copd 환자
유병률이 10%가 넘는다.
그런데 비가역적인 진행성 질환이라고 현대의학에서는 말하고 있음.
건생병사 4과제 + 발효 프로그램으로
가역적 호전이 가능함을 확인하고 있음.

https://www.mayoclinicproceedings.org/article/S0025-6196%2821%2900240-8/fulltext
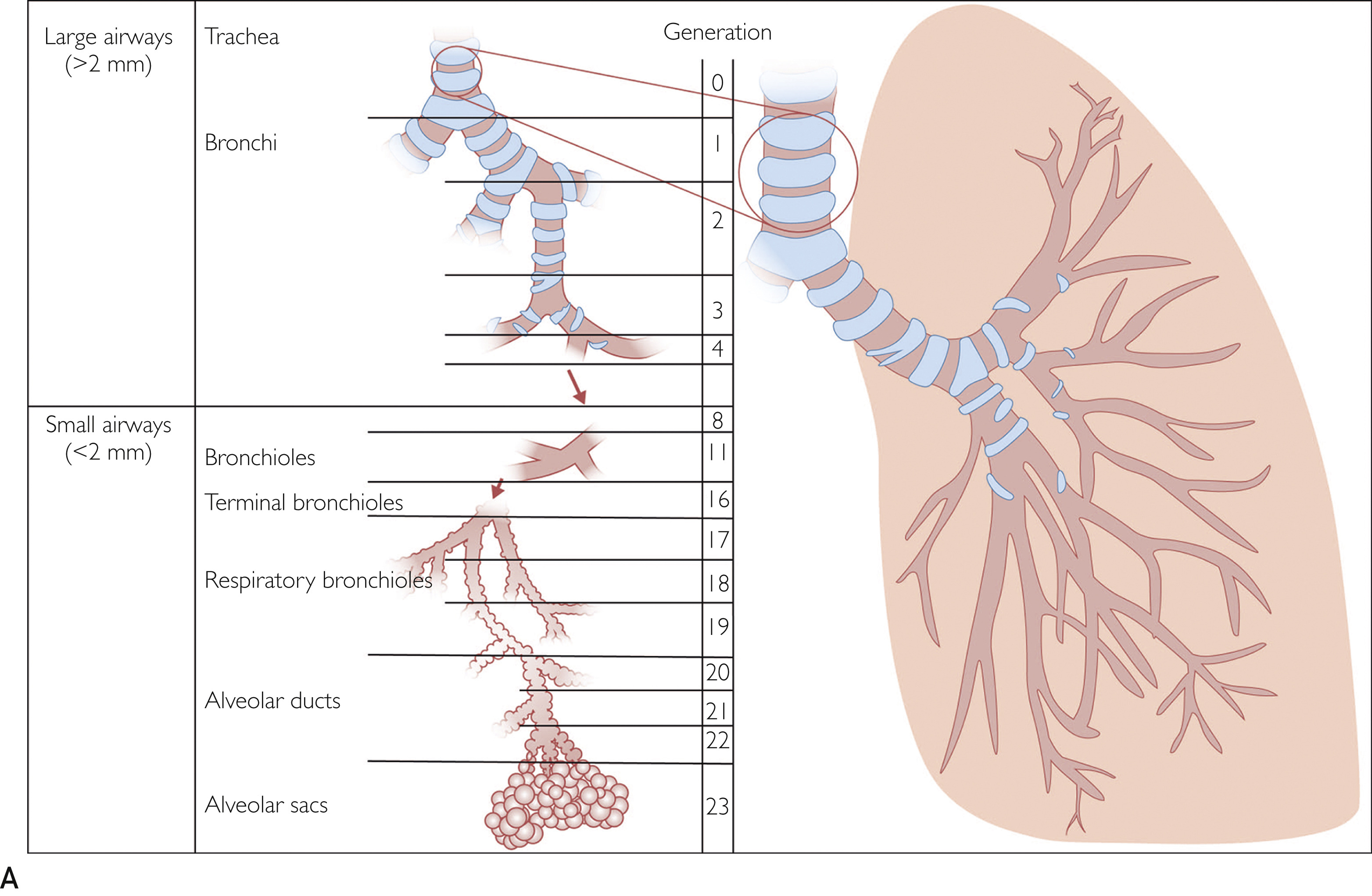
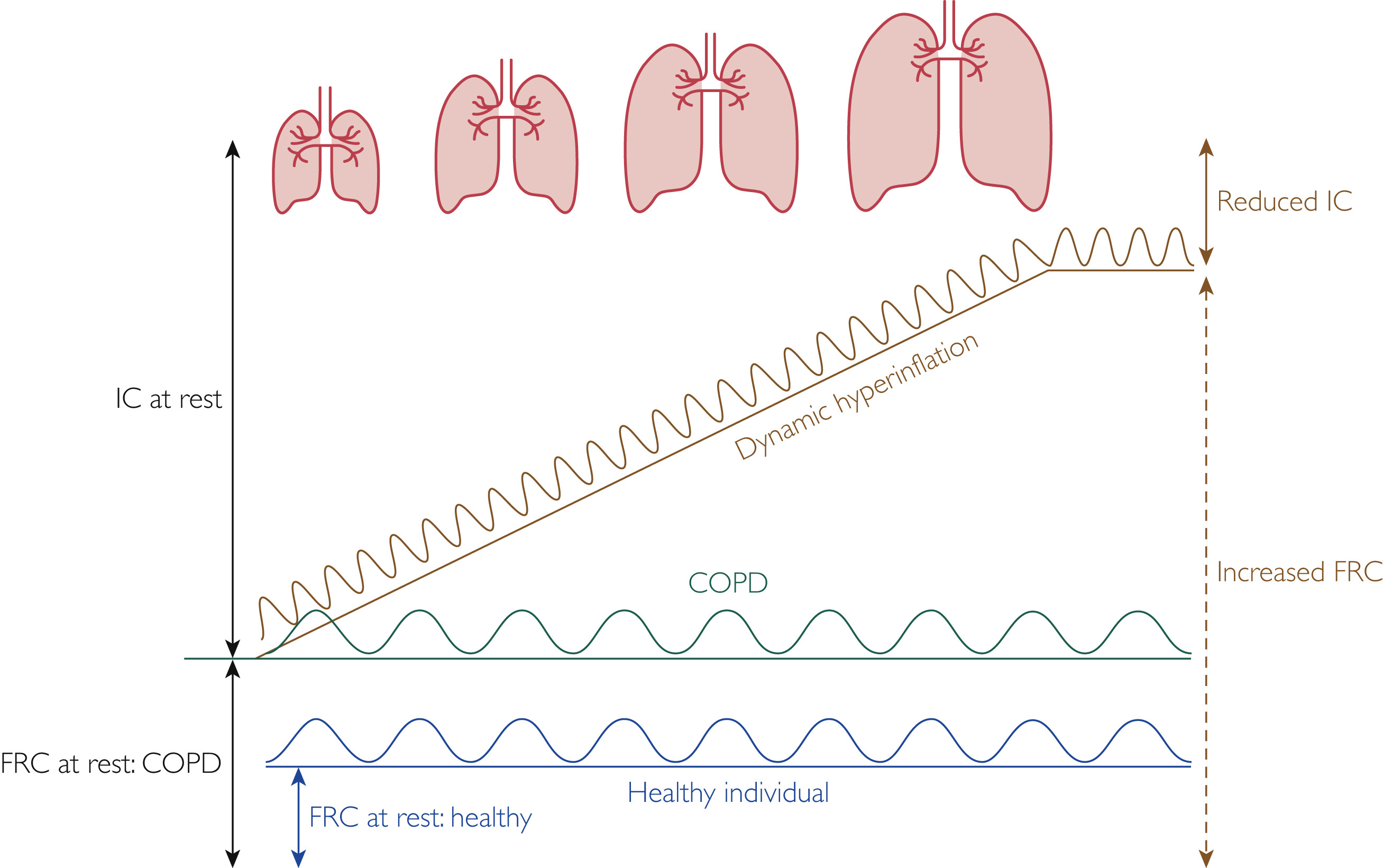
그림 구성 요소 설명
|
(Dynamic hyperinflation 동적 과팽창)
- 정의: 기도 폐쇄로 인해 호기 시 공기가 완전히 배출되지 않고 폐에 갇히는 현상(air trapping). 특히 운동이나 호흡량 증가 시 발생하며, **end-expiratory lung volume (EELV, 호기 말 폐용량)**이 정상 휴식 수준 이상으로 일시적으로 증가합니다.
- 기전: COPD에서 기도 저항 증가와 폐 탄성 감소로 호기 시간이 부족해지며, 빠른 호흡(운동 시)으로 악화됩니다. 이는 static hyperinflation(정적 과팽창, 휴식 시 이미 존재) 위에 추가로 발생합니다.
- 임상적 영향:
- 호흡 근육에 부하 증가 → 호흡 곤란 심화.
- tidal volume(일회호기량) 확장이 제한되어 운동 내성 저하.
- 심혈관계 영향(심박출량 감소 가능).
- 측정: 운동 중 inspiratory capacity(IC) 반복 측정으로 확인 (IC 감소 = dynamic hyperinflation).

IC at rest (휴식 시 Inspiratory Capacity, 흡기 용량)
- 정의: 정상 호기 말(휴식 시)에서 최대 흡기까지 들이마실 수 있는 공기량 (IC = TLC - FRC, 총폐용량 - 기능적 잔기량).
- COPD에서의 특징: 휴식 시 이미 폐 과팽창으로 FRC가 증가해 IC가 정상보다 낮습니다. 이는 static hyperinflation을 간접적으로 반영합니다.
- 임상적 의미: 휴식 IC가 낮을수록 COPD 중증도가 높고, 사망 위험 및 악화 위험이 증가합니다. 운동 시 추가 IC 감소(due to dynamic hyperinflation)를 예측하는 지표입니다.
