


| 크로마토그래피는 혼합물을 분리하는 기술. 고정상과 이동상을 사용하여, 혼합물 속 성분들이 이 두 상에 끌리는 정도의 차이를 이용. 고정상은 움직이지 않는 물질이고, 이동상은 고정상을 따라 흐르는 액체나 기체. 액체 크로마토그래피는 이동상으로 액체를 사용하는 방법. 고성능 액체 크로마토그래피(HPLC)는 이 액체 크로마토그래피의 한 종류로, 고압 펌프를 사용하여 이동상을 빠르게 흘려보내 분리 효율을 높인답니다. 크로마토그래피는 생화학, 화학, 제약 등 다양한 분야에서 사용되며, 혼합물 속 특정 성분을 분석하거나 정제하는 데 유용 |
- nature
- scientific reports
- articles
- article
Application of the HPLC-ELSD technique for the determination of major metabolites of ibuprofen and creatinine in human urine
인간 소변 내 이부프로펜 및 크레아티닌의 주요 대사산물 정량 분석을 위한 HPLC-ELSD 기법의 적용
- Article
- Open access
- Published: 20 November 2023
Application of the HPLC-ELSD technique for the determination of major metabolites of ibuprofen and creatinine in human urine
- Justyna Piechocka,
- Natalia Matwiej,
- Marta Gaweł,
- Michał Matyjaszczyk,
- Rafał Głowacki &
- Grażyna Chwatko
Scientific Reports volume 13, Article number: 20268 (2023) Cite this article
Abstract
The report presents robust and high throughput methods, based on liquid chromatography coupled with evaporative light scattering detection (HPLC-ELSD), for the simultaneous determination of major metabolites of ibuprofen (IBU), namely 2-hydroxyibuprofen and carboxyibuprofen (method A) as well as creatinine (Crn) (method B) in human urine. The assays primarily involve straightforward sample purification. For both methods, the chromatographic separation of the analytes is achieved within 8 min at room temperature on Poroshell 120 SB-C18 (75 × 4.6 mm, 2.7 µm) column using gradient elution. The eluents consisted of 0.1% formic acid in water and acetonitrile (method A) or water and methanol (method B) delivered at a flow rate of 1 or 0.5 mL/min, respectively. In relation to metabolites of IBU, the assay linearity was observed within 0.06–0.5 g/L in urine, while the Crn assay linearity was demonstrated within 0.5–30 mmol/L in urine. The limit of quantification for IBU metabolites was determined to be 0.06 g/L, and 0.5 mmol/L for Crn. These methods were successfully applied to urine samples delivered by ten apparently healthy donors showing that the HPLC-ELSD assays are suitable for human urine screening.
초록
본 보고서는
액체 크로마토그래피와 증발광산란검출기(HPLC-ELSD)를 결합한
견고하고 고속 처리 방식의 방법을 제시하여,
인간 소변 내 이부프로펜(IBU)의 주요 대사산물인
2-하이드록시이부프로펜 및 카르복시이부프로펜(방법 A)과
크레아티닌(Crn)(방법 B)을 동시에 정량 분석한다.
liquid chromatography coupled with
evaporative light scattering detection
분석법은
주로 간단한 시료 정제 과정을 포함한다.
두 방법 모두에서 분석물의 크로마토그래픽 분리는
Poroshell 120 SB-C18 (75×4.6 mm, 2.7 µm) 컬럼을 사용하여
실온에서 8분 이내에 그라디언트 용출로 달성된다.
용출액은
물과 포름산 0.1% 및 아세토니트릴(방법 A) 또는
물과 메탄올 (방법 B)로 구성되었으며,
각각 1 mL/min 또는 0.5 mL/min의 유속으로 공급되었다.
IBU 대사물의 경우,
소변 내 0.06–0.5 g/L 범위에서 분석법의 선형성이 관찰되었으며,
Crn 분석법의 선형성은 소변 내 0.5–30 mmol/L 범위에서 입증되었다.
IBU 대사물의 정량 한계는
0.06 g/L, Crn의 경우 0.5 mmol/L로 결정되었다.
이 방법들은
겉보기 건강한 10명의 기증자로부터 제공된 소변 시료에 성공적으로 적용되어
HPLC-ELSD 분석법이 인간 소변 스크리닝에 적합함을 보여주었다.
Similar content being viewed by others
Article Open access17 October 2023
Article Open access01 March 2025

Article Open access30 April 2024
Introduction
Ibuprofen (IBU) is one of the most commonly used non-steroidal anti-inflammatory, analgesic, and antipyretic drug in human and veterinary medicine. In humans, the drug is rapidly absorbed after ingestion and is extensively metabolized to inactive compounds, namely 1-hydroxyibuprofen, 2-hydroxyibuprofen (2-HIBU), 3-hydroxyibuprofen, carboxyibuprofen (CIBU) and occurring in a trace amounts their glucuronic acid conjugates1,2. Pharmacokinetic studies have shown that the majority of the given dose of IBU is metabolized and eliminated within 24 h in urine. Only a trace amount of the drug is excreted in urine in the unchanged form1,2. Unfortunately, IBU is sometimes misused, most probably as it is an over-the-counter drug. Since adverse effects of IBU are widely known1,2,3,4, facile methods for the determination of the IBU in the body and more importantly its metabolites in urine are necessary. These tools could help in the case of diagnostic exclusion of acute overdose or chronic drug abuse. In parallel, it is also essential to control creatinine (Crn) level in study samples, a compound that is inherently present in all urine samples and widely accepted as a reference for sample normalization in diagnostic testing5,6.
서론
이부프로펜(IBU)은
인간 및 수의학에서 가장 흔히 사용되는
비스테로이드성 항염증제, 진통제 및 해열제 중 하나이다.
인간에서 이 약물은 섭취 후 빠르게 흡수되며,
비활성 화합물인
1-하이드록시이부프로펜,
2-하이드록시이부프로펜(2-HIBU),
3-하이드록시이부프로펜,
카르복시이부프로펜(CIBU) 및 미량으로 존재하는 글루쿠론산 접합체로 광범위하게 대사된다1,2.
1-hydroxyibuprofen,
2-hydroxyibuprofen (2-HIBU),
3-hydroxyibuprofen,
carboxyibuprofen (CIBU) and occurring in a trace amounts their glucuronic acid conjugates
약동학 연구에 따르면
투여된 IBU의 대부분은
24시간 이내에 대사되어 소변으로 배설된다.
미량의 약물만이 변화되지 않은 형태로
안타깝게도 IBU는
처방전 없이 구입 가능한 일반의약품이라는
특성상 오용되는 경우가 종종 있습니다.
IBU의 부작용이 널리 알려져 있음에도 불구하고1,2,3,4,
체내 IBU 및 특히 소변 내 대사산물 검출을 위한
간편한 방법이 필요합니다.
이러한 도구는
급성 과다 복용이나 만성 약물 남용을 진단적으로 배제하는 데
도움이 될 수 있습니다.
동시에,
연구 시료에서 크레아티닌(Crn) 수치를 제어하는 것도 필수적입니다.
크레아티닌은
모든 소변 시료에 본질적으로 존재하는 화합물이며,
진단 검사에서 시료 표준화의 기준으로 널리 인정받고 있습니다5,6.
Numerous methods for determining the above-mentioned compounds have been developed so far, and many review articles on Crn7,8,9 and IBU10,11,12,13 are available. In general, these assays are predominantly based on separation techniques which are used for the determination of such compounds in complex systems. These methods have high-throughput potential, sensitivity, specificity, and excellent resolution, along with high degrees of reproducibility and accuracy. Interestingly, many assays for the quantification of IBU have been elaborated on10,11,12,13, whereas only a few methods allow simultaneous determination of IBU and its metabolites in urine14,15,16,17,18,19,20,21,22,23,24,25.
Among them, methods based on
liquid chromatography (HPLC)16,17,19,21,23,24,
gas chromatography (GC)14,15,18 and
capillary electrophoresis (CE)20,22,25
coupled with ultraviolet (UV)16,17,20,22,23,25,
fluorescence24,
nuclear magnetic resonance21 and
mass spectrometry (MS)14,15,18,19 detection have been proved
to be suitable for fulfilling the purpose.
Interestingly, none of these methods allow for the determination of IBU and its metabolites as well as Crn using HPLC coupled with evaporative light scattering detection (ELSD). ELSD is a low-cost and general-purpose universal detector that can identify semi- and non-volatile analytes over a wide dynamic range with uniform sensitivity regardless of their spectroscopic properties26,27,28,29. In addition, the existing approaches14,15,16,17,18,19,20,21,22,23,24,25 are challenging because they involve relatively labor-intensive, time/energy-consuming, and sophisticated sample processing procedures. In fact, the sample workup is a multistep process consisting primarily of enzymatic or alkaline hydrolysis14,17,19,20,21,22,23,24,25, extensive sample purification by multiple extraction procedures, such as liquid–liquid (LLE)14,15,17,18,24, solid phase (SPE)15,16,20,21,22,23,25, ultrasound-assisted emulsification (USAEME)19 (micro)extraction and chemical derivatization14,15,18,24. The use of these multiple steps can result in an increase in the amount of consumed plastic disposable materials and chemicals as well as considerable risk of losing analyte and precision diminution, among others. Overall, there is undoubtedly a need for streamlining the procedures and providing more effective analytical tools.
지금까지
상기 화합물들을 측정하기 위한 수많은 방법이 개발되었으며,
크레아티닌7,8,9 및 IBU10,11,12,13에 관한
다수의 리뷰 논문들이 존재한다.
일반적으로 이러한 분석법은
주로 복잡한 시스템에서 해당 화합물 측정에 사용되는 분리 기법에 기반합니다.
이러한 방법들은
높은 처리량 잠재력, 민감도, 특이도, 우수한 분해능과 함께
높은 수준의 재현성과 정확도를 갖추고 있습니다.
