH17), and T helper 1 (TH2) cells, as well as CD8+ cytotoxic T cells and CD28null cells, which potentially damage muscle cells. Cytokines produced by these T helper cell subsets induce macrophages to polarize into pro-inflammatory M1 or pro-resolution M2 phenotypes.
Both pro-inflammatory and anti-inflammatory cytokines, as well as CXC-chemokines and CC-chemokines, are expressed in IIM muscle; such mediators include TH1 cytokines (TNF, IFNγ, IL-12, and IL-2), TH2 cytokines (IL-4 and IL-13), TH17 cytokines (IL-17, IL-22, IL-23, TNF-related weak inducer of apoptosis (TWEAK), and IL-6), Treg cytokines (IL-10, transforming growth factor-β (TGFβ)), and innate immune cytokines (IL-1α, IL-β, and type I interferons (IFNα and IFNβ)). These cytokines coordinate various innate and adaptive immune response pathways, and some of them have the potential to cause muscle damage and weakness depending on the stage of the disease119,120. Similarly, both CXC-chemokines (CXC-chemokine ligand 9 (CXCL9), CXCL10) and CC-chemokines (CC-chemokine 2 (CCL2), CCL3, CCL4, CCL19, and CCL21) have a role in sustaining inflammatory responses in IIM muscle121.
In dermatomyositis, evidence from the past decade has demonstrated high levels of type 1 interferon in the muscles of patients, which is associated with perifascicular atrophy122–125. Genes induced by type I interferons (either IFNα or IFNβ) were overexpressed in the muscle, skin, and blood, of patients with dermatomyositis, and in some126–128, but not all129, of those studies the levels of these genes correlated with disease activity. Evidence suggests that myeloid dendritic cells are a major source of type 1 interferons in the muscle of dermatomyositis patients108. Type I interferons affect immune cells either directly, through type I interferon receptor signalling, or indirectly, by inducing the production of chemokines, by inducing the secretion of cytokines such as IL-15 (which regulate natural killer (NK) cell and memory CD8+ T cell proliferation), by stimulating dendritic cells (which, in turn, activate naive T cells), or by inducing the differentiation of monocytes–macrophage lineage cells130.
IFNβ can induce reactive oxygen species and mitochondrial damage in dermatomyositis131, which provides an important link to the cause of the functional impairment in this disorder. Induction of tissue inflammation and autoimmunity by IFNα involves direct toxic effects on tissue as well as provocation of destructive bystander immune responses132. IFNα mediates a long-lasting and preferential MHC class I overexpression in non-immune cells, such as human pancreatic beta cells and thyroid follicular cells, which usually lack MHC class 1 expression. This finding suggests that IFNα might amplify antigen presentation in type 1 diabetes and Hashimoto thyroiditis133,134. The increased expression of MHC class I molecules on skeletal muscle cells of patients with IIM probably leads to increased susceptibility of the cells to cytotoxic T cell attack and endoplasmic reticular (ER) stress–mediated cell death135,136. On the basis on these findings, neutralization of type 1 IFN has been explored as a treatment option in dermatomyositis and polymyositis137.
Non-immune-mediated disease mechanisms
Apart from the inflammatory pathomechanisms discussed in the previous section, mounting evidence suggests that several non-immune-mediated mechanisms also operate in IIM (an overview of the topic has been provided elsewhere138,139). In general, these mechanisms fuel inflammation via a positive feedback loop, affect muscle contraction and cause muscle weakness, imbalance muscular protein homeostasis, and lead to atrophy and mostly irreversible structural damage of muscle fibres. The suggestion that non-immune-mediated mechanisms are clinically relevant in IIM derives from a number of key findings: the muscular inflammation identified by muscle biopsy and MRI does not always correlate with the clinical severity; the effects of immunosuppressive treatments can be limited; and several non-inflammatory mechanisms, including cell stress and degenerative mechanisms, are obvious in many muscle biopsies. The most relevant non-immune mechanisms are discussed in this section.
