Beyond reason
루게릭병(근위축성 축삭경화증)은 미토콘드리아 기능부전질병임
Review articleThe role of mitochondria in amyotrophic lateral sclerosis
Keywords
1. Amyotrophic lateral sclerosis
Motor neuron disease (MND) describes a group of neurological disorders characterised by the selective loss of motor neurons. Amyotrophic lateral sclerosis (ALS), the most common subtype of MND, is characterised by a progressive degeneration of both upper and lower motor neurons, resulting in muscle atrophy, gradual paralysis, and death, usually resulting from respiratory failure. ALS has a worldwide prevalence of 4–6 in 100,000, with differences noted between populations [1], [2]. The average age of onset is between 55 and 65 years of age, however familial cases are frequently associated with an earlier age of onset [3]. The average survival is 2–3 years from diagnosis, with only 25% of patients surviving 5 years and 5–10% surviving 10 years post-diagnosis [4].
ALS clinically overlaps with frontotemporal dementia (FTD). FTD is a common cause of dementia in adults under the age of 65 that is characterised by neurodegeneration of the frontal and temporal cortex [5]. Around 50% of ALS patients display evidence of frontal and temporal lobe dysfunction on detailed neuropsychological testing and up to 5–10% of ALS cases present with clinically diagnosed FTD. Similarly, while approximately 15% of FTD patients go on to develop ALS, about half display some degree of motor involvement [6].
Approximately 10% of ALS cases follow a familial, mostly autosomal dominant inheritance pattern (familial ALS). The remaining 90% of cases have no clear genetic basis (sporadic ALS). To date mutations in over 20 genes have been associated with ALS, including superoxide dismutase 1 (SOD1), TAR DNA binding protein (TARDBP; TDP-43), Fused in sarcoma (FUS), and C9orf72 (Table 1; [7]). Together mutations in these genes account for approximately 60% of familial ALS and 11% of sporadic ALS. In agreement with the clinical overlap between ALS and FTD, the GGGGCC (G4C2) repeat expansion mutation in C9orf72 was found to be the most common genetic cause of both ALS and FTD, accounting for approximately 40 and 25% of familial cases, respectively [8], [9], [10]reviewed in Ref. [11]]. The genes affected in ALS have been implicated in a wide range of cellular pathways, suggesting that ALS is a multi-factorial disease. Possible pathogenic mechanisms underlying motor neuron degeneration include RNA toxicity, excitotoxicity, disruption of proteostasis, defective axonal transport, oxidative stress, and mitochondrial dysfunction [reviewed in Ref. [12]].
Table 1. Impact of ALS-associated genes on mitochondrial function. Pathogenic variants of proteins implicated in ALS have been linked to altered mitochondrial function (Ref. http://alsod.iop.kcl.ac.uk/home.aspx; [7]).
| ALS locus | Gene | Protein | Potential consequence of mutation on mitochondrial function |
|---|---|---|---|
| ALS 1 | SOD1 | Cu, Zn superoxide dismutase 1 | Mutant protein aggregates in IMS; decreased ATP generation; increased cellular ROS and ROS induced cellular damage; imbalance in calcium homeostasis; induction of apoptosis via VDAC inhibition and Bcl-2 binding; disrupted mitochondrial architecture; impaired mitochondrial network dynamics and axonal transport; impaired mitochondrial clearance by mitophagy; disrupted ER–mitochondria contacts |
| ALS 2 | ALS2 | Alsin | Reduced autophagosome formation and decreased mitophagy |
| ALS 6 | FUS | RNA-binding protein FUS | Decreased ATP generation; increased ROS levels; loss of calcium homeostasis and disruption of ER–mitochondria contacts; reduced mitophagy-related gene expression; disrupted mitochondrial architecture; impaired mitochondrial transport via disrupted kinesin gene expression |
| ALS 8 | VAPB | Vesicle-associated membrane protein-associated protein B | Impaired calcium homeostasis; decreased anterograde axonal transport; disrupted ER–mitochondria contacts |
| ALS 10 | TARDBP | TAR DNA-binding protein 43 | TDP-43 aggregates in mitochondria and disrupts mtDNA transcription; decreased ATP generation; impaired calcium homeostasis and disrupted ER–mitochondria contacts; disrupted mitochondrial architecture; altered mitochondrial network dynamics and impaired mitochondrial axonal transport; reduced mitophagy related gene expression; impaired mitochondrial clearance by mitophagy |
| ALS 12 | OPTN | Optineurin | Reduced mitochondrial clearance by mitophagy |
| ALS 14 | VCP | Valosin-containing protein | Decreased ATP levels; mitochondrial uncoupling; reduced mitochondrial clearance by mitophagy |
| ALS 16 | SIGMAR1 | Sigma non-opioid intracellular receptor 1 | Reduced ATP generation; disrupted ER–mitochondria contacts; dysregulated calcium homeostasis; reduced axonal transport |
| ALS-FTD1 | C9orf72 | Chromosome 9 open reading frame 72 | DPR proteins interact with mitochondrial ribosomal proteins; altered MMP; increased cellular ROS levels; poly(GR) DPR induced oxidative stress; impaired autophagy; disrupted mitochondrial architecture and altered mitochondrial network dynamics |
| ALS-FTD2 | CHCHD10 | Coiled-coil-helix-coiled-coil-helix domain containing 10 | Disrupted mitochondrial architecture; decreased electron transport chain activity |
| ALS | SQSTM1 | p62/Sequestosome 1 | Reduced mitochondrial clearance by mitophagy; reduced MMP |
Abbreviations: ATP – adenosine triphosphate; DPR – dipeptide repeat protein; IMS – intermembrane space; MAM – mitochondria associated ER membrane; MMP – mitochondrial membrane potential; ROS – reactive oxygen species.
2. The role of mitochondria in ALS pathogenesis
Mitochondria play a central role in cell survival and metabolism. In addition to their well-known role as producers of ATP via oxidative phosphorylation, mitochondria play important roles in phospholipid biogenesis, calcium homeostasis, and apoptosis. Mitochondria are of particular importance in neurons. Neurons have high metabolic requirements – the brain consumes 20% of the body’s resting ATP production despite being only 2% of its mass [reviewed in Refs. [13], [14]]. Moreover, mitochondria are essential calcium buffering organelles in neurons that modulate local calcium dynamics to, for example, modulate neurotransmitter release [reviewed in Ref. [15]]. Neurons are long-lived cells that persist throughout the lifespan of the individual and as such are more susceptible to the accumulating damage arising from mitochondrial dysfunction [reviewed in Ref. [16]]. Accordingly, the maintenance of a healthy pool of correctly localised mitochondria is essential for neuronal survival and function. It is not surprising therefore that mitochondrial dysfunction has been linked to a large number neurodegenerative disorders including ALS. Many of the identified ALS genes have a role in mitochondrial-associated functions (Table 1) and evidence gathered from in vitro and in vivo disease models and from patient studies strongly implicates the dysfunction of mitochondria as a core ALS disease component.
ALS associated mitochondrial dysfunction comes in many guises, including defective oxidative phosphorylation, production of reactive oxygen species(ROS), impaired calcium buffering capacity and defective mitochondrial dynamics. Furthermore, with the possible exception RNA toxicity, mitochondrial dysfunction appears to be directly or indirectly linked to all of the postulated “non-mitochondrial” mechanisms of toxicity associated with ALS, including excitotoxicity, loss of protein homeostasis and defective axonal transport.
2.1. Structural evidence of damaged mitochondria
Structurally altered and aggregated mitochondria, with a swollen and vacuolated appearance, were one of the first changes observed in ALS patient motor neurons [17], [18] and in Bunina bodies [19]. Sporadic ALS cases occasionally additionally display axonal swellings consisting of neurofilamentaccumulations, swollen mitochondria and secondary lysosomes [20]. Morphologically abnormal mitochondria are also consistently reported in animal and cell models of ALS, with a tendency towards a fragmented mitochondrial population being observed. Expression of wild type or ALS mutant TDP-43 (M337V, Q331K, A315T) resulted in aggregated, fragmented and vacuolated mitochondria [21], [22], [23]. Overexpression of SOD1 G93A had similar effects and mitochondria were significantly less elongated and more spherical in motor neurons isolated from SOD1 G93A transgenic mice [24], [25], [26] and in vivo in motor neurons of early symptomatic SOD1 G37R and SOD1 G85R transgenic mice [27]. Furthermore, in SOD1 G93A transgenic mice mitochondria were found in abnormal clusters along the axon [22].
Overexpression of ALS mutant FUS R521G or R521H in cultured motor neurons resulted in mitochondrial shortening which was exacerbated by the presence of FUS in the cytosol [28]. Similar results were seen in both HT22 cells and primary cortical neurons expressing mutant FUS P525L which causes a juvenile form of ALS, and in a FUS P525L transgenic mouse model [29], [30]. Subtle fragmentation of the mitochondrial network has also been identified in fibroblasts of ALS patients with C9orf72 repeat expansions [31], and swollen mitochondria were reported in an iPSC model of C9orf72-associated ALS [32]. Aggregation of structurally altered mitochondria was reported in cortical neurons from Alsin knockout mice [33]. Similarly, mitochondria co-aggregated with TDP-43 in heterozygous knock-in mice bearing an ALS-associated R155H mutation in valosin-containing protein (VCP; also called p97, cdc48 in yeast – further referred to as VCP/p97) [34], [35].
Direct evidence that disruption of mitochondrial structure (and as a consequence function; see below) may contribute to the aetiology of ALS comes from the discovery of causative mutations in the mitochondrial proteinCHCHD10 which is localised to contact sites between the inner and outer mitochondrial membrane [36]. ALS-associated mutations in CHCHD10 disrupt mitochondrial cristae and have a profound effect on mitochondrial structure [37]. Deformation and loss of mitochondrial cristae have also been reported in C9orf72-related ALS and FTD (C9ALS/FTD) patient fibroblasts, in vitro and in vivo in SOD1 G93A, TDP-43 A513T and FUS P525L ALS models and in Alsin knockout mice [29], [31], [32], [33], [38], [39]. In SOD1 G93A and FUS P525L transgenic mice dilated cristae are already present at disease onset [30], [40].