흥미롭게도,
IBU 정량화를 위한 많은 분석법이 개발되었으나10,11,12,13,
소변 내 IBU와 그 대사물을 동시에 정량할 수 있는 방법은
극소수에 불과하다14,15,16,17,18,19,20,21,22,23,24,25.
그중
액체 크로마토그래피(HPLC)16,17,19,21,23,24,
가스 크로마토그래피(GC)14,15,18 및
모세관 전기영동(CE)20,22,25에 자외선 (UV)16,17,20,22,23,25,
형광24, 핵자기공명21 및 질량 분석법(MS)14,15,18,19 검출을 결합한 방법이
목적 달성에 적합한 것으로 입증되었습니다.
흥미롭게도,
이러한 방법 중 어느 것도 증발광산란검출(ELSD)을 결합한 HPLC를 사용하여
IBU 및 그 대사산물과 Crn을 동시에 정량할 수 없습니다.
ELSD는
저비용의 범용 검출기로,
광범위한 동적 범위에서 반휘발성 및 비휘발성 분석물을
분광학적 특성과 무관하게 균일한 감도로 식별할 수 있다26,27,28,29.
또한 기존 접근법들14,15,16,17,18, 19,20,21,22,23,24,25는
상대적으로 노동 집약적이고
시간/에너지 소모적이며 정교한 시료 처리 절차를 수반하기 때문에 도전적이다.
실제로 시료 전처리 과정은
주로 효소적 또는 알칼리 가수분해14,17,19,20,21,22,23,24,25,
다중 추출 절차(액-액 추출(LLE)14,15,17,18,24,
고상 추출(SPE)15,16,20,21,22,23,25,
초음파 보조 유화(USAEME)19)를 통한 광범위한 시료 정제,
(미세)추출 및 화학적 유도체화14,15,18,24)로 구성됩니다.
이러한 다단계 사용은 소모되는
플라스틱 일회용 재료 및 화학 물질의 양 증가와
더불어 분석물 손실 및 정밀도 저하 등의 상당한 위험을 초래할 수 있습니다.
전반적으로 절차 간소화와 보다
효과적인 분석 도구 제공의 필요성은 의심의 여지가 없습니다.
As a result, the present paper aims to provide evidence that the practical application of HPLC-ELSD technique can be extended to IBU, 2-HIBU, CIBU, and Crn identification. The method enables the simultaneous determination of IBU and its main metabolites, namely 2-HIBU and CIBU (method A) as well as Crn (method B) in human urine. An additional objective of research work was related to greening analytical methodologies and making everything as simple as possible. Urine was selected as the matrix of choice, as it is easily accessible and can be obtained in a non-intrusive and non-invasive way; and because it has been demonstrated that in humans, the IBU and products of its biotransformation are mainly excreted in urine1,2. Important milestones included the development of an effective analytical tools based on HPLC-ELSD and application to real samples in order to demonstrate the methods performance.
따라서
본 논문은 HPLC-ELSD 기법의 실용적 적용이
IBU, 2-HIBU, CIBU 및 Crn 식별으로 확장될 수 있음을 입증하는 것을 목표로 한다.
이 방법은 IBU와 그 주요 대사산물인
2-HIBU 및 CIBU(방법 A)와 Crn (방법 B)를 동시 정량할 수 있다.
연구의 추가 목표는
분석 방법의 친환경화와 최대한의 단순화에 있었다.
소변은 접근성이 용이하고 비침습적·비간섭적 방식으로 채취 가능하며,
또한 인간에서 IBU 및 그 생체변환 생성물이
주로 소변으로 배설된다는 사실이 입증되었기 때문이다1,2.
주요 성과로는
HPLC-ELSD 기반의 효과적인 분석 도구 개발과 방법 성능 입증을 위한
실제 시료 적용이 포함된다.
Materials and methods
Reagents and materials
All chemicals were commercially available and at least of analytical-reagent grade. Crn, IBU sodium salt, 2-HIBU, CIBU as well as HPLC-gradient grade acetonitrile (ACN) and methanol (MeOH) were obtained from Sigma-Aldrich (St. Louis, MO, USA). Mobile phase additive suitable for HPLC–MS technique, namely formic acid (FA) was purchased from Merck KGaA (Darmstadt, Germany), while perchloric acid (PCA) was from J.T. Baker (Deventer, The Netherlands). Deionized water was produced in the laboratory. Commercially available 400 mg or 600 mg tablets containing active substance IBU, were used.
Instrumentation
All analyses were performed using an Agilent 1220 Infinity LC system equipped with a binary pump integrated with a two-channel degasser, autosampler, column oven, UV detector, and ELSD detector 1260 Infinity II series from Agilent Technologies (Waldbronn, Germany). Instrument control, data acquisition, and analysis were carried out using OpenLAB CDS software. Analytes were separated on Poroshell 120 SB-C18 (75 × 4.6 mm, 2.7 µm) column from Agilent Technologies (Waldbronn, Germany). During the study, a Mikro 220R centrifuge with a fast cool function (Hettich Zentrifugen, Tuttlingen, Germany), and Multi-Speed Vortex MSV-3500 (Biosan, Riga, Latvia) were used. Samples were stored in an ultra-low-temperature freezer (Panasonic Healthcare Co., Ltd., Sakata, Japan). Water was purified using a Direct-Q 3 UV water purification system (Millipore, Vienna, Austria).
시약 및 재료
모든 화학 물질은
시판 제품으로 분석용 시약 등급 이상이었다.
Crn, IBU 나트륨 염, 2-HIBU, CIBU 및 HPLC-그라디언트 등급 아세토니트릴(ACN)과 메탄올(MeOH)은 Sigma-Aldrich(미국 세인트루이스)에서 구입하였다. 미국 미주리주 세인트루이스)에서 구입하였다. HPLC-MS 기법에 적합한 이동상 첨가제인 포름산(FA)은 Merck KGaA(독일 다름슈타트)에서 구입하였으며, 과염소산(PCA)은 J.T. Baker(네덜란드 데벤터)에서 구입하였다. 탈이온수는 실험실에서 제조하였다. 활성 성분 IBU를 함유한 시판용 400mg 또는 600mg 정제를 사용하였다.
기기
모든 분석은 이원 펌프와 2채널 탈기 장치, 자동 시료 주입기, 컬럼 오븐, UV 검출기, ELSD 검출기가 통합된 Agilent 1220 Infinity LC 시스템(Agilent Technologies, 독일 발트브론)을 사용하여 수행하였다. 기기 제어, 데이터 수집 및 분석은 OpenLAB CDS 소프트웨어를 사용하여 수행하였다. 분석 물질은 Agilent Technologies(독일 Waldbronn)의 Poroshell 120 SB-C18(75×4.6 mm, 2.7 µm) 컬럼에서 분리되었다. 연구 과정에서는 고속 냉각 기능이 탑재된 Mikro 220R 원심분리기(Hettich Zentrifugen, 독일 투틀링겐)와 Multi-Speed Vortex MSV-3500(Biosan, 라트비아 리가)을 사용하였다. 시료는 초저온 냉동고(Panasonic Healthcare Co., Ltd., 일본 사카타)에 보관하였다. 물은 Direct-Q 3 UV 정수 시스템(Millipore, 오스트리아 빈)을 사용하여 정제하였습니다.
Stock solutions
The stock solutions of IBU (10 g/L), 2-HIBU (1 g/L), and CIBU (1 g/L) were prepared by dissolving an appropriate amount of the powder in a mixture of MeOH and water (50:50, v/v). These solutions were kept at − 20 °C for no longer than 7 days without a noticeable change in the analyte content. The working solutions were prepared daily by diluting a particular standard solution with a mixture of MeOH in water (50:50, v/v) as needed and processed without delay.
The stock solution of Crn (0.15 mol/L) was prepared as needed in deionized water. The solution was kept at 4 °C for a maximum of 7 days without a noticeable change in the analyte content. The working solutions of Crn were prepared daily by diluting a particular standard solution with deionized water as needed and processed without delay.
The remaining solutions, including PCA (3 mol/L) and mobile phase component consisting of 0.1% FA in water were prepared by diluting a particular standard solution with deionized water and stored in tight glass flasks at ambient temperature.
원액
IBU(10 g/L), 2-HIBU(1 g/L), CIBU(1 g/L) 원액은 적정량의 분말을 메탄올과 물 혼합액(50:50, v/v)에 용해하여 조제하였습니다. 이 용액들은 분석물 함량에 뚜렷한 변화 없이 −20°C에서 7일 이내로 보관되었다. 작업용 용액은 필요 시 특정 표준 용액을 물과 메탄올 혼합액(50:50, v/v)으로 희석하여 매일 제조하였으며 지체 없이 처리하였다.
Crn(0.15 mol/L)의 저장용액은 필요 시 탈이온수로 조제하였다. 이 용액은 분석물 함량에 뚜렷한 변화 없이 최대 7일간 4°C에서 보관되었다. Crn의 작업용액은 필요 시 특정 표준용액을 탈이온수로 희석하여 매일 조제하고 지체 없이 처리하였다.
PCA(3 mol/L) 및 물에 0.1% FA를 함유한 이동상 성분을 포함한 나머지 용액들은 특정 표준 용액을 탈이온수로 희석하여 제조하고 실온에서 밀폐된 유리 플라스크에 보관하였다.
Biological samples collection
First, early morning urine samples (about 10 mL) were collected from individuals after overnight fasting using a standard method5. Samples of “mid-stream” urine were obtained by asking donors to put fluid into a sterile container. Then, samples were cooled on ice, delivered to the laboratory within 3 h after collection, and stored at − 80 °C until analysis. In each case, samples were processed without delay, immediately after defrosting at room temperature, using the procedures described in "Urine samples preparation" and "Chromatographic conditions" sections.
