
Abstract
While timely reperfusion of acute ischemic myocardium is essential for myocardial salvage, reperfusion results in a unique form of myocardial damage. Functional alterations occur, including depressed contractile function and decreased coronary flow as well as altered vascular reactivity. Both myocardial stunning and infarction are seen. Over the last two decades, it has become increasingly clear that oxidant and oxygen radical formation is greatly increased in the post-ischemic heart and serves as a critical central mechanism of post-ischemic injury. This oxidant formation is generated through a series of interacting pathways in cardiac myocytes and endothelial cells and triggers subsequent leukocyte chemotaxis and inflammation. Nitric oxide (NO) production and NO levels are also greatly increased in ischemic and post-ischemic myocardium, and this occurs through NO synthase (NOS)-dependent NO formation and NOS-independent nitrite reduction.
Recently, it has been shown that the pathways of oxygen radical and NO generation interact and can modulate each other. Under conditions of oxidant stress, NOS can switch from NO to oxygen radical generation. Under ischemic conditions, xanthine oxidase can reduce nitrite to generate NO. NO and peroxynitrite can inhibit pathways of oxygen radical generation, and, in turn, oxidants can inhibit NO synthesis from NOS. Ischemic preconditioning markedly decreases NO and oxidant generation, and this appears to be an important mechanism contributing to preconditioning-induced myocardial protection.
Ischemic heart disease secondary to acute myocardial infarction is among the most prevalent health problems in the world, and is a major cause of morbidity and mortality. In view of this, one of the major research efforts in cardiovascular medicine has been to develop approaches to salvage heart muscle at risk of necrosis in patients with acute myocardial infarction. Over the last two decades acute revascularization with thrombolytic drugs or interventional procedures has emerged as the standard treatment for patients with acute myocardial infarction [1,2]. While timely reperfusion of acute ischemic myocardium is essential for myocardial salvage, reperfusion may also result in a unique form of myocardial damage. This reperfusion injury is characterized by a unique histologic picture with the formation of contraction bands in the contractile proteins, calcific granules within mitochondria as well as cell swelling and the disruption of sarcoplasmic and mitochondrial membranes [3-6]. Functional alterations including depressed contractile function, decreased coronary flow as well as altered vascular reactivity also occur [7-9]. Two major types of myocardial injury occur following myocardial reperfusion: (1) stunning–manifested by reversible contractile dysfunction, and (2) infarction–with irreversible cell death [1,2,10-12]. In addition, reperfusion is associated with potentially lethal arrhythmias that are rapidly and predictably induced within seconds of the onset of reflow [13].
A number of mechanisms have been proposed to mediate reperfusion injury. These include: cellular calcium loading; the occurrence of a no reflow phenomenon due to cell swelling, impaired vascular relaxation or the formation of white cell plugs; and perhaps most importantly the formation of oxygen radicals. While low levels of oxygen radicals and oxidants are normally formed in cells and play important roles in cellular homeostasis, mitosis, differentiation, and signaling [14], following ischemia and reperfusion radical formation is greatly increased triggering cellular injury. Although mammalian cells including cardiomyocytes express endogenous free radical scavenging enzymes [15], such as superoxide dismutase (SOD), catalase, and glutathione peroxidase, these antioxidative defenses are overwhelmed after ischemia and reperfusion. The oxygen radical hypothesis is of particular importance because it can potentially explain each of the other mechanisms of reperfusion injury. It has been demonstrated that exogenous free radicals cause cellular calcium loading with inhibition of the sarcoplasmic reticulum calcium ATPase and inhibition of the sodium potassium ATPase leading to sodium mediated calcium gain [16]. Oxygen radicals cause lipid peroxidation that can result in cell membrane breakdown causing cell swelling. It has been suggested that oxygen radicals result in the chemotaxis of neutrophils that in turn can lead to white cell plugging of capillaries and microvascular compression. In addition, white cells which are chemotaxed and activated are potent sources of further oxygen radical generation [17]. Over the last two decades, a large number of studies in animal models have shown enhanced recovery of contractile function or reduction of infarct size following reflow in the presence of drugs or enzymes which either prevent or scavenge damaging oxidants and free radicals [7,18-26]. Direct methods have also been developed to measure these critical mediators of cellular injury.