Reduced IC (감소된 IC)
- 원인: Dynamic hyperinflation으로 EELV/FRC 증가 → 흡기 가능한 공간(IC)이 줄어듭니다. 운동 중 IC가 더 감소하면 tidal volume 확장이 제한되어 "no room to breathe" 상태가 됩니다.
- 임상적 영향:
- 호흡 곤란 급증 (dyspnea의 주요 원인).
- 운동 능력 저하 (peak ventilation 제한).
- 기도 폐쇄가 심할수록 IC 감소가 뚜렷합니다.
- 치료 효과: 기관지확장제(예: tiotropium)로 IC 증가 → dynamic hyperinflation 감소 및 증상 개선.
Increased FRC (증가된 Functional Residual Capacity, 기능적 잔기량)
- 정의: 정상 호기 말에 폐에 남아 있는 공기량 (FRC = RV + ERV, 잔기량 + 예비호기량).
- COPD에서의 특징: 폐 탄성 감소(emphysema)와 air trapping으로 FRC가 증가합니다.
- 휴식 시: static hyperinflation.
- 운동 시: dynamic hyperinflation으로 추가 증가.
- 임상적 의미: FRC 증가 → 폐 과팽창 → 흡기 근육 불리한 위치(기능 저하) → 호흡 노력 증가.
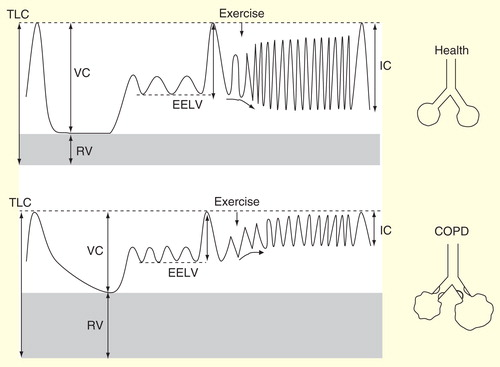
용량 정상 COPD (과팽창 시 ) 설명
| TLC (총폐용량) | 정상 | 증가 또는 정상 | 폐 파괴로 증가 가능 |
| FRC (기능적 잔기량) | 정상 | 증가 (static + dynamic) | air trapping 주요 원인 |
| IC (흡기 용량) | 정상 | 감소 (휴식 및 운동 시) | 흡기 여유 공간 줄음 |
| RV (잔기량) | 정상 | 증가 | 공기 배출 불완전 |

COPD의 유병률 (Prevalence)
COPD의 유병률은 지역, 연령, 흡연 및 환경 요인에 따라 다양하며,
최근 연구에서는 저소득 및 중소득 국가(LMICs)와 여성에서의 증가 추세가 강조되고 있다.
전 세계적으로 노화 인구와 지속적인 위험 요인 노출로 인해
2050년까지 환자 수가 크게 증가할 것으로 예상된다.
- 2019 JAMA Review (Diagnosis and Outpatient Management of COPD): 미국 성인 3천만 명(12%)이 COPD를 앓고 있으며, 매년 의사 방문의 3.2%를 차지한다. 이는 진단 및 치료의 변화가 빠르게 일어나는 가운데, 주로 1차 진료 의사에 의해 진단된다.
- 2023 JAMA Network Open (Global Burden of COPD Through 2050): 2020년 기준 전 세계 25세 이상 성인 중 유병률 10.6%(4억 8천만 명), 남성 13.4%, 여성 7.8%. 2050년까지 23% 증가하여 5억 9천 2백만 명(유병률 9.5%)으로 예상되며, 여성(47.1% 증가)과 LMICs(두 배 이상 증가)에서 가장 큰 성장. 사하라 이남 아프리카에서 2050년 유병률 15.1%로 최고.
- 2024 GOLD Report (Global Initiative for Chronic Obstructive Lung Disease 2025 Update): 성인 40세 이상에서 글로벌 유병률 10.3%(95% CI 8.2–12.8%), 남성 11.8%, 여성 8.5%, 비흡연자 3–11%. LMICs에서 비흡연 요인(대기 오염 등)이 50% 이상 차지. 2050년까지 23% 증가하여 약 6억 명 예상, 여성 및 LMICs에서 더 급증. 지역별: 라틴 아메리카 도시 7.8–19.7%, PRISm(보존비율 저하 spirometry) 7.1–11%.