A group of ten apparently healthy anonymous individuals was involved in the study. The control subjects, belonging to an ethnically homogeneous group, were neither supplemented with the analytes before sample collection, or their precursors for at least 7 days prior to sampling. In addition, no medications were allowed. Regarding donors who have administered one dose of IBU (400 mg or 600 mg) in the form of commercially available tablets, urine samples were collected just before, at least 1 h, and no later than 24 h after ingestion of IBU-containing pharmaceutical preparation as described above. All subjects involved in the study have also declared that, to the best of their knowledge, none of them suffer from any disease.
Urine samples preparation2-HIBU and CIBU determination
The urine sample (100 µL) was mixed with 10 µL of 3 mol/L PCA. The mixture was shaken by hand for 1 min and then kept in a centrifuge at 12,000g for 5 min at 4 °C. A 5 µL aliquot of the upper layer of the resulting solution was assayed according to the procedure described in "2-HIBU and CIBU determination" section. Samples with concentrations that did not fall within the calibration range were diluted with ACN (the ratio of ACN to sample was predominantly 1:1) and re-assayed as described above.
Crn determination
Samples were prepared according to the slightly modified method of Kuśmierek et al.30. The urine sample (50 μL) was diluted by 500 times with deionized water. 10 µL of the resulting solution was injected into the HPLC system and assayed according to the procedure described in "Crn determination" section.
Chromatographic conditions2-HIBU and CIBU determination
The IBU metabolites in urine samples, prepared according to the procedure described in "2-HIBU and CIBU determination" section, were separated using a mobile phase composed of 0.1% FA in water (solvent A) and ACN (solvent B) delivered at 1 mL/min the flow rate. The chromatographic separation was performed at 25 °C using the gradient elution: 0–4 min 30–45% B, 4–5 min 45–90% B, 5–6 min 90–30% B. The column was re-equilibrated between analyses by setting the post-run conditioning at 30% B for 2 min. The effluent was monitored with an ELSD detector operated with the following set of operation parameters: nebulizer temperature 90 °C, evaporator temperature 50 °C, gas (nitrogen) flow rate 1.3 SLM, data rate 10 Hz, photomultiplier tubes (PMT) gain of 10, smoothing 30 (3 s).
Crn determination
The chromatographic separation of the Crn in urine samples, prepared according to the procedure described in "Crn determination" section, was accomplished using the mobile phase consisting of water (solvent A) and MeOH (solvent B), delivered at the flow rate of 0.5 mL/min. The chromatographic separation was performed at 25 °C using gradient elution: 0–1 min 5% B, 1–2 min 5–30% B, 2–4 min 30–5% B, 4–8 min 5% B. The effluent was monitored with an ELSD detector operated with the following set of operation parameters: evaporator temperature 85 °C, nebulizer temperature 90 °C, gas (nitrogen) flow rate 1.15 SLM, data rate 10 Hz, PMT gain of 9, smoothing 30 (3 s).
Institutional review board statement
The study was conducted according to the guidelines of the Declaration of Helsinki, and approved by the Bioethics Committee of the University of Lodz (decision identification code: 3/KBBN-UŁ/III/2020-21, date of approval 27.04.2021) as well as the Bioethics Committee of the Institute of the Polish Mother’s Memorial Hospital in Łódź (protocol code 35/2022, date of approval 12.04.2022).
Informed consent
Written informed consent was obtained from all subjects involved in the study.
생물학적 시료 채취
먼저, 표준 방법5에 따라 개인들로부터 밤새 금식 후
이른 아침 소변 시료(약 10 mL)를 채취하였다.
기증자에게 무균 용기에 소변을 배출하도록 요청하여
“중간 소변” 샘플을 채취하였다.
이후 샘플은 얼음 위에 냉각된 후 채취 후
3시간 이내에 실험실로 전달되었으며,
분석 시까지 −80°C에서 보관되었다.
모든 경우에 샘플은 실온에서 해동 직후 지체 없이 “
소변 샘플 준비” 및 “크로마토그래피 조건” 섹션에 기술된 절차에 따라 처리되었다.
외관상 건강한 익명의 개인 10명으로 구성된 집단이 연구에 참여하였다.
인종적으로 동질적인 집단에 속한 대조군 대상자들은
시료 채취 전 최소 7일간 분석 대상 물질 또는 그 전구체를 보충하지 않았으며,
약물 복용도 허용되지 않았다.
시판되는 정제 형태로 IBU(400mg 또는 600mg) 1회 용량을 투여받은 기증자의 경우,
위에서 설명한 바와 같이 IBU 함유 의약품 복용 직전, 최소 1시간 후, 최대 24시간 이내에 소변 샘플을 채취하였습니다.
연구에 참여한 모든 피험자는 또한 자신이 아는 한 어떠한 질환도 앓고 있지 않다고 진술하였습니다.
소변 시료 전처리2-HIBU 및 CIBU 측정
소변 시료(100 µL)에 3 mol/L PCA 10 µL를 혼합하였다. 혼합물을 1분간 손으로 흔든 후 4°C에서 12,000g로 5분간 원심분리하였다. 생성된 용액의 상층부에서 5 µL를 취해 “2-HIBU 및 CIBU 측정” 절에 기술된 절차에 따라 분석하였다. 교정 범위에 속하지 않는 농도의 시료는 ACN으로 희석(ACN과 시료의 비율은 주로 1:1)한 후 위와 동일한 방법으로 재분석하였다.
Crn 정량
Kuśmierek 등30의 방법을 약간 수정하여 시료를 준비하였다.
소변 시료(50μL)를 탈이온수로 500배 희석하였다.
얻어진 용액 10μL를 HPLC 시스템에 주입하고
“Crn 정량” 절에 기술된 절차에 따라 분석하였다.
크로마토그래피 조건2-HIBU 및 CIBU 측정
“2-HIBU 및 CIBU 측정” 섹션에 기술된 절차에 따라 준비된 소변 시료 내 IBU 대사산물은 0.1% 포름산 수용액(용매 A)과 아세토니트릴(용매 B)로 구성된 이동상을 사용하여 분리한 후, 분당 1mL의 유속으로 공급하였다. 크로마토그래피 분리는 25°C에서 그라디언트 용출을 사용하여 수행되었습니다: 0–4분 30–45% B, 4–5분 45–90% B, 5–6분 90–30% B. 분석 사이마다 컬럼은 2분 동안 30% B로 설정된 후 처리 조건을 통해 재균형화되었습니다. 유출액은 다음 작동 매개변수로 구동되는 ELSD 검출기로 모니터링되었습니다: 분무기 온도 90°C, 증발기 온도 50°C, 가스(질소) 유량 1.3 SLM, 데이터 속도 10Hz, 광전증배관(PMT) 이득 10, 평활화 30(3초).
Crn 측정
“Crn 측정” 섹션에 설명된 절차에 따라 준비된 소변 시료의 Crn 크로마토그래피 분리는 용매 A(물)과 용매 B(메탄올)로 구성된 이동상을 유속 0.5 mL/min으로 공급하여 수행되었습니다. 크로마토그래피 분리는 25°C에서 그라디언트 용출법으로 수행: 0–1분 5% B, 1–2분 5–30% B, 2–4분 30–5% B, 4–8분 5% B. 유출액은 다음 작동 매개변수로 구동되는 ELSD 검출기로 모니터링되었습니다: 증발기 온도 85°C, 분무기 온도 90°C, 가스(질소) 유속 1.15 SLM, 데이터 속도 10Hz, PMT 이득 9, 평활화 30(3초).
Result and discussion
It is generally known that proper sample handling and management combined with separation and detection conditions play a pivotal role in the quality of generated results. In this study, experiments were carried out to provide the information regarding reliability of HPLC-ELSD assays. In particular, considerable attention was given to optimizing procedures and conditions related to selective separation and detection of the analytes. While designing of the methodology, a strong emphasis was put on greener analytical procedures considering each of the twelve principles of Green Analytical Chemistry (GAC)31.
During the study, the chemical and flow variables influencing each of the implemented sample preparation steps, and the chromatographic separation of analytes and their detection, were optimized in detail. All investigations were performed using the procedures described herein. In each case, the appearance of a particular analyte-delivered peak on the chromatogram and a comparison of its area/height were used to determine the process efficiency. All experiments concerning the optimization of sample preparation procedure as well as chromatographic and detection conditions were run at least in triplicate. The following (sub)sections of the article provide all necessary information regarding the development, validation, and in-study use of the described herein HPLC-ELSD based methods for the determination of primary metabolites of IBU, namely 2-HIBU and CIBU (method A) as well as Crn content (method B) in human urine.
결과 및 논의
일반적으로 적절한 시료 취급 및 관리와 분리 및 검출 조건이
생성된 결과의 품질에 중추적인 역할을 한다는 것은 잘 알려져 있습니다.
본 연구에서는
HPLC-ELSD 분석법의 신뢰성에 관한 정보를 제공하기 위한 실험을 수행하였습니다.
특히, 분석물의 선택적 분리 및 검출과 관련된 절차 및 조건의 최적화에 상당한 주의를 기울였습니다.
방법론 설계 시,
녹색 분석 화학(GAC)의 12가지 원칙31 각각을 고려한
친환경 분석 절차에 중점을 두었다.
연구 과정에서,
시행된 각 시료 전처리 단계와 분석물의 크로마토그래피 분별 및 검출에 영향을 미치는
화학적 변수와 유량 변수를 상세히 최적화하였다.
모든 연구는 본문에 기술된 절차에 따라 수행되었다.
각 경우에서 크로마토그램 상 특정 분석물이 생성한 피크의 출현과 그 면적/높이 비교를 통해 공정 효율성을 판단하였다.
시료 전처리 절차 및 크로마토그래피·검출 조건 최적화와 관련된 모든 실험은
최소 3회 이상 반복 수행되었다.
본 논문의 다음 (하위)절에서는
인간 소변 내 IBU의 주요 대사산물인 2-HIBU 및 CIBU(방법 A)와 크롬(Crn) 함량(방법 B)을 정량하기 위한
본문에 기술된 HPLC-ELSD 기반 방법의 개발, 검증 및 연구 내 사용에 관한 모든 필수 정보를 제공한다.
