beyond reason
미토콘드리아 기능을 위해 황을 함유한 채소를 많이 먹어야 하는 이유(클릭클릭)
황과 철의 관계에 대하여...
철, 황, 미토콘드리아 기능과 관계
Mitochondria and Iron: Current Questions
Abstract
Introduction
Mitochondria are cellular organelles that perform numerous bioenergetic, biosynthetic, and regulatory functions and play a central role in iron metabolism. Extracellular iron is taken up by cells and transported to the mitochondria, where it is utilized for synthesis of cofactors essential to the function of enzymes involved in oxidation-reduction reactions, DNA synthesis and repair, and a variety of other cellular processes.
Areas Covered
This article reviews the trafficking of iron to the mitochondria and normal mitochondrial iron metabolism, including heme synthesis and iron-sulfur cluster biogenesis. Much of our understanding of mitochondrial iron metabolism has been revealed by pathologies that disrupt normal iron metabolism. These conditions affect not only iron metabolism but mitochondrial function and systemic health. Therefore, this article also discusses these pathologies, including conditions of systemic and mitochondrial iron dysregulation as well as cancer. Literature covering these areas was identified via PubMed searches using keywords: Iron, mitochondria, Heme Synthesis, Iron-sulfur Cluster, and Cancer. References cited by publications retrieved using this search strategy were also consulted.
Expert Commentary
While much has been learned about mitochondrial iron, key questions remain. Developing a better understanding of mitochondrial iron regulation will be paramount in developing therapies for syndromes that affect mitochondrial iron.
1. Introduction
Mitochondria are ancient organelles thought to have originated from a symbiotic association between alpha-proteobacteria and a eukaryotic progenitor [1]. Mitochondria possess a circular DNA genome that is approximately 16 Kb. During co-evolution with host cells, the mitochondrial genome has become truncated, with the result that some mitochondrial proteins are encoded by nuclear genes [1]. Similar to their bacterial ancestors, mitochondria are enclosed by a double membrane consisting of an inner (IMM) and outer membrane (OMM). The OMM is highly permeable to ions and small uncharged metabolites due to the presence of large porin channels [2]. In contrast, the IMM forms a tight diffusion barrier to ions and metabolites and is folded into a complex form with invaginations known as cristae. Transfer of specific metabolites across the IMM is facilitated by hydrophobic mitochondrial carrier proteins [3]. The IMM and OMM delineate two compartments, the intermembrane space (IMS) between the IMM and OMM, and the mitochondrial matrix within the inner membrane (Figure 1, ,2).2). Differences in the permeability of the IMM and OMM result in distinct environments within these compartments.

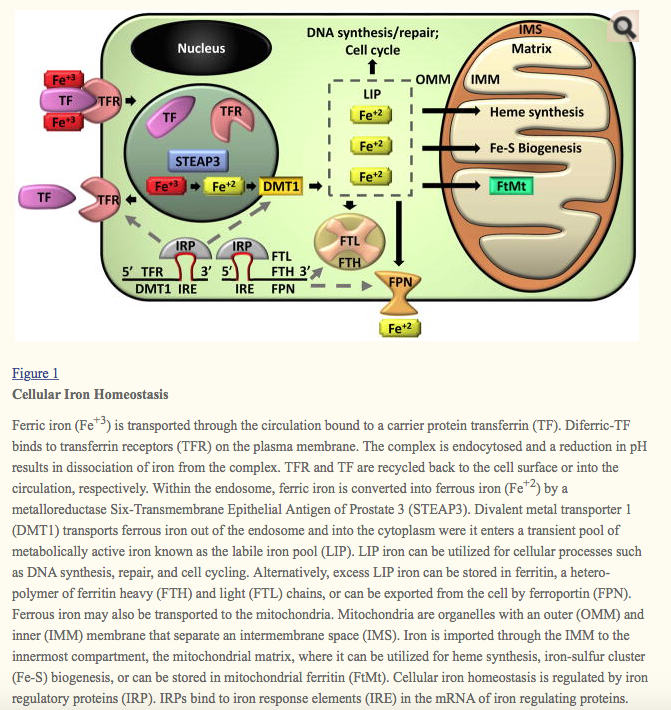
Ferric iron (Fe+3) is transported through the circulation bound to a carrier protein transferrin (TF). Diferric-TF binds to transferrin receptors (TFR) on the plasma membrane. The complex is endocytosed and a reduction in pH results in dissociation of iron from the complex. TFR and TF are recycled back to the cell surface or into the circulation, respectively. Within the endosome, ferric iron is converted into ferrous iron (Fe+2) by a metalloreductase Six-Transmembrane Epithelial Antigen of Prostate 3 (STEAP3). Divalent metal transporter 1 (DMT1) transports ferrous iron out of the endosome and into the cytoplasm were it enters a transient pool of metabolically active iron known as the labile iron pool (LIP). LIP iron can be utilized for cellular processes such as DNA synthesis, repair, and cell cycling. Alternatively, excess LIP iron can be stored in ferritin, a hetero-polymer of ferritin heavy (FTH) and light (FTL) chains, or can be exported from the cell by ferroportin (FPN). Ferrous iron may also be transported to the mitochondria. Mitochondria are organelles with an outer (OMM) and inner (IMM) membrane that separate an intermembrane space (IMS). Iron is imported through the IMM to the innermost compartment, the mitochondrial matrix, where it can be utilized for heme synthesis, iron-sulfur cluster (Fe-S) biogenesis, or can be stored in mitochondrial ferritin (FtMt). Cellular iron homeostasis is regulated by iron regulatory proteins (IRP). IRPs bind to iron response elements (IRE) in the mRNA of iron regulating proteins. IRP/IRE binding to the 3′ end of mRNA, which occurs in TFR and DMT1, stabilizes the mRNA and enhances translation. IRP/IRE binding to the 5′ end of mRNA, which occurs in FTH, FTL, and FPN, blocks the binding of translational machinery and reduces mRNA translation.