- 2025 Lancet Respiratory Medicine (The Global Burden of COPD: Epidemiology and Effect of Prevention Strategies): 여성 유병률 증가, 남성 감소 추세로, 여성 흡연 증가가 원인. 예방 전략(금연, 대기 오염 감소)이 중요.
https://jamanetwork.com/journals/jamanetworkopen/fullarticle/2812622

COPD(만성 폐쇄성 폐질환)는 전 세계적으로 주요 건강 및 경제적 부담을 초래하는 질환입니다. 기존 연구는 현재 유병률에 초점을 맞췄으나, 장기 예측이 자원 계획에 필요합니다. 이 연구는 위험 요인(흡연, 대기 오염 등)을 고려해 2050년까지 글로벌 COPD 부담을 예측합니다. 모델링 연구 결과, 2020년부터 2050년까지 25세 이상 인구에서 COPD 사례가 23% 증가해 약 6억 명에 달할 것으로 예상되며, 여성(47.1% 증가)과 저·중소득 국가(LMICs)에서 가장 큰 증가가 나타납니다. 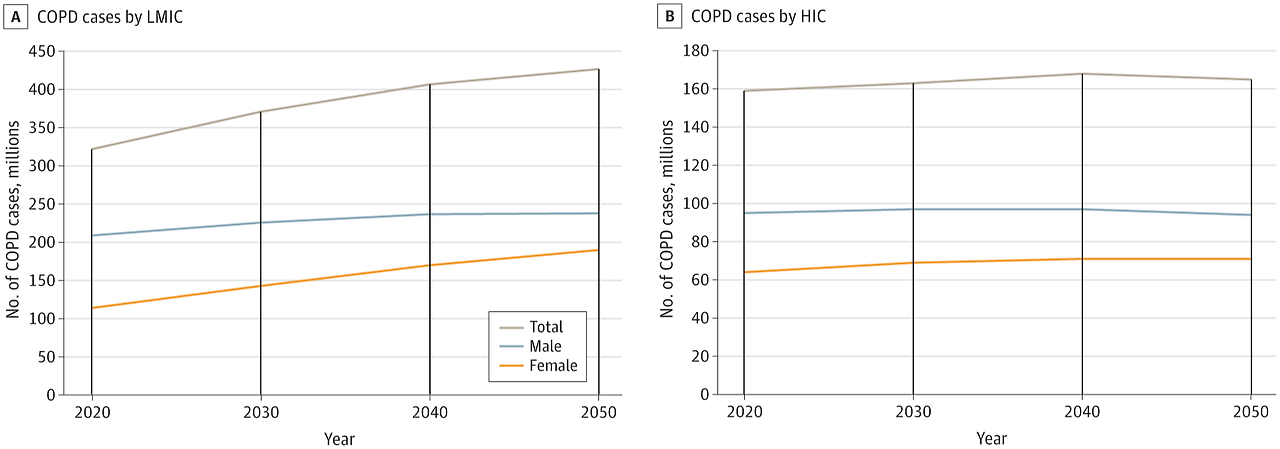 |
| 게재일: 2025년 2월 (Volume 13, Issue 2, Pages 148-160) DOI: 10.1016/S2213-2600(24)00413-2 저자: Chao Li, Huihuan Luo, et al. (중국 및 국제 연구팀) 배경 (Background) 만성 폐쇄성 폐질환(COPD)은 전 세계적으로 주요 사망 및 장애 원인으로, 흡연 외에 장기적인 대기 오존(O₃) 노출이 COPD 위험을 증가시킨다는 증거가 축적되고 있습니다. 그러나 오존으로 인한 COPD의 글로벌 부담을 체계적으로 평가한 연구는 부족했습니다. 이 연구는 Global Burden of Disease (GBD) 2019 데이터를 활용해 1990~2019년 동안 대기 오존 오염으로 인한 COPD의 사망, 장애조정생존연수(DALYs), 유병률 등을 추정합니다.  |

만성 폐쇄성 폐질환(COPD)은 주요 사망 원인으로, 고체 연료(장작, 숯 등)로 인한 가정 내 대기 오염(Household Air Pollution, HAP)이 중요한 위험 요인입니다. 특히 개발도상국에서 여성과 어린이에게 큰 영향을 미치지만, 1990~2019년 동안 HAP 귀인 COPD 부담의 글로벌 추이를 체계적으로 분석한 연구는 부족했습니다. 이 연구는 GBD 2019 데이터를 활용해 HAP(고체 연료 사용)로 인한 COPD 사망, DALYs(장애조정생존연수), 유병률 등을 평가합니다.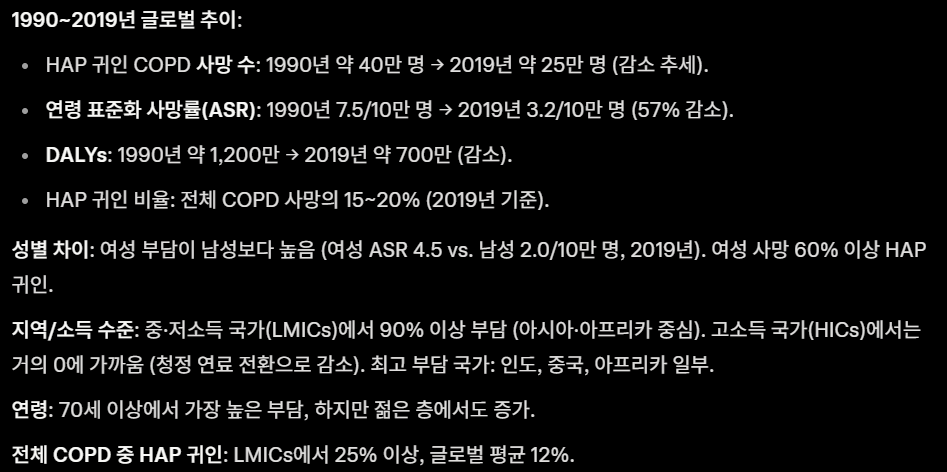 |
https://www.nature.com/articles/s41598-025-20056-z