Sample preparation
Human urine primarily consists of 95% water, while the rest is urea (2%), Crn (0.1%), uric acid (0.03%), chloride, sodium, potassium, sulfate, ammonium, phosphate, and other ions and molecules in lesser amounts. Under normal conditions, protein is only found in trace amounts compared to their values in blood plasma32,33,34. In general, the urine of healthy individuals contains up to 150 mg of protein in the urine 24-h volume. In contrast, urinary protein excretion of more than 3.5 g per 24 h can occur in patients with proteinuria, according to the American Association for Clinical Chemistry. Since the HPLC-ELSD system cannot accommodate such kind of biomolecules, it has been concluded that sample deproteinization is needed in order to protect the analytical system against a decrease in its performance.
Urine samples were assayed for Crn according to a previously published procedure, based on HPLC–UV measurements30, involving sample dilution with deionized water. Importantly, this approach reduced the concentration of all interfering substances to an undetectable level. Regarding IBU metabolites assay, extensive sample dilution was excluded, taking into account the sensitivity of the elaborated method as well as the expected unknown 2-HIBU and CIBU concentration in study samples. In this case, sample preparation involved treatment with 3 mol/L PCA followed by centrifugation to remove urinary proteins. This is one of the most commonly used techniques for effectively eliminating proteins from biological samples, by the addition of water-miscible organic solvent and (ultra)filtration35,36,37,38. Based on our earlier findings, sample acidification with a popular protein precipitating agent, namely PCA, was taken into account. Importantly, the approaches utilizing centrifugal concentrators or membrane filters were excluded to minimize plastic consumption and reduce the quantity of the samples. The use of ACN, typically recognized as the most effective protein precipitating agent among organic solvents, was also eliminated due to a large excess of ACN needed in relation to biological sample, comparing to 3 mol/L PCA which is needed for complete removal of proteins35,36,37,38. Taking into account the trace concentration of urinary proteins, the efficient protein precipitation is achieved by mixing the urine sample with 3 mol/L PCA and crashing at 10:1 ratio by volumes. Since such an approach was beneficial for workflow simplification and results were satisfactory, no additional experimental work was undertaken to further optimize sample preparation step. Significantly, the duration and complexity of the sample pretreatment procedure was reduced in comparison with other methods14,15,16,17,18,19,20,21,22,23,24,25. The sample preparation time in our method only takes 5 min of centrifugation while in published methods, around 20 h20,25, 2.48 h (without evaporation to dryness)17, 2.05 h23, 2 h14,15,18, 46 min19, 10 min (without SPE)21. Longer sample preparation time is a consequence of using multi-step procedures involving SPE15,16,20,21,22,25, LLE14,17,18,24, solid phase microextraction23, USAEME19, hydrolysis17,19,20,21,22,23,25, evaporation to dryness14,15,17,18,22,24 and derivatization14,15,17,18.
Since chemical compounds can be decomposed prior to chromatographic analysis under different circumstances, the stability of the analytes under experimental conditions was evaluated. The problem has been approached qualitatively to measure the intactness of the analytes in a given matrix at room temperature for preselected time intervals. In the stability experiments, calibration standards at the lower/upper limit of quantification (L/U LOQ) were assayed according to the procedure of choice described in "Urine samples preparation" and "Chromatographic conditions" sections. Notably, it was found that Crn content remains stable for at least 7 days since the drop to 97.67% of the initial concentration is achieved when samples were left in the not temperature controlled autosampler. In parallel, there was no noticeable change in 2-HIBU and CIBU delivered peak area/height which remained stable for at least 1 working day at ambient temperature. Importantly, these results agree with the literature data and support information that Crn and IBU metabolites, namely 2-HIBU and CIBU in water (acidic) solutions, are stable19,23,39,40. In this way, sample handling and management effort can be significantly minimized due to the excellent stability of the analytes under experimental conditions. The analyte stability allows for preparation of a large batch of samples without the need of speed through the HPLC-ELSD method.
시료 전처리
인간 소변은 주로 95%의 물로 구성되며,
나머지는 요소(2%), 크레아티닌(Crn, 0.1%), 요산(0.03%),
염화물, 나트륨, 칼륨, 황산염, 암모늄, 인산염 및
기타 이온과 분자가 소량 포함됩니다.
정상적인 조건에서 단백질은
혈장 내 수치에 비해 극미량만 검출됩니다32,33,34.
일반적으로 건강한 사람의 24시간 소변량에는
최대 150mg의 단백질이 포함됩니다.
반면 미국임상화학협회에 따르면
단백뇨 환자의 경우
24시간당 3.5g 이상의 소변 단백질 배설이 발생할 수 있습니다.
HPLC-ELSD 시스템은
이러한 종류의 생체 분자를 수용할 수 없기 때문에,
분석 시스템의 성능 저하를 방지하기 위해 시료의 탈단백질화가 필요하다는 결론이 내려졌다.
소변 시료는
이온화수(deionized water)로 시료를 희석하는 것을 포함하는,
HPLC–UV 측정에 기반한 이전에 발표된 절차에 따라 크론Crn)을 분석하였다.30
중요한 점은 이 접근법이
모든 간섭 물질의 농도를 검출 불가능 수준으로 감소시켰다는 것이다.
IBU 대사체 분석의 경우,
정교화된 방법의 민감도와 연구 시료 내 예상되는 미지 2-HIBU 및 CIBU 농도를 고려하여
과도한 시료 희석은 배제되었다.
이 경우 시료 전처리에는
3 mol/L PCA 처리 후 원심분리를 통한 요단백질 제거가 포함되었다.
이는 생물학적 시료에서
단백질을 효과적으로 제거하기 위해 가장 흔히 사용되는 기법 중 하나로,
물과 혼합 가능한 유기 용매를 첨가한 후 (초)여과를 수행하는 방식이다35,36,37,38. 우
리의 이전 연구 결과를 바탕으로,
널리 사용되는 단백질 침전제인 PCA를 이용한 시료 산성화가 고려되었다.
특히,
플라스틱 소비를 최소화하고 시료 양을 줄이기 위해
원심 농축기나 막 여과기를 활용하는 접근법은 배제되었다.
유기 용매 중 가장 효과적인 단백질 침전제로 인정받는 아세토니트릴(ACN)의 사용도 배제되었습니다. 이는 생물학적 시료 대비 ACN의 과도한 사용량이 필요하기 때문이며, 단백질 완전 제거에 필요한 3 mol/L PCA와 비교됩니다35,36,37,38. 소변 단백질의 극미량 농도를 고려할 때, 효율적인 단백질 침전은 3 mol/L PCA와 소변 시료를 혼합하고 부피 기준 10:1 비율로 분쇄함으로써 달성됩니다. 이러한 접근법이 작업 흐름 간소화에 유리하고 결과가 만족스러웠기 때문에, 시료 전처리 단계를 추가로 최적화하기 위한 실험 작업은 수행하지 않았습니다. 특히, 다른 방법들14,15,16,17, 18,19,20,21,22,23,24,25. 본 방법의 시료 전처리 시간은 원심분리 5분만 소요되는 반면, 기존 방법에서는 약 20시간20,25, 2.48시간(완전 건조 증발 제외)17, 2.05시간23, 2시간14,15,18, 46분19, 10분(SPE 제외)21이 소요됩니다. 더 긴 시료 전처리 시간은 SPE15,16,20,21,22,25, LLE14,17,18,24, 고체상 미세 추출23,
USAEME19, 가수분해17,19,20,21,22,23,25, 완전 증발14,15,17,18,22,24 및 유도체화14,15,17,18을 포함하는 다단계 절차의 사용으로 인한 결과이다. 화합물은 다양한 조건 하에서 크로마토그래피 분석 전에 분해될 수 있으므로, 실험 조건 하에서의 분석물의 안정성을 평가하였다.
이 문제는 실온에서 특정 매트릭스 내 분석물의 무결성을 사전 선정된 시간 간격으로 정성적으로 측정하는 방식으로 접근하였다. 안정성 실험에서는 “소변 시료 준비” 및 “크로마토그래피 조건” 섹션에 기술된 선택된 절차에 따라 정량 하한/상한(L/U LOQ)의 교정 표준물을 분석하였다. 특히, Crn 함량은 비온도 제어 자동주입기에 시료를 방치했을 때 초기 농도의 97.67%까지 감소하는 데 최소 7일이 소요되어 안정적임을 확인하였다. 동시에, 2-HIBU 및 CIBU의 피크 면적/높이는 실온에서 최소 1일 동안 안정적으로 유지되어 뚜렷한 변화가 관찰되지 않았다.
중요한 점은
이러한 결과가 문헌 데이터와 일치하며,
Crn 및 IBU 대사산물인 2-HIBU와 CIBU가 물(산성) 용액에서 안정적이라는 정보를 뒷받침한다는 것입니다19,23,39,40.
이러한 방식으로,
실험 조건 하에서 분석물의 우수한 안정성 덕분에 시료 취급 및 관리 노력을 크게 최소화할 수 있습니다.
분석물의 안정성은 HPLC-ELSD 방법을 통해 신속하게 처리할 필요 없이 대량의 시료를 준비할 수 있게 합니다.
Chromatographic and detection conditions
Among several separation techniques, HPLC has been the most frequently used technique to separate IBU, its metabolites, and Crn in complex systems7,8,9,16,17,19,21,23,24. Importantly, the above-mentioned compounds exhibit high compatibility with liquid phase separation techniques, due to their good solubility in commonly used mobile phases. In addition, these methods have taken advantage of separation in traditional reverse phase mode (RP-HPLC). In the present study, InfinityLab Poroshell 120 superficially porous column for RP-HPLC separations was chosen among those available in our laboratory. Importantly, we aimed to develop two different methods using the same analytical column in order to reduce expenses associated with running the analyses.