Thus, mitochondrial structure and the mitochondrial network appears to be disrupted in most if not all models of familial ALS and in ALS patients. Moreover, the structural damage to mitochondria and fragmentation of the mitochondrial network was reported to occur in early disease stages in in vivomodels of ALS indicating that mitochondrial morphological alterations occur potentially as an upstream source of degeneration rather than a consequence [22], [23], [27].
2.2. ALS-associated proteins interact with mitochondria
A number of proteins that have been linked to familial and sporadic ALS, including SOD1, TDP-43, FUS, C9orf72, and the C9orf72 GGGGCC repeat expansion-associated glycine/arginine (GR) dipeptide repeat protein (DPR), have been shown to interact with mitochondria [29], [41], [42], [43], [44], [45]. The interaction of these ALS-associated proteins with the mitochondria appears to be instrumental to the induction of mitochondrial damage associated with ALS.
Mutant SOD1 localises to the intermembrane space (IMS), where it has been shown to aggregate, and reduce the activity of the electron transport chain(ETC) complexes (see below) [46], [47]. Furthermore, SOD1 aggregates have been proposed to interfere with the activity of voltage-dependent anion channel 1 (VDAC1) which is responsible for the exchange of ATP, ADP and other respiratory substrates across the outer mitochondrial membrane (OMM) [reviewed in Ref. [48]]. Direct interaction of ALS mutant SOD1 with VDAC1inhibits channel conductance and reduces its permeability to ADP at both presymptomatic and symptomatic disease stages in the spinal cord of SOD1 G93A transgenic rats. Furthermore, an approximately 25% reduction in VDAC1 activity accelerated disease in SOD1 G37R transgenic mice [49]. Mutant misfolded SOD1 has also been shown to interact with Bcl-2 family proteins on the OMM, leading to pro-apoptotic changes (see below) [50].
TDP-43 and to a greater extent ALS mutant TDP-43 accumulates in mitochondria where it preferentially binds the mRNAs of the mtDNA-encoded complex I subunits ND3 and ND6 and causes complex I disassembly by impairing their transcription. Accumulation of ALS mutant TDP-43 in mitochondria appears to be mediated by internal mitochondrial targeting sequences in TDP-43 [42]. Since cytoplasmic TDP-43 accumulation is a hallmark pathology in most ALS (but not SOD1 or FUS-related ALS), this may also explain the mitochondrial defects observed in sporadic ALS and other familial ALS cases.
Both wild type FUS and ALS mutant FUS P525L co-purify with mitochondria and this is at least in part due to their interaction with the mitochondrial chaperone heat shock protein (HSP) of 60 kDa (HSP60) [29]. Mitochondrial localisation of FUS correlated with augmented ROS levels [29], and overexpression of FUS has been shown to reduce mitochondrial ATP production [51]. Reducing the association of FUS with mitochondria by knockdown of HSP60 expression ameliorated neurodegeneration in FUS transgenic Drosophila [29].
Several mitochondrial proteins have been identified as possible C9orf72-interacting proteins in a BioIP proteomics screen, including members of the IMM solute carrier family, VDAC3 and translocase of the inner mitochondrial membrane 50 (TIMM50). Furthermore, C9orf72 was detected in mitochondria-enriched fractions [44]. The relevance of this association is not yet clear. Another link between C9ALS/FTD and mitochondria comes from the finding that poly(GR) DPRs preferentially bind to mitochondrial ribosomal proteins[43].
2.3. Defective mitochondrial respiration and ATP production
Reductions in cellular respiration and ATP production are well documented in ALS (Fig. 1). In post-mortem spinal cord of sporadic ALS patients the activity of all ETC complexes, complex I, II, III, and IV was found to be reduced [52], [53]. In addition, the activity of complexes I and IV were reported to be impaired in skeletal muscle [54], [55], [56] while complex I activity and ATP levels were reduced in lymphocytes of sporadic ALS patients [57]. Counterintuitively, in fibroblasts obtained from skin biopsies of sporadic ALS patients the mitochondrial membrane potential (MMP) was increased compared to healthy controls. Possibly the increased MMP observed in fibroblasts reflects an attempt to rescue inefficient ATP synthesis or the metabolic differences between skin fibroblasts and central nervous system (CNS) cells [58]. Indeed, similar to fibroblasts obtained from sporadic patients, MMP was increased in C9orf72 patient fibroblasts [31] but was significantly decreased in iPSC-derived motor neurons reprogrammed from C9orf72 patient fibroblasts [32].
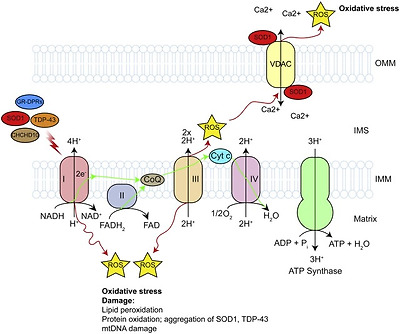
Fig. 1. Defective mitochondrial respiration, ATP production and oxidative stress.
The activity of the electron transport chain is required for the generation of cellular ATP. The activity of the complexes involved in the electron transport chain have been shown to be decreased in SOD1, TDP-43 and CHCHD10-related ALS. This results in decreased MMP and ATP generation, and increased generation of ROS. See text for details.
In SOD1 G93A transgenic mice impaired ATP synthesis and depressed mitochondrial respiration rates in the brain and spinal cord were found well before disease onset and they persist throughout the course of the disease [39], [45]. Associated with these changes, decreased complex I + III, II + III and IV activity was observed in the spinal cord of 17-week old symptomatic SOD1 G93A transgenic mice [45] and complex IV activity was reduced in forebrain mitochondria isolated from presymptomatic, symptomatic and end-stage SOD1 G93A transgenic mice [39], [45]. Analysis of regional changes in ETC complex activities further showed a selective decrease of complex I and IV activity in the ventral horn of the spinal cord of these mice prior to the onset of disease which further declined as disease progressed [59]. In these cases, the reduced activity of complex IV may relate to impaired association of cytochrome c with the IMM [39], [45]. A decrease in complex I activity was also confirmed in ex vivo motor cortex slices from SOD1 G93A mice [60] (Fig. 1).
Analysis of mutant SOD1 ALS cell models recaptured the defects found in SOD1 G93A mice. SOD1 I113T patient fibroblast lines showed reduced oxidative phosphorylation compared to non-disease controls [61]. Stable low level expression of SOD1 G93A or G37R in NSC-34 motor neuron-like cell lines reduced the activity of complex II and IV [62], [63] accompanied by reduced MMP [64] while doxycycline-induced expression of SOD1 G93A, A4V, H46R, H80R, D90A, or D123H in the same cell line caused a decrease in complex I, II + III and IV activity which was accompanied by reduced cellular ATP levels [46]. In N2A neuroblastoma cells overexpression of SOD1 G37R resulted in reduced complex I activity which correlated with a drop in MMP and reduced levels of cytosolic ATP, but complex II, III and IV activities were unaffected [63].
In contrast to these studies, Bowling et al. found increased complex I activity in post-mortem frontal cortex of familial ALS patients with an SOD1 A4V mutation [65] and the same group reported an increase in complex I activity in the forebrain of SOD1 G93A transgenic mice [66]. Furthermore, when they assessed mitochondrial respiration solely via complex II, the mitochondrial respiration rate was not different in SOD1 G93A transgenic mice compared to wild type SOD1 transgenic mice [67]. Similarly, others found no evidence for reduced mitochondrial respiration via complex I or II in presymptomatic SOD1 G93A transgenic mice [68]. It is not readily apparent what accounts for the discrepancy between these studies, but these measurements are particularly sensitive to variations in experimental conditions and differences between the SOD1 G93A mouse strains cannot be excluded.
Nevertheless, despite differences between studies and cell models, the overarching trend is that ALS mutant SOD1 decreases ETC activity and causes impaired ATP production. Moreover, the presymptomatic occurrence of oxidative phosphorylation disruption in SOD1 G93A transgenic mice suggests it may have a causative role in this ALS model.
Defective mitochondrial respiration and ATP production have also been observed in models of non-SOD1 related familial ALS. Depolarisation of mitochondria was described in NSC-34 cells expressing wild type or mutant TDP-43 (Q331K and M337V) and primary motor neurons expressing TDP-43 M337V [21], [23], [69], [70]. In NSC-34 cells TDP-43-associated MMP reduction was accompanied by decreased complex I activity [70]. Similarly, in TDP-43 G298S and A382T patient fibroblasts reduced MMP was accompanied by decreased complex I activity, reduced oxygen consumption and decreased ATP levels [42]. However, this may not always be the case as others reported reduced MMP, but no changes in mitochondrial respiration or ATP content in 3 independent TDP-43 A382T patient fibroblast lines [31].
As was the case for TDP-43, overexpression of wild type or mutant FUS P525L caused a reduction in MMP in HEK293 cells [29] and ATP production was impaired upon expression of wild type or ALS mutant FUS R521C or R518K in NSC-34 cells [51]. Similarly overexpression of the sigma non-opioid intracellular receptor 1 (Sig1R) E102Q mutant in N2A cells reduced ATP production [71]. Decreased MMP was further observed in SQSTM1 knockout MEFs and in SH-SY5Y, mouse cortical neurons and astrocytes in which VCP/p97 expression was ablated using siRNA to model ALS-associated loss of function mutants in p62/Sequestosome1 and VCP/p97, respectively [72], [73]. In case of VCP/p97 the same results were obtained in VCP/p97 R155C, R155H and R191Q ALS patient fibroblast lines [73]. In these cell models of VCP/p97-associated ALS the decrease in MMP was accompanied by a decrease in ATP levels but an increase in the rate of mitochondrial respiration and oxygen consumption, suggesting that depletion of MMP is due to uncoupling rather than ETC activity deficits [73]. Finally, analysis of fibroblasts from patients with mutations in the mitochondrial protein CHCHD10 revealed impaired ETC activity at complexes I, II, III and IV which was accompanied by a severe bioenergetics deficit [36], [37].