| 이 논문은 **가정 내 대기 오염(Household Air Pollution, HAP)**의 건강 영향을 재고하는 에디토리얼로, Scientific Reports의 특집 컬렉션("Household Air Pollution" 컬렉션)을 소개합니다. HAP는 환경 보건 정책에서 오랫동안 간과되었으나, 특히 저·중소득 국가(LMICs)에서 가정이 가장 큰 대기 오염 노출 장소라는 증거가 명확해졌습니다. 선진국 사람들의 경우 하루 90% 이상을 실내에서 보내고, 그중 2/3 이상이 가정에서 보내며, 실내 공기에는 미세먼지(PM), 휘발성 유기화합물(VOCs), 일산화탄소, 이산화질소, 생물학적 오염물 등이 야외보다 높은 농도로 존재합니다. 건강 영향으로는 천식, COPD, 심혈관 질환, 폐암 위험이 증가하며, 어린이·노인·기저질환자 등 취약 계층이 더 위험합니다. COVID-19 팬데믹은 환기 부족과 장기 실내 체류가 공기 전파 위험을 악화시킨다는 점을 강조했습니다. 이 컬렉션은 6개의 최근 연구를 모아 가정 활동(요리, 청소, 난방 등)에서 발생하는 배출, 실내 오염물 농도, 건강 위험을 조사합니다. 환경 공학, 에어로졸 과학, 역학, 공중 보건을 아우르며, 실내 공기 질 개선이 필요하고 실현 가능하다는 점을 보여줍니다.
|
COPD의 예후 (Prognosis)
COPD 예후는 중증도, 동반질환, 악화 빈도, 폐 기능 저하 등에 의해 좌우되며, 연간 3백만 명 사망(전 세계 6%)을 초래한다. GOLD 분류에 따라 사망률과 악화 위험이 크게 다르며, 흡연 중단, 재활, 산소 요법 등이 생존을 개선한다.
- 2019 JAMA Review: 사망 원인 4위(미국 연간 12만 6천 명). 휴식 시 저산소증(SpO₂ <89%) 환자에서 보충 산소가 생존 향상. 장기 기관지 확장제, 흡입 스테로이드, 폐 재활이 증상 감소, 기능 최적화, 악화 빈도 저하.
- 2025 JoGH (Prognostic Risk Profiling in COPD Using GOLD 2023 ABE): 대만 3만 8천 928명 코호트에서 GOLD A군 30.2%, B군 46.4%, E군 23.5%. E군에서 중증 기류 제한(11.8%), 동반질환 점수 높음(CCI ≥4: 15.6%), 악화 빈도 높음(44% ≥2회). 5년 전체 사망률 14.3%(A군 8.4%, E군 22.6%). A군 대비 B군 HR 1.332, E군 HR 1.727. 중증 악화 위험: B군 HR 1.407, E군 HR 2.127. 주요 동반질환(암, 심부전, 치매 등)이 사망 위험 증가.
- 2024 GOLD Report: 연간 사망 3백만 명(90% LMICs), DALYs 7천 4백 4십만(1990–2019 25.7% 증가). 예측 인자: 연령, 동반질환(CVD, 당뇨, 폐암 등), 저하 FEV1, 급속 폐 기능 저하, BODE 점수, 폐 고혈압, 저체중, 빈번 악화(≥2회/년). 흡연 중단(5년 위험 저하), 장기 산소 요법(중증 저산소증 개선), NIV(만성 고이산화탄소혈증 저하), 삼중 요법(악화 감소). 악화: 감염, 오염 등 유발, 빈번 시 사망 위험 ↑(입원 후 50% 5년 사망).
COPD의 질병기전 (Disease Mechanisms/Pathogenesis)
COPD는 유전자-환경 상호작용(GETomics)으로 인한
만성 염증, 산화 스트레스, 폐 노화 가속화로 특징지어지며,
기도 제한, 만성 기관지염, 폐기종을 유발한다.
염증 세포 하위유형(호중구, 호산구 등)이 주요 역할을 하며, 노화 관련 경로가 새로운 치료 타겟으로 부상.
- 2019 JAMA Review: 불완전 가역적 호기 기류 제한으로, spirometry로 진단. 중증도, 증상, 악화 빈도에 따라 치료.