To successfully complete the project, a standard approach was employed to specify optimal conditions by assessing the influence of many operating parameters of the HPLC-ELSD system on the methods’ performance. Firstly, careful optimization of separation conditions was performed by taking into account operational guidelines provided by the column manufacturer. Many variables have been studied to find optimal conditions affording satisfactory separation performance. In principle, the best quality of results produced a satisfactory method selectivity by selecting the composition of the mobile phase (type of organic modifier, pH), its flow rate, and elution mode. As a result, crucial rules have been developed in the method to perform successful analysis. In particular, it was found that gradient elution was necessary to maintain the efficient resolution of the particular analyte peak from other sample components, properly equilibrate the chromatographic system between analyses, and reduce carry-over to the minimum level. Regarding Crn, it was also noted that an initial MeOH, instead of ACN, with a content no higher than 5%, and a flow rate of mobile phase set to 0.5 mL/min were essential to retain the analyte. In relation to IBU and its primary metabolites, successful separation of all sample constituents has been achieved using a typical mobile phase for HPLC-ELSD technique, consisting of ACN and water with 0.1% FA. Importantly, it has been found that separation had to be accomplished under acidic conditions in order to enhance the hydrophobicity of the target compounds to make them more compatible with stationary phase. Moreover, it has been recognized that the FA concentration, which was tested in the range of 0.1–1%, had a negligible effect on retention behaviors of solutes as well as resolution. Thus, ACN and water with 0.1% FA were selected as the eluent as they helped to decrease the noise level at high gain settings. Importantly, it has been recognized that the particular target compound peak produced a single symmetrical peak, which was well-resolved from other peaks on the column under set conditions. Therefore, no further experimental work was undertaken to optimize separation conditions during new methods development.
In the next stage of the method development process, ELSD detection conditions were optimized in order to increase sensitivity and selectivity in trace analysis. In the beginning, the evaporator temperature, nebulizer temperature, and gas flow rate parameters that greatly affects the detector sensitivity, were evaluated. Subsequently, frequency of data collection, applied signal amplification factor, and smoothing factor on signal quality were carefully studied. The initial experiments were conducted using the default settings of the ELSD detector. Then, each of the parameters mentioned above was adjusted to achieve the best performance. Importantly, only one parameter settings varied in the range specified by manufacturer with the constant value of other parameters. All experiments were run at least in triplicate, and the repeatability of the results, expressed as the coefficient of variation (CV) of peak height, was satisfactory. CV varied from 0.45 to 5.41% and 0.03 to 4.61% for Crn assay and IBU metabolites, respectively under any evaluated detection conditions. In general, predictable relationships of the data were obtained while optimizing the performance of the ELSD detector (Supplementary Materials, Figure S1 a–e and Figure S2 a–f). Finally, the specified operating conditions resulted in not only improvement of batch to batch reproducibility, but also sensitivity of the methods.
Under optimized conditions described in "Chromatographic conditions" section, the 2-HIBU, CIBU, and Crn peak was approximately 65-times, 127-times, and 25-times higher in comparison with that registered with non-optimized ELSD settings, respectively. In addition, the peak of Crn (2.40 min), 2-HIBU (2.78 min), CIBU (3.13 min), and IBU (6.61 min) eluted within 8 min, and their corresponding peaks were easy to distinguish from the responses of all the concomitant components (Figs. 1b–d, 2b). Importantly, every time the elution profile of blank samples was free from any interference at the retention time of the analytes (Fig. 1a–c, 2a). The analysis time 8 min is more favorable than in other methods such as GC14,15,18, HPLC16,17,21,23 and CE22,25 where it is 17 min14,18,22, 21.8 min15, 30 min23,25, 35 min16,21, and 80 min17. The analysis time is the same as in CE method20 and worse than in HPLC method (1.8 min19 and 5 min30).
크로마토그래피 및 검출 조건
여러 분리 기술 중 HPLC는
복잡한 시스템에서 IBU,
그 대사산물 및 Crn을 분리하는 데 가장 빈번히 사용된 기술이다7,8,9,16,17,19,21,23,24.
중요한 점은,
상기 화합물들이 일반적으로 사용되는 이동상에 대한
우수한 용해성으로 인해 액상 분리 기술과 높은 호환성을 보인다는 것이다.
또한, 이러한 방법들은
전통적인 역상 모드(RP-HPLC)에서의 분리를 활용해 왔다.
본 연구에서는 실험실에 구비된 컬럼 중 RP-HPLC 분리를 위한 InfinityLab Poroshell 120 표면 다공성 컬럼을 선택하였다. 특히, 분석 실행과 관련된 비용을 절감하기 위해 동일한 분석 컬럼을 사용하여 두 가지 다른 방법을 개발하는 것을 목표로 했습니다.
프로젝트를 성공적으로 완료하기 위해, HPLC-ELSD 시스템의 여러 작동 매개변수가 방법 성능에 미치는 영향을 평가하여 최적 조건을 규명하는 표준 접근법을 채택했습니다. 먼저, 컬럼 제조업체가 제공한 운영 지침을 고려하여 분리 조건을 신중하게 최적화했습니다. 만족스러운 분리 성능을 제공하는 최적 조건을 찾기 위해 다양한 변수를 연구하였다. 원칙적으로, 이동상 조성(유기 용매 유형, pH), 유속, 용출 모드를 선택함으로써 최상의 결과 품질과 만족스러운 방법 선택성을 확보하였다. 그 결과, 성공적인 분석 수행을 위한 핵심 규칙이 방법에 개발되었다. 특히, 특정 분석물 피크를 다른 시료 성분으로부터 효율적으로 분해능 유지, 분석 간 크로마토그래피 시스템의 적절한 평형화, 캐리오버 최소화를 위해 그라디언트 용출이 필수적임이 확인되었다. 크롬(Cr)의 경우, 분석물 유지에는 ACN 대신 5% 이하의 메탄올을 초기 용매로 사용하고 이동상 유속을 0.5 mL/min으로 설정하는 것이 필수적임이 확인되었다. IBU 및 그 주요 대사산물과 관련하여, ACN과 0.1% 포스포르산(FA)을 함유한 물을 혼합한 HPLC-ELSD 기법용 표준 이동상을 사용하여 모든 시료 성분의 성공적인 분리가 달성되었다. 중요한 점은, 표적 화합물의 소수성을 향상시켜 고정상과의 친화성을 높이기 위해 분리를 산성 조건에서 수행해야 한다는 사실이 확인되었다는 것이다. 또한, 0.1~1% 범위에서 시험된 FA 농도는 용질의 유지 행동 및 분해능에 미미한 영향을 미치는 것으로 파악되었다. 따라서 고이득 설정 시 노이즈 수준을 감소시키는 데 도움이 되는 0.1% FA를 함유한 ACN과 물을 용출액으로 선택하였다. 특히, 특정 표적 화합물 피크가 설정된 조건 하에서 컬럼 상의 다른 피크들과 잘 분해되는 단일 대칭 피크를 생성한다는 점이 확인되었다. 따라서 새로운 방법 개발 과정에서 분리 조건을 최적화하기 위한 추가 실험 작업은 수행되지 않았다.
방법 개발 과정의 다음 단계에서는 미량 분석의 감도와 선택성을 높이기 위해 ELSD 검출 조건을 최적화하였다. 초기에는 검출기 감도에 큰 영향을 미치는 증발기 온도, 분무기 온도, 가스 유량 파라미터를 평가하였다. 이후 데이터 수집 주파수, 적용 신호 증폭 계수, 신호 품질에 대한 평활화 계수를 신중히 연구하였다. 초기 실험은 ELSD 검출기의 기본 설정값을 사용하여 수행되었습니다. 이후 최상의 성능을 달성하기 위해 상기 각 매개변수를 조정하였습니다. 특히, 다른 매개변수 값을 일정하게 유지한 상태에서 단일 매개변수 설정값만을 제조사 지정 범위 내에서 변화시켰습니다. 모든 실험은 최소 3회 반복 수행되었으며, 피크 높이의 변동계수(CV)로 표현된 결과의 반복성은 만족스러운 수준이었습니다. 평가된 모든 검출 조건에서 Crn 분석과 IBU 대사물의 CV는 각각 0.45~5.41%, 0.03~4.61% 범위를 보였다. 일반적으로 ELSD 검출기 성능 최적화 과정에서 데이터의 예측 가능한 관계가 확인되었다(보충 자료, 그림 S1 a–e 및 그림 S2 a–f). 결국, 지정된 운영 조건은 배치 간 재현성 향상뿐만 아니라 방법의 감도 향상으로도 이어졌다.
“크로마토그래피 조건” 섹션에 설명된 최적화된 조건 하에서, 2-HIBU, CIBU 및 Crn 피크는 각각 비최적화 ELSD 설정으로 기록된 피크에 비해 약 65배, 127배, 25배 더 높았다. 또한 Crn(2.40분), 2-HIBU(2.78분), CIBU(3.13분), IBU(6.61분) 피크는 8분 이내에 용출되었으며, 이들 피크는 동시 분석된 모든 성분의 반응과 쉽게 구별되었다(그림 1b–d, 2b). 중요한 점은, 분석물의 유지 시간에서 백색 시료의 용출 프로파일이 항상 간섭 없이 깨끗했다는 것이다(그림 1a–c, 2a). 8분의 분석 시간은 GC14,15,18, HPLC16,17,21,23 및 CE22,25와 같은 다른 방법들(각각 17분14,18,22, 21.8분15, 30분23,25, 35분16,21, 80분17과 비교하여 유리하다. 분석 시간은 CE법20과 동일하며 HPLC법(1.8분19 및 5분30)보다 불리하다.