Neurons do not operate as isolated units, but depend on the surrounding cellular environment and supporting cells such as microglia and astrocytes for survival and function. Accordingly, it is now clear that ALS is a non-cell-autonomous disease with astrocytes in particular associated with disease progression [74, reviewed in Refs. [12], [75]]. One way in which glia support neurons is by shuttling of lactate, produced by conversion of the glycolysisproduct pyruvate, to the neighbouring neuron where it is converted back to pyruvate which is subsequently converted to acetyl-CoA and enters the Krebs cycle [76, reviewed in Ref. [77]]. In fact, evidence suggests that glial lactate is the main energy source of neurons (i.e. the Astrocyte-Neuron Lactate ShuttleHypothesis (ANLSH)). Several lines of evidence suggest that impairment of the ANLSH may be involved in the bioenergetics deficits observed in ALS. The expression of phosphoglycerate kinase 1 (PGK1) which transfers phosphate from 1,3-bisphosphoglycerate (1,3-BPG) to ADP to produce 3-phosphoglycerate (3-PG) and ATP during glycolysis, as well as of the monocarboxylate transporter 4 (MCT4; Slc16a4), which is involved in the transport of lactate are downregulated in SOD1 G93A astrocytes [78]. Similarly, MCT1 (Slc16a1) expression is downregulated in oligodendrocytes in the spinal cord of SOD1 G93A transgenic mice. Furthermore, consistent with a possible role in neurodegeneration knock down of MTC1 caused motor neuron death in vivo [79]. Hence reductions in neuronal respiration and ATP production may be caused by failure of glia to provide respiratory substrates.
Despite differences in mechanisms and specific observations depending on the models studied, decreased ETC activity and ATP levels emerge as a common feature in ALS. It is conceivable that, in line with the high energy demands of neurons, gradual depletion of ATP due to reduced respiration may trigger degeneration.
2.4. Oxidative stress
ROS are a natural by-product of oxidative phosphorylation. The ETC is responsible for the majority of ROS produced in a cell, with free radicals, mainly superoxide anion (O2
Increased levels of ROS and ROS-associated damage have been widely reported in ALS [reviewed in Ref. [86]]. Increased markers of ROS damage have been found in biofluids of patients with sporadic ALS [87], [88], [89], [90] as well as in post-mortem tissue [91], [92], [93], [94]. Similarly, increased ROS levels were reported in lymphoblasts of familial ALS cases with SOD1 mutations [95] and fibroblasts of patients with C9orf72 G4C2 repeat expansions [31]. In contrast, analysis of lymphoblasts and fibroblast cell lines from sporadic ALS patients or familial ALS patients with a TDP-43 A382T mutation did not show evidence of increased ROS production or oxidative damage [31], [95], [96], [97], but some were more sensitive to NO
Oxidative damage to DNA, RNA, proteins and lipids has been widely reported in SOD1 G93A rodent models and cell models [reviewed in Ref. [86]]. DNA and RNA appear to be especially vulnerable to oxidation, and mRNA oxidation has been shown to precede motor neuron degeneration and cause reduced expression of the encoded proteins in SOD1 G93A transgenic mice [94]. Interestingly mRNAs coding for ETC complexes and ATP synthase were selectively susceptible to oxidation [94]. Furthermore, SOD1 itself is a target of oxidative damage [99] and this has been linked to its misfolding and aggregation [100]. Since misfolded, aggregated SOD1 has been shown to disrupt mitochondrial function and increase superoxide production [49], [101], [102] a vicious cycle mechanism emerges in which mitochondrial damage and oxidative stress caused by misfolded SOD1 leads to exacerbation of SOD1 misfolding and downstream mitochondrial damage. Interestingly using monoclonal antibodies to misfolded SOD1, misfolded wild type SOD1 species have been shown to be present in sporadic ALS patients [103], [104] although this has been disputed by others [105].
Oxidative damage has also been proposed to promote aggregation of TDP-43 via cysteine oxidation and disulphide bond formation and acetylation [106], [107]. In agreement, treatment of COS-7 cells with 4-hydroxynonenal (HNE), which is produced in cells by lipid peroxidation, was shown to cause insolubilisation, phosphorylation, and partial cytosolic localisation of TDP-43 [108]. Overexpression of wild type and mutant TDP-43 M337V and Q331K or its C-terminal fragments in NSC-34 have been shown to increase ROS and cause oxidative damage [21]. In addition to these deleterious effects TDP-43 also appears to have a protective function in response to oxidative stress. Indeed, oxidative stress has been shown to induce recruitment of TDP-43 to stress granules [109], [110], [111].
Other ALS-associated proteins that have been linked to oxidative stress include FUS P525L which has been shown to augment ROS levels when overexpressed in HEK293 cells [29], and poly(GR) DPRs which have been shown to increase oxidative stress [43]. Interestingly as was the case for TDP-43, an oxidative environment increased FUS inclusions, again pointing toward a detrimental feed-forward loop [112].
2.5. Calcium mishandling
Loss of calcium homeostasis has been observed in in vitro and in vivo models of mutant SOD1, vesicle-associated membrane protein-associated protein B (VAPB), TDP-43, and FUS-related ALS and in the motor nerve terminals of ALS patients [51], [68], [113], [114], [115], [116], [117], [118], [119]. In SOD1 G93A transgenic mice a significant decrease in mitochondrial calcium loading capacity in the CNS was observed long before disease onset, suggesting an early loss of calcium buffering may be causal in disease [68]. Similarly, analysis of calcium handling in vulnerable hypoglossal motor neurons identified a reduction of uniporter-dependent mitochondrial calcium uptake which was not observed in resistant oculomotor neurons [120]. How these defects in mitochondrial calcium uptake affect motor neurons is not completely clear, but SOD1 G93A motor neurons show a delay in the recovery to basal level of the calcium increase following α-amino-5-methyl-3-hydroxisoxazolone-4-propionate (AMPA) receptor activation [121].
Loss of ER–mitochondria communication has emerged as a major cause of loss of calcium homeostasis in ALS. An estimated 5–20% of mitochondria are closely associated with the ER at contact sites called mitochondria-associated ER membranes (MAM). Several protein complexes that tether ER to mitochondria have been proposed, including homo and heterotypic interactions between mitofusin (Mfn) 1 and 2, interaction of inositol 1,4,5-trisphosphate receptor (IP3R) with VDAC via glucose-regulated protein 75 (Grp75), and the integral ER protein VAPB that binds to protein tyrosinephosphatase-interacting protein 51 (PTPIP51) on the OMM (Fig. 2). Among other functions, ER–mitochondria contact sites allow calcium exchange between the two organelles [reviewed in Refs. [122], [123]]. Disruption of ER–mitochondria interactions have been reported in mutant SOD1, Sig1R, TDP-43, and FUS-related ALS [51], [115], [124]. In the case of mutant TDP-43 and FUS-related ALS, reduced ER–mitochondria communication was caused by a GSK3ß-dependent reduction in VAPB-PTPIP51 interaction [51], [115]. In contrast, ALS mutant VAPB P56S was shown to have greater affinity for PTPIP51 and to increase ER–mitochondria association [118]. However, since in ALS8 patient-derived iPSC neurons VAPB expression is down-regulated because of reduced expression of the VAPB P56S mutant allele [125], it is likely that in VAPB P56S-related ALS ER–mitochondria contacts are actually decreased as well. Whether VAPB–PTPIP51 interaction is also disrupted in mutant SOD1 and Sig1R-related ALS is not known, but in SIGMAR1 knockout mice which model loss of Sig1R function, the interaction between IP3R and VDAC was decreased in motor neurons [124]. Interestingly VAPB expression levels in the spinal cord of sporadic ALS cases are significantly lower compared to healthy controls [126]. Thus, disrupted ER–mitochondria communication may be a general feature in ALS.
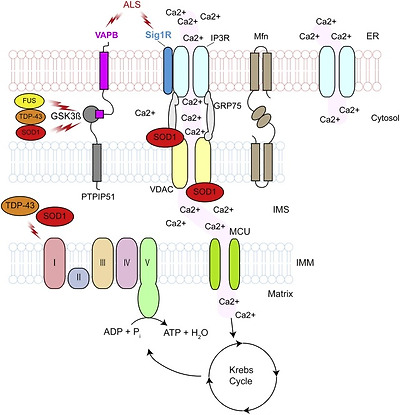
Fig. 2. Loss of calcium homeostasis.
Calcium homeostasis is achieved though the exchange of calcium between the ER and mitochondria at ER–mitochondria contacts. ER–mitochondria contacts are disrupted in SOD1, TDP-43, VAPB, Sig1R and FUS ALS. Disruption of the contacts may lead to increases in cytosolic calcium levels and disruption of calcium dependent cellular processes, including axonal transport, ATP generation and protein homeostasis. MCU, mitochondrial calcium uniporter. See text for details.
In ALS mutant FUS and TDP-43 models, impaired ER–mitochondria communication leads to reduced calcium uptake in mitochondria and an associated rise in cytosolic calcium upon triggering of calcium release from the ER [51], [115]. Similarly, in SIGMAR1 knockout mice cytosolic calcium levels were elevated and the time to return to basal levels was significantly longer [124]. As mentioned above, similar loss of calcium homeostasis has been reported in SOD1 G93A transgenic mice. Whether these are due to reduced ER–mitochondria communication has not been formally shown, but neuronal overexpression of wild type human VAPB has been shown to slow disease and increase survival in SOD1 G93A transgenic mice [127]. It is tempting to speculate, that restoring ER–mitochondria contacts is at the basis of this protective effect.
Dysregulation of calcium, possibly by miscommunication between ER and mitochondria, may be a primary cause of motor neuron death in ALS. Indeed, motor neurons possess several properties that make them more vulnerable to calcium dysregulation compared to other neuronal populations. They express a high number of calcium permeable AMPA receptors at the postsynaptic terminal, which results in a greater vulnerability to excitotoxicity thorough excessive calcium influx during excitatory neurotransmission events [117], [128], [129]. In addition, motor neurons have reduced cytosolic buffering capacity because they express low levels of calcium buffering proteins such as parvalbumin and calbindin D-28k and this makes them more dependent on mitochondria for calcium buffering [[130], [131], [132], reviewed in Refs. [133], [134]]. Furthermore, loss of calcium homeostasis may contribute to a number of other ALS-associated toxic mechanisms, such as axonal transport defects and oxidative stress, and loss of protein homeostasis [[135], [136], reviewed in Ref. [86]]. Mitochondrial calcium regulates ATP production by activating the rate-limiting enzymes of the Krebs cycle and regulates oxidative phosphorylation and ATP synthesis to match local energy demand. As discussed above, reduced ATP production is a common feature in ALS. Diminished ATP levels may directly impact on axonal transport of mitochondria, vesicles and other cargoes by starving molecular motors of ATP. At the same time, elevated levels of cytosolic calcium may disrupt axonal transport of mitochondria by interacting with the mitochondrial kinesin-1receptor Miro1 and in turn exacerbate axonal transport deficits and local ATP levels. Indeed, elevated cytosolic calcium levels were shown to disrupt axonal transport of mitochondria in ALS mutant VAPB P56S expressing neurons [114, reviewed in Ref. [135]].