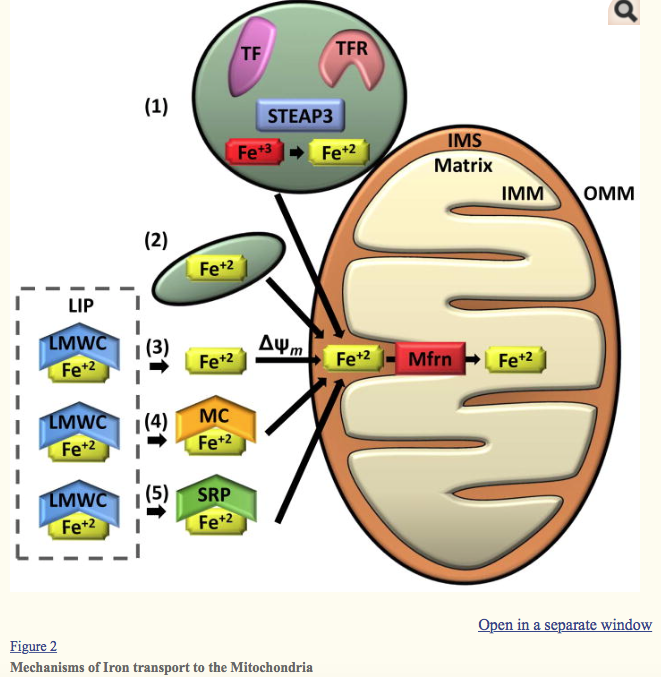
Several mechanisms for transporting iron into the mitochondria have been suggested. (1) “Kiss and Run” mechanism: Ferric iron (Fe+3) taken up by endocytosis of the transferrin receptor (TFR)-transferrin (TF) complex is likely reduced to ferrous iron (Fe+2) by a metalloreductase Six-Transmembrane Epithelial Antigen of Prostate 3 (STEAP3) and directly transported from the endosome to the mitochondria without entering the cytosol. The “Kiss and Run” mechanism has only been demonstrated in erythroid cells. (2) Non-transferrin bound iron may be taken up via fluid-phase endocytosis by a partially solvent occluded mechanism. This method of iron transport was proposed from data from murine cardiac cells. (3–6) Cytosolic labile iron (LIP) is thought to be bound by low molecular weight complexes (LMWC) and is presumed to be the source of iron delivered to the mitochondria through the cytosol. (3) Iron has been shown to be transported into the mitochondria via a mitochondrial membrane potential-dependent manner (Δψm) in yeast and rat hepatocytes. (4) Alternatively, iron may be trafficked to the mitochondria by high molecular weight complexes such as metallochaperones (MC) or (5) siderophores (SRP). Once iron is transported through the outer mitochondrial membrane (OMM) and into the intermembrane space (IMS), it is transferred through the inner mitochondrial membrane (IMM) and into the mitochondrial matrix by mitoferrins (Mfrn) and associated regulatory proteins (i.e. ATP Binding Cassette Subfamily B Member 10 [Abcb10] in erythroid cells).
Mitochondria are highly dynamic organelles, and the number of mitochondria per cell is dependent on cellular metabolic requirements and tissue types [4]. For example, erythrocytes do not have mitochondria, whereas liver and muscle cells may contain hundreds to thousands of mitochondria [5,6]. Cells maintain a functional pool of mitochondria by constant fission and fusion. Depending on physiological requirements, the process of fission, fusion, and mitochondrial morphology are regulated by a dedicated protein machinery (recently reviewed [4]).
2. Mitochondrial Function
Mitochondria carry out diverse cellular functions and play crucial roles in cellular respiration and energy production. Metabolic pathways such as β-oxidation of fatty acids and the tricarboxylic acid cycle (TCA) occur in the mitochondrial matrix. These pathways generate reducing equivalents (NADH and FADH2) that participate in energy (ATP) generation by donating electrons to the electron transport chain and generating an electrochemical gradient across the IMM. Mitochondria are also involved in the biosynthesis of lipids, amino acids, pyrimidine and various other metabolic intermediates [7]. In addition, mitochondria exert control over a wide range of cellular functions by shaping the spatiotemporal distribution of cellular Ca2+ [8]. One such Ca2+-dependent process regulated by mitochondria is the control of cellular fate (survival or death) through apoptotic signaling [9].
Apart from the functions mentioned above, mitochondria play a pivotal role in metabolism of trace elements, especially the metabolism of iron. Iron is a key player in many important cellular processes, including energy metabolism, respiration, and DNA synthesis. The ability of iron to influence diverse biologic processes is a consequence of its chemical properties. Iron has unpaired electrons and is capable of accepting or donating electrons. As a result, iron exhibits flexible coordination chemistry and redox activity, and is often incorporated as a prosthetic group in enzymes and structural proteins [10]. The reactive properties that allow iron to facilitate diverse biological reactions can, however, allow iron to participate in potentially harmful reactions, such as the generation of reactive oxygen species (ROS) which can lead to cell death [10,11]. Therefore, cells have evolved sophisticated mechanisms to control acquisition, usage, and detoxification of iron to ensure that iron metabolic needs are met and the risk of iron toxicity is minimized [12]. Mitochondria are key to this carefully controlled regulation and metabolism of iron.
3. Cellular Iron Metabolism
Iron metabolism is a tightly controlled process (Figure 1). To prevent unintended reactions, iron absorbed from the diet or recycled from dying cells is bound to a carrier protein, transferrin (TF), for transport through the circulation. TF is capable of binding two ferric (Fe+3) iron atoms. Under normal physiological conditions, circulating iron-bound TF, or holo-TF, is delivered to all tissues of the body by binding to the membrane-bound transferrin receptor 1 (TFR1). The resulting holo-TF-TFR1 complex is internalized by receptor-mediated endocytosis. Low pH (approximately pH 6) within the endosome is generated by the action of proton pumps and results in dissolution of the holo-TF-TFR1 complex and release of ferric iron. Free ferric iron (Fe3+) in the endosome is reduced to ferrous iron (Fe2+) by the ferric reductase six-transmembrane epithelial antigen of prostate 3 (STEAP3) present in the endosomal membrane. Ferrous iron is then transported across the endosomal membrane by divalent metal transporter 1 (DMT1) to enter the metabolically available transient cytosolic labile iron pool (LIP). LIP iron is available for immediate cellular use. Excess iron is stored in ferritin, a multimeric protein composed of 24 subunits that forms a hollow sphere capable of storing as many as 4500 iron atoms. In mammals ferritin is a hetero-polymer of two isoferritins: H-ferritin (FTH1) and L-ferritin (FTL) [13]. Iron not stored in ferritin can be exported back to the circulation by the iron efflux protein ferroportin (FPN).
Cellular iron homeostasis is tightly controlled by a posttranscriptional regulatory mechanism involving iron regulatory proteins (IRP1 and IRP2) and iron responsive elements (IRE) in target mRNAs. IREs are short stem loop structures present in the 5′ and 3′ untranslated regions (UTR) of mRNAs of key proteins involved in iron metabolism. The binding of IRPs to mRNAs with 5′ IREs, such as FTH1, FTL, and FPN1, suppress translation. In contrast, mRNAs containing 3′ IRE elements, such as TFR1 and DMT1, are stabilized by IRP binding, resulting in increased translation. The activity of IRPs is controlled by iron: when intracellular levels of iron are low, IRPs are active and suppress translation of mRNAs of proteins such as FTH1, FTL and FPN, which are involved in iron storage and export. Simultaneously, mRNAs of proteins involved in iron uptake, such as TFR1, are stabilized. The converse occurs when intracellular iron levels are high (reviewed in [10]).
4. Iron and Mitochondria
Mitochondria are major hubs of iron utilization and accumulation. For example, in lung carcinoma cells, approximately 20% of total cellular iron was found in mitochondria [14]. In rat hepatocytes, the concentration of metabolically available iron in mitochondria was twice that in the cytosol, as measured by selective fluorescent chelators [15]. Iron in the mitochondria functions as a cofactor in iron-sulfur cluster-containing proteins, heme-containing proteins, and iron ion-containing proteins. Important mitochondrial iron sulfur cluster-containing proteins include electron-transfer flavoproteins, NADH:ubiquinone oxidoreductase, Rieske iron-sulfur protein, and subunits of succinate dehydrogenase, all of which are involved in respiratory chain function, as well as enzymes involved in other processes, such as aconitase, lipoic acid synthase, biotin synthase, and radical S-adenosylmethionine (SAM) enzymes. Heme-containing proteins include cytochrome bc1, cytochrome c, cytochrome c oxidase and succinate dehydrogenase, all of which are involved in the electron transport chain. In addition to proteins with iron-containing prosthetic groups, proteins with iron-ion cofactors are found in the mitochondria. These include diiron monooxygenases and dioxygenases, such as members of the AlkB family of Fe(II)-and 2-oxyglutarate dioxygenases ALKBH1 and ALKBH7 which are involved in mitochondrial-tRNA modification [16] and regulation of cellular necrosis [17], respectively. The prevalence of iron-containing proteins in the mitochondria attests to the importance of iron in normal mitochondrial function.