Nitric oxide (NO), a relatively stable free radical and signaling molecule, features diverse physiological and pathological actions. While low levels of NO exert a number of regulatory and cytoprotective effects, higher levels are potentially toxic. Furthermore, many of the toxic effects of NO are not directly due to NO itself but are mediated by its oxidative reaction products.
In this review, we summarize the mechanisms shown to give rise to myocardial free radical generation and oxidative stress in the post-ischemic heart, and alterations in myocardial NO generation. We review the evidence that these alterations lead to myocardial ischemia–reperfusion injury and consider how ischemic preconditioning modifies this process of radical generation.
1 Historical perspective on the free radical hypothesis of reperfusion injury
The cardiovascular system is continuously exposed to both reactive oxygen and nitrogen species. Oxygen, although essential for tissue survival, can be injurious during reperfusion of previous ischemic myocardium. Compelling evidence for a role for radical injury comes from a number of sources. The oxygen paradox hypothesis was first introduced by Hearse et al. [27] where they demonstrated that reoxygenation of the hypoxic rat heart resulted in significant damage rather than improvement. Second, Lucas et al. [28], Stewart et al. [29], and Jolly et al. [19] substantiated this concept in the canine and the rabbit model of global ischemia–reperfusion using known oxygen radical scavengers. In these studies, the ability of oxygen radical scavengers to improve mechanical, sarcoplasmic reticulum, and mitochondrial function suggested that oxygen free radicals participate in experimental ischemia–reperfusion injury. Third, by perfusing an oxygen-radical generating system in an isolated rabbit heart preparation, Burton et al. [30] directly demonstrated oxygen radical-induced myocardial damage. Subsequently, Ytrehus et al. [31] also demonstrated deleterious effects of oxygen radicals in isolated rat hearts. Fourth, by developing sensitive and specific electron paramagnetic resonance (EPR) methods, Zweier et al. [32] directly measured and characterized the free radical generation following reperfusion of the ischemic myocardium. Both direct EPR measurements on rapidly frozen tissue and spin trapping measurements have demonstrated that a prominent burst of oxygen radical generation occurs during the early minutes of reperfusion. Importantly, these powerful techniques along with more recent innovations allowing in vivo measurement and imaging of free radicals have provided rigorous approaches to measure and characterize the process of free radical generation in the ischemic heart, and have established that oxygen free radical-mediated injury is a central mechanism of reperfusion injury [33-35]. It was specifically shown in isolated hearts that superoxide-derived free radical generation occurs upon myocardial reperfusion, and leads to functional injury with impaired contractile function [36]. Finally, Bolli et al. [37], Grill et al. [38], and Mergner et al. [22] measured oxygen radical generation in in vivo models of regional myocardial ischemia and reperfusion, and showed that potent reactive oxygen species which are produced during the early minutes of reperfusion play a critical role in myocardial injury in models of myocardial stunning and infarction. At this time, it was demonstrated that exogenous administration of the magnitude of oxygen radical generation measured in the post-ischemic heart is sufficient to cause functional injury mirroring that seen on reperfusion [39,40].
The importance of limiting myocardial ischemia–reperfusion injury in clinical practice has been appreciated for more than three decades. Unfortunately, to date no therapeutic approach has been demonstrated clinically effective in salvaging heart muscle at risk of necrosis or improving contractile function. Interestingly, clinical studies have shown that oxygen radical scavenging is effective in preventing reperfusion-induced arrhythmias [41]. A number of factors have contributed to the difficulty in translating potential therapies to the clinic, as reviewed [41,42].
2 Free radicals, oxidants and cellular injury
The term “free radical” is used to define an atom or molecule that can exist independently with one or more unpaired electrons. By virtue of their unpaired electron, free radicals are typically unstable, highly reactive, and short-lived. Oxygen-derived free radicals and oxidants (reactive oxygen species, ROS) are formed continuously in small amounts during the normal metabolism of cells and are normally inactivated by endogenous scavenging mechanisms.