In ALS motor neurons, mitochondrial calcium overload may result from the physiological activity of AMPA receptors with pathologically increased calcium permeability. The resulting chronic calcium overload in mitochondria and concomitant abnormal oxidative phosphorylation has been shown to increase ROS production and oxidative stress [45], [137]. Mitochondrial calcium overload is predicted to result in depletion of calcium in the ER which causes protein misfolding and induces ER stress [reviewed in Refs. [133], [136]]. Elevated cytosolic calcium levels activate calpain, which has been shown to cleave TDP-43 to generate aggregation-prone fragments and could account for the TDP-43 pathology observed in ALS [138], [139]. Finally, chronic mitochondrial calcium overload and protein misfolding both induce apoptosis.
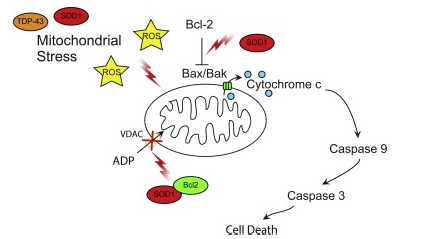
2.6. Pro-apoptotic signalling
Apoptosis is a last resort mechanism employed by cells upon suffering irreparable damage [reviewed in Ref. [140]]. Apoptosis serves to remove a damaged cell from the environment in a controlled manner so as to not induce large-scale degeneration. Mitochondria are integral to apoptosis. Upstream apoptotic signalling cascades converge on mitochondria to bring about cytochrome c release, which activates downstream executioner caspases[reviewed in Ref. [140]] (Fig. 3). Apoptosis is regulated by pro- and anti-apoptotic proteins of the Bcl-2 family, which control release of caspase-activating factors from mitochondria [reviewed in Refs. [140], [141]]. Activation of apoptotic signalling cascades has been observed in most ALS models, but this is mostly an indirect consequence of other toxic events. However, ALS mutant SOD1 has been shown to directly influence apoptotic signalling by interaction with Bcl-2. Wild-type and ALS mutant SOD1 (A4V, G37R, G41D and G85R) have been shown to bind the anti-apoptotic factor Bcl-2 in spinal cord samples [50]. When bound to mutant SOD1, the BH3 domain of Bcl-2 is exposed and this causes a pro-apoptotic gain of function of the Bcl-2 proteinin both cell and animal models of mutant SOD1 G93A ALS and in mutant SOD1 A4V patient spinal cord [142] (Fig. 3). The toxic mutant SOD1–Bcl-2 complex inhibits mitochondrial permeability to ADP and induces mitochondrial hyperpolarization due to reducing the interaction of Bcl-2 and VDAC1 [143] (Fig. 3).
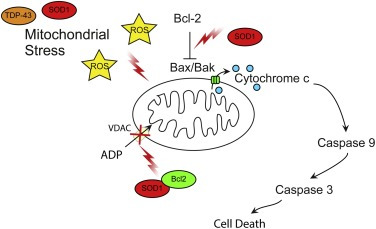
Fig. 3. Pro-apoptotic signalling.
Mitochondrial stress is increased due to dysfunctional cellular pathways resulting in increased ROS generation, and mitochondrial damage. Mutant SOD1 contributes to apoptotic signalling in ALS by binding the anti-apoptotic factor Bcl-2 and promoting a pro-apoptotic conformation of the protein. The Bcl2-SOD1 complex inhibits VDAC ADP permeability and induces mitochondrial hyperpolarisation. See text for details.
2.7. Impaired mitochondrial dynamics
Mitochondria are dynamic organelles that undergo fusion and fission events and directed transport. Several lines of research indicate that impaired mitochondrial dynamics may contribute to the aetiology of ALS.
2.7.1. Aberrant mitochondrial fission and fusion
Mitochondria form a dynamic network that evolves according to the energetic requirements of the cell. An individual mitochondrion can fuse to or fission from the mitochondrial network in response to various cues [144]. Fusion allows mitochondria to share mitochondrial metabolites, DNA and proteins, and allows for dissipation of small changes in MMP. Fission facilitates mitochondrial motility and allows the isolation of damaged parts of the network prior to disposal by mitophagy. In healthy cells, mitochondrial network morphology is governed by the interplay between Dynamin-related protein 1 (Drp1) and Fission 1 (Fis1) that promote fission, and Mfn1, Mfn2 and Optic atrophy 1 (Opa1) that promote fusion [reviewed in Ref. [145]] (Fig. 4). Evidence points towards imbalances in these two opposing pathways in ALS.
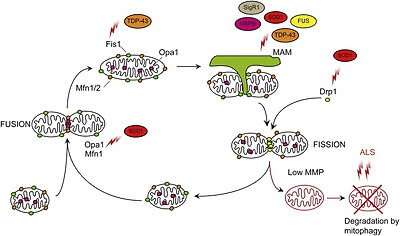
Fig. 4. Aberrant mitochondrial fission and fusion.
Mitochondrial network dynamics are required for correct mitochondrial function. Fission is mediated by Drp1, which is recruited to mitochondria by Fis1. Fusion of the OMM is mediated by Mfn1/2 and the fusion of the IMM by Opa1. Mitochondrial fissionis promoted in SOD1 and TDP-43 ALS by increases in the fission factors Fis1 and Drp1 and a decrease in the fusion factors Mfn1 and Opa1. Decreased ER–mitochondria contact sites may also lead to decreased mitochondrial fission events. Finally, dysfunctional removal of damaged organelles through the mitophagy pathway may also contribute to accumulation of fragmented mitochondria. See text for details.
In SOD1 G93A expressing SH-SY5Y and NSC-34 cell models, a decrease in Opa1 and increase in Drp1 levels resulting in a fragmented mitochondrial network were reported [146]. In vivo, the levels of Mfn1 and Opa1 progressively decreased while the levels of activated Drp1 phosphorylated at Ser616 and Fis1 remained stable in the spinal cord of SOD1 G93A transgenic mice [147] (Fig. 4). In contrast, in SOD1 G93A mouse muscle fibres there was no reported change in Mfn1, Mfn2 or Drp1 levels [148].
Overexpression of wild type TDP-43 and to a greater extend ALS mutant TDP-43 Q331K and M337V resulted in reduced mitochondrial length in primary motor neurons which was sensitive to Mfn2 levels [23]. Transgenic mice expressing wild type TDP-43 exhibited increased levels of Fis1 and activated phospho-Ser616 Drp1 and a reduction in Mfn1, which correlated with aberrant mitochondrial morphology and clustering [149]. In contrast, the same group reported no changes in the levels of phospho-Ser616 Drp1, Fis1 or Mfn1 in mutant TDP-43 M337V transgenic mice, despite a similar mitochondrial phenotype [150]. Possibly different pathways lead to fragmentation of the mitochondrial network in wild type and mutant TDP-43 transgenic mice, or, alternatively, external factors linked to disease such as changes in apoptotic factors result in mitochondrial fragmentation. Nevertheless, analysis of TDP-43 A382T patient fibroblasts showed that Fis1 levels were significantly increased compared to control cells [31] (Fig. 4). Counterintuitively, mitochondrial network fragmentation in C9ALS/FTD patient fibroblasts was associated with an increase in Mfn1 levels [31]. Possibly Mfn1 expression is increased to compensate for dysregulation of other fusion and fission factors.
ER–mitochondria contact sites have been shown to regulate mitochondrial morphology. Mitochondrial fission occurs at ER–mitochondria contact sites where ER tubules wrap around and constrict mitochondria to aid division [151]. As discussed above reduced ER–mitochondria contacts are a common observation in various ALS models. Consistent with reduced fission, decreased ER–mitochondria contacts in motor neurons of SIGMAR1 knockout correlated with elongated mitochondria compared to controls [124] whereas, in VAPB P56S overexpressing cells increased ER–mitochondria contacts correlated with fragmented mitochondria [118] (Fig. 4). However, in SOD1, TDP-43 and FUS ALS models reduced ER–mitochondria contacts appear to correlate with fragmented mitochondria [23], [26], [51], [115], [152], [153]. Therefore, there is no straightforward correlation between ALS-associated disruption of ER–mitochondria contacts and changes in mitochondrial network morphology.
The relevance of exacerbated mitochondrial fission in the aetiology of ALS is not clear. Perhaps, smaller mitochondria, which tend to be less energetically favourable and more prone to accumulating ROS induced damage if they cannot fuse back to the network [154], may exacerbate mitochondrial damage. Thus, the fragmented mitochondrial network observed may correlate directly with the reduced MMP, ATP levels and increased ROS reported in ALS. Alternatively, mitochondrial fragmentation may merely reflect mitophagy, a physiological quality control response to mitochondrial damage (see below). Indeed, it is well established that one of the first steps of mitophagy is to isolate damaged mitochondria from the mitochondrial network [reviewed in Ref. [155]]. However, as disease progresses and mitochondrial damage accumulates, mitophagy may become unable to cope and as a consequence mitochondrial fragmentation could become self-propagating. In any case, evidence suggests that mitochondrial fragmentation participates in ALS. Inhibition of mitochondrial fission by overexpression of dominant negative Drp1 K38A rescued SOD1 G93A induced mitochondrial fragmentation and trafficking defects and increased motor neuron viability [156]. Similarly, promotion of mitochondrial fusion by co-expressing Mfn2 in wild type or TDP-43 M333V expressing primary motor neurons rescued mitochondrial fragmentation [23]. Whether rescuing mitochondrial network morphology is sufficient to improve neuron survival in vivo remains unclear.
