Re:Therapeutic targeting of glutaminolysis as an essential strategy to combat cancer
작성자문형철작성시간20.02.17조회수970 목록 댓글 0beyond reason

Therapeutic targeting of glutaminolysis as an essential strategy to combat cancer
Abreviations
Keywords
1. Introduction
Glutamine (Gln) is the most abundant amino acid in blood and muscle and it has a pleiotropic function for carcinogenesis [1]. Its critical roles in cancer cells include generation of energy, biosynthesis of essential molecules (amino acids, purines, pyrimidines, fatty acids), control of redox homeostasis, and fine regulation of cell signaling [2]. Mitochondria, the cell’s energy factories, are directly involved in the pathogenesis of cancer since these organelles control most reprogrammed oncometabolic circuits, including signaling pathways, as well as synthesis of amino acids, nucleic acids, lipids, and regulatory molecules like NADH, NADPH, and reactive oxygen species (ROS) [3]. There are two main processes to provide anaplerotic fluxes in cancer: glutaminolysis and pyruvate carboxylation [4]. Glutaminolysis has been established as a hallmark of cancer metabolism [2].
In this review, we describe oncogenes, tumor suppressors, and tumor microenvironment (TME) as key regulators of Gln metabolism in cancer (Fig. 1). Gln and Gln-related enzymes have become useful tools in therapeutic targeting of neoplasms [5]. Simultaneously, clinical applications of in vivo assessment of glutaminolysis are essential for discovering novel strategies for the therapy and diagnosis of many types of cancer [6]. Anyhow, cancer heterogeneity, chemoresistance and metastasis keep as tough barriers for the entire success of metabolic therapy against cancer.
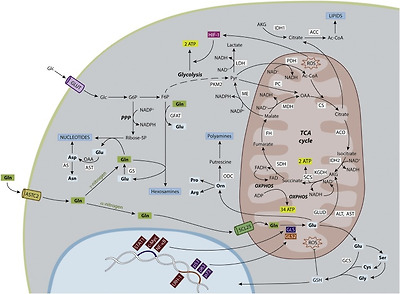
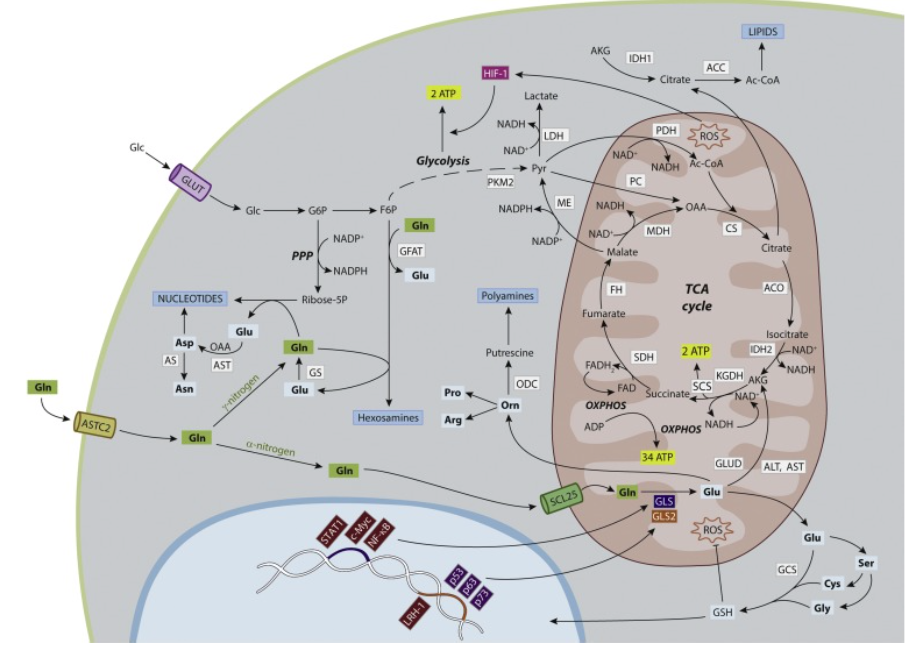
Fig. 1. Glutamine and glutaminolysis in the metabolic network regulating cancer growth. Gln (in bold, green squares) provides its α-nitrogen, γ-nitrogen and carbon skeleton for bioenergetics and biosynthetic purposes. Key reactions in nitrogen and carbon metabolism are shown. Glutaminolysis starts with glutaminase (GLS, GLS2) reaction to generate Glu and ammonium. Glu can be used for the synthesis of many NEAAs (in bold, cyan squares) for protein biosynthesis. γ-nitrogen is used for synthesis of aspartate, asparagine, hexosamines and nucleotides. ALT and AST catalyze the transformation of glutamate’s α-nitrogen into pyruvate and oxaloacetate (OAA) forming alanine and aspartate, respectively. GLUD transform Glu in AKG. α-Nitrogen from Glu is also used to synthetize Ser. Glycine and cysteine are generated from Ser. Glutamate, cysteine and glycine integrate GSH. GSH maintains redox homeostasis fighting against mitochondrial ROS. GSH is also recruited into the nucleus to regulate chromatincompaction, cell cycle and DNA repair. Glu can be also converted to ornithine (which in turn forms polyamines) and then into proline and/or arginine. Carbon backbone from Gln is oxidized in mitochondria, leading to ATP production by OXPHOS. Mitochondrial metabolism produces ROS, that activate glycolysis via HIF-1. Mutations in some genes encoding several TCA cycle enzymes (IDH, SDH, FH, CS and ACO) evoke a metabolic reshuffle of cells to produce oncogenic transformation. Cytosolic IDH1 transforms AKG to citrate which is subsequently converted into Ac-CoA by ACC for lipids biosynthesis. On the other hand, PKM2 regulates the last step of glycolysis to produce pyruvate, which is consecutively transformed in lactate by LDH. Both PKM2 and LDH, together with ME and PC, contribute to higher metabolic regime of cancer cells. ME converts malate into the TCA carbon source pyruvate and NADPH. In glycolysis, G6P is diverted to PPP shunt for production of extra NADPH and ribose-5-phosphate, which reacts with Gln to build nucleotides. Mutant IDH1 cells significantly increased fractional flux through PC to utilize pyruvate instead of Gln to feeding TCA cycle. In nucleus, oncogenic transcription factors (burgundy red) such as c-Myc, STAT1 and NF-κB can regulate GLS (dark blue), while oncogenic (burgundy red) LRH-1 and tumor suppressor factors (purple) such as p53, p63 and p73 can context-dependent regulate GLS2 (orange). ACC, Acetyl-CoA carboxylase; Ac-CoA, acetyl-coenzyme A; ACO, aconitase; AKG, α-ketoglutarate; ALT, alanine transaminase; AS, asparagine synthethase; AST, aspartate transaminase; CS, citrate synthase; ERK, extracellular signal-regulated kinases; F6P, fructose-6-phosphate; FH, fumarate hydratase; G6P, glucose-6-phosphate; GCS, gamma-glutamylcysteine synthetase; GFAT, glutamine:fructose-6-phosphate aminotransferase; Gln, glutamine; GLS, glutaminase isoenzyme; GLS2, glutaminase 2 isoenzyme; GLUD, glutamate dehydrogenase; Glc, glucose; Gln, glutamine; GLS, glutaminase isoenzyme; GLS2, glutaminase 2 isoenzyme; GLUD: glumamate dehydrogenase; GS, glutamine synthetase; GSH, glutathione; HIF-1, hypoxia-inducible factor-1; IDH-1, cytosolic isocitrate dehydrogenase; IDH-2, mitochondrial isocitrate dehydrogenase; KGDH: ketoglutarate dehydrogenase; KRAS, V-Ki-ras2 Kirsten rat sarcoma; LDH: lactate dehydrogenase; LRH-1, nuclear receptor liver receptor homolog 1; MDH, malate dehydrogenase; ME, malic enzyme; mTOR, mammalian target of rapamycine; NF-κB, nuclear factor-kappa B; OAA, oxaloacetate; ODC, ornithine decarboxylase; OXPHOS, oxidative phosphorylation; PC, pyruvate carboxylase; PDH, pyruvate dehydrogenase; PKM2, pyruvate kinase M2 isoform; PPP, pentose phosphate pathway; ROS, reactive oxygen species; SCS, succynil-Coenzyme A-synthetase; SDH, succinate dehydrogenase; STAT1, signal transducer and activator of transcription 1.