Figure 1
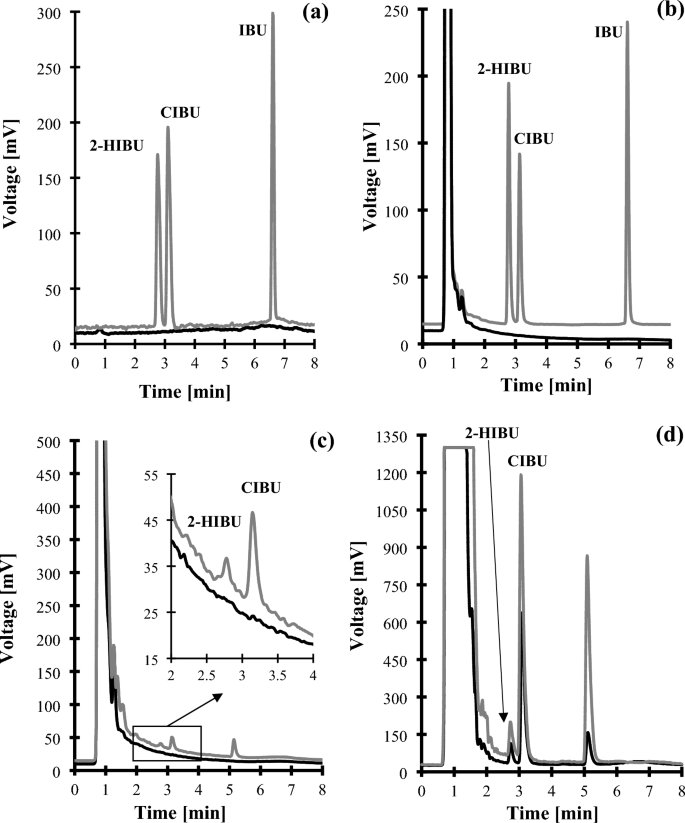
Representative chromatograms of standard solutions and human urine were prepared according to the procedure described in "2-HIBU and CIBU determination" section. Chromatographic conditions were as described in "2-HIBU and CIBU determination" section. (a) Blank standard solution (black line) and standard solution of IBU, 2-HIBU and CIBU (0.4 g/L in urine) (grey line); (b) normal human urine sample (black line) and the same sample spiked with the IBU, 2-HIBU and CIBU (0.4 g/L in urine) (grey line); (c) Normal human urine collected before (black line) and after 3 h oral ingestion of IBU-containing 400 mg pharmaceutical formulation (grey line); (d) Normal human urine collected after 3 h oral ingestion of IBU-containing 400 mg (black line) and 600 mg (grey line) pharmaceutical formulation (grey line). Under these conditions, the 2-HIBU, CIBU, and IBU peaks appear at 2.78 min, 3.13 min, and 6.61 min, respectively.
Figure 2
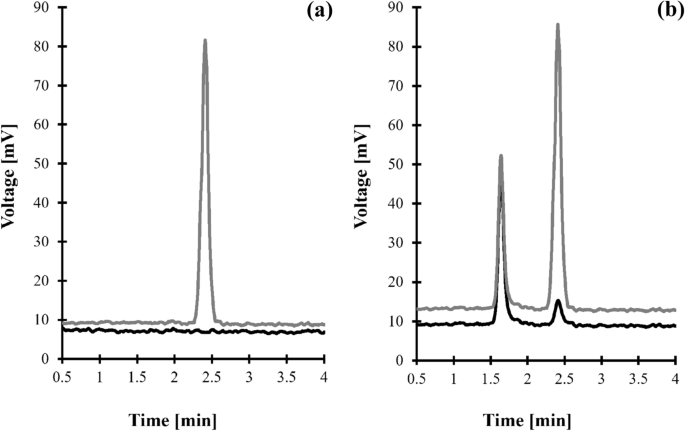
Representative chromatogram of standard solutions and human urine were prepared according to the procedure described in "Crn determination" section. Chromatographic conditions were as described in "Crn determination" section. (a) Blank standard solution (black line) and standard solution of Crn (15 mmol/L in urine) (grey line); (b) normal human urine sample (black line) and the same sample spiked with Crn (15 mmol/L in urine) (grey line). Under these conditions, the peak of Crn appears at 2.40 min.
The identification and confirmation of the target compounds were performed by analyzing the standard solution of analytes processed according to the procedure described in "Urine samples preparation" and "Chromatographic conditions" sections. Each solution of a particular compound of interest was prepared separately and then was processed according to the method of choice to ascertain that a single analyte did not yield more than one chromatographic peak. In addition, the particular analyte peak was evaluated for purity by carrying out the analyses in a HPLC system coupled with both ELSD and UV detector. The UV detector was set to collect time and spectral information throughout the entire chromatogram. The spectra obtained during the elution of the target compound peak were compared. Importantly, the same spectra were acquired in different sections of the particular analyte peak, indicating its purity. Finally, the confirmation of the origin of each analyte peak and quantification of the compound of interest in real samples were based upon the comparison of retention time with the corresponding set of data obtained by analyzing an authentic compound.
Greenness assessment of delivered HPLC-ELSD methods
In recent years, the assessment of an analytical procedure’s greenness is growing in popularity. There are several approaches which enable us to measure the degree of greenness of analytical methods. In the present study, AGREE—Analytical GREEnness metric approach and software (version 0.5 beta)31,41,42,43, which evaluates analytical procedures considering each of the twelve principles of GAC, was used to assess the greenness of the delivered HPLC-ELSD methods for urinary 2-HIBU and CIBU (method A) and Crn (method B) determination. Equal weights have been set for all twelve principles evaluated.
Regarding to the IBU metabolites assay, the following assumptions have been made in order to assess the analytical procedure’s greenness: the procedure involves an off-line analysis (principle 1); the volume of urine is 100 µL (principle 2); the analytical device is positioned off-line (principle 3); the number of distinctive analytical steps is two, including deproteinization combined with centrifugation, and chromatographic analysis (principle 4); the procedure is semi-automated and involves a miniaturized sample preparation methods (principle 5); derivatization step is not required (principle 6); the total amount of waste is 10.61 (g and mL combined), consisting of the sample itself, chemicals, and plastic disposable ware used to prepare the sample as well as mixture of solvents used during HPLC analysis (principle 7); two analytes are determined in a single run, and the sample throughput is seven samples per hour (principle 8); the most demanding technique is HPLC-ELSD (principle 9); none of the reagents are from renewable sources (principle 10), and finally the procedure requires no more than 3.5 mL of toxic chemical reagents (mobile phase ingredients) (principle 11) of which ACN is perceived as highly flammable and toxic to humans (principle 12). Since trace amounts of FA and PCA were used, they were not taken into account at the stage of assessing the operator`s safety.
In relation to Crn assay, it has been assumed that the procedure involves an off-line analysis (principle 1); the volume of urine is 50 µL (principle 2); the analytical device is positioned off-line (principle 3); the number of distinctive analytical steps is two, including sample dilution and HPLC-ELSD analysis (principle 4); the procedure is semi-automated and involves a miniaturized sample preparation methods (principle 5); Crn was not derivatized (principle 6); the total amount of waste is 10.25 (g and mL combined), consisting of the sample itself, chemicals, and plastic disposable ware used to prepare sample (principle 7); one analyte is determined in a single run, and the sample throughput is seven samples per hour (principle 8); the most demanding technique is HPLC-ELSD (principle 9); none of the reagents can be obtained from bio-based sources (principle 10), and finally the procedure requires approximately 0.35 mL of toxic chemical reagents (principle 11), while MeOH is perceived as highly flammable and toxic to humans (principle 12). The AGREE results for the methods under consideration are presented in Fig. 3.
Figure 3
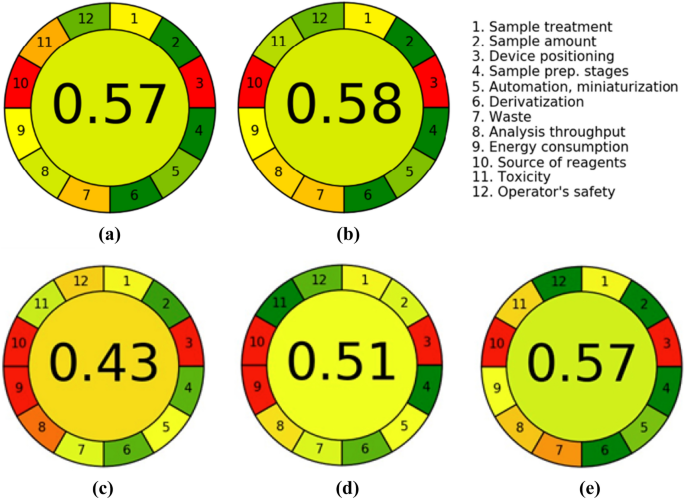
The assessment results with AGREE—Analytical GREEnness analysis of procedures for determination of (a) IBU metabolites, namely 2-HIBU and CIBU (method A), (b) Crn (method B) in human urine, (c) IBU metabolites in urine by GC–MS method18, (d) IBU metabolites in urine by HPLC–MS method19 and (e) Crn in urine by HPLC–UV method30.
Overall, it was concluded that the HPLC-ELSD assays fall into principles of GAC. The overall score, which is shown in the middle of the colored pictograms, is 0.57 and 0.58 in relation to IBU metabolites and Crn assays, respectively. Generally, values close to one and dark green in color indicate that the assessed procedure is greener. In addition, the use of AGREE—Analytical GREEnness metric approach helped identify the strong and weak points of our analytical procedures, whose distribution is comparable in both cases (Fig. 3). In particular, it has been assumed that the methods can be considered environmentally-friendly thanks to carrying out the chemical analysis on a very small scale combined with a low consumption of hazardous chemicals and laboratory disposable plastics. The assays have a relatively high-throughput potential and simplicity in sample preparation. Undoubtedly, a serious limitation of the presented methods is the use of energy-intensive measurement technique without the possibility of carrying out the analysis in situ.