5. Iron Trafficking to the Mitochondria
Despite the critical role played by iron in mitochondrial function and the important role of mitochondria in cellular iron metabolism, mechanisms by which mitochondria acquire iron are incompletely understood. It is likely that several mechanisms contribute to iron delivery to mitochondria including direct delivery of extracellular iron to the mitochondria and mitochondrial uptake of cytosolic iron. Evidence for these methods of iron trafficking to mitochondria has been derived from diverse cell types. It is unclear whether specific transport methods are generalizable or cell-type specific (Figure 2).
Delivery of TF-bound iron via direct interorganellar transfer of iron from holo-TF-containing endosomes to mitochondria has been demonstrated in reticulocytes, developing erythroid cells that utilize vast amounts of iron for hemoglobin synthesis [18,19]. In this “kiss and run” model, endosomes form transient contacts with the OMM to facilitate the direct transfer of iron into the mitochondria [18]. A proposed benefit of direct iron transfer is the ability to avoid unwanted reactions of iron in the oxygen rich cytosol. This may be particularly important in reticulocytes, which process large amounts of iron, as this method of mitochondrial iron delivery has not yet been demonstrated in other cell types. Instead, in cardiac cells, non-transferrin-bound iron was suggested to be delivered to mitochondria via fluid-phase endocytosis by a partially solvent occluded mechanism [20]. In this model non-transferrin-bound iron is delivered to mitochondria without passing through the cytosolic labile iron pool, unlike transferrin-bound iron [21,22]. This was supported by the observation that mitochondrial iron uptake was not affected by treatment with cytosol-specific iron chelators.
Iron has also been proposed to enter the mitochondria from the cytosolic labile iron pool (LIP). Iron imported into the cytosol by the TF/TFR1 pathway enters a transient pool of labile iron composed predominately of ferrous iron, as determined by experiments showing that iron (II) chelators are more efficient in chelating LIP than iron (III) chelators [23]. It has been shown in rat hepatocytes and yeast that ferrous iron from the cytosol can be taken up by the mitochondria in a mitochondrial membrane potential-dependent manner [24,25]. Ferrous iron in the LIP is presumed to form low molecular weight iron complexes (e.g., iron (ATP)2, iron-citrate etc.) [23,26] which act as intermediates between absorbed extracellular iron and intracellular iron-utilization. However the nature of intracellular iron complexes is not fully established [27], and incompletely characterized high molecular weight iron intermediates were also identified in K562 chronic myelogenous leukemia cells [28].
Interacting or chaperone proteins may also play a role in intracellular iron trafficking [27]. Metallochaperones are proteins that bind to metal ions and deliver them to cognate enzymes and transporters by protein-protein interaction. Metallochaperones were identified for copper transport more than a decade ago [29]. While the identification of iron chaperones is still unfolding, a few candidate proteins such as poly(rC)-binding proteins and Grx3-type monothiol glutaredoxins have been identified [30,31]. Poly(rC)-binding proteins (PCBP1) are RNA/DNA binding proteins with diverse functions in splicing, transcript stabilization, and translational regulation [32]. Glutaredoxins are a class of glutathione-dependent redox enzymes involved deoxyribonucleotide formation, protein folding, signal transduction, and ROS defense [33]. These posited iron chaperones mediate the uptake of a “chelator-inaccessible” cytosolic iron pool distinct from the LIP [20]. Differences in phenotypes of knockouts suggest that iron chaperones may exhibit specificity in iron trafficking. Thus, while PCBP1 is capable of delivering iron to ferritin in yeast and the hepatocellular carcinoma cell line Huh7 [30], PCBP1 had minimal effects on mitochondrial iron levels in HEK293T cells [34]. In addition, a yeast complex consisting of glutaredoxin 3 and bovine lymphocyte antigen (BolA)-like protein has been shown to deliver iron to mitochondria [35]. Yeast cells depleted of glutaredoxin 3/4 exhibited defects in iron import to mitochondria and impaired iron-sulfur cluster and heme biogenesis [31]. In addition to iron chaperones, the mammalian siderophore 2,5-dihydroxybenzoic acid (2,5-DHBA) has been postulated to mediate delivery of iron to mitochondria. B-hydroxybutyrate dehydrogenase (BDH2) is the enzyme that catalyzes the production of 2,5-DHBA. Depletion of BDH2 in mouse erythroleukemia cells resulted in high amounts of cytoplasmic iron and decreased levels of mitochondrial iron [36].
Whichever delivery mechanism cells adopt to acquire mitochondrial iron, to gain access the mitochondrial matrix the iron must cross the outer and inner membrane of mitochondria (Figure 2). The exact mechanism by which iron traverses the OMM is not fully understood. It is postulated that porins may allow the passage of iron across the OMM [37]. However, direct evidence for the movement of divalent ferrous iron across the OMM via porins remains elusive. An alternate hypothesis is that a mitochondrial isoform of DMT1, which was found to localize to the OMM in HEK293 cells, imports ferrous iron into the intermembrane space [38,39].
A majority of mitochondrial iron metabolism occurs in the mitochondrial matrix. As such, iron must further be transported across the IMM. Transport of iron across the IMM is an active process and dependent on membrane transporters. Mitoferrin (Mfrn1) and its homolog mitoferrin 2 (Mfrn2) are the principal importers of iron across the IMM in a variety of tissues, although the exact mechanism remains obscure [40,41]. Mfrn1 is known to interact with Abcb10, an IMM ATP-binding cassette transporter, which enhances Mfrn1 stability and mitochondrial iron import in erythroid cells [42]. Furthermore, ferrochelatase, an important enzyme for heme synthesis in the mitochondria (described in more detail below), has been shown to complex with Mfrn1 and Abcb10 in mouse erythroleukemia (MEL) cells [43]. This complex has been proposed to direct iron to ferrochelatase and enhance heme synthesis. Another member of the ABC transporter family, ABCB7, has also been shown to interact with ferrochelatase in MEL cells [44] and may participate in two important mitochondrial functions: heme synthesis and iron-sulphur cluster biogenesis. ABCB7 has further been shown to be essential for hematopoiesis in vivo and was suggested to facilitate iron-sulfur cluster (Fe-S) export from the mitochondria [45]. A third family member, ABCB8, appears to facilitate iron export from the mitochondria, specifically in cardiac cells, either in Fe-S or bound to an undetermined partner [46].