2. Glutamine as a multifunctional agent in cancer
2.1. Glutamine as energy substrate
Following the publication of famous Warburg’s paper [7] pointing out the essential role of glucose (Glc) in cancer metabolism, Weinhouse published [8] that cancer cells have a normal mitochondrial oxidative phosphorylation(OXPHOS) capacity to metabolize Glc to carbon dioxide (CO2) and water (H2O) to render 36 molecules of ATP [8]. In 1979, it was demonstrated that more than half of the ATP requirement comes from Gln by aerobic oxidation from the TCA cycle. These experiments were carried out in Reitzer’s lab using HeLa cells, with high Glc concentrations in the medium [9].
In last years, highly cited articles state the idea that Gln is a major nutrient in neoplastic tissues [10]. In addition, it has been proved that Gln is a perfect substrate for oxidative metabolism in many tumor cells [11]. In fact, catabolismof Gln into lactate results on NADPH via the activity of malic enzyme [12]. Oxidation of glutamine's carbon backbone in the mitochondria requires transformation of Gln to α-ketoglutarate (AKG), via glutaminase (GA, EC 3.5.1.2) activity followed by conversion of glutamate (Glu) to AKG by either transaminases or glutamate dehydrogenase (GLUD) [10].
An additional important OXPHOS-independent energy source is the ATP regeneration by the glycolytic enzyme pyruvate kinase [13]. In proliferating cells and especially in tumor cells the pyruvate kinase isoenzyme M2 (M2-PK, PKM2) is expressed, which may occur in a highly active tetrameric form as well as a nearly inactive dimeric form [14]. Depending upon the metabolic demands of the cells the tetramer:dimer ratio of PKM2 regulates whether Glc carbons are channeled into synthetic pathways of cell buildings blocks (nearly inactive dimeric form) or are degraded to pyruvate with regeneration of energy (highly active tetrameric form). In most tumors the dimeric form is predominant due to interaction with oncoproteins [13,14]. Accordingly, in many tumors a high metabolic flexibility exists which can alter metabolic fluxes following normoxic or hypoxic conditions [4]. In tumor cells, the proportion of glycolytic pyruvate infiltrated into the TCA cycle after decarboxylation to acetyl coenzyme A (AcCoA) is low. This is due to an inhibition of pyruvate dehydrogenase (PDH) by PDH kinase, as well as the expression of the lactate dehydrogenase (LDH) isoenzyme M4 which favors lactate production from pyruvate [15]. Because the major anaplerotic sources in fast tumor growing cells are pyruvate and Gln, pyruvate carboxylase (PC) and GA are key metabolic enzymes for the generation of ATP in cancer [16]. Hence, PC protein expression and activity are increased in human non-small-cell lung cancer (NSCLC) and ex vivo tissue slices, whereas silencing of PC attenuates NSCLC cell growth, reducing equivalents and energy (ATP and GTP), as well as leading to a lower biosynthesis of glutathione (GSH), lipids and nucleotides [17].
2.2. Glutamine as biosynthetic precursor
Glycolysis, OXPHOS, the pentose phosphate pathway (PPP), and Gln metabolism are interconnected in proliferating cells [12]. Feeding the TCA cycle through amino acids and fatty acid is essential for oncogene-induced tumorigenicity and the synthesis of nucleotides, proteins and lipids [18]. Gln, via Glu, is the most important source of amino groups for nonessential amino acids, such as alanine, aspartate, glycine, proline, and serine (Fig. 1). On the other hand, Gln-derived nitrogen is critical for N-glycosylation reactions, controlling nucleic acid synthesis [19].
Either way, glutaminolysis has some characteristics for being such efficient biosynthetic source: (i) Gln can be used for de novo protein synthesis, as well as other amino acids derived from Gln (Glu, proline, histidine, alanine, aspartic acid, and arginine); (ii) Gln can facilitate the uptake of other amino acids; (iii) it is critical for the biosynthesis of purine nucleosides; (iv) GLUD-mediated transformation of Glu to AKG produces NADH; and (v) Gln-derived AKG can take part in a reverse carboxylation process to render citrate for the generation of AcCoA, essential to form fatty acid and cholesterol [20].