We attempted to compare our methods in terms of environmental friendliness with other published assays for determination of IBU metabolites in urine by GC–MS18 and HPLC–MS19, and Crn by HPLC–UV30. Unfortunately, in these methods, the authors did not provide the degree of ecological friendliness of their procedures. Therefore, we estimated it using a dedicated calculator and data available in the articles. As can be seen in Fig. 3, our method for determining Crn (score 0.58) is comparable in terms of greenness to the HPLC–UV method (score 0.57)30, but for IBU metabolites (score 0.57) it is better than the GC–MS method (score 0.43)18 and HPLC–MS (score 0.51)19. This is due to the fact that the GC–MS and HPLC–MS techniques are more energy-consuming (principle 9) than HPLC-ELSD, which can be observed by comparing Fig. 3a with Fig. 3c, d. Moreover, both GC–MS and HPLC–MS methods have a much lower analysis throughput (principle 8) due to the long sample preparation time, requiring extraction in both methods and derivatization (90 min) in the GC–MS method18.
Validation of the methods
Full validation of the new methods for determining 2-HIBU and CIBU (method A) and Crn (method B) concentration in human urine was conducted to establish that the performance characteristic of each procedure meets the requirements for the intended analytical application. All studies were planned and performed per the United States Food and Drug Administration (U.S. FDA) guidance for validation of bioanalytical methods44. Particularly, the process included evaluation of selectivity, linearity, intra-/inter-assay precision, accuracy, and LOQ. These essential parameters were measured in combined experiments. In addition, the matrix, dilution, and carry-over effects were evaluated during method validation. Some parameters were checked among system suitability testing. Detailed data regarding all evaluated validation parameters are presented herein.
System suitability
System suitability parameters, such as repeatability of chromatographic retention, expressed as CV of retention time, asymmetry factor, and number of theoretical plates, were selected during a particular validation method to determine instrument performance under optimized conditions. System suitability test calculations were performed as a part of linearity assessment by analyzing the calibration standards at the ULOQ in ten replicate injections. Good system suitability was demonstrated, ensuring that the system performs in an accurate and reproducible way. The CV value of retention time was 0.15%, 0.14% and 0.43% (acceptance criteria ≤ 1%), the mean asymmetry factor was 0.83, 0.81 and 0.99 (acceptance criteria 0.8–1.5), and number of theoretical plates was 6163, 7856 and 2818 (acceptance criteria ≥ 2000) for 2-HIBU, CIBU, and Crn, respectively.
Selectivity
The selectivity of a particular analyte in the presence of any other ELSD-producing signal endogenous components in a sample was verified during studies concerning the identification and confirmation of the origin of a particular analyte peak as described in "Chromatographic and detection conditions" section. Crn selectivity studies also assessed interferences originating from urea as it is the most abundant component of urine apart from water32,33,34. The blank standard solution (water) and standard solution of urea were assayed according to the procedure described in "Crn determination" and "Crn determination" sections. As shown in Fig. 2a, the elution profile is free from any interferences at the Crn peak's retention time. Under these conditions, urea is not retained, and its corresponding peak is eluted before the column dead volume. To confirm the finding, normal urine samples from six individual sources and the same samples spiked with urea were assayed according to the procedures described herein ("Crn determination" and "Crn determination" sections). No increase in the peak area/height of Crn was observed.
Regarding 2-HIBU and CIBU, the selectivity studies evaluated interferences originating from all (un)known endogenous components of urine and the precursor of metabolites mentioned above, namely IBU. Firstly, a blank standard solution [a mixture of MeOH in water (50:50, v/v)] and a standard solution of IBU were prepared and analyzed according to the procedure described in "2-HIBU and CIBU determination"and "2-HIBU and CIBU determination" sections, respectively. Normal urine samples from six control subjects and the same samples spiked with IBU were assayed according to the procedures described herein. As shown in Fig. 1a–c, no response attributable to interfering components was observed at the retention time of 2-HIBU and CIBU, denoting the ability of HPLC-ELSD method to differentiate and measure the analytes in the presence of interfering substances in urine samples.
Linearity
An external standard calibration method was used to verify the calibration range of the methods. For this purpose, the multilevel calibration curves consisted of a blank sample, and six calibrators, including LOQ, were generated for each analyte and were run in triplicate over 3 subsequent working days. Each set contained calibration standards at the level of 0.5, 1, 5, 10, 15, 30 mmol/L regarding Crn assay, and 0.06; 0.1; 0.2; 0.3; 0.4; 0.5 g/L concerning 2-HIBU and CIBU assay. Importantly, the calibration curve for Crn consisted of points that cover the entire range of expected analyte concentrations in the test samples. In contrast, the relatively narrow calibration range of the IBU metabolites method was experimentally defined by assessing the lowest and highest calibration standards whose nominal 2-HIBU and CIBU concentration and the response of the analytical platform to the particular analyte were well fitted as described below. Significantly, such an approach has been employed to evaluate the calibration range of the IBU metabolites assay. It is almost impossible to specify the entire expected analytes concentration range in the test urine samples which are highly dependent on the dose of the IBU-containing painkillers. Calibration standards were prepared in our laboratory pooled urine by spiking with known quantities of the particular analyte. Since urine samples free of Crn were unavailable, the endogenous concentration of the analyte was evaluated before the calibration curves preparation by triplicate analysis.
The linearity was initially evaluated graphically by visually inspecting a plot of the peak area/height as a function of the particular analyte concentration and by using the least-squares regression model to describe the concentration–response relationship. Since it has been recognized that ELSD response increased with an increase in any analyte concentration in a non-linear manner, a few calibration curve fitting methods were tested to fit experimental data to a linear calibration curve. Overall, a very good linear fit of log ELSD response against log analyte concentration was observed in the case of each method. In addition, the calibration standards fitted well into the linear regime, which gave correlation coefficient values of at least 0.9970, showing that the instrument response was proportional to the analytes’ concentration within the experimentally defined quantitation range. Data dealing with validation parameters, evaluated as a part of the linearity assessment using an external standard calibration method, and calculated from the peak height values, are shown in Table 1.
Table 1 Validation data corresponding to intra-assay measurements (n = 3).
Conducted experiments also indicated that only the particular analyte-delivered signal peak area/height increased with its growing concentration. Substantial changes in the slope of any regression line obtained across the day, as well as over 3 subsequent days were not observed, suggesting that the presented analytical methods are not affected by matrix components.
Accuracy and precision
The accuracy and precision of the assay, referring to intra- and inter-day measurements, were evaluated as a part of a linearity assessment. The precision was expressed as the CV of measurement repeatability, whereas accuracy was the percentage of analyte recovery. In particular, accuracy was calculated by expressing the mean measured amount as a percentage of added amount of a particular analyte. In the Crn assay, as the analyte is also an endogenous molecule, the concentration of the endogenous molecule in the blank matrix was determined and subtracted from the total concentrations observed in the spiked samples. As recommended by U.S. FDA44, the accuracy of the Crn assay was explicitly calculated using of the following formula Accuracy (%) = [(measured amount − endogenous content)/added amount] × 100. Intra-assay precision and accuracy were demonstrated by triplicate analysis of freshly prepared calibrators, which referred to pooled urine samples containing known amounts of the particular analyte at three different levels corresponding to values close to LLOQ, the middle of the quantitation range, and ULOQ, respectively. Experiments concerning estimating the intermediate accuracy and precision were repeated in the same manner over 3 subsequent days. All concentrations were tested using the calibration curves prepared on that occasion. Importantly, the obtained results from analytical runs met the acceptance criteria. In relation to Crn, the accuracy ranged from 81.19 to 114.72% and 81.19 to 108.72% for intra- and inter-day variation, respectively. In parallel, the precision varied from 0.67 to 4.29% and 2.20 to 13.15% for intra- and inter-day measurements, respectively. Regarding IBU metabolites, the precision did not exceed 14.83% of CV at any examined concentration level. It varied from 1.06 to 14.83% and 6.39 to 14.53% for intra- and inter-day measurements, respectively. In parallel, accuracy ranged from 85.10 to 105.92% and 86.96 to 108.08% for intra- and inter-day variation, respectively. Detailed data on precision and accuracy from the 3 day experiments, compared with intra-assay precision and accuracy, are shown in Table 2.
Table 2 Precision and accuracy of the data (n = 3).
The limit of quantification
LOQ was evaluated in parallel with the calibration range’s intra-assay precision and accuracy assessment. The LLOQs, are equal to 0.06 g/L and 0.5 mmol/L in urine for the IBU metabolites and Crn, respectively, were accepted as LOQ. In each case, this concentration of the particular analyte produced easy to distinguish from the background noise and reproducible detector response with a precision that did not exceed 11.16%, and accuracy ranged from 81.19 to 120.16%. In addition, the estimated LOQ values closely corresponded to the LOQs determined experimentally by the signal-to-noise method. In this method, a blank urine sample or a surrogate matrix (water) was enriched with decreasing concentrations of the particular analyte regarding IBU metabolites and Crn, respectively. The surrogate matrix was used to evaluate the LOQ of Crn assay because obtain a urine sample free from Crn was impossible. Samples were then handled according to the procedures described in "Urine samples preparation" and "Chromatographic conditions" sections until the injected amount resulted in a peak 10 times as high as the baseline signal. Notable, the obtained LOQ values were satisfactory and allowed the determination of the compounds of interest content in human urine.
Matrix effect
The effect of the matrix between different independent sources, defined as an alteration in analyte(s) response due to interfering and usually unidentified sample components, was assessed in a relevant volunteer population. The volunteer population refers to control subjects who were apparently healthy volunteers involved in the study. Apart from selectivity and dilution integrity studies, the matrix effect assessment involved comparing calibration curves of the six individual sources of urine samples against a calibration curve of the pooled matrix and surrogate matrix (water). Importantly, it was recognized that the CV of the slope of the regression lines did not deviate by more than 3.44%, 2.71%, and 0.86% in relation to Crn, 2-HIBU, and CIBU, respectively, denoting the absence of any matrix effect. In fact, with this difference in slopes, the maximum error in analytical results of Crn, 2-HIBU, and CIBU would be 8.74%, 5.20%, and 1.46% respectively. Thus, an external standard addition method was used to establish the levels of the particular analyte in urine samples as it provides the procedure’s reliability along with effort minimization.