Another family of proteins with a potential role in maintaining mitochondrial iron homeostasis and function are the sideroflexins. In humans, the sideroflexin family comprises five paralogous proteins (SFXN1-5) with a high degree of sequence similarity and overlapping expression patterns [47,48]. Sideroflexins are inner mitochondrial membrane proteins predicted to have five transmembrane domains [49], suggesting that these proteins may act as channel or carrier molecules. Functional reduction in SFXN4 expression led to mitochondriopathy and impaired erythropoiesis in two human subjects [49]. Similarly, a mutation in SFXN1 was implicated in transient embryonic and neonatal sideroblastic anemic in flexed-tail mice [48]. The mechanisms of action of these proteins are not fully understood. However, it is postulated that they mediate the transport of factor(s) that is/are important for mitochondrial function, possibly including metabolites important in iron utilization [12].
6. Mitochondrial Iron Utilization
Following import, mitochondrial iron is primarily utilized in three metabolic pathways: (i) heme synthesis; (ii) iron-sulphur cluster biogenesis; and (iii) mitochondrial iron storage.
6.1 Heme synthesis
The synthesis of heme is absolutely dependent on mitochondria (Figure 3). Heme is a complex of ferrous iron and protoporphyrin IX. It is an important prosthetic group for numerous vital proteins such as hemoglobin, myoglobin, cytochrome c, cytochrome p450, catalase, peroxidase and others. Prosthetic heme is involved in oxygen transport and storage, electron transfer for enzymatic redox reactions, signal transduction, ligand binding, and control of gene expression [50]. In mammals, heme synthesis involves the sequential action of eight enzymes. These eight enzymes comprise four distinct stages of heme biosynthesis: 1) synthesis of an individual pyrrole ring; 2) polymerization of four pyrroles to a tetrapyrrole ring; 3) side chain modification and ring closure; and 4) insertion of Fe2+ into the ring (Figure 3). Synthesis of heme is initiated in the mitochondrial matrix by the formation of 5-aminolevulinic acid (ALA). Aminolevulinic acid synthase (ALAS) mediates ALA synthesis by catalyzing the condensation of glycine, thought to be supplied by the transporter SLC25A38, and succinyl-CoA [51,52]. Mammals have two isoforms of ALAS. The first isoform, ALAS1, is expressed in all tissues and provides a housekeeping function. The second isoform, ALAS2, is expressed only in erythroid cells [53]. ALA is then exported to the cytosol to undergo four more enzymatic conversions. In the cytosol, two molecules of ALA are condensed to form porphobilinogen (PBG) by porphobilinogen synthase (PBGS) [54]. Subsequently, four molecules of porphobilinogen are polymerized by porphobilinogen deaminase (PBGD) to form the linear tetrapyrrole 1-hydroxymethlbilane (HMBS) [55]. HMBS is then acted upon by uroporphyrinogen synthase to complete ring closure and yield closed tetrapyrrole uroporphyrinogen III [56]. The last enzymatic step in the cytosol is the decarboxylation of uroporphyrinogen III to yield coproporphyrinogen III, a step that is carried out by uroporphyrinogen decarboxylase [57]. Import of coproporphyrinogen III into mitochondria is thought to be mediated by ATP-binding cassette transporter ABCB6 in K562 chronic myelogenous leukemia cells and mouse erythroleukemia (MEL) cells [58]. However, the role of ABCB6 in the import of coproporphyrinogen III remains incompletely understood. In fact, other proteins, such as 2-oxoglutarate carrier (OGC) in HeLa, HepG2, and A127 cells [59] and peripheral-type benzodiazepine receptor (PBR) in MEL cells [60], have been reported to perform the same function.

Enzymatic steps and intermediates formed during the biosynthesis of Heme from 5-aminolevelunic acid. Heme synthesis in the mitochondria begins in the mitochondrial matrix. Glycine and succinyl CoA are condensed by 5-aminolevulinate synthase to 5-aminolevelunic acid (ALA). ALA is exported to the cytosol through the inner mitochondrial membrane (IMM), the intermembrane space (IMS), and the outer mitochondrial membrane (OMM). In the cytosol, ALA undergoes four steps of enzymatic modification to generate coproporphyrinogen III (COPRO III). COPRO III is transported back to mitochondrial matrix where the last three enzymatic reactions occur. Abbreviations: ALA, 5-aminolevelunic acid; ALAS, 5-Aminolevulinate synthase; PBG, Porphobilinogen; PBGS, Porphobilinogen synthase; HMB, Hydroxymethylbilane; HMBS, Hydroxymethylbilane synthase; UROIII, Uroporphyrinogen III; UROS, Uroporphyrinogen III synthase; COPRO III, Coproporphyrinogen III; UROD, Uroporphyrinogen decarboxylase; PPG IX, Protoporphyrinogen IX; CPOX, Coproporphyrinogen oxidase; PP IX, Protoporphyrin IX; PPOX, Protoporphyrinogen oxidase; FECH, Ferochelatase.
Once in the mitochondria, coproporphyrinogen III undergoes oxidative decarboxylation of its propionyl side chains on two pyrrole rings to generate protoporphyrinogen IX. The enzyme involved in this step is coproporphyrinogen III oxidase (CPO), an oxygen-dependent enzyme located in the mitochondrial intermembrane space [61]. In the next step, six hydrogen atoms are removed from the porphyrinogen ring to form protoporphyrin IX. This oxidation is catalyzed by the oxygen-dependent enzyme porphyrinogen IX oxidase (PPO) present in the IMM [62]. The last step in heme synthesis is mediated by ferrochelatase (FECH), a homodimer with two 2Fe-2S cluster moieties. FECH in the IMM catalyzes insertion of ferrous iron into protoporphyrin IX to generate Fe-protoporphyrin IX, or heme, in the mitochondrial matrix [63]. Feline leukemia virus subgroup C receptor 1b (FLVCR1b) has been shown to regulate mitochondrial heme export into the cytosol [64].
Condensation of glycine and succinyl-CoA by ALAS1/2, is the rate-limiting step in heme synthesis. Heme negatively regulates the expression of ALAS1 through feedback inhibition. A number of mechanisms have been described for heme-mediated repression of ALAS1, including transcriptional repression [65], destabilization of ALAS1 mRNA[66], and inhibition of translocation of the ALAS1 precursor from the cytosol to mitochondria [67]. In contrast, the expression of ALAS2, which is predominantly expressed in erythroid cells, is not suppressed by heme, thereby maintaining a steady supply of heme for the production of a large amount of hemoglobin [68]. ALAS2 is instead post-transcriptionally regulated by the IRE-IRP system. IRE-IRP regulation allows heme synthesis in differentiating red cells to fluctuate based on cellular iron availability and mitochondrial function [69].