Free radicals are generated by one electron reduction or oxidation of molecules creating an unpaired electron. In normal mitochondrial oxidative phosphorylation, O2 is reduced by four electrons to form H2O. The energy derived from this reduction of O2 serves to meet the energy demands of the cell. Paradoxically, it is also the actual process of oxygen reduction that leads to the formation of oxygen free radicals. Incomplete reduction of O2 leads to the generation of superoxide anion (O2−), hydrogen peroxide (H2O2), and hydroxyl radical (OH). O2− is unstable with a lifetime of milliseconds at neutral pH, and in aqueous solution it spontaneously reacts or dismutates to yield H2O2 and O2. The OH is an extremely reactive and short-lived free radical produced in biological systems. In the Haber-Weiss reaction, O2 and two OH are formed when O2− reacts spontaneously with H2O2. In the Fenton reaction (also known as the iron-catalyzed Haber-Weiss reaction), reduction or oxidation of a trace metal in the presence O2− and H2O2 gives rise to·OH. The chemical mechanisms of free radical reactions in biological systems have been previously described [43] and recently extensively reviewed [44].
The increased generation of free radicals and oxidants during ischemia and reperfusion has been demonstrated using EPR spectroscopy [32,35,36] and chemiluminescence [45] techniques. ROS may injure cells by causing peroxidation of membrane lipid, denaturation of proteins including enzymes and ion channels, and strand breaks in DNA. Lipid peroxidation triggers loss of membrane integrity, necrosis and cell death [46]. Lipid peroxidation occurs upon reperfusion, and can be prevented when oxygen radicals are inactivated by specific scavengers [13,44]. Oxidative damage by ROS has been documented in a number of experimental studies from subcellular and cellular to in vitro and in vivo models [47], and also in humans [48,49].
3 Measurement of oxidants and free radicals
There are a number of methods that can be used to measure oxidant and free radical generation. These include chemical assays for superoxide, hydrogen peroxide, or hydroxyl radical generation, direct chemiluminescence or that in the presence of chemiluminigenic probes, fluorescence detection in the presence of redox sensitive probes, and either direct or spin trapping based electron paramagnetic resonance spectroscopy (EPR). The luminescence and fluorescence techniques are highly sensitive but can lack specificity. A particular concern of both of these techniques in the presence of exogenous probes is that the probes used can in themselves induce ROS generation, so that caution and appropriate controls are essential to interpret the results obtained [50,51]. Only EPR enables the direct detection of free radicals. It provides high sensitivity and specificity of radical detection and can enable quantitation and chemical identification of the radical formed [44]. A number of reviews have appeared detailing these methods and their use for the measurement of ROS [44,52].
4 How can ROS have dual roles in signaling and injury?
One may ask how ROS can have dual roles as homeostatic signaling molecules and mediators of injury. The basis for this duality appears to be primarily due to the much larger magnitude of ROS generation following ischemia and reperfusion as well as the fact that ischemic and post-ischemic conditions can result in the conversion of less reactive ROS to stronger more reactive oxidant species. We have directly observed that the levels of oxygen radicals detectable in post-ischemic hearts during the early period of reperfusion are 1 to 2 orders of magnitude higher than those detected in nonischemic control hearts [32,35,36]. In addition, the conditions occurring during ischemia and early reperfusion with marked acidosis and strong reducing state favor release of ferric or ferrous iron from metalloproteins, in turn facilitating iron mediated Fenton chemistry with conversion of the less potent oxidants superoxide and hydrogen peroxide to the highly reactive strong oxidant, hydroxyl radical. Indeed, we have observed that iron is released in the post-ischemic heart and that treatment with high affinity iron chelators such as deferoxamine can result in strong myocardial protection [53,54]. Beyond the release of iron or other redox cycling transition metal ions, we have also observed that the highly reduced and markedly hypoxic conditions occurring during ischemia and early reperfusion also shift ROS generating enzymes to produce more highly reduced and reactive oxidant species. For example, we have observed that under marked hypoxic conditions as occur in the myocardium during ischemia, xanthine oxidase directly reduces hydrogen peroxide to form the hydroxyl radical [55]. One would also expect that other enzymes such as aldehyde oxidase or heme based redox centers could also similarly reduce hydrogen peroxide to the hydroxyl radical. In addition, the concurrent formation of high levels of superoxide and NO favor their reaction to form the potent oxidant peroxynitrite [56]. This reaction proceeds with a high rate constant approaching the diffusion limit, so with the high levels of both superoxide and NO formed, the protection normally conferred by SOD would be lost.