- 2024 Respiratory Investigation (Unveiling Mechanisms of Lung Aging in COPD): COPD는 폐 노화 가속화로, 60세 이상 유병률 높음. 산화 스트레스(담배 연기, 만성 염증 유발), 세포 노화(p21, p16, p53 경로로 세포 주기 정지) 핵심. 노화 세포가 SASP(염증 단백질 분비) 생산, 면역 반응 변화. 폐 구조/기능/재생 저하, 동반질환 유발. 노화 타겟 치료가 새로운 치료 가능성 제시.
- 2025 International Journal of Chronic Obstructive Pulmonary Disease (Unraveling the Immune Landscape of COPD): 만성 자극(연기, 오염)으로 상피 손상, TSLP/IL-33/IL-25/ROS/CXCL8 방출. 염증 세포: 호중구(CXC 케모카인 유도, NETs/ROS/프로테아제 방출, 조직 손상/폐기종); 림프구(CD8⁺ T 세포 IFN-γ/TNF-α, Th17 IL-17, B 세포 자가항체); 단핵구(대식세포 분화, IL-6/TNF-α/MMPs); 호산구(IL-5/13/33/CCR3, 리모델링/악화, ≥300 cells/μL 예측); 기저구(CCL5, IL-4/MMP-12, 폐기종 촉진). 이형성: 호중구성(스테로이드 저항), 호산구성(Th2), 림프구성(면역 조절 장애). 프로테아제-항프로테아제 불균형, 산화 스트레스, 리모델링 유발.
- 2024 GOLD Report: GETomics(유전자-환경, 생애 주기), 산화 스트레스(H2O2, 8-isoprostane), 프로테아제 불균형(엘라스틴 파괴, AATD), 섬유증, 혈관 변화, 만성 점액 과분비(MUC5AC/B, 기침/객담/악화). 소기도 질환(저항) + 실질(폐기종, 반동 상실); 지속 염증(대식세포/호중구/림프구), 자동항원/미생물군. 조기 요인(저체중 출생, 어린 시 감염, dysanapsis), 천식/과민성(위험 증가).

| 초록 및 배경 만성 폐쇄성 폐질환(COPD)은 기류 제한과 기도 구조 변화(만성 기관지염, 소기도 질환, 폐기종)를 특징으로 하는 만성 염증성 폐 질환입니다. 전 세계 사망 원인 3위로, 현재 치료법으로는 완치가 불가능합니다. COPD 유병률은 60세 이상에서 높아 **가속화된 폐 노화(accelerated lung aging)**로 간주됩니다. 자연스러운 폐 노화는 분자·세포·생리적 변화로 폐 구조·기능·재생 능력 저하와 면역 반응 약화를 초래하며, 이는 COPD 같은 질환으로 이어질 수 있습니다. 가속화된 폐 노화 메커니즘은 복잡하며, 담배 연기 등에 의한 산화 스트레스 증가, 만성 염증, 기도 내 노화 세포(senescent cells) 증가로 구성됩니다. 세포 노화(cellular senescence)는 세포 분열 정지로, p21CIP1, p16INK4, p53 활성화와 함께 염증성 분비물(SASP: senescence-associated secretory phenotype)을 유발합니다. 이 리뷰는 폐 노화 메커니즘의 최근 발전을 설명하고, COPD 병태생리 및 동반질환에 미치는 영향을 논의합니다. 노화 메커니즘 이해와 타겟팅은 COPD 및 다른 노화 관련 질환의 신규 치료 개발로 이어질 수 있습니다. 방법 이 논문은 리뷰 기사로, 기존 문헌을 종합하여 노화 바이오마커, 정상 폐 노화 메커니즘, COPD에서의 역할을 분석합니다. 실험적 방법은 없음. 주요 결과
COPD의 가속화된 폐 노화는 유전·후성유전·환경 요인에 의해 발생합니다. 노화 바이오마커와 생물학적 나이 정의로 COPD 및 동반질환 조기 탐지 가능. 장기 간 상호작용(폐 노화가 심혈관 노화 영향) 연구 필요. 치료 타겟: senotherapeutics – senomorphics(mTOR 억제제 rapamycin: COPD 폐 세포에서 노화/SASP 방지), senolytics(navitoclax: 노화 세포 제거, 마우스 모델 유익하나 위험성 있음). 추가 연구로 재생 및 동반질환 효과 검증 필요. 그림 설명
 DDR은 DNA Damage Response의 약자로, DNA 손상 반응을 의미합니다 이 다이어그램은 산화 스트레스(oxidative stress)가 세포 노화(cellular senescence)를 유발하는 경로를 보여주고 있으며, DDR은 산화 스트레스로 인한 DNA 손상이 지속되면 활성화되어 세포 주기 정지(cell cycle arrest, 예: ↑p53, ↑p21, ↑p16)를 유발하고, 결국 세포 노화의 핵심 특징인 SA-β-gal 활성 증가, SASP 분비, NF-κB 활성화 등을 초래합니다. DDR 경로는 세포 노화 연구에서 매우 중요한 메커니즘으로, DNA 손상을 감지하고 복구를 시도하다가 실패하면 영구적인 세포 주기 정지를 유도하는 역할을 합니다. 다이어그램의 왼쪽 아래 부분에서 Oxidative stress → ↓Nrf2 → DDR → FOXO3, p53/p21/p16 등으로 이어지는 흐름이 이를 나타냅니다. 정밀 의학 및 바이오마커 함의 노화 바이오마커(텔로미어 길이, p16/p21 등)와 multi-omics 접근으로 건강 노화 vs. 병적 노화 구분 가능. 이는 COPD 조기 진단과 개인화 치료를 촉진하며, 세포 노화 타겟팅(senolytics 등)이 COPD 및 노화 관련 질환의 정밀 의학으로 발전할 잠재력 있습니다. 이 리뷰는 COPD를 노화 질환으로 재정의하며, 이전 COPD proteomics 논문과 연결해 노화 메커니즘 타겟팅의 치료 가능성을 강조합니다. |
Chin Med J Pulm Crit Care Med
. 2024 Sep 17;2(3):133–141. doi: 10.1016/j.pccm.2024.08.007
Unveiling mechanisms of lung aging in COPD: A promising target for therapeutics development
- Author information
- Article notes