ER stress
ER stress is one of the best-studied elements of non-immune-mediated damage to skeletal muscle in all forms of IIM (overview in140). ER stress mechanisms include the unfolded protein response (UPR) and the ER overload response (EOR). UPR is characterised by upregulation of cyclic AMP-dependent transcription factor 6α (ATF6α), eukaryotic translation initiation factor 2α-kinase 3 (EIF2α kinase, also known as PERK), serine/threonine-protein kinase/endoribonuclease IRE1 (IRE1α), and the ER chaperones endoplasmin (also known as GRP94) and 78-kDa glucose-regulated protein (GRP78, also known as BiP). The collective function of these molecules is to reduce the protein overload and subsequent accumulation of unfolded proteins in the ER. The second ER stress pathway, EOR, modulates inflammation by upregulating NF-κB signalling. Both ER stress pathways are activated in the muscle in all forms of IIM, including IBM135,141,142. Data from the past years suggest that ER stress might even contribute directly to muscular weakness in IIM (overview in143). The NF-κB pathway has been shown to be activated in IIM144,145. At the same time, relevant molecules of the immunoproteasome such as β1i and β5i were present in the muscle of patients with IIM146. The NOD-, LRR- and pyrin domain containing protein 3 (NLRP3) inflammasome has also been shown to be upregulated in dermatomyositis and polymyositis, and this was associated with elevated levels of IL-1β and IL-18147. Since it is known that ER stress can induce the NLRP3 inflammasome in other cell systems148, it is possible that ER stress is a crucial factor of muscle pathology in IIM by inducing molecules of the inflammasome and immunoproteasome pathways.
Other key factors
Free radicals are key factors in muscle fibre damage in all forms of IIM149,150, and these molecules are speculated to contribute directly to muscle weakness143. In a mouse model of chronic inflammation (mice overexpressing TGFβ), muscular atrophy was mediated by inflammation, production of reactive oxygen species, mitochondrial damage, and caspase activation. The mitochondrial damage and muscle atrophy were efficiently downmodulated following red grape polyphenols supplementation despite continuous muscle inflammation151.
TNF-related apoptosis-inducing ligand (TRAIL) expression is upregulated and associated with autophagy and cell death in the skeletal muscle of patients with IIM152. In IIM muscle fibres, the expression of heat-shock proteins HSP70 and HSP90153 and the alarmin high mobility group box protein 1 (HMGB1)154 is increased compared with healthy controls; HMGB1 functions via Toll-like receptor-4 and is thought to mediate muscular inflammation and weakness155–157. Consistent findings relating to increased HMGB1 expression have been reported in experimental models of autoimmune myositis in rodents158. Dysregulation of HMGB1 might also be relevant for the regenerative potential of skeletal muscle in IIM, because HMGB1 is an important factor during recovery and is required for the patient to regain muscle function after severe damage159 and, thus, could serve as a prognostic marker in severe IIM160.
Inclusion body myositis
The largest body of evidence for non-immune-mediated mechanisms in IIM is available for IBM161. The pathogenesis of IBM includes many pathways involved in protein homeostasis and cell stress mechanisms, and several of these pathways seem to be linked directly to inflammation.
Protein homeostasis and the heat shock response
A variety of unwanted and defective proteins that should be removed from the cell, including β-amyloid and its associated proteins, can accumulate in the muscle of patients with IBM162–164. In IBM, intracellular accumulation of abnormal or no-longer-needed proteins in muscle fibres is hypothesized to cause or aggravate cell stress pathways, thus leading to structural damage and weakness of the fibres. In patients with IBM, proteomic analysis of vacuolated fibres identified FYVE domains and coiled-coil domain–containing protein 1 (FYCO1) as a novel component in the rimmed vacuoles27. This finding was associated with a missense variant of the FYCO1 gene in 11% of IBM patients (see above), which supports the hypothesis of impaired autophagic activity in IBM (see below for details).
HSP expression has been demonstrated in the muscles of patients with IBM and in human muscle fibres cultured in experimental conditions that mimic either inflammatory or degenerative aspects of IBM pathology165,166. One of the most commonly expressed HSPs in IBM, crystallin α-B chain (also known as αB-crystallin or CRYAB or HSPB5), seems to be an early element in the pathological cascade, as this protein is upregulated in healthy muscle fibres165,167. Heat-shock factor protein 1 (HSF1) can ameliorate cell stress caused by aggregation of TAR DNA binding protein 43 (TDP43) in muscle cells168. In cell culture and animal models with pathologic features of IBM, treatment with arimoclomol, a modulator of the heat-shock response, protected skeletal muscle cells against protein accumulation and cell stress169. The same study showed that arimoclomol was safe and well-tolerated in patients with IBM. Collectively, these data suggest that protein dyshomeostasis is an important non-immune element in the pathology of IBM and that this process is associated with a heat-shock response that could be a suitable target for future clinical trials.