6.2 Iron-sulfur cluster biogenesis
One of the indispensable functions of mitochondria is the biogenesis of iron-sulfur (Fe-S) clusters, an important functional form of iron (Figure 4). Fe-S clusters have remarkable structural versatility as they can exist in different configurations depending on the number of iron and sulfur atoms present. The common forms are rhomboid [2Fe-2S], cuboidal [3Fe-4S], and cubane [4Fe-4S] clusters. More complex Fe-S cluster configurations have also been identified that contain other metal ions such as molybdenum [70]. The iron atoms present in the Fe-S cluster can exist either as ferric or ferrous iron and cycle between the redox states, allowing the Fe-S cluster to participate in redox reactions. Fe-S clusters are usually ligated to their respective apoproteins via ionic interactions between iron and cysteine residues present in the peptide. In some cases, other amino acids, such as histidine, arginine, and serine, may also serve as ligands [71]. Fe-S clusters act as cofactors in a diverse array of biological processes. They are essential to the function of aconitase and succinate dehydrogenase, metabolic enzymes of the citric acid cycle. Fe-S clusters mediate electron transfer within and between the respiratory complexes of the electron transport chain [72,73]. Several nucleic acid processing enzymes such as helicases, glycosylases and primase contain Fe-S clusters. DNA polymerase and DNA repair enzymes (Fanconi anemia group J protein (FANCJ) and Xeroderma pigmentosum group D protein (XPD)) require Fe-S clusters for their function [74,75]. ABCE1, a cytosolic and nuclear Fe-S protein, plays a role in ribosome biogenesis/maturation and protein synthesis [76].

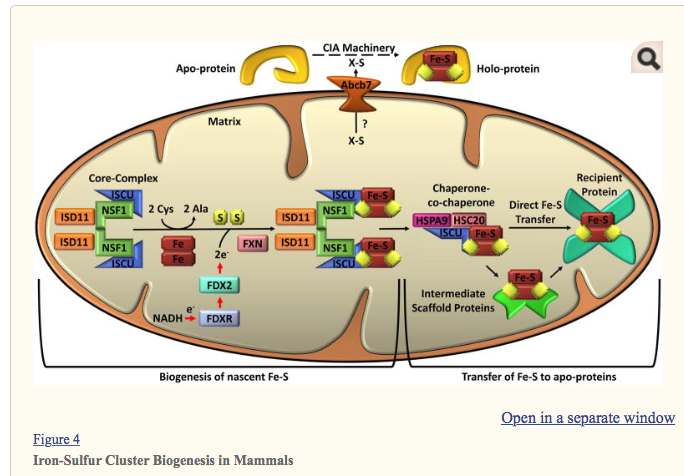
Separate pathways are involved in mitochondrial iron-sulfur cluster biogenesis and cytosolic iron-sulfur cluster biogenesis, but both depend on mitochondria. Mitochondrial iron-sulfur cluster biogenesis begins in the mitochondrial matrix with the nascent iron-sulfur cluster (Fe-S) assembly core-complex. This core-complex consists of a cysteine desulfurase (NFS1) dimer, two monomers of the scaffold protein ISCU, two regulatory proteins ISD11, and frataxin (FXN). NFS1 abstracts sulfur from cysteine (Cys), converting it to alanine (Ala), for nascent Fe-S cluster assembly. The stability of NFS1 depends on its binding partner ISD11. Iron entry and cysteine desulfurase activity are possibly regulated by FXN. Iron and electrons (e-) provided by NADH, ferredoxin reductase (FDXR), and ferredoxin (FDX2) facilitate the assembly of the nascent ISC on the ISCU. The ISCU-bound nascent Fe-S is transferred to recipient apo-proteins or intermediate carrier proteins by a dedicated chaperone-co-chaperone (HSPA9/HSC20) system. For cytosolic iron-sulfur cluster biogenesis, an undetermined sulfur-containing compound (X-S) is exported from the mitochondrial to the cytosol by ATP-binding cassette sub-family B member 7 (ABCB7). Cytoplasmic Fe-S assembly (CIA) machinery then facilitates the loading of Fe-S onto apo-proteins to form holo-proteins. Numerous proteins are involved in the transfer of Fe-S to specific proteins; see text for discussion.
Fe-S cluster biogenesis is a complex, multistep process that involves a set of dedicated multimeric protein complexes. Fe-S cluster biogenesis involves two basic steps: nascent cluster assembly followed by its transfer into the apoprotein (Figure 4). Mammalian Fe-S cluster biogenesis consists of mitochondrial iron-sulfur cluster assembly (ISC) and cytosolic iron-sulfur assembly (CIA). In addition to synthesizing Fe-S clusters destined for mitochondrial Fe-S proteins, mitochondria also export an unidentified sulfur-containing compound to the cytosol for cytosolic and nuclear Fe-S cluster formation [77,78]. Thus, the core step of Fe-S cluster biogenesis in the mitochondria (ISC) is essential for all cellular Fe-S proteins in mammals.
6.2.1 Mitochondrial Iron-sulfur Cluster Assembly (ISC)
In mammals, Fe-S clusters are assembled on a dedicated multimeric protein core complex with the cluster transiently bound on a scaffold protein (ISCU). ISCU provides the structural backbone for cluster assembly and the cysteine residues required for cluster coordination. The core complex is composed of cysteine desulfurase (NFS1), NFS1 binding partner ISD11, and frataxin (FXN). NFS1 forms a homodimer to which monomers of ISCU bind. Sulfur is provided by cysteine desulfurase, which abstracts sulfane sulfur (S0) from free cysteine through a persulfide intermediate with the help of the cofactor pyridoxal phosphate (PLP). Sulfane sulfur is reduced to sulfide (S2-), in a process that likely involves the concerted action of ferredoxin (FDX2) and ferredoxin reductase (FDXR) [79]. Cysteine desulfurase activity and stability is critically dependent on the dedicated protein co-factor ISD11 [80,81]. The physiological source of iron for the nascent cluster remains to be clearly identified. FXN, due to the presence of acidic surface patches that can bind iron, was initially suggested as a possible iron donor [82]. Subsequently, FXN was suggested to regulate the entry of iron during Fe-S assembly and act as an allosteric effector that can either enhance, stabilize, or repress the activity of the NFS1/ISD11/ISCU core complex [83]. More recent studies have shown that FXN enhances the rate of sulfur transfer to ISCU and the release of sulfide by small free thiol reduction of NFS1 persulfide, and accelerates [2Fe-2S] formation [84,85]. Although these recent results shed light on the molecular functions of mammalian FXN, the precise mechanism of iron delivery to the nascent Fe-S clusters awaits further clarification.
Once formed on ISCU, nascent Fe-S clusters are released and transferred to recipient proteins in a process mediated by a specialized chaperone-co-chaperone system. In mammals, the chaperones are mainly members of the mitochondrial HSP70 heat shock protein family, such as HSPA9 [86,87]. The HSP70 cognate chaperone has inherent ATPase activity which is stimulated by a histidine-proline-aspartate (HPD) motif present in co-chaperones such as HSC20 [71,88,89]. The co-chaperone HSC20 binds holo-ISCU via three hydrophobic C-terminal residues and delivers holo-ISCU to the substrate binding domain the HSPA9 chaperone [71,87]. HSPA9 then uses the energy released from ATP hydrolysis to drive conformational changes which facilitate Fe-S cluster release and refolding of the target protein [87]. Exactly how Fe-S clusters are released from the ISCU and transferred to target proteins remains unclear. This process may rely on competitive binding of the scaffold protein (ISCU) between the cysteine desulfurase (NFS1) and the co-chaperone (HSC20) as has been demonstrated in yeast [71,90].