2.7.2. Disrupted mitochondrial quality control
As discussed above, accumulation of fragmented, rounded mitochondria is a common feature in ALS, and may indicate failure of mitophagy to clear damaged mitochondria. The molecular mechanisms of mitophagy have been reviewed in detail elsewhere [157], but briefly, loss of MMP results in accumulation of full length PINK1 on the OMM where it phosphorylates ubiquitin, which in turn promotes the recruitment of the E3 ligase Parkin. Parkin ubiquitinates and promotes the VCP/p97-dependent degradation of various OMM proteins such as Mfn1/2 and Miro1 to isolate and immobilise the damaged mitochondrion. Ubiquitinated mitochondria are then recognised by the canonical autophagy machinery and are delivered to the lysosome for degradation (Fig. 5).
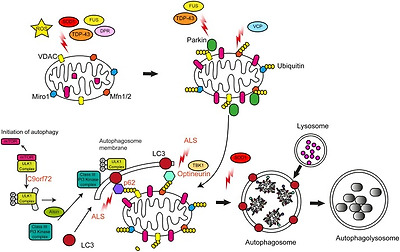
Fig. 5. Disrupted autophagy/mitophagy.
Mitophagy removes dysfunctional mitochondria from the cell. Mitochondria are exposed to high levels of stress due to elevations in ROS levels and aggregates of mutant ALS protein associated with mitochondria. Mitophagy is impaired at multiple stages in ALS. Mutant TDP-43 and FUS mislocalisation may affect Parkin levels. The removal of ubiquitinated Mfn1/2 may be impaired by mutant VCP/p97. Recognition of ubiquinated mitochondrial cargos by nascent autophagosomes may be impaired by mutations in p62/Sequestosome-1, Optineurin and TBK1. Other ALS genes such as SOD1, Alsin, and C9orf72 have also been linked to autophagy. See text for details.
TDP-43 and FUS directly regulate the expression of Parkin [158], [159]. Loss of TDP-43 or FUS was shown to decrease Parkin levels [158], and consistent with loss of TDP-43 function in the nucleus decreased Parkin protein levels were observed in sporadic ALS spinal cord motor neurons containing cytoplasmic TDP-43 aggregates [158]. Parkin protein levels were found to be decreased in TDP-43 A513T transgenic mice [38] but this was not observed by others [160]. Thus, since cytosolic TDP-43 aggregation is a near universal phenomenon in ALS, loss of Parkin may impair mitophagy in most ALS cases. Furthermore, since, as discussed above, TDP-43 accumulates in mitochondria and causes mitochondrial damage, a two-hit scenario emerges in which loss of mitophagy exacerbates the consequences of mitochondrial damage. Of note, Parkin has been shown to ubiquitinate TDP-43 and to promote its translocation from the nucleus to the cytosol in an HDAC6-dependent process [160]. Even though the latter is likely to be a reflection of the role of Parkin and HDAC6 in aggrephagy rather than mitophagy, it does emphasise a close link between Parkin and TDP-43 that merits further examination.
The Type II AAA+ ATPase VCP/p97 is involved in a multitude of processes, but its main function is to dislodge damaged ubiquitinated proteins from large protein complexes and membranes for degradation by the proteasome[reviewed in Refs. [161], [162]]. VCP/p97 is recruited to the OMM of damaged mitochondria following Parkin recruitment and extracts ubiquitinated Mfn1 and 2 from the OMM to allow their degradation by the proteasome [163]. As such VCP/p97 is essential for mitophagy [164]. Missense mutations in VCP/p97 cause of 1–2% of familial ALS cases as well as inclusion body myopathy (IBM) with Paget's disease (PDB) and FTD [165], [166]. Although it is not known exactly how disease-associated mutations in VCP/p97 alter its cellular functions, most mutations appear to convey varying degrees of loss of function [reviewed in Ref. [167]]. Consistent with loss of function, MEFs expressing mutant VCP/p97 A232E do not clear mitochondria following damage, despite labelling with Parkin [164]. As discussed above, pathogenic mutations in VCP/p97 induce mitochondrial uncoupling and impair ATP production [73]. Possibly, this phenotype is caused by accumulation of damaged, uncoupled mitochondria that should have been degraded.
Interestingly, not unlike Parkin, VCP/p97 has been linked to TDP-43 metabolism. In SHSY-5Y cells expression of mutant VCP/p97 (R95G, R155H/C, R191Q, A232E) caused TDP-43 redistribution from the nucleus to the cytoplasm [168]. Similarly, in VCP/p97 R152H or A229E transgenic Drosophilaand spinal cord motor neurons of VCP/p97 R155H or A232E transgenic mice expression of mutant VCP/p97 induced TDP-43 pathology [169]. Furthermore, in Drosophila, TBPH, the fly homolog of TDP-43 was identified as a genetic modifier that suppressed mutant VCP/p97 toxicity, indicating that VCP/p97 toxicity is at least in part mediated by a toxic gain of function of TDP-43 [169]. We now know that TDP-43 accumulates in mitochondria and causes damage to the ETC (see above; [42]). Hence a possible explanation for this genetic interaction is that accumulation of cytoplasmic TDP-43 exacerbates loss of mitochondrial quality control by mutations in VCP/p97.
Mitophagy requires autophagy receptors such as optineurin and p62/Sequestosome-1 that accumulate on ubiquitinated mitochondria and deliver them to the autophagosome by interaction with LC3-II on autophagic membranes. Optineurin and p62/Sequestosome-1 are substrates of Tank-binding kinase 1 (TBK1); phosphorylation by TBK1 promotes ubiquitin and LC3 binding and enhances mitophagy [reviewed in Refs. [170], [171], [172]].
Mutations in the OPTN, SQSTM1 and TBK1 genes which encode Optineurin, p62/Sequestosome-1, and TBK1, respectively, have been associated with ALS [[173], [174], [175], reviewed in Ref. [11]] (Table 1). ALS-associated mutations in these genes appear to cause loss of function phenotypes. The ALS-associated Optineurin E478G mutation maps to the ubiquitin binding domain of Optineurin and the mutant protein is no longer recruited to damaged mitochondria [176], [177]. Along the same lines, the ALS-associated L341V mutation in p62/Sequestosome-1 maps to the LC3-interacting region (LIR) domain and disrupts interaction with LC3 [178]. The ALS mutant forms of TBK1, TBK1 del690-713 and E696K, prevent the interaction between TBK1 and Optineurin and impair clearance of LC3 labelled autophagic cargos [179], [180]. If ALS-associated TBK1 mutants also affect mitophagy remains to be determined, but the role of TBK1 in mitophagy is well established. Furthermore, the activation of TBK1 is dependent on the activity of the upstream activity of PINK1/Parkin which as discussed above is affected in ALS [179], [180]. Consistent with failure to deliver damaged mitochondria to the lysosome, LC3-II positive mitochondrial aggregates have been observed in wild type and ALS mutant TDP-43 Q331K and M337V expressing NSC-34 cells as well as in SOD1 G93A transgenic mouse spinal cord [21], [181]. In case of SOD1 G93A the observed failure to clear damaged mitochondria appears to be caused by lysosomal deficits caused by impairment of retrograde trafficking of late endosomes rather than specific mitophagy deficits [182].
Interestingly TBK1 has also been found to interact with C9orf72 in complex with SMCR8 and WDR41, and C9orf72 controls autophagy via regulation of the small Rab GTPases Rab1a, Rab8a and Rab39b [[183], [184], [185], [186], reviewed in Ref. [187]]. Another ALS and autophagy-associated regulator of small Rab GTPases is Alsin, a guanine nucleotide exchange factor (GEF) for Rab5. Loss-of-function mutations in Alsin cause a recessive juvenile form of ALS, ALS2 [188], [189]. Loss of Alsin in mice impairs autophagy and corticospinal motor neurons exhibit fused mitochondria engulfed by vacuole structures consistent with failure to remove damaged mitochondria by mitophagy [33], [190]. Interestingly loss of Alsin in corticospinal motor neurons correlated with a progressive decline in Rab1a levels suggesting a direct link to C9orf72 [33]. Thus, a number of ALS-associated insults appear to converge on autophagy and mitophagy.
2.7.3. Impaired axonal transport of mitochondria
Motor neurons are highly polarised cells and require membrane-bound vesicles, organelles, proteins, lipids and RNA to be transported from the soma to the axon terminal and vice versa. Impaired axonal transport of mitochondria is a well-documented phenomenon in ALS. In fact, axonal transport defects are one of the earliest pathophysiological events in ALS motor neurons, indicating that they may be a primary cause of motor neuron loss [reviewed in Refs. [135], [191]].
Long distance transport of mitochondria is mediated by the molecular motors kinesin-1 (previously referred to as conventional kinesin or KIF5) and cytoplasmic dynein that move along microtubules. Kinesin-1 moves toward the faster growing plus end of microtubules whereas cytoplasmic dyneinmoves toward the opposite minus end. Because axonal microtubules are uniformly orientated with their plus ends pointing away from the cell body, kinesin-1 transports mitochondria toward the axon terminal (anterograde transport) and cytoplasmic dynein ferries mitochondria to the cell body (retrograde transport) (Fig. 6).
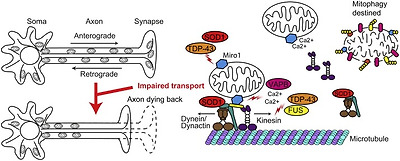
Fig. 6. Disrupted axonal transport.
Neurons depend on axonal transport of mitochondria for their ATP requirement and calcium buffering. Axonal transport of mitochondria is impaired in ALS. Miro1, the OMM protein involved in mitochondrial trafficking, is reduced in SOD1 and TDP-43-associated ALS. Mutant SOD1 binds the dynein/dynactin complex required for retrograde transport sequestering it in the cytosol. FUS and TDP-43 are proposed to regulate kinesin expression levels. Increases in cytosolic calcium levels due to decreased ER–mitochondria contacts in SOD1, TDP-43, FUS, Sig1R and VAPB-related ALS may contribute to detachment of the mitochondria from kinesin, through aberrant Miro1 regulation. Loss of mitochondria from the axon terminal may lead to the dying back of the axon. See text for details.