2.3. Glutamine as redox modulator
GSH is the most important intracellular antioxidant, critical to fight against oxidative stress generated by rapid metabolism in cancer [21]. Gln is essential for maintaining GSH homeostasis not only because the tripeptide consists in Glu (which comes from Gln), cysteine and glycine, but because Glu is also necessary for the uptake of cistine, precursor of cysteine, which is the limiting molecule for GSH production [10].
Vitamin C can decrease the growth of colorectal cancers that express membrane Glc transporter (GLUT1) by diminishing the NADPH and GSH levels that, in turn, augment ROS to provoke apoptosis. In fact, oxidized form of vitamin C, dehydroascorbate, is imported into cells through GLUT1. When the cell takes up dehydroascorbate, it is reduced back to vitamin C by GSH, which becomes GSSG. Following, GSSG is converted back to GSH by NADPH. Both GSH and NADPH expense elicits oxidative stress and cancer cell death [4]. On the other hand, because oxidative stress occurs in tumors, ROS can inhibit the aconitase-catalyzed reaction of citrate to isocitrate, thereby reducing the provision of AKG from glycolysis, then favoring glutaminolysis [20] that eventually generates GSH to combat oxidative stress [5].
Gln, GSH and their related metabolic enzymes regulate cancer redox homeostasis [21]. Importantly, by screening the effect of GA and gamma-glutamylcysteine synthetase (GCS) inhibitors in a panel of 407 tumor cell lines it has been found a high degree of co-dependency on GA and de novo GSH synthesis, linking GA and redox status [22].
2.4. Glutamine as signaling molecule
Gln has multiple roles for both anabolic and regulatory processes, including signaling pathways [23]. Metabolic reprogramming is the result of mutations in oncogenes and tumor suppressors, eliciting the activation of phosphoinositide 3-kinase (PI3K), protein kinase B (PKB)/AKT, viral oncogeneV-Ki-ras2 Kirsten rat sarcoma (KRAS), and mammalian target of rapamycin(mTOR) signaling networks, as well as transcriptional pathways involving Myc, hypoxia-inducible factors (HIFs), PKM2, and steroid receptors bindingproteins [4,6,13]. Simultaneously, Myc regulates glycolysis, OXPHOS, glutaminolysis, and fatty acid metabolism to coordinate energy balance in cancer cells [20]. Very recently, it has been demonstrated that Myc is directly regulated by mTOR to actively modulate the link between glycolysis and glutaminolysis in glioblastoma (GBM) cells, regulating glucosamine-6-phosphate synthesis through glutamine:fructose-6-phosphate aminotransferase (GFAT) [24]. Transcriptional targets of Myc include enzymes required for glutaminolysis and biosynthesis [25], and increased Gln catabolism in Myc-induced liver tumors is associated with a switch of GA isoenzymes ratio towards those more related with enhanced proliferation (see Section 4) [26]. One of the consequences of Myc overexpression is the induction of apoptosis by shifting the balance between pro-survival (bcl-2) to pro-death (bid) signals [27].
KRAS stimulates cancer growth through increased generation of AKG via the TCA cycle and glutamate pyruvate transaminase [28]. Likewise, KRAS-mutant lung adenocarcinoma is dependent on increased glutaminolysis, stimulating the Nrf2 antioxidant program and cooperating with mutant KRAS to activate tumor progression [29]. Accordingly, induction of mitochondrial metabolismand ROS formation are pivotal for KRAS-induced cancer growth [18]. Gln levels, GA expression and KRAS mutation are essential to predict response to radiotherapy in NSCLC. The effect of GA inhibition on higher oxidative stress (lower concentrations in GSH and NADPH) and radiosensitivity has also been confirmed for cervical and pancreatic cancer cells [30]. Over 90% of patients with pancreatic ductal adenocarcinoma (PDAC) have KRAS mutations and show a critical dependency on Gln for survival and proliferation despite high levels of Glc [31].
Gln is a basic signal metabolite in the mTOR signaling pathway in some set of cancers [5]. Gln surplus is a signal to induce tumor proliferation and to inhibit catabolism [10]. Although the mechanism is not clear, it has been demonstrated that high levels of Glc or Gln promote mTOR protein activity [24]. On the other hand, the production of reducing equivalents for the generation of mitochondrial ROS by complex I, II, and III of the electron transport chain are signaling molecules to moderate the ERK1/2 MAPKpathway to compatible levels with cancer proliferation [18].
Since Gln influences cell signaling, its impact in gene expression has been described in several cancer models [4,5,10,11]: (i) addition of Gln triggers expression of oncogenic factors as JUN and Myc; (ii) Gln enhances cell survival through inhibition of CHOP, GADD45, Fas and ATF5; (iii) Gln's involvement in MnSOD expression was locked by lowering the TCA cycle, ERK1/2 or mTOR; (iv) silencing of GA elicited higher phosphorylation and transcriptional activity of Sp1 [21]; (v) pre-treatment of animals with Gln reduced tissue inflammation and expression of nuclear factor kappa-B (NF-κB), a mediator linking Gln availability to stress responses. Conversely, other reports found an inverse correlation between Gln availability and NF-κB-mediated gene expression [10].
Gln regulates immune responses through the modulation of redox homeostasis, bioenergetics, nitrogen balance and HIF [1,6,32]. HIF drives metabolic adaptation to hypoxic conditions in many solid tumors characterized by limited oxygen availability through upregulation of Glc transporters and glycolytic enzymes [33]. HIF also regulates human PDAC carcinogenesis and modulates non-canonical Gln metabolism by inducing of PI3K/mTOR pathway [34]. The role of HIF as a therapeutic target for patients with PDAC was confirmed in Singh’s laboratory [35]. They inhibited tumor growth by targeting HIF-1alpha with digoxina and de novo pyrimidine biosynthesis with leflunomide to block metabolic reshuffle evoked by gemcitabine resistence.