- Copyright and License information

PMCID: PMC11471098 PMID: 39403409
Abstract
Chronic obstructive pulmonary disease (COPD) is a chronic inflammatory lung disease characterized by airflow limitation and changes in airway structures that can lead to chronic bronchitis, small airway diseases, and emphysema. COPD is the 3rd leading cause of death worldwide and despite current research, there are no curative disease treatments for COPD. As the prevalence of COPD is higher in people over 60 years old than in younger age groups, COPD is considered a condition of accelerated lung aging. Natural lung aging is associated with molecular, cellular, and physiological changes that cause alteration in lung structure, in lung function and regeneration, and decreased immune system response that could lead to lung disease like COPD. Mechanisms of accelerated lung aging are complex and composed by increased oxidative stress induced by exposure to cigarette smoke, by chronic inflammatory processes, and increased number of senescent cells within the airways. Cellular senescence is the cessation of cell division after a finite number of proliferation cycles or in response to cell stressors, such as oxidative stress. Senescent cells show activation of the cell cycle regulators p21CIP1 (cyclin-dependent kinase inhibitor-1), p16INK4 (cyclin-dependent kinase inhibitor-2A), and p53 (cellular tumor antigen p53) that lead to cell cycle arrest. Senescent cells exhibit a change in their phenotype and their metabolic activity, along with the production of proinflammatory proteins collectively known as senescence-associated secretory phenotype (SASP). This review aims to describe recent developments in our understanding of aging mechanisms and how the acceleration of lung aging participates in COPD pathophysiology and comorbidities. Understanding and targeting aging mechanisms may result in the development of new therapeutics that could be effective for COPD and also for other age-related diseases.
만성 폐쇄성 폐질환(COPD)은
만성 기관지염, 소기도 질환 및 폐기종으로 이어질 수 있는
기류 제한 및 기도 구조 변화를 특징으로 하는 만성 염증성 폐질환이다.
chronic inflammatory lung disease characterized by
airflow limitation and changes in airway structures
that can lead to chronic bronchitis, small airway diseases, and emphysema
COPD는
전 세계적으로 세 번째 주요 사망 원인이며,
현재의 연구에도 불구하고 COPD에 대한 완치적 치료법은 존재하지 않습니다.
COPD 유병률은
젊은 연령층보다 60세 이상에서 더 높기 때문에,
COPD는 가속화된 폐 노화 상태로 간주됩니다.
자연적인 폐 노화는 분자적, 세포적, 생리적 변화를 동반하며,
이는 폐 구조와 기능, 재생 능력의 변화 및 면역 체계 반응 감소를 초래하여
COPD와 같은 폐 질환으로 이어질 수 있습니다.
가속화된 폐 노화의 기전은 복잡하며, 담배 연기 노출로 인한
산화 스트레스 증가, 만성 염증 과정, 기도 내 노화 세포 수 증가 등으로 구성됩니다.
세포 노화는
유한한 증식 주기 후 또는 산화 스트레스와 같은 세포 스트레스 요인에 대한 반응으로
세포 분열이 중단되는 현상이다.
노화 세포는
세포 주기 조절인자인
p21CIP1(사이클린 의존성 키나제 억제제-1),
p16INK4(사이클린 의존성 키나제 억제제-2A),
p53(세포 종양 항원 p53)의 활성화를 보여 세포 주기 정지를 초래한다.
노화 세포는 표현형과 대사 활동의 변화를 보이며,
노화 관련 분비 표현형(SASP)으로 통칭되는 전염증성 단백질을 생성합니다.
본 리뷰는
노화 메커니즘에 대한 최신 이해와
폐 노화 가속화가 만성폐쇄성폐질환(COPD) 병리생리학 및 동반 질환에 어떻게 관여하는지
설명하는 것을 목표로 합니다.