Defects in both anterograde and retrograde transport of mitochondria have been reported in a number of in vitro and in vivo experimental models. ALS mutant SOD1 G93A, A4V, G85R or G37R caused reduced anterograde but not retrograde axonal transport of mitochondria in cultured cortical neurons and an identical phenotype was observed in embryonic motor neurons isolated from SOD1 G93A transgenic mice [26]. Similarly, overexpression of the ALS mutant VAPB P56S caused a selective block in anterograde transport of mitochondria [114]. Reduced overall mitochondrial transport was observed in primary motor neurons expressing wild-type TDP-43 and to a greater extent ALS mutant TDP-43 Q331K or M337V [23] and in Sig1R deficient motor neurons [124]. Interestingly, normal levels of axonal transport of mitochondria were reported in cortical neurons expressing wild type or mutant TDP-43 M337V or A315T at 5–7 days in culture, suggesting a possible cell type specificity [192]. Time-lapse recordings in single axons in the intact sciatic nerve of presymptomatic SOD1 G93A and TDP-43 A315T transgenic mice and rats confirmed deficits in axonal transport of mitochondria in vivo [22], [193], [194], [195].
Mitochondrial transport defects result in redistribution of mitochondrial in axons. In vitro, the number of axonal mitochondria was significantly reduced and the remaining mitochondria were spaced further apart in primary neurons expressing ALS mutant SOD1 [26]. In vivo the number of axonal mitochondria was reduced in motor neurons of early symptomatic SOD1 G37R and SOD1 G85R transgenic mice and the motor axon terminals of SOD1 G93A transgenic rats and TDP-43 transgenic mice [27], [194], [196]. Furthermore, the distribution of the remaining mitochondria was no longer homogeneous throughout the axons, with abnormal mitochondrial clusters observed along the axon [22], [27], [196], [197]. Similar mislocalisation and aggregation of mitochondria has been observed in the soma, dendrites and proximal axons of motor neurons in spinal cord sections from ALS patients, suggesting that disruption to mitochondrial transport is a general phenomenon in ALS [18].
A number of possible mechanisms underlying mitochondrial axonal transport defects have been proposed. In rare cases, pathogenic mutations in the axonal transport machinery, such as cytoplasmic dynein, kinesin-1 and α-tubulindirectly disrupt axonal transport. In most cases however the disruption of axonal transport of mitochondria appears to be indirect; proposed mechanisms include microtubule destabilisation, pathogenic kinase signalling, protein aggregation, and mitochondrial damage [reviewed in Ref. [135]]. Axonal transport of mitochondria is controlled by the integral OMM protein Miro1 [reviewed in Ref. [198]]. Miro1 is an atypical Rho GTPasecomprised of two GTPase domains separated by two calcium-binding E-helix-loop-F-helix (EF)-hand motifs [199]. Miro1 is anchored in the OMM and connects mitochondria to kinesin-1 and cytoplasmic dynein via the adapter proteins TRAK1 and 2 [200], [201], [202], [203], [204], [205].
Miro1 regulates mitochondrial trafficking in response to physiological calcium stimuli and mitochondrial damage. Binding of calcium to the Miro1 EF-hand motifs modulates the interaction of kinesin-1 with Miro1 and impedes anterograde transport of mitochondria [206], [207]. During mitophagy, PINK1 phosphorylates Miro1 in response to mitochondrial damage [208], [209], [210]. Phosphorylation marks Miro1 for Parkin-dependent proteasomal degradation and results in detachment of kinesin-1 from mitochondria and arrested mitochondrial movement [209].
As discussed above, mitochondrial calcium handling is dysregulated in ALS. Hence ALS-associated mitochondrial axonal transport defects may be the direct result of aberrant calcium-mediated regulation of Miro1. In agreement with such a mechanism, cytosolic calcium levels are elevated in cellular ALS models and in motor neurons from transgenic ALS models [114], [119] and, at least in case of VAPB P56S there is direct evidence that elevated calcium disrupts transport of mitochondria via Miro1 [114] (Fig. 6). Similarly, ALS mutant SOD1, TDP-43, and Sig1R-associated reductions in ER–mitochondria contacts result in transiently elevated cytosolic calcium levels due to reduced calcium-uptake in mitochondria that coexist with axonal transport deficits, but the possible involvement of Miro1 remains to be determined [115], [124], [152].
Decreased levels of Miro1 have been reported in SOD1 G93A and TDP-43 M337V transgenic mice, as well as in the spinal cord of ALS patients [211]. Since both SOD1 G93A and TDP-43 M337V damage mitochondria (see above), these results suggest that ALS-associated mitochondrial damage leads to halting of mitochondrial transport via PINK1/Parkin-dependent degradation of Miro1.
In some cases, ALS-associated proteins may directly affect the axonal transport machinery. Both TDP-43 and FUS have been shown to regulate the expression of several kinesins, including kinesin-1, and TDP-43 binds TRAK1 mRNA [159], [212], [213]. ALS-mutant SOD1 directly binds to cytoplasmic dynein in SOD1 G93A and G37R cell and mouse models and the interaction becomes more prevalent throughout disease progression [214], [215], [216]. Thus, in case of mutant SOD1, axonal transport defects may in part be due to sequestration of dynein.
There are several ways in which defective axonal transport of mitochondria may contribute to disease. Defective axonal transport of mitochondria may lead to imbalances in ATP generation and calcium buffering at synaptic terminals. In conjunction with mitochondrial damage this may cause the dying back of the axon, a feature implicated in ALS [217] (Fig. 6). Retrograde transport defects are associated with defects in the removal of damaged organelles by mitophagy, which could explain the mitochondrial aggregates found in the axons of ALS patients [20], [218]. Furthermore, defective axonal transport of mitochondria may also affect the transport of other axonal cargoes such as signalling endosomes which appear closely linked to ALS pathology [reviewed in Ref. [135]].
3. Mitochondria as therapeutic targets in ALS
Finding a cure for ALS has thus far been unsuccessful. Targeting mitochondrial dysfunction presents an attractive treatment option due to the widespread prevalence of mitochondrial dysfunction in disease. Potential therapeutic strategies have aimed to decrease ROS generation, increase mitochondrial biogenesis, inhibit apoptotic pathways or dampen excitotoxicity.
The only FDA drug approved for use in ALS is Riluzole. However, Riluzole only extends survival by approximately 3months. The mechanism by which Riluzole acts to improve survival is unclear. Some evidence suggests that Riluzole decreases ROS through induction of glutathione synthesis [219]. However little effect was observed on basal levels of ROS, and therefore Riluzole may only act in this way in the context of elevated ROS levels. Riluzole has been found to display a range of effects on glutamatergic signalling at the synapse, leading ultimately to attenuation in calcium handling requirements. Indeed, treatment in vitro with Riluzole prevents motor and cortical neuron loss induced by sustained glutamate increase and elevated ROS levels [220]. Furthermore, Riluzole has been demonstrated to reduce inward calcium currents and rescue the axonal transport of neurofilaments [221], [222]. Attenuation of inward calcium currents is predicted to alleviate stresses on cytosolic and mitochondrial calcium buffering mechanisms and potentially decrease cytosolic calcium levels with knock-on effects on ROS generation and mitochondrial function. However, with the limited effect on prognosis offered by Riluzole, alternative therapies are required for the treatment of ALS.
Despite many drug-based and gene therapy approaches targeting mitochondrial dysfunction in ALS, neither treatments aimed at increasing mitochondrial function and survival nor those aimed at reducing oxidative stress have yielded significant results in clinical trials, despite promising trials in animal models [223]. For example, several drugs targeting mitochondrial function and/or ROS such as Coenzyme Q10, Dexpramipexole, Olesoxime and Creatine all showed initial success in animal models but were unsuccessful in human clinical trials [224], [225], [226], [227], [228], [229], [230], [231]. Reducing ROS using Edaravone, a free radical scavenger was successful in mouse models but had only a small effect on human disease progression [232], [233]. Similarly, minocyline, an anti-apoptotic and anti-inflammatory drug which extended survival in mouse models, failed in a human phase III randomised trial [234]. The discrepancy between the results from mouse models and human trials highlights the disparity between models and the human disease, and the need for better disease models [235]. In addition, the wide range of mitochondrial dysfunction in ALS means that a single therapy is unlikely to attenuate all aspects of dysfunction. Therefore, identifying which of the mitochondrial dysfunctions are relevant to disease causation and progression will continue to be important for the development of neuroprotective therapies in ALS. Targeting specific proteins dysregulated in ALS may provide alternative therapy avenues. Indeed a blocking peptide that prevents the abnormal interaction between mutant SOD1 and Bcl-2 has been shown to prevent apoptosis and improve mitochondrial function in SOD1 G93A transgenic mice [143].
4. Conclusions
Despite the surge in the number genes found to be associated with ALS since the turn of the millennium, the aetiology ALS remains largely unknown. Mitochondrial dysfunction has emerged as a common, early phenomenon in ALS. The appearance of deficits in oxidative phosphorylation, calcium buffering and mitochondrial transport prior to the onset of disease symptoms in vivo in disease models suggests an important role for loss of mitochondrial integrity in the aetiology of ALS.
Nevertheless, clinical trials targeting mitochondria have been disappointing, indicating that mitochondrial dysfunction alone may not be a primary cause of disease. Possibly the widespread presence of mitochondrial dysfunction in ALS models and patients, may be secondary to other disease-related insults such as protein aggregation and excitotoxicity. In such a scenario, mitochondrial function would gradually decay until a point-of-no-return threshold is reached and the motor neuron dies. Such a mechanism would be predicted to conspire with the age-associated decline in mitochondrial function and may explain the late onset of the majority of ALS cases. In any case, since, with the possible exception of Riluzole, treatments targeting other proposed causes of ALS have been equally unsuccessful in clinical trials, it transpires that any successful therapeutic strategy may have to address multiple targets in a combination therapy.
Importantly, mitochondrial dysfunction has also been widely reported in other neurodegenerative diseases such as Parkinson’s, Huntington’s and Alzheimer’s disease [236], [237], [238]. Similarities in defects in mitochondrial clearance and bioenergetic functions exist between these diseases and ALS despite different clinical presentation. Therefore, neurodegenerative conditions appear to be group of diseases with common dysfunction but with varying endpoints. Possibly discovery and study of common mechanisms underlying these diseases, including mitochondrial dysfunction, will increase our understanding of the essential requirements for neuronal survival that can inform future neuroprotective therapies.