2.4.1. TME as major regulator of Gln metabolism in cancer
Under hypoxic stress, cells use Gln to generate citrate and support proliferation thorough lipids synthesis [36,37]. Therefore, hypoxia is an inducer of reductive metabolism in cancer [38,39]. Reductive carboxylation is increased during hypoxia through a HIF1-dependent mechanism that provides Gln carbon utilization in order to produce AcCoA and support fatty acid biosynthesis [36]. In fact, environmental hypoxia has a key effect on gene expression, mainly by stabilization of HIF1 as well as by degradation of one of the components of ketoglutarate dehydrogenase (KGDH) complex [37]. Besides, hypoxia causes a reduction in Glc-derived citrate because of diminished PDH activity (Fig. 1). Nevertheless, an increased reductive metabolism of Gln was also described in the absence of hypoxia during anchorage-independent growth of human lung cancer cells. Thus, when cells are detached from monolayer culture (2D) and grow as tumor spheroids (cell culture model in 3D) important changes in Gln metabolism occurs, including an increased reductive formation of citrate from Gln through cytosolic isocitrate dehydrogenase 1 (IDH1) [39]. In this study, isotope tracing showed that in spheroids, isocitrate/citrate produced reductively in the cytosol could enter the mitochondria to participate in oxidative metabolism, including oxidation by IDH2. Mitochondrial IDH2 is essential for the increase in reductive carboxylation flux of Gln-derived AKG to maintain citrate levels and proliferation in hypoxia (Fig. 1). Cancer-associated mutations in the active sites of either IDH1 or IDH2 facilitate the production of oncometabolite 2-hydroxyglutarate (2-HG) [38].
Moreover, a metabolic shift from a glycolytic metabolism toward the reductive Gln metabolism was observed in response to chronic acidic conditions in cancer [40]. Resistance to acidosis was associated by these authors with an increase in sirtuins, leading to HIF1 decrease and a parallel HIF2 enhancement. Simultaneously, Gln transporter SLC7A5 and glutaminase (GLS) were upregulated.
3. Transport of glutamine across the mitochondrial membrane
Gln has to cross the plasma and the mitochondrial membranes before its deamidation to Glu by mitochondrial GA can take place (Fig. 1). For the transport of Gln across the plasma membrane several Gln transporters have been characterized [41]. In contrast, the way of transporting Gln across the mitochondrial membrane has not yet been elucidated partly due to the fact that also the submitochondrial localization of GA is still a controversial issue. One hypothesis suggests that there must be a Gln transporter when GA occurs in the mitochondrial matrix in order to direct Gln to the active site of GA (Fig. 1). In contrast, if GA is localized in the mitochondrial intermembrane space, a Glu transporter is necessary [42]. Using isolated mitochondria evidences concerning the existence of a mitochondrial Gln uniport were first provided in 1970 [43]. In 1995 Molina et al. described the transport of Gln in vesicles isolated from tumor-cell mitochondrial inner membrane [44]. Later a Gln transporter was isolated from rat kidney mitochondria and purified [45]. Recently, for three members of the mitochondrial transporter SLC25 family putative Gln binding sites have been identified through in silico analysis [46]. Whether Gln uptake to mitochondria is linked with its degradation by GA is not clear yet, although biochemical experiments with inner mitochondrial membrane vesicles isolated from Ehrlich tumor cells do not support that the Gln carrier and GA may form part of the same protein (see [1] for an in-depth discussion). Nevertheless, in brain mitochondria the aminoacids histidine, homocysteine, leucine and a newly synthesized alanine analogue MRC01 induced an inhibition of Gln uptake, probably either by inhibition at the site of Gln entry or by heteroexchange stimulation, as well as decrease of GA activity. This data supports that GA is located in the inner face of the inner mitochondrial membrane, and part of it in the mitochondrial matrix [44,47]. On the other hand, factors such as calcium plus inorganic phosphate (Pi), which stimulate GA activity, inhibited Gln uptake, while taurine and N-acetyl-aspartic acid inhibited GA but did not affect Gln uptake [48].
4. Glutaminase isoenzymes
GA plays an essential role in the metabolism of amino acids by catalyzing the hydrolytic deamidation of Gln to stoichiometric amounts of Glu and ammonium ions. The enzyme is widely distributed in mammalian tissues and fulfils essential tasks related to tissue-specific function; for example, control of the acid-base balance in kidney, coupling of ammonia production with urea synthesis in liver and synthesis of neurotransmitter Glu in brain [43,49]. Mammalian GAs are encoded by two paralogous genes, Gls and Gls2, presumably derived by gene duplication of a common ancestor [5,50]. Both genes code for two different isoforms, which means that a total of four distinct GA proteins can be expressed in mammals, each of them with markedly different molecular, kinetic, protein interacting partners and regulatory properties. This complex pattern of isoenzyme expression endow cells and tissues with flexible and context-dependent mechanisms for regulation of glutaminolysis and Gln/Glu pools in response to different physiological circumstances, as well as in pathological states like cancer.
Obviously, being GA the first committed step of glutaminolysis and a key pathway for energy and nitrogen metabolism in many types of cancer cells, considerable effort in tumor biology has been devoted from long time ago to inhibit either Gln supply or GA isoenzymes as important therapeutic targets, in an attempt to block tumors’ uncontrolled proliferation, growth and metastatic capacity. Pioneer studies of tumor metabolism early noticed a high rate of Gln consumption by cancer cells normally exceeding their biosynthetic and energetic needs [51,52]. Previously, Mider labeled tumors as “nitrogen traps” indicating their ability to compete with advantage for host nitrogen compounds [53]. Since then, a considerable number of studies dealing with human and experimental tumors confirmed this anomalous Gln uptake as a common metabolic feature of many, but not all, cancer cells; in fact a new term, “Gln addiction”, is now widely used to reflect the strong dependence shown by most cancer cells for this essential nitrogen substrate after metabolic reprogramming [54]. In parallel, a correlation is usually found between GA expression and malignancy; however, as more findings are unveiled about the molecular portrait of GA expression in cancer, a completely different picture seems to emerge for the two main GA isoenzymes, GLS and GLS2, which apparently play opposing roles in cancer growth and proliferation.
4.1. GLS isoforms
With the renewed interest on Gln metabolism in cancer cells, a great deal of effort is being dedicated to understand the mechanisms that regulate GLS gene expression and activity. Fifty years ago, Linder-Horowitz and colleagues already observed a correlation between kidney-type GA activity, growth rate and dedifferentiation in rat hepatomas and other non-hepatic tumors [55]. Since then, numerous studies exposed the critical role played by GLS isoenzymes in growth and proliferation of many types of cancer and their potential as therapeutic targets [56]. A key discovery was the reversion of malignant phenotype and loss of tumorigenic capacity in vivo of Ehrlich ascites tumor cells after knocking down murine Gls with antisense technology [57]. Subsequent studies in which GLS expression was reduced by RNA interference confirmed the important function of this isoenzymes in cancer [58,59].