5 NO and its role in myocardial function and injury
It has been shown that endothelial cells produce a factor, EDRF, which promotes vascular smooth muscle relaxation [57,58]. This factor was chemically identified as NO, a labile free radical [58-60]. Vascular endothelial cells contain an enzyme, nitric oxide synthase (NOS), which synthesizes NO [61]. To date, three major NOS isoforms have been identified. Two of these are constitutive and calcium and calmodulin dependent, NOS1 primarily in neurons, and NOS3 primarily in endothelial cells. The third isoform, NOS2, is inducible and calcium independent and is primarily involved in inflammation. Each of the three NOS isoforms convert L-arginine to L-citrulline and NO, and require the substrates NADPH and oxygen as well as the co-factor tetrahydrobiopterin (BH4). Coronary endothelium, endocardial endothelium, cardiac nerves, and cardiomyocytes of the normal heart all express NOS and have basal production of NO [62]. Only small quantities of NO are produced for brief periods when intracellular Ca2+ levels are elevated. This process of vascular and myocardial NOS generation is greatly altered by ischemia [63]. During ischemia, the rise in myocardial calcium leads to initial activation, however, subsequently oxygen levels fall limiting its availability as a substrate. In addition, marked intracellular acidosis occurs with prolonged ischemia rendering the enzyme inactive [63,64].
NO has been shown to react with superoxide to form the highly reactive oxidant, peroxynitrite, ONOO−, which can cause tissue injury [56,65]. Based on this, it has been suggested that NO formation may be of critical importance in modulating the toxicity of superoxide or other oxidants [65]. Over the last several years, it has been suggested that alterations in NO formation in ischemic myocardium result in post-ischemic injury. It was shown that vascular reactivity is decreased in the post-ischemic heart [8,9,66] and inferred that this was due to altered NO production or breakdown; however, it was not known if NO generation was increased or decreased during and after myocardial ischemia. Direct EPR methods have been developed to measure and quantitate NO in the heart [67]. We have observed that a large increase in NO production occurs during ischemia. This was shown to be due to both NOS-dependent formation during the early period of ischemia, and increasingly due to the reduction of tissue nitrite with prolonged ischemia [67-69].
6 Mechanisms of oxygen free radical generation in the post-ischemic heart
A number of mechanisms have been proposed to cause oxygen radical generation in reperfused myocardium. These include: the enzyme xanthine oxidase, mitochondrial oxidation, cyclooxygenase mediated unsaturated fatty acid oxidation, catecholamine oxidation, P450-mediated oxidation, activation of leukocyte NADPH oxidase, and iron release and redox cycling. A number of changes occur in ischemic tissues that predispose the tissue to subsequent oxidative reperfusion injury. It has been demonstrated that the enzyme xanthine dehydrogenase (XD) is converted to xanthine oxidase (XO), a potent generator of O2− and hydrogen peroxide [70]. During ischemia the substrates for this reaction, hypoxanthine and xanthine, accumulate. In addition, during ischemia cellular defenses against oxidative injury are impaired with lower activities of superoxide dismutase (SOD) and glutathione peroxidase [71].
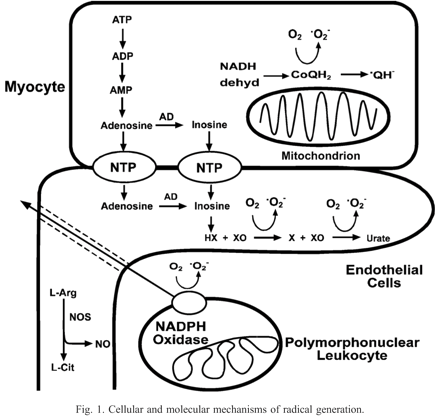
In prior studies, three major enzyme pathways were directly identified as important sources of radical generation: (1) xanthine oxidase (XO), primarily within endothelial cells; (2) the mitochondrial electron transport chain, primarily from the myocytes; and (3) NADPH oxidase, primarily within leukocytes (Fig. 1). In addition, it has been shown that under conditions of arginine or BH4 depletion, as induced by oxidant stress, NOS becomes uncoupled and switches from NO to superoxide generation.

{kind=link}
Cellular and molecular mechanisms of radical generation.