Acknowledgements
This work was funded by grants from the UK Medical Research Council (MRC; MR/K005146/1 and MR/M013251/1 to KJDV). EFS is supported by a Motor Neurone Disease Association Prize Studentship (DeVos/Oct13/870-892 to KJDV). PJS is supported as an NIHR Senior Investigator and by funding awards from the UK Medical Research Council, the National Institute for Health Research, the Motor Neurone Disease Association and the EU Horizon 2020 programme.
References
- [1]
- A. Chiò, G. Logroscino, B.J. Traynor, J. Collins, J.C. Simeone, L.A. Goldstein, L.A.WhiteGlobal epidemiology of amyotrophic lateral sclerosis: a systematic review of the published literatureNeuroepidemiology, 41 (2013), pp. 118-130
- [2]
- P. Mehta, W. Kaye, L. Bryan, T. Larson, T. Copeland, J. Wu, O. Muravov, K. HortonPrevalence of amyotrophic lateral sclerosis – United States, 2012–2013MMWR Surveill. Summ., 65 (2016), pp. 1-12
- [3]
- M. Orsini, A.B. Oliveira, O.J. Nascimento, C.H. Reis, M.A. Leite, J.A. de Souza, C.Pupe, O.G. de Souza, V.H. Bastos, M.R. de Freitas, S. Teixeira, C. Bruno, E.Davidovich, B. SmidtAmyotrophic lateral sclerosis: new perpectives and updateNeurol. Int., 7 (2015), p. 5885
- [4]
- M.C. Kiernan, S. Vucic, B.C. Cheah, M.R. Turner, A. Eisen, O. Hardiman, J.R.Burrell, M.C. ZoingAmyotrophic lateral sclerosisLancet, 377 (2011), pp. 942-955
- [5]
- A.L. Boxer, M. Gold, E. Huey, F.-B. Gao, E.A. Burton, T. Chow, A. Kao, B. Leavitt, B.Lamb, M. Grether, D. Knopman, N.J. Cairns, I.R. Mackenzie, L. Mitic, E.D.Roberson, D. Van Kammen, M. Cantillon, K. Zahs, S. Salloway, J. Morris, G. Tong, H. Feldman, H. Fillit, S. Dickinson, Z. Khachaturian, M. Sutherland, R. Farese, B.L. Miller, J. CummingsFrontotemporal degeneration, the next therapeutic frontier: molecules and animal models for FTD drug developmentAlzheimer's Dement., 9 (2013), pp. 176-188
- [6]
- B. Swinnen, W. RobberechtThe phenotypic variability of amyotrophic lateral sclerosisNat. Rev. Neurol., 10 (2014), pp. 661-670
- [7]
- O. Abel, J.F. Powell, P.M. Andersen, A. Al-ChalabiALSoD. A user-friendly online bioinformatics tool for amyotrophic lateral sclerosis geneticsHum. Mutat., 33 (2012), pp. 1345-1351
- [8]
- M. DeJesus-Hernandez, I. Mackenzie, B. Boeve, A. Boxer, M. Baker, N. Rutherford, A. Nicholson, N. Finch, H. Flynn, J. Adamson, N. Kouri, A. Wojtas, P. Sengdy, G.Y.Hsiung, A. Karydas, W. Seeley, K. Josephs, G. Coppola, D. Geschwind, Z. Wszolek, H. Feldman, D. Knopman, R. Petersen, B. Miller, D. Dickson, K. Boylan, N. Graff-Radford, R. RademakersExpanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALSNeuron, 72 (2011), pp. 245-256
- [9]
- A. Renton, E. Majounie, A. Waite, J. Simón-Sánchez, S. Rollinson, J. Gibbs, J.Schymick, H. Laaksovirta, J. van Swieten, L. Myllykangas, H. Kalimo, A. Paetau, Y.Abramzon, A. Remes, A. Kaganovich, S. Scholz, J. Duckworth, J. Ding, D. Harmer, D. Hernandez, J. Johnson, K. Mok, M. Ryten, D. Trabzuni, R. Guerreiro, R. Orrell, J. Neal, A. Murray, J. Pearson, I. Jansen, D. Sondervan, H. Seelaar, D. Blake, K.Young, N. Halliwell, J. Callister, G. Toulson, A. Richardson, A. Gerhard, J.Snowden, D. Mann, D. Neary, M. Nalls, T. Peuralinna, L. Jansson, V.M. Isoviita, A.L. Kaivorinne, M. Hölttä-Vuori, E. Ikonen, R. Sulkava, M. Benatar, J. Wuu, A.Chiò, G. Restagno, G. Borghero, M. Sabatelli, D. Heckerman, E. Rogaeva, L.Zinman, J. Rothstein, M. Sendtner, C. Drepper, E. Eichler, C. Alkan, Z. Abdullaev, S. Pack, A. Dutra, E. Pak, J. Hardy, A. Singleton, N. Williams, P. Heutink, S.Pickering-Brown, H. Morris, P. Tienari, B. TraynorA hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTDNeuron, 72 (2011), pp. 257-268
- [10]
- M. Van Blitterswijk, M. DeJesus-Hernandez, R. RademakersHow do C9ORF72 repeat expansions cause ALS and FTD: can we learn from other non-coding repeat expansion disorders?Curr. Opin. Neurol., 25 (2014), pp. 689-700
- [11]
- A.E. Renton, A. Chiò, B.J. TraynorState of play in amyotrophic lateral sclerosis geneticsNat. Neurosci., 17 (2014), pp. 17-23
- [12]
- L. Ferraiuolo, J. Kirby, A.J. Grierson, M. Sendtner, P.J. ShawMolecular pathways of motor neuron injury in amyotrophic lateral sclerosisNat. Rev. Neurol., 7 (2011), pp. 616-630
- [13]
- D.G. Nicholls, S.L. BuddMitochondria and neuronal survivalPhysiol. Rev., 80 (2000), pp. 315-360
- [14]
- E. Engl, D. AttwellNon-signalling energy use in the brainJ. Physiol. (Lond.), 593 (2015), pp. 3417-3429
- [15]
- R. Rizzuto, D. De Stefani, A. Raffaello, C. MammucariMitochondria as sensors and regulators of calcium signallingNat. Rev. Mol. Cell Biol., 13 (2012), pp. 566-578
- [16]
- B.A. Payne, P.F. ChinneryMitochondrial dysfunction in aging: much progress but many unresolved questionsBiochim. Biophys. Acta, 1847 (2015), pp. 1347-1353
- [17]
- T. AtsumiThe ultrastructure of intramuscular nerves in amyotrophic lateral sclerosisActa Neuropathol. (Berl.), 55 (1981), pp. 193-198
- [18]
- S. Sasaki, M. IwataMitochondrial alterations in the spinal cord of patients with sporadic amyotrophic lateral sclerosisJ. Neuropathol. Exp. Neurol., 66 (2007), pp. 10-16
- [19]
- M.N. Hart, P.A. Cancilla, S. Frommes, A. HiranoAnterior horn cell degeneration and Bunina-type inclusions associated with dementiaActa Neuropathol. (Berl.), 38 (1977), pp. 225-228
- [20]
- K. Okamoto, S. Hirai, M. Shoji, Y. Senoh, T. YamazakiAxonal swellings in the corticospinal tracts in amyotrophic lateral sclerosisActa Neuropathol. (Berl.), 80 (1990), pp. 222-226
- [21]
- K. Hong, Y. Li, W. Duan, Y. Guo, H. Jiang, W. Li, C. LiFull-length TDP-43 and its C-terminal fragments activate mitophagy in NSC34 cell lineNeurosci. Lett., 530 (2012), pp. 144-149
- [22]
- J. Magrané, C. Cortez, W. Gan, G. ManfrediAbnormal mitochondrial transport and morphology are common pathological denominators in SOD1 and TDP43 ALS mouse modelsHum. Mol. Genet., 23 (2014), pp. 1413-1424
- [23]
- W. Wang, L. Li, W.L. Lin, D.W. Dickson, L. Petrucelli, T. Zhang, X. WangThe ALS disease-associated mutant TDP-43 impairs mitochondrial dynamics and function in motor neuronsHum. Mol. Genet., 22 (2013), pp. 4706-4719
- [24]
- M.C. Dal Canto, M.E. GurneyDevelopment of central nervous system pathology in a murine transgenic model of human amyotrophic lateral sclerosisAm. J. Pathol., 145 (1994), pp. 1271-1279
- [25]
- C.M.J. Higgins, C. Jung, Z. XuALS-associated mutant SODIG93A causes mitochondrial vacuolation by expansion of the intermembrane space by involvement of SODI aggregation and peroxisomesBMC Neurosci., 4 (2003)
- [26]
- K.J. De Vos, A.L. Chapman, M.E. Tennant, C. Manser, E.L. Tudor, K.F. Lau, J.Brownlees, S. Ackerley, P.J. Shaw, D.M. McLoughlin, C.E. Shaw, P.N. Leigh, C.C.J.Miller, A.J. GriersonFamilial amyotrophic lateral sclerosis-linked SOD1 mutants perturb fast axonal transport to reduce axonal mitochondria contentHum. Mol. Genet., 16 (2007), pp. 2720-2728
- [27]
- C. Vande Velde, K.K. McDonald, Y. Boukhedimi, M. McAlonis-Downes, C.S.Lobsiger, S. Bel Hadj, A. Zandona, J.-P. Julien, S.B. Shah, D.W. ClevelandMisfolded SOD1 associated with motor neuron mitochondria alters mitochondrial shape and distribution prior to clinical onsetPLoS One, 6 (2011), p. e22031
- [28]
- M.L. Tradewell, Z. Yu, M. Tibshirani, M. Boulanger, H.D. Durham, S. RichardArginine methylation by prmt1 regulates nuclear-cytoplasmic localization and toxicity of FUS/TLS harbouring ALS-linked mutationsHum. Mol. Genet., 21 (2012), pp. 136-149
- [29]
- J. Deng, M. Yang, Y. Chen, X. Chen, J. Liu, S. Sun, H. Cheng, Y. Li, E.H. Bigio, M.Mesulam, Q. Xu, S. Du, K. Fushimi, L. Zhu, J.Y. WuFUS interacts with HSP60 to promote mitochondrial damagePLoS Genet., 11 (2015), p. e1005357