In humans, the GLS gene is located in chromosome 2 [50] and encodes isozymes termed KGA and GAC, originated by alternative splicing [60]. GAC shows higher catalytic activity than KGA in the presence of their activator Pi, indicating that the unique C-terminal region in GAC encoded by exon 15, that replaces the last 4 exons present in the KGA transcript, must be important for this greater efficiency, as N-terminal and GA domains are identical in both GLS isoforms [61]. In addition, GAC is the predominant GLS isoform in tumors [19,62,63] and its high levels predict a poor prognosis [64]. In fact, GAC was cloned from a human colon carcinoma cell line and was the prevailing GLS isoform in a breast cancer cell line that exhibited high GA activity [60]. The oncogenic transcription factor c-Myc, involved in the regulation of Glc metabolism, is also implicated in Gln metabolism in Myc-transformed cells (Fig. 1). Thus, by downregulation of miRNAs miR-23a and miR-23b, which target GLS 3´untranslated region (UTR), c-Myc indirectly relieves repression of GLS [58]. The mTORC1 signaling pathway positively regulates GLS by enhancing Myc translation efficiency [65]. Like c-Myc, NF-κB p65 subunit suppresses miR-23a expression, resulting in higher levels of GLS [66]. Other miRNAs that are downregulated in several types of cancer also directly target GLS expression, such as miR-153 [67], miR-1-3p [68] and miR-137, the latter being suppressed by heat shock factor 1 (HSF1) [69]. Although these data indicate that miRNAs may have an important role in the regulation of GLS expression, the GAC isoform lacks any miRNA-binding site within its 3´ UTR [70]. A mechanism that regulate the alternative splicing of GLS has been identified in colorectal cancer cells. The G allele of the long non-coding RNA(lncRNA) colon cancer-associated transcript 2 (CCAT2), associated with greater risk of colorectal neoplasia than the T allele, interacts with cleavage factor I (CFIm) complex, inducing the preferential expression of GAC by selecting the poly(A) site in intron 14 of the precursor GLS mRNA [71]. Another oncogenic transcription factor, c-Jun, when activated downstream of Rho GTPasesignaling, enhances GLS expression by directly binding to its promoter region [72].
Besides regulation of GLS at transcriptional and post-transcriptional level, several post-translational modifications also affect GLS activity. Epidermal growth factor (EGF)-mediated RAF/MEK/ERK signaling activates GLS and this activation is phosphorylation-dependent: the inhibition of kinases or co-expression of phosphatase PP2A abolish the enhanced GLS activity [73]. Signaling by Rho GTPases also elevates GLS activity via NF-κB by promoting its phosphorylation [59]. Recently, the phosphorylated residue (Ser314) responsible for the enhanced activity of GAC has been identified: NF-kB regulates GAC through phosphorylation mediated by one of the ten protein kinase C (PKC) isoforms, PKCε [74]. Thus, cancer cells may use a plethora of regulatory mechanisms to induce GLS isoforms and activate the glutaminolytic pathway, essential for growth and proliferation.
4.2. GLS2 isoforms
The mammalian Gls2 gene codes for two different GA transcripts [75], named LGA and GAB. The canonical long transcript GAB is composed by the full 18 exons of the Gls2 gene and was first isolated from human breast cancer cells [80]. The short transcript LGA was originally cloned from rat liver [79] and lacks exon 1; it arises by alternative transcription initiation and possesses an alternative promoter located on intron 1 of the Gls2 gene [75].
In sharp contrast with GLS isoforms, the role of GLS2 in tumor cells is far from being well understood. We have summarized the history of the major findings for GLS2 in cell growth, survival, differentiation and proliferation in Fig. 2. Many evidences suggest that it behaves as a tumor suppressor gene in certain types of malignancies such as glioblastoma, liver, and colon cancers (Fig. 2), where GLS2 expression is repressed as a result of DNA methylation [90,95] and GLS become the predominant GA isoforms [84,99]. Hence, repression of GLS2 might have a causative role in the malignization process as part of the transcriptional program of transformation. Analysis of GA expression profiles in human leukemia, breast cancer cells and rat hepatoma showed a pervasive pattern associated to the malignant transformation: upregulation of GLS isoenzymes and simultaneous silencing of GLS2 isoforms [6,83]. Thus, we postulated that GA enzymes showed opposing roles in cancer: GLS expression correlated with increased rates of proliferation while predominance of GLS2 was related to differentiated and quiescent cell states [83].
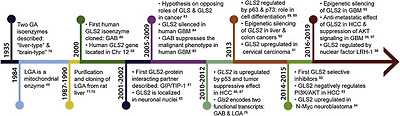
Fig. 2. Timeline of milestones in the history of glutaminase GLS2 isoenzymes to elucidate their roles in cell growth, survival, differentiation and proliferation. Relevant findings supporting a segregation of functional roles between GLS and GLS2 isoforms in tumor biology are highlighted. Other main scientific achievements regarding GLS2 roles in mammalian tissues, particularly in liver, were omitted for the purpose of this review. GAB, long glutaminase 2 isoform; GBM, glioblastoma; GLS, glutaminase isoenzyme; GLS2, glutaminase 2 isoenzyme; HCC, hepatocellular carcinoma; LGA, short glutaminase 2 isoform; PI3K, phosphatidylinositide 3-kinase; TSS, transcription start site [[76], [77], [78],81,82,92,98].