Experiments performed in isolated rat hearts have demonstrated that radical generation and functional injury are decreased by inhibition of XO with oxypurinol [25]. Similarly in human aortic or venous endothelial cells, xanthine oxidase mediated radical generation has been shown to be a central mechanism of oxygen radical generation upon post-ischemic reoxygenation [72]. However, questions remained regarding the role of XO formation or that of its substrates, xanthine and hypoxanthine, in triggering this process. In isolated ischemic hearts and hypoxic endothelial cells, ATP is metabolized to ADP, AMP, adenosine, inosine, and finally the XO substrates hypoxanthine (HX) and xanthine (X), which react with XO upon reoxygenation or reflow resulting in O2− generation [25,73]. We have observed that this substrate availability is of critical importance in controlling the magnitude and time course of endothelial radical generation [74,75]. Further studies with inhibition of adenosine deaminase have confirmed the importance of XO substrate formation [73]. Thus in the post-ischemic heart, increased XO formation occurs due to conversion of XD to XO and there is marked XO substrate formation due to intra-ischemic ATP degradation. Studies with a polyclonal XO antibody demonstrated that XO is primarily localized within the endothelium of the heart. Although the presence of XO in human heart and vasculature had been previously questioned [76], subsequent studies have shown that XO is present in human arterial and venous endothelial cells and can generate sufficient levels of oxygen radicals to trigger endothelial injury [72,74]. While the activity of XO was reported to be low in human heart compared to other species [77], most recently, XO has been shown to be upregulated in human dilated cardiomyopathy patients [78]. Allopurinol, a potent inhibitor of XO, has been shown to improve postoperative recovery [79,80] and reduce lipid peroxidation [79] in open-heart surgery patients. Reduction of reperfusion injury in human myocardium has also been reported [81]. Despite the presence of numerous supporting studies, there have also been negative reports, so that the role of XO in human cardiovascular disease remains controversial and merits further investigation [82].
In the heart, it has been shown that myocytes are the main source of these XO substrates and that either blocking substrate formation with inhibition of adenosine deaminase (AD) or blocking nucleotide transport (NT) from the myocytes to the endothelium would be effective at blocking radical generation [73]. It has also been observed in isolated hearts and myocytes that the mitochondrial electron transport chain distal to site one, NADH dehydrogenase, is an important source of radical generation with the formation of ubiquinone (CoQ) radicals as well as O2− derived radicals [32,33,35]. More recently, it has been shown that mitochondrial permeability transition is induced by ROS and in turn triggers mitochondrial ROS generation. This phenomenon has been termed mitochondrial ROS-induced ROS release[83].
There is evidence that post-ischemic oxidants may alter NOS function and endothelial reactivity [8]. It has been observed that NOS can switch from NO to O2− generation. All 3 NOS isoforms (nNOS, eNOS, or iNOS) can become potent sources of O2− with depletion of either the substrate L-arginine or the co-factor BH4 triggering this fundamental alteration in NOS function [84-86]. Since BH4 can be readily depleted by oxidants, oxidation of BH4 could result in a switch of NOS from NO to O2− generation. There are also other mechanisms that could trigger uncontrolled O2− generation including release of FAD from the enzyme, disruption of the active dimer, structural changes resulting in uncoupling of the reductase and oxygenase sites [60].
Endothelial and myocyte oxidant formation along with complement activation results in chemotaxis of polymorphonuclear leukocytes (PMNs) in the post-ischemic heart, markedly amplifying oxidant formation and injury [87,88]. It was observed that O2− derived radicals can trigger PMN adhesion molecule expression and PMN adhesion [89,90], thus, endothelial or myocyte derived radicals trigger PMN-mediated injury with adhesion to the endothelium followed by transmigration to the cardiac myocytes. Beyond these studies with isolated hearts in the presence of leukocytes and plasma, a large number of studies of in vivo regional ischemia/reperfusion models in a number of species have demonstrated protection with therapeutics designed to prevent leukocyte adhesion or complement activation [90,91]. However, in view of several negative clinical studies, the importance of leukocytes in clinical reperfusion injury has been questioned [91]. Based on the complexity and limitations of these clinical studies, it is unclear if these unsuccessful efforts truly indicate a lack of a critical role of leukocytes in clinical reperfusion injury or specific failures in study design or the properties of the therapeutics tested. In view of this discordance, it remains to be seen if therapies directed at preventing leukocyte adhesion or activation can effectively prevent clinical reperfusion injury. Thus, several interacting mechanisms have been shown to trigger oxidant and oxygen radical formation in post-ischemic myocardium (Fig. 1). Regardless of the source of oxidants and oxygen radicals, cellular injury can occur during reperfusion if the level of oxidative stress exceeds the capacity of endogenous free radical scavenging mechanisms such as SOD, catalase, glutathione peroxidase, glutathione, and uric acid.