Dysregulation of autophagy
Several lines of evidence have demonstrated malfunction of the autophagic machinery in IBM170,171. Macroautophagy is active during the accumulation of β-amyloid in vacuoles (although it is not known yet whether macroautophagy causes β-amyloid to accumulate)172, and this process depends on extracellular signal-regulated kinase (ERK) signalling173. Other autophagy adaptor molecules have been implicated in IBM, including sequestosome 1 (SQSTM1; also known as p62)174, NBR1175,176, and nuclear factor erythroid 2-related factor 2 (NRF2; also known as NFE2L2)177. Collectively, these data indicate that autophagy is a relevant mechanism in IBM pathology and provide the rationale for a recently completed, placebo-controlled clinical trial of rapamycin, an immunosuppressant that activates macroautophagic activity, in patients with IBM (results not yet published)178.
Mitochondrial abnormalities and free radicals
Apart from inflammation and protein accumulation, mitochondrial abnormalities, such as cyclo-oxygenase-deficient muscle fibres, are hallmarks of IBM. Several mitochondrial defects have been demonstrated in the muscle of patients with IBM179. Such mitochondrial changes are associated with oxidative damage180, inflammatory mediators181, and functional impairment of muscle strength182. Signs of mitochondrial dysfunction have also been shown in a mouse model of IBM, in which mice overexpressing human amyloid precursor protein develop IBM-like pathological features owing to the accumulation of amyloid precursor protein in the skeletal muscle183 and in human muscle cells following adenovirus-mediated upregulation of amyloid precursor protein in vitro.
In addition to the reports of free radical generation in IIM, production of nitric oxide has been demonstrated in IBM in association with accumulation of β-amyloid and inflammation in skeletal muscle as well as in different cell culture systems that mimic relevant aspects of IBM184,185.
Targeting non-immune-mediated pathways
Non-immune-mediated mechanisms are relevant in all forms of IIM and can lead to structural damage of muscle fibres, cause direct weakness of the muscle, or reciprocally fuel inflammatory cell stress pathways. Many of these pathways are far from being understood and present possibly novel areas of mechanistic studies and therapeutic approaches. One of the novel treatment strategies in IBM is to regulate the function of protective HSPs. Future treatment directions should include therapies that scavenge free radicals and/or target other damage signals, such as alarmins and non-inflammatory mediators. Such therapies could reduce structural damage to muscle fibres and ameliorate weakness, particularly when these features are mediated by a functional impairment of muscle homeostasis and energy metabolism, such as mitochondrial dysfunction.
Conclusions
Studies in the past decade have elucidated possible genetic and environmental risk factors, as well as possible immune and non-immune mechanisms, that result in the development of IIM phenotypes. Yet, current paradigms are often biased by certain assumptions and use non-standardised disease or phenotype definitions that can limit our understanding and result in the assessment of different entities by different investigators. Current studies have emphasised the importance of using mutually exclusive and stable phenotypes to minimize confounding factors and to allow for greater power by using more homogeneous groups and smaller sample sizes, as are needed for rare diseases such as the IIMs. Multidisciplinary IIM collaborative study groups have played a key role in developing consensus on how to define and study IIM phenotypes (see the Reviews by Lundberg et al. <citation> and Rider et al. <citation> on classification and outcome assessment, respectively, in this Issue).
In the future, emphasis needs to be placed on multidisciplinary collaborative investigations of genetic and environmental risk factors and their interactions, as well as pathogenic mechanisms in homogeneous, well-defined phenotypes, utilizing the many IIM registries and repositories that have been developed to allow for the most cost-effective strategies186. More investment in these areas seems appropriate to develop preventative strategies and to allow for innovative approaches to treatment as new pathways to disease are discovered.
Acknowledgments
We thank Drs. Lisa Rider, Ingrid Lundberg, Andrew Mammen, and Christine Parks for many useful discussions and concepts in this area and helpful comments on the manuscript, and we are grateful to Lisa Maroski for her technical assistance. This research was supported in part by the Intramural Research Program of the NIH, National Institute of Environmental Health Sciences. KN is supported by the National Institutes of Health (1R21AI128248–01, K26OD011171), The Myositis Association, and the US Department of Defense (W81XWH-11-1-0809 (KN and FM), W81XWH-11-1-0782 (KN). JL is supported by the Medical Research Council, UK (MR/N003322/1) and The Myositis Association.
Glossary terms
| Rimmed vacuoles | a space within the cytoplasm of a muscle cell with a purplish staining rim on trichrome staining |
| Functional annotation | characterizing the function assigned to each gene product or genetic variant |
| Expression quantitative trait locus (eQTL) | a genomic locus that regulates gene expression |
| Macroautophagy | a process in which cellular contents are degraded by lysosomes or vacuoles and recycled |
Footnotes
Competing interests
The authors have declared no competing interests.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.