In agreement with this hypothesis, constitutive overexpression of GLS2 in human glioblastoma cells of different tumorigenic potentials and genetic backgrounds suppressed the malignant phenotype by inducing strong inhibitions in cell migration and proliferation [85,97]. Furthermore, GLS2 was confirmed as a target gene of the tumor suppressor p53, in such a way that p53-controlled enhanced GLS2 expression was linked to a tumor-suppressive response, including reduced growth and colony formation of tumor cells [86,87]. Later on, direct regulation of GLS2 was demonstrated by other transcription factors belonging to the p53 family: p63 and p73 [88,89]. Interestingly, these authors described a potential role of GLS2 in the cellular differentiation of human keratinocytes and neuroblastoma cells, respectively. The antiproliferative effect elicited by GLS2 may involve deep transcriptomealterations shifting the oncogene/tumor suppressor gene expression balance [85], but may also induce changes in signaling pathways controlling proliferation. In this regard, recent studies are now shedding light on potential antiproliferative mechanisms elicited by GLS2. Thus, GLS2 showed tumor suppression activity in HCC and negatively regulated the PI3K/AKT signaling, frequently activated in HCC; hence, authors concluded that GLS2-induced PI3K/AKT impairment greatly contributed to the function of GLS2 in tumor suppression [93]. Of note, a similar result was found in human glioblastoma cell lines of different tumorigenic potentials and genetic backgrounds [97]. Other GLS2-induced effects favoring a more-differentiated and less malignant phenotype in human GBM cells with ectopic GLS2 expression were an increased sensitivity to oxidative stress [33] and enhanced susceptibility towards an alkylating agent temozolomide (TMZ) often used in GBM therapy [97,100]. Finally, another study implicated GLS2 as a powerful anti-metastatic factor in HCC by a mechanism involving miRNA regulation and repression of the transcriptional repressor of E-Cadherin SNAIL [96].
Even though repression of GLS2 is a frequent trait observed in many tumors, this behavior is not universal and also overexpression of GLS2 isoforms has been reported in a few types of human cancers. For example, the expression of GLS2 was significantly enhanced in cervical carcinoma; even more, this upregulation was related to therapeutic resistance [91]. Also, a robust GLS2 expression was demonstrated in human neuroblastoma with enhanced expression of N-Myc (in sharp contrasts with c-Myc neuroblastomas); in these tumor cells, the increase of GLS2 was associated with an enhancement of aerobic glycolysis and glutaminolysis in order to maintain cell survival and high rates of proliferation [94]. Nevertheless, despite these few evidences, a strikingly different expression profile is becoming evident for GLS and GLS2 in many malignancies usually showing a pervasive pattern: upregulation of GLS isoforms and silencing of GLS2 ones [101]. Furthermore, anti-tumor strategies based on GLS2 upregulation have demonstrated therapeutic efficacy at least in human cancer cell lines [27,85,97]. In summary, these results strongly suggest that GLS2 upregulation has tumor suppression activity and could help to rewire cellular metabolism toward a normal non-proliferative phenotype, providing a new strategy to combat some types of cancer where GLS2 is frequently silenced (Fig. 2).
5. Combination therapies to target mitochondrial cancer metabolism
Targeting mitochondrial metabolism has gained importance in oncology in last years [6]. However, a hallmark of cancer cells is their plasticity, which triggers changes in the regulatory pathways compromising the effectiveness of targeted therapies and rendering cancer cells to be dependent on compensatory metabolic circuits [102]. Most successful strategies consist of chemotherapy in combination with antiglycolytics, antiglutaminolytics and inhibitors of the Krebs cycle [6]. Drugs targeting metabolic enzymes gain effect by synergising with other targeted therapies to selectively kill cancer cells [35,103,104], and to enhance the therapeutic outcome or, at least, sensitize tumor cells to emerging therapies [3].
Active-site Gln analogs (6-diazo-5-oxy-l-norleucine, azaserine and acivicin) inhibit GA and show antitumor activity but with severe neuro- and gastrotoxicity; hence, less damaging inhibitors targeting the oligomerizationprocess required for GLS activation have been reported (Fig. 3). GLS inhibitors include allosteric inhibitors, i.e: compound 968 (5-(3-Bromo-4-(dimethylamino)phenyl)-2,2-dimethyl-2,3,5,6-tetrahydrobenzo[a]phenanthridin-4(1 H)-one), BPTES (bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl)ethyl sulfide), and molecule CB-839 (2-(pyridin-2-yl)-N-(5-(4-(6-(2-(3-(trifluoromethoxy)phenyl)acetamido)pyridazin-3-yl)butyl)-1,3,4-thiadiazol-2-yl)acetamide), which is more stable and soluble and has been used in clinical trials [63].
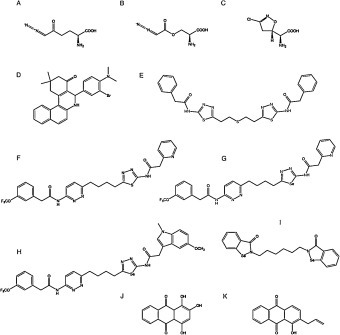
Fig. 3. Main glutaminase-related pharmacological inhibitors to block glutaminolysis. (A) 6-diazo-5-oxy-L-norleucine, (B) azaserine, (C) acivicin, (D) compound 968, (E) BPTES, (F) CB-839, (G) CPD-20, (H) CPD-23, (I) CPD-3B, (J) purpurin, (K) R-162.
More recently, new specific GLS inhibitors were described, including CB-839 selenadiazole-derivatives CPD-20 and CPD-23 [105]. These compounds showed better cellular and tumor accumulation, increased GLS inhibition, higher induction of ROS, and improved effect eliminating cancer cells [106]. For Gln addicted cancers, a dual specific glutaminolysis inhibitor has been designed, named Hexylselen (compound CPD-3B), that efficiently disrupted the mitochondrial membrane potential, inducing apoptosis, without inhibition of the normal cell growth and minimal toxicity [107]. Hexylselen is a KGA/GLUD inhibitor which showed no toxicity to normal cells up to a 10 μM concentration and could completely inhibit the growth of many aggressive cancer cell lines. This compound targets not only KGA and GLUD but also thioredoxin reductase (TrxR) and amidotransferase (GatCAB), which results in corresponding regulation of Akt/Erk/caspase-9 signaling pathways [108].