7 Mechanisms of myocardial NO formation in the post-ischemic heart
Alterations in NO generation had been hypothesized to be a critical cause of injury in the ischemic and post-ischemic heart, however, these alterations in NO which occur were unknown. Based on the observed loss of endothelium-mediated vasodilation in post-ischemic hearts, it was initially proposed that NO levels were decreased in the post-ischemic heart [66]. Therefore, there was a need for techniques of directly measuring NO in the ischemic heart. Since NO is a free radical and reacts to form high affinity complexes with a variety of metal chelates and metalloproteins, the distinctive EPR spectra of these NO-complexes can provide a quantitative measure of NO generation [92-95]. While nitrosyl-heme formation is an intrinsic NO trap, these complexes are labile in the presence of O2. The Fe2+-diethyldithiocarbamate (DETC) complex has been proposed as a more stable and O2-independent trap suitable for measuring NO [94]. This complex has limited solubility in water, therefore, the ferrous iron complex of N-methyl-D-glucamine-dithiocarbamate (MGD), Fe2+-MGD2 (Fe-MGD), was also developed and has been applied for measuring NO in living tissues [67,69,95].
Measurements of NO in isolated rat ischemic hearts were performed using Fe-MGD, which binds NO giving rise to a characteristic triplet EPR spectrum. While only a small triplet signal was observed in normally perfused hearts (Fig. 2A), a 10-fold increase occurred after 30min ischemia indicating a marked increase in NO (Fig. 2B). NO formation increased as a function of the duration of ischemia [56,67,92,96]. With short ischemic durations of 30min or less, NO generation was decreased by the NOS blockers L-NAME or L-NMMA. Blockade of NO generation with L-NAME also resulted in increased recovery of contractile function after reperfusion [56,67,96]. While infusion of sufficient L-NAME or L-NMMA totally blocked NOS-mediated NO formation, only partial inhibition of the NO triplet signal was observed with a 60–80% decrease. This lack of total inhibition of NO formation with blockade of NOS suggested the existence of a NOS-independent pathway of NO formation [69,93]. To confirm this, direct measurements of NO formation via its binding to intrinsic metallo-heme centers within the tissue were also performed. The binding of NO to heme proteins gives rise to a unique EPR spectrum with axial symmetry and with the 5-coordinate complex, a characteristic inverted triplet is seen due to the hyperfine coupling of the nitrogen nucleus of bound NO [69]. These complexes are inherently O2 labile and are best observed at low O2 tensions as occur with prolonged ischemia. In control hearts, no NO-heme signal was seen, however, in ischemic tissue after 2–12h, increasingly prominent NO-heme signals were seen, confirming that increased NO formation occurs during myocardial ischemia. Pretreatment of hearts with L-NAME partially decreased these NO signals with only a 30–50% decrease after 4–8h of ischemia, further supporting the existence of a NOS-independent pathway of NO generation secondary to nitrite reduction.

{kind=link}
EPR spectra of NO trapping in isolated rat hearts with Fe-MGD. (A) Control heart. (B) Heart after 30min ischemia. Recorded at 77°K with a frequency of 9.32GHz using 1.0mW power (reproduced with permission from the Journal of Biological Chemistry 1995; 270(1): 304–307).