Dilution integrity
The dilution integrity of IBU metabolites has been assessed as the method measures diluted samples. In order to evaluate the impact of the sample dilution procedure on the measured concentration of the analyte, calibration standards at the concentration above the ULOQ (0.8 g/L in urine) were prepared in urine from six individual sources and the surrogate matrix. The samples were diluted with ACN and analyzed according to the procedure described in "2-HIBU and CIBU determination"and "2-HIBU and CIBU determination" sections. Five different dilution factors, corresponding to the expected dilutions in the study, were evaluated that amounted to 1–5 times the volume of the (urine) sample. Notably, the mean accuracy and precision of these diluted calibration standards were 96.30% and 9.56%, respectively. In parallel, it has been recognized that instrument response was directly proportional to the 2-HIBU, and CIBU concentration and the CV of the slope of the regression lines did not deviate by more than 3.44% concerning any matrix under consideration. In addition, 2-HIBU and CIBU detector signals of the same peak height/area were registered regardless of the matrix type. Importantly, these results have proved that the measured concentrations are not affected by the magnitude by which the samples were diluted within the calibration range. Along with the documented high accuracy and precision, this demonstrates that the method can analyze samples at a concentration exceeding the ULOQ of the calibration curve without the influence of a matrix.
Carry-over
Carry-over between samples, meaning the appearance of an analyte in a sample from a preceding sample, can occur in analytical methods. As it may impact on the precision and accuracy of the study sample concentrations, the potential of carry-over was thus investigated in the study as a part of the linearity assessment. In each case, the standard blank solution sample(s) were placed after the calibration standard at the ULOQ to evaluate the carry-over. Importantly, each time the response of blank samples was as high as the background signal, indicating that the carry-over effect did not occur. The registered chromatograms were similar to the elution profile of blank samples presented in Figs. 1a–c and 2a.
In conclusion, it has been shown that upon validation the performance of the presented methods is suited to the analysis of study samples. Importantly, it has been demonstrated that the methods are sensitive enough and have suitable precision, accuracy, and linearity levels, falling within acceptable tolerance limits44. Importantly, it has been recognized throughout the application of the methods that carry-over between samples did not occur in any analytical method and matrix as well as sample dilution have a negligible impact on these assays results. Based on the analysis of validation data, it has been concluded that the performance of the presented HPLC-ELSD based methods is sufficient to allow them to be used in diagnostic testing.
Application of the methods
The validated HPLC-ELSD assays were used to quantitatively determine IBU metabolites, namely 2-HIBU, CIBU, and Crn in urine samples from apparently healthy human subjects. Since it has been recognized that the assays are not affected by matrix components, an external standard addition method was used to establish urinary levels of the analytes in study samples, handled according to the procedures described in "Urine samples preparation" and "Chromatographic conditions" sections. Importantly, 2-HIBU and CIBU were only detected in study samples delivered by donors who have ingested orally IBU-containing pharmaceutical preparation, while Crn was found in all urine samples. The concentration of the analytes in each sample was calculated using the mathematical formula corresponding to the equation of the calibration line generated on that occasion. Importantly, the results for urinary 2-HIBU and CIBU content were adjusted for Crn in order to facilitate comparison of different individuals. The estimated concentrations of urinary 2-HIBU, CIBU, and Crn, based on data obtained by triplicate analysis of a particular sample from an individual source, varied from 0 to 0.266 g/L (0–34.68 g/mol Crn), from 0 to 0.495 g/L (0–77.68 g/mol Crn) and from 0.64 to 23.88 mmol/L in urine respectively. Importantly, these values were similar to those previously reported, using different technical approaches7,8,9,16,17,19,20,21,22,23,24,25, denoting the data’s reliability from the presented HPLC-ELSD assays.
In addition, the HPLC-ELSD assay was used to perform clinical pharmacokinetic studies of IBU. One apparently healthy adult volunteer was involved in the experiment. The donor has administered one dose of IBU-containing 400 mg tablet orally, which did not exceed the recommended drug dose. Urine samples were collected just before and several times within 24 h after ingestion of the drug as described in "Biological samples collection" section, and then handled according to the procedure described in "Urine samples preparation" and "Chromatographic conditions" sections. First, these in vivo experiments have confirmed the identity of the peaks eluting at 2.78 min and 3.13 min indicating that they are derived from 2-HIBU and CIBU present in samples after urine donors ingested a drug orally (Fig. 1c). Moreover, it has been recognized that IBU is excreted by the kidney within no later than 24 h after ingestion of the drug (Fig. 4), and the concentration of CIBU was higher than the concentration of 2-HIBU (Fig. 1d), in agreement with literature data1,2. Moreover, it has been demonstrated that the presented HPLC-ELSD assay is suitable for screening human urine in terms of drug metabolites.
Figure 4
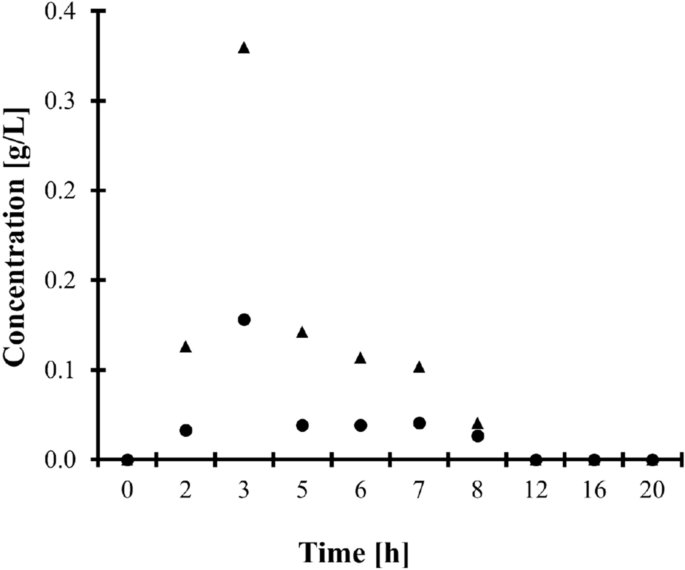
Renal excretion of IBU metabolites, namely 2-HIBU (dots) and CIBU (triangles), after oral administration of one dose of IBU-containing 400 mg tablets.
Conclusions
To the best of our knowledge, the article presents the first HPLC-ELSD based methods for simultaneous assessment of IBU and its main metabolites (2-HIBU and CIBU) and Crn content in human urine. In particular, the attractiveness of the presented HPLC-ELSD assays relies on (1) a streamlined one-step sample preparation procedure followed by (2) a short chromatographic analysis as well as (3) a possibility of carrying out chemical analysis on a small scale combined with (4) low consumption of hazardous chemicals and laboratory disposable plastic, and (5) greater greenness resulting using less energy than HPLC–MS and GC–MS methods. For instance, using the presented assays for preparing of a set of twenty four samples and their HPLC analysis for IBU metabolites and Crn takes no more than 45 min and 6.5 h, respectively, considering all the operations that need to be performed. This achievement in terms of the sample preparation step is the same for Crn (45 min) compared to HPLC–UV method30 and is better for IBU metabolites compared to GC–MS14,15,18 and HPLC–MS19 methods, where this process takes 2.5 h and 53 min, respectively, for twenty four samples. However, the time of HPLC analysis of the same number of samples is better or similar to GC–MS methods (8.72 h15 and 6.84 h14,18), and worse than HPLC–MS method (43.2 min19). In addition, sample handling and preparation is accompanied by consumption of as little as 0.01 mL 3 mol/L PCA and approximately 2.40 mL deionized water per one sample, which represent inexpensive and non-toxic chemicals. In contrast, the GC–MS14,18 and HPLC–MS19 methods use greater volumes of additives such as diethyl ether (2 mL), ethyl acetate (0.1 mL), methyl iodide (0.05 mL), and anhydrous potassium carbonate (50 mg) in GC–MS method, and 1 mol/L sodium hydroxide (0.4 mL), 1 mol/L hydrochloric acid (0.4 mL), 1-octanol (0.1 mL), and MeOH (0.09 mL) in HPLC–MS method. Moreover, the semi-automation of the analytical procedures brought beneficial consequences as it reduces labor intensity, maximizes sample throughput and improves the accuracy and reproducibility of the methods. Interestingly, presented assays have produced compelling evidence supporting the conclusion that 2-HIBU and CIBU are main metabolites of IBU, as reported elsewhere1,2. Importantly, the HPLC-ELSD based methods provide new analytical tools that can facilitate studies of the compounds mentioned above in health and disease, for example, by providing information about acute overdose or chronic abuse of IBU-containing drugs. In our opinion, these methods are free of restrictions. It nonetheless needs to be emphasized that a successful analysis using the proposed methods can only be achieved when the recommended sample handling and management procedures are followed.
Data availability
Essential data is contained within the article and its Supplementary Materials file. In addition, the dataset generated and analyzed during this study, which contributed to the article, can be made available by the corresponding authors (J.P. and G.C.) upon reasonable request as long as the request does not compromise intellectual property interests. Urine samples are not available from the authors.
Abbreviations
ACN:
Acetonitrile
CE:
Capillary electrophoresis
CIBU:
Carboxyibuprofen
Crn:
Creatinine
CV:
Coefficient of variation
ELSD:
Evaporative light scattering detection
FA:
Formic acid
GAC:
Green Analytical Chemistry
GC:
Gas chromatography
IBU:
Ibuprofen
LLE:
Liquid–liquid extraction
(L/U) LOQ:
(Lower/upper) limit of quantification
MeOH:
Methanol
MS:
Mass spectrometry
PCA:
Perchloric acid
PMT:
Photomultiplier tubes
(RP)-HPLC:
(Reverse-phase) liquid chromatography
SPE:
Solid phase extraction
U.S. FDA:
United States Food and Drug Administration
USAEME:
Ultrasound-assisted emulsification microextraction
UV:
Ultraviolet detection
2-HIBU:
2-Hydroxyibuprofen
References