Because a single strategy is not enough to achieve strong anticancer results, dual or combination therapy are gaining interest in metabolic therapy [6]. Both knockdown of GLS expression and pharmacologic GLS inhibition by the compound CB-839 reduces OXPHOS and has synergistic effect with the Bcl-2inhibitor ABT-199, showing high antileukemic activity in acute myeloid leukemia [19]. Similarly, inhibition of GLS using BPTES impaired leukemic cell growth and sensitized T-cell acute lymphoblastic leukemia cells to NOTCH1 inhibition therapies [103]. Efficacy of nanoparticles encapsulated BPTES was similar to CB-839 but without undesirable side effects in liver enzyme levels. Furthermore, combination therapy using metformin produced significantly greater tumor growth inhibition compared with monotherapy with either encapsulated BPTES or metformin [31]. These authors state that this treatment can be applicable to other Gln addicted tumors, including those with deficiency in fumarate hydratase (FH) or succinate dehydrogenase (SDH), as well as the ones bearing mutant IDH1/2.
Squamous cell carcinomas display both a high glycolytic profile and an increased glutaminolytic pattern (higher amounts of Glu and GSH). To combat this kind of cancer combination therapy consisted in lonidamine (1-(2,4-dichlorobenzyl)-1H-indazole-3-carboxylic acid) and compound 968 [30]. Lonidamine inhibits glycolysis through interference with hexokinase II, inducing lactate accumulation, inhibiting the formation of fumarate and malate, and disturbing the mitochondrial membrane potential (inducing ROS through SDH inhibition, and reducing NADPH and GSH by inhibition the PPP) [109]. Of note, only NSCLC patients harboring KRAS mutation benefit from the combination of radiation therapy plus metabolic therapy (glycolysis and glutaminolysis inhibition) [30]. On the other hand, rapid tumor regression in vivo in mouse NSCLC xenografts was obtained with a dual therapy using CB-839 in combination with erlotinib to simultaneously impair cancer Gln and Glc utilization, compromising redox homeostasis and inducing autophagy to defeat cancer [110]. Similarly, CB-839 displayed significant antitumor activity in two xenograft models of triple-negative breast cancer (TNBC) cell lines, both as a single agent and in combination with paclitaxel [63].
Other investigation target GLUD using purpurin and its cell permeable derivative R162 [104]. Purpurin directly binds and specifically inhibits GLUD activity in vitro, showing no effect on the activity of other NADPH-dependent enzymes such as FH [3]. Interestingly, 2-deoxyglucose inhibits glycolytic and glutaminolytic metabolisms in GBM cells, accompanied by reduced protein activities of mTOR and Myc [24]. Future studies might consider a plural inhibition using also GFAT and/or GLS specific inhibitors. Moreover, V-9302, a potent inhibitor of Gln transport, selectively targets the amino acid transporterASCT2 (SLC1A5), reducing cancer cell growth and proliferation in vitro and in vivo [111]. V-9302 increased apoptosis and autophagy, and enhanced ROS, through a GSH-dependent mechanism. This antagonist of Gln transmembrane flux is now in preclinical development [111].
6. Applied metabolomics and fluxomics for cancer tracking
Glutamine-related metabolomics methods and isotope tracing, using long-term steady-state labeling, have been used for characterizing anaplerotic fluxes in tumor cells and to establish new concepts in cancer metabolism, or to identify new targets for diagnostic imaging and therapy [6]. Using gas chromatography (GC)-mass spectrometry (MS) and liquid chromatography (LC) tandem MS, 338 metabolites were analyzed in vivo to find Glu, cysteine, AKG, citrate, adenine dinucleotide cofactors, GSH, oxidized GSH, polyunsaturated fatty acids, and ROS as biomarkers for a GLS knockdown xenograft model of breast cancer [22]. In previous studies, after pharmacological inhibition of GLS by molecule CB-839, the metabolites Gln, Glu, GSH, citrate, malate and aspartate were designed as predictive biomarkers in xenograft models of TNBC cancer [63]. In human GBMs orthotopically-transplanted in the mouse brain in vivo, Gln, Glu, aspartate and gamma-aminobutyric acid (GABA) were validated as biomarkers of aggressive GBM, showing how these gliomas with diffuse infiltration, and good access to oxygen and nutrients, can produce Gln from Glc [16].
Metabolomic analysis in vivo is a key tool to integrate the influence of both driver oncogenes and TME on Gln metabolism in human cancer patients and mouse models [112,113]. To optimize reliable tumor patterns it is desirable to design ex vivo models of cancer that better mirror the in vivo TME. Emerging technologies include MS imaging, which will allow multiplex analysis of hundreds to thousands of molecules in the very same tissue section simultaneously [114]. Of note, all tissue parts can be examined at once but separately. Matrix-assisted laser desorption ionization (MALDI) and desorption electrospray ionisation (DESI) MS allow the spatial identification of metabolites [33]. In addition, new computational tools allow to measure intercellular metabolite allocation between different cell types using stable isotope tracing. Using 13C metabolic flux analysis (13C-MFA) of co-culturesystems, no physical separation of cells is needed to quantify metabolic exchanges because total biomass labeling is used for flux elucidation, estimating relative population sizes, and improving accuracy in co-cultures where cells have similar metabolic characteristics [115]. Metabolomics approaches find changes in TCA cycle intermediates in cancer by studying the reductive carboxylation network of tumor cells showing low levels of pyruvate capture into mitochondria. Additionally, isotope tracing assays using 13C-Gln as the tracer, are the best way to characterize adjustments in TCA cycle [16]. For example, after GLS inhibition in human breast cancer cell xenographs, a decreased incorporation of Gln into TCA cycle metabolites, as well as into GSH, was demonstrated in vitro using 13C-mass isotopomer distribution analysis (MIDA) [22].