- [30]
- A. Sharma, A.K. Lyashchenko, L. Lu, S.E. Nasrabady, M. Elmaleh, M. Mendelsohn, A. Nemes, J.C. Tapia, G.Z. Mentis, N.A. ShneiderALS-associated mutant FUS induces selective motor neuron degeneration through toxic gain of functionNat. Commun., 7 (2016), p. 10465
- [31]
- E. Onesto, C. Colombrita, V. Gumina, M.O. Borghi, S. Dusi, A. Doretti, G.Fagiolari, F. Invernizzi, M. Moggio, V. Tiranti, V. Silani, A. RattiGene-specific mitochondria dysfunctions in human TARDBP and C9ORF72 fibroblastsActa Neuropathol. Commun., 4 (2016), p. 47
- [32]
- R. Dafinca, J. Scaber, N.A. Ababneh, T. Lalic, G. Weir, H. Christian, J. Vowles, A.G.L. Douglas, A. Fletcher-Jones, C. Browne, M. Nakanishi, M.R. Turner, R.Wade-Martins, S.A. Cowley, K. TalbotC9orf72 hexanucleotide expansions are associated with altered endoplasmic reticulum calcium homeostasis and stress granule formation in induced pluripotent stem cell-derived neurons from patients with amyotrophic lateral sclerosis and frontotemporal dementiaStem Cells, 34 (2016), pp. 2063-2078
- [33]
- M. Gautam, J.H. Jara, G. Sekerkova, M.V. Yasvoina, M. Martina, P.H. OzdinlerAbsence of alsin function leads to corticospinal motor neuron vulnerability via novel disease mechanismsHum. Mol. Genet., 25 (2016), pp. 1074-1087
- [34]
- H.Z. Yin, A. Nalbandian, C.I. Hsu, S. Li, K.J. Llewellyn, T. Mozaffar, V.E. Kimonis, J.H. WeissSlow development of ALS-like spinal cord pathology in mutant valosin-containing protein gene knock-in miceCell Death Dis., 3 (2012), p. e374
- [35]
- A. Nalbandian, K.J. Llewellyn, M. Badadani, H.Z. Yin, C. Nguyen, V. Katheria, G.Watts, J. Mukherjee, J. Vesa, V. Caiozzo, T. Mozaffar, J.H. Weiss, V.E. KimonisA progressive translational mouse model of human valosin-containing protein disease: the VCPR155H/+ mouseMuscle Nerve, 47 (2013), pp. 260-270
- [36]
- S. Bannwarth, S. Ait-El-Mkadem, A. Chaussenot, E.C. Genin, S. Lacas-Gervais, K.Fragaki, L. Berg-Alonso, Y. Kageyama, V. Serre, D.G. Moore, A. Verschueren, C.Rouzier, I. Le Ber, G. Auge, C. Cochaud, F. Lespinasse, K. N'Guyen, A. de Septenville, A. Brice, P. Yu-Wai-Man, H. Sesaki, J. Pouget, V. Paquis-FlucklingerA mitochondrial origin for frontotemporal dementia and amyotrophic lateral sclerosis through CHCHD10 involvementBrain, 137 (2014), pp. 2329-2345
- [37]
- E.C. Genin, M. Plutino, S. Bannwarth, E. Villa, E. Cisneros-Barroso, M. Roy, B.Ortega-Vila, K. Fragaki, F. Lespinasse, E. Pinero-Martos, G. Augé, D. Moore, F.Burté, S. Lacas-Gervais, Y. Kageyama, K. Itoh, P. Yu-Wai-Man, H. Sesaki, J.E. Ricci, C. Vives-Bauza, V. Paquis-FlucklingerCHCHD10 mutations promote loss of mitochondrial cristae junctions with impaired mitochondrial genome maintenance and inhibition of apoptosisEMBO Mol. Med., 8 (2016), pp. 58-72
- [38]
- C. Stribl, A. Samara, D. Trümbach, R. Peis, M. Neumann, H. Fuchs, V. Gailus-Durner, M. Hrabě de Angelis, B. Rathkolb, E. Wolf, J. Beckers, M. Horsch, F. Neff, E. Kremmer, S. Koob, A.S. Reichert, W. Hans, J. Rozman, M. Klingenspor, M.Aichler, A.K. Walch, L. Becker, T. Klopstock, L. Glasl, S.M. Hölter, W. Wurst, T.FlossMitochondrial dysfunction and decrease in body weight of a transgenic knock-in mouse model for TDP-43J. Biol. Chem., 289 (2014), pp. 10769-10784
- [39]
- I.G. Kirkinezos, S.R. Bacman, D. Hernandez, J. Oca-Cossio, L.J. Arias, M.A. Perez-Pinzon, W.G. Bradley, C.T. MoraesCytochrome c association with the inner mitochondrial membrane is impaired in the CNS of G93A-SOD1 miceJ. Neurosci., 25 (2005), pp. 164-172
- [40]
- J. Kong, Z. XuMassive mitochondrial degeneration in motor neurons triggers the onset of amyotrophic lateral sclerosis in mice expressing a mutant SOD1J. Neurosci., 18 (1998), pp. 3241-3250
- [41]
- C.M. Higgins, C. Jung, H. Ding, Z. XuMutant Cu, Zn superoxide dismutase that causes motoneuron degeneration is present in mitochondria in the CNSJ. Neurosci., 22 (2002)
- [42]
- W. Wang, L. Wang, J. Lu, S.L. Siedlak, H. Fujioka, J. Liang, S. Jiang, X. Ma, Z. Jiang, E.L. da Rocha, M. Sheng, H. Choi, P.H. Lerou, H. Li, X. WangThe inhibition of TDP-43 mitochondrial localization blocks its neuronal toxicityNat. Med., 22 (2016), pp. 869-878
- [43]
- R. Lopez-Gonzalez, Y. Lu, T.F. Gendron, A. Karydas, H. Tran, D. Yang, L. Petrucelli, B.L. Miller, S. Almeida, F.B. GaoPoly(GR) in C9ORF72-related ALS/FTD compromises mitochondrial function and increases oxidative stress and DNA damage in iPSC-derived motor neuronsNeuron, 92 (2016), pp. 383-391
- [44]
- A.M. Blokhuis, M. Koppers, E.J. Groen, D.M. van den Heuvel, S. Dini Modigliani, J.J. Anink, K. Fumoto, F. van Diggelen, A. Snelting, P. Sodaar, B.M. Verheijen, J.A.Demmers, J.H. Veldink, E. Aronica, I. Bozzoni, J. den Hertogn, L.H. van den Berg, R.J. PasterkampComparative interactomics analysis of different ALS-associated proteins identifies converging molecular pathwaysActa Neuropathol. (Berl.), 132 (2016), pp. 175-196
- [45]
- M. Mattiazzi, M. D'Aurelio, C.D. Gajewski, K. Martushova, M. Kiaei, M. Flint Beal, G. ManfrediMutated human SOD1 causes dysfunction of oxidative phosphorylation in mitochondria of transgenic miceJ. Biol. Chem., 277 (2002), pp. 29626-29633
- [46]
- A. Ferri, M. Cozzolino, C. Crosio, M. Nencini, A. Casciati, E.B. Gralla, G. Rotilio, J.S. Valentine, M.T. CarriFamilial ALS-superoxide dismutases associate with mitochondria and shift their redox potentialsProc. Natl. Acad. Sci. U. S. A., 103 (2006), pp. 13860-13865
- [47]
- C. Vijayvergiya, M.F. Beal, J. Buck, G. ManfrediMutant superoxide dismutase 1 forms aggregates in the brain mitochondrial matrix of amyotrophic lateral sclerosis miceJ. Neurosci., 25 (2005), pp. 2463-2470
- [48]
- V. Shoshan-Barmatz, D. Ben-HailVDAC, a multi-functional mitochondrial protein as a pharmacological targetMitochondrion, 12 (2012), pp. 24-34
- [49]
- A. Israelson, N. Arbel, S. Da Cruz, H. Ilieva, K. Yamanaka, V. Shoshan-Barmatz, D.W. ClevelandMisfolded mutant SOD1 directly inhibits VDAC1 conductance in a mouse model of inherited ALSNeuron, 67 (2010), pp. 575-587
- [50]
- P. Pasinelli, M.E. Belford, N. Lennon, B.J. Bacskai, B.T. Hyman, D. Trotti, R.H.Brown Jr.Amyotrophic lateral sclerosis-associated SOD1 mutant proteins bind and aggregate with Bcl-2 in spinal cord mitochondriaNeuron, 43 (2004), pp. 19-30
- [51]
- R. Stoica, S. Paillusson, P. Gomez-Suaga, J.C. Mitchell, D.H. Lau, E.H. Gray, R.M.Sancho, G. Vizcay-Barrena, K.J. De Vos, C.E. Shaw, D.P. Hanger, W. Noble, C.C.MillerALS/FTD-associated FUS activates GSK-3beta to disrupt the VAPB-PTPIP51 interaction and ER-mitochondria associationsEMBO Rep., 17 (2016), pp. 1326-1342
- [52]
- F.R. Wiedemann, G. Manfredi, C. Mawrin, M. Flint Beal, E.A. SchonMitochondrial DNA and respiratory chain function in spinal cords of ALS patientsJ. Neurochem., 80 (2002), pp. 616-625
- [53]
- G.M. Borthwick, M.A. Johnson, P.G. Ince, P.J. Shaw, D.M. TurnbullMitochondrial enzyme activity in amyotrophic lateral sclerosis: implications for the role of mitochondria in neuronal cell deathAnn. Neurol., 46 (1999), pp. 787-790
- [54]
- F.R. Wiedemann, K. Winkler, A.V. Kuznetsov, C. Bartels, S. Vielhaber, H. Feistner, W.S. KunzImpairment of mitochondrial function in skeletal muscle of patients with amyotrophic lateral sclerosisJ. Neurol. Sci., 156 (1998), pp. 65-72
- [55]
- S. Vielhaber, D. Kunz, K. Winkler, F.R. Wiedemann, E. Kirches, H. Feistner, H.J.Heinze, C.E. Elger, W. Schubert, W.S. KunzMitochondrial DNA abnormalities in skeletal muscle of patients with sporadic amyotrophic lateral sclerosisBrain, 123 (2000), pp. 1339-1348