The detailed molecular mechanisms that guide delivery of Fe-S clusters to specific recipient proteins are not fully understood. It has recently been shown that human co-chaperone HSC20 controls the selectivity of Fe-S cluster delivery for some proteins by binding to a conserved leucine-tyrosine-arginine (LYR) motif present in the recipient Fe-S protein or in the accessory proteins that mediate the insertion of Fe-S cluster into apoproteins [91]. Protein-Protein interaction studies indicate that many HSC20 binding partners contain the LYR motif, including succinate dehydrogenase subunit b (SDHB), an iron-sulfur cluster-containing subunit of complex II [91]. The LYR motif present in SDHB or in the succinate dehydrogenase complex assembly factor 1 (SDHAF1) mediates the interaction with co-chaperone HSC20 to guide the insertion of Fe-S cluster into the iron-sulfur containing subunit of complex II [91]. Similarly, LYRM7 (Rieske Fe-S protein chaperone of complex III), another LYR motif-containing protein, interacts with both Rieske Fe-S protein of complex III (UQCRFS1) and HSC20 to mediate the insertion of [Fe2-S2] cluster into UQCRFS1 [71,92]. A third LYR motif containing protein, LYRM6 (also known as NDUFA6), was shown to a play an important role in the incorporation of Fe4-S4 into the iron-sulfur containing subunit of complex 1 [93]. These examples provide insight into the complex mechanisms by which recipient proteins can be directly targeted for Fe-S cluster delivery.
In addition to direct delivery of Fe-S clusters to apo-proteins, Fe-S clusters may be delivered indirectly through intermediate carriers or secondary scaffolds. For example, glutaredoxin 5 (GLRX5) is an intermediate carrier that can accept [2Fe-2S] clusters and interact with the chaperone-co-chaperone system to facilitate insertion of the Fe-S cluster into target proteins [94]. Glutaredoxin 5 contains a consensus sequence of lysines (K139K140x10K151K151) through which it likely interacts with HSC20 [91]. Additional intermediate carrier proteins, including ISCA1, ISCA2, IBA57, BOLA3, NFU1, and NUBPL, have also been identified. These carrier proteins facilitate Fe-S cluster transfer to a specific subset of Fe-S cluster-containing proteins. For example, the A-type scaffold proteins ISCA1 and ISCA2 play an important role in the maturation of a subset of mitochondrial [Fe4-S4]-containing proteins including respiratory chain complex I and II, aconitase, lipoic acid-bound pyruvate dehydrogenase (PDH), and α-ketogluterate dehydrogenase complex [95,96]. Another carrier protein, IBA57, interacts with A-type proteins (ISCA1 and ISCA2) and facilitates incorporation of Fe-S clusters to mitochondrial aconitase, radical SAM Fe-S enzyme, and lipoic acid synthase [95,97]. BOLA3 and NFU1 have been implicated in the generation and maturation of lipoic acid synthase (LIAS), a Fe-S cluster containing enzyme [98,99]. Finally, NUBPL is an intermediate carrier protein implicated in the transfer of Fe-S cluster to complex I [100]. Despite impressive progress in elucidating specific processes in Fe-S cluster transfer, the complete roster of proteins involved in this process has yet to be fully defined.
6.2.2 Cytosolic Iron-sulfur Assembly (CIA)
There is little evidence that intact Fe-S clusters synthesized in the mitochondria are able to cross the mitochondrial membrane. Rather, recent studies have identified and characterized a distinct pathway of cytosolic iron-sulfur assembly (CIA) [101]. This pathway is postulated to rely on mitochondrial export of an uncharacterized sulfur-containing compound (X-S) via the mitochondrial export protein ABCB7 [102]. In humans, cytosolic nascent [4Fe-4S] clusters are assembled on a tetrameric scaffold composed of two members of the P-loop NTPases CFD1 and NBP35 [103]. This initial step involves transfer of electrons from NADPH to the cluster via NDOR1 and CIAPIN1 [104]. The transfer of nascent [4Fe-4S] clusters from the CFD1-NBP35 complex is accomplished by another Fe-S protein, IOP1 (iron-only hydrogenase-like protein) [105], and a multi-component CIA targeting complex. The CIA targeting complex is composed of CIAO1, CIA2B, CIA2A, and MMS19 proteins. The CIA targeting complex interacts with cytosolic recipient Fe-S proteins for delivery of Fe-S cluster [106–109].
6.3 Mitochondrial iron storage
Certain mammalian tissues express a mitochondria-specific ferritin termed mitochondrial ferritin (FtMt). FtMt is encoded by an intronless gene located at human chromosome 5q23 [110]. Synthesized as a precursor peptide, FtMt possesses a mitochondrial targeting sequence (MTS) that is cleaved following the arrival of FtMt at the mitochondria. The mature peptide is highly similar to H-ferritin in sequence, structure, and in the presence of ferroxidase activity [111]. Unlike cytosolic ferritins, however, FtMt lacks post-transcriptional regulation by the IRP-IRE system. The expression of FtMt is tissue-specific and is limited to certain cell types and organs[112]. In humans, FtMt protein is detectable in testis, brain, spinal cord, heart, kidney and islet of Langherans of pancreas. It is interesting to note that no expression of FtMt was identified in hepatocytes and splenoctes, sites important in the regulation of systemic iron levels [112]. One of the primary functions of FtMt appears to be to sequester free iron to prevent iron-induced ROS generation in the mitochondria [113]. Overexpression of FtMt disrupts cellular iron homeostasis by depleting cytosolic iron and enhancing accumulation of iron in mitochondria [14].
7. Iron perturbation and Mitochondria
Iron perturbations at the systemic or cellular level can negatively impact mitochondrial health and function.
7.1 Systemic Iron Disorders and Mitochondria
Iron deficiency has several etiologies, including low dietary iron intake, heredity iron dysregulation, and chronic diseases such as cancer [114–116]. At the systemic level, anemia is the most common manifestation of iron deficiency [117]. Iron deficiency anemias are characterized by insufficient erythropoiesis, reduced blood oxygenation, and subsequent tissue hypoxia. Iron deficiency anemia in vivohas been shown to decrease activity of iron-containing enzymes in skeletal muscle, cardiac, and liver mitochondria resulting in impaired oxygen metabolism [118,119]. Furthermore, mitochondria of iron-deficient cells exhibit decreased cytochrome concentration and gluconeogenesis [120,121]. In addition to functional deficiencies, iron deficiency alters mitochondrial morphology, causing an increase in size, rounded shape, and a decrease in cristae [122,123]. Ultimately, these functional and morphological deficiencies lead to mitochondrial damage, such as DNA fragmentation [124].
Like iron deficiency, iron overload can negatively impact mitochondrial health and function. Iron overload is principally problematic due to the susceptibility of loosely bound iron to catalyze production of ROS [10]. ROS accumulation can damage cellular components including lipids which interferes with cellular integrity, membrane fluidity, and permeability [125]. ROS are known to damage mitochondria as well [126,127]. Iron loading of rat cardiac myocytes in vitro resulted in mitochondrial DNA damage, decreased expression of mitochondrial-encoded respiratory chain subunits, and diminished respiratory function [128]. These effects can cause cell death, including iron-dependent ferroptosis, leading to tissue damage and eventually organ failure [11,129,130].