Metastasis, the main cause of death in patients with cancer, is favored when tumor cells show high glycolysis, OXPHOS, and β-oxidation of fatty acids, together with inhibition of apoptosis. Fluxomics experiments are needed to validate how these pathways lead to a metastatic phenotype of cancer cells [116]. Higher uptake of Glc in tumors and metastasis have been fully characterized by positron emission tomography (PET) imaging [117]. The most frequently used PET tracer in oncology is 2-[18F]fluoro-2-deoxy-d-glucose (18FDG) [28]. Nevertheless, [18F]-(2S,4R)-4-fluoroglutamine (18FGln), a Gln analog for radiologic imaging, has been used in PET trials to provide clinical validation of abnormal Gln metabolism in different cancer types, including breast, pancreas, renal, neuroendocrine, lung, colon, lymphoma, bile duct, glioma or neuroblastoma, without adverse effects and without a requirement for patient fasting (unlike 18FDG PET) [118,119]. PET imaging with 18FDG and l-[5-11C]-glutamine (11C-Gln) tracers has also been used to measure metabolic response to dual therapy against glycolysis and glutaminolysis in NSCLC xenografts mice [109]. Additionally, patients suffering from early-stage NSCLC were infused with 13C6-Glc or 13C5,15N2-Gln tracers to study the activation of PC and GLS. Characterization was performed by GC–MS, nuclear magnetic resonance (NMR), and Fourier-Transform Ion Cyclotron Resonance MS (FT-ICR-MS) [17]. NSCLC is heterogeneous in both the genetic and tissue perfusionwhich influence cell metabolism [35]. Combining clinical imaging and intra-operative 13C-Glc infusion allowed the in vivo identification of local alterations of metabolism [112]. Metabolic heterogeneity in NSCLC included different degrees of Glc, lactate and other fuels consumption (highly TME-dependet) predicted by magnetic resonance imaging (MRI) marker of perfusion. Surprisingly, Gln carbon contributes minimally to the TCA cycle in KRASdriven lung tumors and adjacent lung [113]. These results point out the importance of considering TME together with genetics to best target cancer metabolism.
Very recently, an ex vivo and in vivo targeted tandem LC–MS/MS fluxomics method has been developed to trace the metabolism of 13C-Glc, 13C-Gln, and 15N-Gln, using a hybrid triple quadrupole mass spectrometer [120]. A novel, non-invasive and in real-time hyperpolarized MRI method has demonstrated that Gln is a carbon-source for the generation of the oncometabolite 2-HG in vivo, utilizing patient-derived chondrosarcoma cells harboring endogenous mutations in IDH1/2 [121]. Of note, 7 T magnetic resonance spectroscopic imaging (7 T-MRSI) revealed millimetric high-resolution multi-metabolite (Gln, Glu, choline, N-acetyl-aspartate, inositol, and creatine) mapping of gliomas uncovering the metabolic activity of grade II, III and IV gliomas, and finding an outstanding increase of Gln in all ten patients [122].
7. Conclusions and perspectives
Substrates used to generate ATP or to synthesize cell building blocks can differ between cancer and healthy cells [116]. Gln is a key molecule involved in all metabolic circuits to be targeted for cancer treatment: glycolysis, TCA, OXPHOS, glutaminolysis, fatty acid oxidation, nucleic acid synthesis, lipid synthesis, and amino acid metabolism [28].
The design of synergic strategies through combination therapy is appearing as a strong tool to struggle one of the most dangerous cancer weapons: metabolic heterogeneity [30,31]. Although glycolysis and glutaminolysis are common processes in cancer, many studies characterizing tumor metabolism have found multiple metabolic strategies for therapy, consistent with metabolic heterogeneity in different types and subtypes of cancers [6]. While several types of brain and lung cancers increase oxidation of Glc-derived carbons feeding TCA cycle, renal cell carcinomas are distinguished by aerobic glycolysis. GBM and NSCLC may synthesize Gln from Glc-derived carbons, other brain and liver tumors use acetate to fuel TCA, and NSCLCs utilize circulating branched chain amino acids as biosynthetic precursors [23]. On the other hand, inhibition of GCS and GLS isoenzymes in more than 400 tumor cell lines demonstrate a strong correlation between tumor sensitivity and downregulation of both enzyme targets [22]. Of relevance, this study also identify the metabolic state associated with GLS-dependence for anaplerotic and redox purposes as the mesenchymal subtype. This signature was validated in vivo using four lung patient-derived xenograft models, which exhibited reduced capacity for OXPHOS and increased susceptibility to oxidative stress. In this model, Glu, citrate and GSH were characterized as key metabolic biomarkers [22]. Consequently, nutrient availability, mutations in oncogenesand tumors suppressor factors, as well as tissue of origin can determine metabolic preferences and the better therapeutic strategy [26].
Other challenge in Gln-dependent metabolic cancer therapy is chemoresistance. More studies are required to confirm whether delivery of mitochondrial nucleic acids and proteins through cancer-derived exosomes may attenuate tumor growth and modulate cancer chemoresistance [3]. Since pharmacological evidence suggests that the TME modifies neoplasm metabolism, another important issue for experimental characterization is to evaluate the better cancer model system to be used, as it can affect metabolic phenotypes, i.e.: when cancer cells are implanted in mice, higher contribution of Glc-derived carbon and a reduced input of Gln carbon to the TCA cycle are observed [25]. Moreover, it is necessary to underpin how interactions with nutrients can affect sensitivity to drugs in cancer therapy [123]. In addition, symbiotic relationship may exist within tumors that might alter metabolite levels [25]. Otherwise, tumors can secrete metabolites (i.e. lactate) to create a hostile metabolic environment for immune cells [33].
Personalized oncology is an additional challenge to optimize metabolic sensitivities and therapeutic requirements [25]. Future research is needed to better understand whether patient’s metabolic patterns are causes or consequences of individual cellular programs, and should give important opportunities for anticancer metabolic therapies [23]. Among them, targeting glutaminolysis stays as a promising chance to defeat cancer [124].
Acknowledgements
This work was supported by Ministerio de Ciencia y Tecnología of Spain, SAF 2015-64501-R (to JMM, JACS and JM), and by the European Union's Horizon 2020 research and innovation programme under grant agreement No 722605(to FJDP and SM).
References
- [1]
- J.M. Matés, et al.Glutamine homeostasis and mitochondrial dynamicsInt. J. Biochem. Cell Biol., 41 (2009), pp. 2051-2061
- [2]
- L. Yang, S. Venneti, D. NagrathGlutaminolysis: a hallmark of cancer metabolismAnnu. Rev. Biomed. Eng., 19 (2017), pp. 163-194
- [3]
- F. Guerra, A.A. Arbini, L. MoroMitochondria and cancer chemoresistanceBiochim. Biophys. Acta Bioenerg., 1858 (2017), pp. 686-699
- [4]
- R.J. DeBerardinis, N.S. ChandelFundamentals of cancer metabolismSci. Adv., 2 (2016), Article e1600200
- [5]
- J.M. Matés, J.A. Campos-Sandoval, J. MárquezGlutaminase isoenzymes in the metabolic therapy of cancerBiochim. Biophys. Acta Rev. Cancer, 1870 (2018), pp. 158-164
- [6]