Myocardial ischemia results in intracellular acidosis and severe hypoxia leading to a highly reduced state that subsequently leads to nitrite reduction. To determine if nitrite, NO2−, was reduced during ischemia to form NO, experiments were performed in hearts subjected to ischemia in the presence of isotopically labeled 15NO2−[69]. Since 15N has a nuclear spin of 1/2, doublet hyperfine splitting was observed in the EPR spectra of NO–Fe2+–MGD complexes instead of the triplet splitting observed for the natural abundance 14N that has a nuclear spin of 1. In normally perfused control hearts, 15NO2− did not result in significant NO formation (Fig. 3A, left). In hearts which were labeled and then subjected to 30min of ischemia, however, marked NO formation was seen with the appearance of a large doublet signal (Fig. 3B, left). In matched experiments with natural abundance 14NO2−, large NO triplet signals were seen (Fig. 3C, left). Thus, NO2− was shown to be reduced to NO in the ischemic heart. Nitrosyl-heme formation was also demonstrated in ischemic hearts labeled with 15NO2−. A prominent doublet nitrosyl-heme signal was seen due to the formation and binding of 15NO to these proteins, further confirming that NO is generated from NO2− (Fig. 3B, right). With 14NO2− a similar magnitude triplet NO signal was observed (Fig. 3A, right). Experiments measuring the concentration of nitrite within the heart prior to ischemia showed that relatively large nitrite concentrations of 12μM were present [69,93]. Subsequently, it has been appreciated that nitrite can be an important source of NO in the cardiovascular system. Recently, the molybdenum enzyme XO that has structural similarity to bacterial nitrite reductases has been shown to reduce nitrite to NO under both anaerobic and aerobic conditions [97,98].

{kind=link}
EPR spectra of NO formation in the rat heart. Hearts were prelabeled with 1mM 15N or 14N nitrite (NO2−). The left panel shows spectra from hearts with Fe-MGD; (A) before ischemia; (B) 30min ischemia with 15NO2−; (C) 30min ischemia with 14NO2−. The right panel shows nitrosyl-heme formation; (A) with 14NO2−; (B) with 15NO2− (reproduced with permission from Nature Medicine 1995; 1(8): 804–809).
8 Interactions between the pathways of oxygen radical and NO formation
As summarized above, it is clear that in the ischemic and reperfused heart a number of pathways of oxygen radical and NO formation are activated giving rise to high levels of oxidants and NO. Recent reports have shown that NO and peroxynitrite can inhibit several critical pathways of oxygen radical generation including XO and leukocyte NADPH oxidase [99,100]. Thus, exogenous NO from pharmacological NO-donors or endogenous NO or secondary peroxynitrite could inhibit subsequent oxygen radical generation, with this contributing to a process limiting myocardial injury. Similarly, as noted above, it is clear that oxidants also modulate the function of NOS [101].
9 Preconditioning induced alterations in nitric oxide and oxygen radical generation
There is evidence that ischemic preconditioning (IPC) may be induced by low level oxidant formation during the brief periods of preconditioning ischemia and reflow [102]. From earlier studies evaluating the effect of ischemic duration on the magnitude of oxygen radical generation, it is clear that even short periods of ischemia produce a measurable but small oxidant burst upon reperfusion [35]. In addition, it would be expected that IPC would alter the subsequent processes of oxygen radical and NO production during prolonged ischemia and reperfusion.
While there has been considerable controversy regarding the role of NO and NOS in the process of acute IPC, we and others have observed that IPC markedly decreases NO generation during subsequent prolonged ischemia [103,104]. Since NO inhibits in vivo mitochondrial oxygen consumption in post-ischemic myocardium resulting in marked myocardial hyperoxygenation and impaired mitochondrial function [105], IPC-induced decrease of post-ischemic NO production would prevent myocardial hyperoxygenation upon post-ischemic reperfusion, in turn diminishing oxygen radical and peroxynitrite generation with preservation of mitochondrial function.
10 Conclusion
Over the last three decades, it has become clear that oxidants and free radicals are generated in the ischemic and reperfused heart, and are important mediators of post-ischemic injury. Both oxygen radical and NO formation are greatly increased through a series of interacting enzymes and cellular pathways. Nitric oxide generation is increased in the ischemic heart through both NOS-dependent formation and NOS-independent nitrite reduction. It has become clear that there is a critical balance of oxygen radical formation and NO formation and their catabolism in normal myocardial physiology and signaling. In the setting of myocardial ischemia and reperfusion with the marked increases in oxygen radical and NO generation, this balance is disrupted resulting in oxidative injury. Ischemic preconditioning markedly decreases subsequent NO generation and there is recent evidence that oxidant formation is also decreased. More work in the future will be required to fully understand and characterize the critical role of oxidants and NO in post-ischemic injury and how to optimally translate this understanding to clinical treatments that will salvage heart muscle at risk in acute coronary syndromes.
Acknowledgements
This work was supported by the National Institutes of Health Grants HL63744, HL65608, HL38324.