Systemic iron overload can be acquired or result from hereditary diseases, such as hereditary hemochromatosis (HH) [115,131]. It has been shown that acquired iron overload in rats leads to impaired hepatic mitochondrial function [132]. Interestingly, iron-overload from HH primarily results in accumulation of cytosolic iron and not mitochondrial iron in hepatocytes. Despite this finding, iron overload from HH nevertheless resulted in mitochondrial dysfunction, including reduced mitochondrial oxygen consumption [133]. Excessive cytosolic iron accumulation in HH was found to impair mitochondrial uptake of manganese (Mn). Subsequently, reduced mitochondrial Mn resulted in mitochondrial dysfunctional, likely due to decreased metalation and activity of mitochondrial manganese-dependent superoxide dismutase, an enzyme that protects mitochondria from respiration-generated free radicals [133,134].
7.2 Localized Mitochondrial Iron Disorders
As opposed to systemic iron overload, rare iron overload diseases can result in localized iron accumulation in mitochondria. Diseases resulting in severe mitochondrial iron overload can be grouped into 2 categories: (i) defective iron-sulfur cluster biogenesis; and (ii) defective heme synthesis. In both cases, mitochondrial iron overload is likely the result of diminished negative feedback from an undetermined product.
7.2.1 Defective Iron-sulfur Cluster Biogenesis
Several genetic diseases can result in disrupted iron-sulfur cluster biogenesis and severe mitochondrial iron overload. For example, point mutations or homozygous unstable GAA trinucleotide expansion in the FXN gene can result in Friedreich’s ataxia (FRDA) [135], an autosomal recessive disease characterized by severe neurodegeneration and cardiomyopathy. These manifestations of FRDA are caused by an accumulation of intra-mitochondrial iron [136,137], which decreases mitochondrial function and increases sensitivity to oxidative stress. Another example is infantile mitochondrial complex II/III deficiency, an autosomal recessive disease characterized by hypotonia, lactic acidemia, and multisystem organ failure. Infantile mitochondrial complex II/III deficiency is caused by an NFS1 mutation resulting in mitochondrial iron accumulation, respiratory chain complex II and III deficiency, and abnormal mitochondria [138]. Neonatal oxidative phosphorylation deficiency is a mitochondrial disease that presents with potentially fatal neonatal liver failure [139]. Mutations in the LYRM4 gene which encodes ISD11 have been shown to cause neonatal oxidative phosphorylation deficiency with mitochondria iron overload in yeast models [81,140,141]. Loss-of-function mutations in ISCU also result in mitochondrial iron overload and myopathy with severe exercise intolerance [142–144]. A novel mitochondrial muscle myopathy was recently shown to be caused by a homozygous mutation of FDX1L, the gene encoding FDX2 [145]. Mutations of another protein involved in Fe-S transfer, NUBPL1, cause complex I deficiency, mitochondrial dysfunction, and encephalopathy [146,147]. Furthermore, mutations in three proteins, NFU1 [99], BOLA3 [98,99], and IBA57 [97], cause lipoic acid defects and result in multiple mitochondrial dysfunctions syndrome (MMDS 1–3 respectively). MMDS syndromes present early in life and are often characterized by encephalopathy, hypotonia, seizures, developmental delay, failure to thrive, and lactic acidosis.
7.2.2 Defective Heme Synthesis
Disrupted heme synthesis occurs in sideroblastic anemia, a heterogenous disease characterized by ring sideroblasts in the bone marrow. Sideroblastic anemias result in iron accumulation in the mitochondria with an increase in FtMt [148,149]. Sideroblastic anemias can be acquired or hereditary. Acquired sideroblastic anemia can result from myelodysplastic syndromes, therapeutics, and nutritional deficiencies [150–153]. Hereditary gene mutations in ABCB7, ABCB10, ALAS2, HSPA9, FECH, GLRX5, and SLC25A38 can also result in mitochondrial iron overload and sideroblastic anemia [52,149,154–158]. Importantly, ABCB7, HSPA9, and GLRX5 mutations primarily disrupt iron-sulfur cluster biogenesis but also affect heme-synthesis. Sideroblastic anemia may present with myopathy and lactic acidosis in a syndrome known as MLASA. Originally, MLASA appeared to be associated with nuclear-encoded defects resulting in deficient mitochondrial translation; namely mutations in pseudouridine synthase 1 (PUS1) and mitochondrial tyrosyl-tRNA synthetase (YARS2) [159,160]. Recently however, a mutation in a mitochondrial gene, ATP6, which resulted in a complex V respiratory defect was proposed to cause MLASA [161].
8. Mitochondrial iron metabolism in cancer
It was long thought that cancer cell metabolism is independent of mitochondrial metabolism and that the metabolic shift from mitochondrial oxidative phosphorylation (OXPHOS) to aerobic glycolysis (“the Warburg effect”) occurs in cancer cells as a result of dysfunctional mitochondria. However, recent investigations have disproved this theory, and suggest that mitochondria play an important role in cancer development and progression [162]. Several studies have shown that depletion of the mitochondrial genome (mtDNA) (ρ0 cells) reduces or inhibits tumorigenicity [163–165]. Further supporting this notion is a recent report that mtDNA-depleted cancer cells had increased tumor latency when implanted in vivo. Moreover, ρ0 cells acquired mtDNA from the host to grow aggressively and to metastasize, consistent with an important role of oxidative phosphorylation in tumor progression [164]. Intriguingly, ISCU, mitoferrin1 (SLC25A37), SFXN1, and SFXN5, genes involved in ISC biogenesis and mitochondrial iron trafficking, were among 16 genes identified as an Iron Regulatory Gene Signature [166]. Expression of genes comprising this signature was predictive of survival in breast cancer patients, suggesting a link between mitochondrial function and patient outcome and underlining the potential importance of mitochondrial iron in cancer.
The specific role(s) of mitochondrial iron metabolism in cancer progression are poorly understood. It is well documented that cancer cells exhibit higher iron retention than their non-malignant counterparts [11,167]. It is, however, uncertain whether cytosolic iron accumulation results in increased mitochondrial iron. Nevertheless, increased mitochondrial ROS in cancer is suggestive of increased mitochondrial iron [168–170]. Furthermore, data suggest that head and neck cancers exhibit differential expression of mitoferrin 2 which alters mitochondrial iron uptake and response to treatment [171]. In addition, mitochondria of cancer cells may import more iron to generate functional forms of iron such as heme and Fe-S cluster to satisfy increasing demands for these cofactors during rapid cell proliferation. Recently it has been shown that non-small-cell lung cancer cells (NSCLC) exhibited elevated rates of heme synthesis compared to normal nonmalignant lung cells [172]. The expression of ALAS1 in NSCLC was significantly higher than in normal cells. Moreover, inhibition of heme synthesis significantly affected colony forming ability of these cells [172]. Thus, one of the ways by which mitochondria aid tumor progression may be by generating more of these essential iron cofactors.
9. Conclusion
Mitochondria play a critical role in iron metabolism, serving as a nexus for heme and FeS cluster biogenesis. Several mechanisms have been described for the delivery of iron to mitochondria following the import of iron into the cell. These include direct transfer of transferrin-bound iron from endosomes to mitochondria, uptake of low/high molecular weight cytosolic iron complexes, or transfer of iron bound to metallochaperones or siderophores. Upon reaching the mitochondria, iron is imported into the mitochondrial matrix by mitoferrins and primarily utilized for three functions: heme synthesis, iron-sulfur cluster biogenesis, and storage in mitochondrial ferritin. Heme and iron-sulfur clusters facilitate oxidation-reduction reactions and are important cofactors for enzymes involved in a variety of cellular and systemic processes. Due to the importance of these factors, disruption of cellular iron levels or mitochondrial iron metabolism can result in cellular stress or death. Systemic iron deficiency, which has numerous causes including chronic disease such as cancer, can lead to reduced mitochondrial function and anemia. Similarly, systemic iron overload can impair mitochondrial function through regulation of mitochondrial manganese levels. Localized iron overload in the mitochondria occurs in several diseases characterized by disrupted heme synthesis or iron-sulfur cluster biogenesis. Mitochondrial iron overload in these disorders likely results from a lack of negative feedback from undetermined products. Aberrantly high mitochondrial iron can lead to reduced mitochondrial function, cell death, and potentially organ dysfunction/failure, as is seen with Friedreich’s Ataxia.
10. Expert Commentary
Mitochondria serve a key role in the synthesis and assembly of heme and Fe-S clusters, and are therefore essential for the delivery of iron to client proteins. Appropriate levels of iron, however, are also important for the proper function of the mitochondria. Conditions of iron deficiency or excess disrupt a myriad of mitochondrial functions. This interdependence between iron and mitochondria is best demonstrated by diseases that simultaneously disrupt mitochondrial iron and mitochondrial function, such as Frederich’s Ataxia, infantile mitochondrial complex II/III deficiency, neonatal oxidative phosphorylation deficiency, and sideroblastic anemia.
Despite compelling evidence for the reciprocal importance of iron to mitochondria and mitochondria to iron, our understanding of this relationship remains fundamentally limited. Complicated by an incomplete understanding of how cytosolic iron is handled, it is still not clear how iron is delivered to the mitochondria. A number of possible mechanisms by which iron may be delivered to mitochondria have been shown (reviewed above). However, definitive evidence for some of these mechanisms remains limited. Furthermore, as these studies have been performed in various cell types, it is unclear if these alternative mechanisms operate within the same cell. It is possible that they function as redundant means to supply the mitochondria with iron. It is equally possible that the mechanism of iron delivery to the mitochondria is cell specific and possibly dependent on the metabolic needs of the cell. A thorough characterization of iron delivery to the mitochondria in diverse cell types would help to answer these questions.
In addition to uncertainties regarding the mode of mitochondrial iron delivery, mechanisms that regulate proteins involved in mitochondrial iron homeostasis remain incompletely understood. For example, transcription factors GATA-1 and GATA-2 have been shown to regulate transcription of mitoferrin 1 in erthyroid cells [173]. GATA-1 is a critical regulator of erythroid differentiation [174] and may account for differential expression of mitoferrin 1 and mitoferrin 2 in erythroid and nonerythroid tissues [40]. However, mechanisms underlying transcriptional regulation of mitoferrin 2, which has been observed following mitochondrial stress [175] and in cancer [171], remains unknown. Further complicating this picture is the substantial contribution of posttranslational control to mitoferrin regulation [41]. Posttranslational regulation of mitoferrins is likely mediated by accessory proteins, such as members of the ABC transporter family [42].
Fundamental gaps in our knowledge remain, notably in the identification of the complete array of proteins that participate in Fe-S assembly and transfer to client proteins. In fact, the mere identification of Fe-S proteins remains something of a challenge [176]. These basic issues will need to be resolved before our understanding of mitochondrial iron metabolism can be complete.
While we have gained some insights into mitochondrial iron import and utilization, mitochondrial iron export is largely undetermined. There is some question as to whether iron is exported from mammalian mitochondria, as vertebrates lack homologs of purported yeast mitochondrial iron exporters [177]. However, members of the ABC transporter family, ABCB7 [178] and ABCB8 [46], have been suggested to mediate iron export from the mitochondria. The exact mechanisms by which these proteins might mediate iron export remain undetermined, as is the form of iron which is exported (i.e. as part of iron-sulfur clusters or atomic chelator-bound iron). FLVCR1b has been shown to facilitate the export of mitochondrial iron through heme, although mechanistic details remain to be elucidated [64].
Understanding the regulation of mitochondrial iron homeostasis is of great significance, particularly as it relates to cytosolic iron levels. It is highly likely that crosstalk exists between mitochondrial iron regulation and cytosolic iron regulation [12]. Evidence suggests that cytosolic iron homeostasis is altered to meet the metabolic-iron demands of the mitochondria (i.e. an increase in transferrin receptor when heme synthesis is inhibited [179]). The mechanism facilitating this crosstalk remains obscure, but its elucidation is of keen interest to further our understanding iron biology and diseases of iron dysregulation, including cancer. Interestingly, mitochondrial iron levels also seem to be insulated, to a certain extent, from disruptions in cytosolic iron levels, as evidenced by normal mitochondrial iron levels in Hereditary Hemochromatosis [133]. This finding suggests that while mitochondrial iron may crosstalk with cytosolic iron regulators, the mitochondria may also maintain independent iron regulation.
11. 5-Year View
The study of rare genetic diseases resulting in mitochondrial iron disruption has been an effective means of identifying regulators of mitochondria iron homeostasis. A continued push in this area may help to fill gaps in our current understanding. Considering the important role for iron [10] and mitochondrial function [162] in cancer progression, it is possible that de novo mutations that affect mitochondrial iron metabolism may be identified in cancer, and provide clues that can complement information gained from the study of rare genetic diseases. Currently, characterization of mitochondrial iron and its metabolism in cancer remains understudied. Utilizing publicly available datasets, such as The Cancer Genome Atlas, may provide a good initial avenue to screen for candidate mitochondrial iron regulatory proteins that contribute to cancer progression.
A major goal of understanding mitochondrial iron metabolism is to identify novel methods of treating pathological conditions characterized by mitochondrial iron dysregulation, such as sideroblastic anemia and Friedreich’s Ataxia. In recent years, significant strides have been made in developing therapies to treat systemic iron overload, one of the causes of mitochondrial dysfunction [132]. These novel approaches, such as increasing hepcidin, a systemic iron regulatory hormone, may be effective in preventing mitochondrial dysfunction secondary to systemic iron overload [115]. However, based on our current understanding of mitochondrial iron trafficking and utilization, such global approaches may have limited application to syndromes that cause localized iron accumulation in mitochondria. To develop effective treatment for pathologies leading to localized mitochondrial iron overload or deficiency, we will first need to develop a more detailed understanding of mitochondrial iron regulation. We anticipate substantial progress towards this goal in the next five years.
Acknowledgments
Funding
This paper was supported in part by grants R01 CA188025 (S.V.T), R01 CA171101 (F.M.T), and T90 DE021989 (D.H.M.) from the US National Institutes of Health.
Footnotes
Declaration of Interest
DH Manz received grant T90 DE021989, SV Torti has received grant R01 CA188025, and FM Torti has received grant R01 CA171101, from the US National Institutes of Health. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
References
*Article of interest
** Article of considerable interest