노화 메커니즘을 이해하고 표적화하면
COPD뿐만 아니라 다른 노화 관련 질환에도
효과적인 새로운 치료제 개발로 이어질 수 있다.
Keywords: Chronic obstructive pulmonary disease, Aging, Cellular senescence, Senescence-associated secretory phenotype, MicroRNAs
Introduction
Chronic obstructive pulmonary disease (COPD) is a progressive respiratory condition characterized by airflow limitation, persistent inflammation, and irreversible parenchymal lung tissue destruction.1 COPD is the 3rd leading cause of death worldwide2 and despite many years of research, there are currently no curative treatments for COPD. The acceleration of lung aging has been identified as an essential driver of COPD pathophysiology. The molecular drivers of aging in COPD are multifactorial and they still need to be fully identified. In this review, we provide an overview of the aging biomarkers, of the mechanisms of normal aging in the lungs, and how these mechanisms play a role in COPD pathophysiology.
만성 폐쇄성 폐질환(COPD)은
공기 흐름 제한, 지속적인 염증,
그리고 돌이킬 수 없는 폐 실질 조직 파괴를 특징으로 하는 진행성 호흡기 질환이다.1
COPD는
전 세계적으로 3번째 주요 사망 원인이다.2
수년간의 연구에도 불구하고,
현재 COPD에 대한 치료법은 존재하지 않는다.
폐 노화 가속화는
COPD 병리생리학의 핵심 유발 요인으로 확인되었다.
COPD에서 노화를 유발하는 분자적 요인은
다인성이며 아직 완전히 규명되지 않았다.
본 리뷰에서는
노화 생체표지자, 폐의 정상 노화 메커니즘,
그리고 이러한 메커니즘이 COPD 병리생리학에서 어떻게 작용하는지에 대한 개요를 제시한다.
Mechanisms of normal aging
Aging is currently defined as “the process of accumulation of consequences of life, such as molecular and cellular damages, that leads to functional decline, chronic diseases, and ultimately mortality”.3 Aging not only affects humans, animals, plants, but also unicellular organisms subjected to environmental stresses such as bacteria, protozoa, and fungi.4 Aging is a physiological process, however, its mechanisms contribute to the development of diseases,5 and has major consequences for the burden of health care.6 In Australia, over 80% of patients over 75 years old have multimorbidity,7 and in a Mediterranean cohort of people over 85 years, 95% have multimorbidity.8 Thus, multidisciplinary studies and new models are critically needed to uncover the mechanisms of aging and age-related diseases.
정상 노화의 메커니즘
노화는
현재 “분자 및 세포 손상과 같은 생명의 결과들이 축적되어
기능 저하, 만성 질환, 그리고 궁극적으로 사망으로 이어지는 과정”으로 정의됩니다. 3
노화는
인간, 동물, 식물뿐만 아니라
박테리아, 원생동물, 곰팡이와 같은 환경 스트레스를 받는 단세포 생물에도 영향을 미친다.4
노화는 생리적 과정이지만,
그 기전은 질병 발생에 기여하며,5
의료 부담에 중대한 결과를 초래한다. 6
호주에서는 75세 이상 환자의 80% 이상이 다중질환을 앓고 있으며,7
지중해 지역 85세 이상 코호트에서는 95%가 다중질환을 가지고 있다.8
따라서
노화 및 노화 관련 질환의 기전을 규명하기 위해서는
다학제적 연구와 새로운 모델이 절실히 필요하다.
Biomarkers of aging
As the number of people over 80 years of age will quadruple in the next few decades, reliable biomarkers for age-related diseases have become critical tools to target aging mechanisms and, to develop new therapeutics.9 Aging mechanisms are highly complex and heterogeneous. Not all individuals age in the same way or at the same rate. These inter-individual differences have led to the definition of chronological age—the individual's age defined by the time elapsed since birth, and biological age—the level of biological changes, such as molecular and cellular damages accumulation.3 Numerous studies have attempted to discover biomarkers of aging on different levels: molecular and cellular biomarkers, systemic markers, multi-omics approaches, or non-molecular biomarkers.10 In 2023, López-Otin et al11,12 have defined 12 hallmarks of aging classified into three categories [Fig. 1]: the primary hallmarks are the primary damages that accumulate within the genome and organelles of the cells, the antagonistic hallmarks that reflect the consequences of the damages accumulated within the cells, and the integrative hallmarks that arise when the accumulation of the primary and antagonistic damages cannot be compensated within the organ.12
노화의 생체표지자
향후 수십 년간 80세 이상 인구가 4배로 증가함에 따라,
노화 관련 질환에 대한 신뢰할 수 있는 생체표지자는
노화 메커니즘을 표적화하고 새로운 치료법을 개발하는 데 핵심적인 도구가 되었습니다.9
노화 메커니즘은
매우 복잡하고 이질적입니다.
모든 개인이
동일한 방식으로 또는 동일한 속도로 노화하는 것은 아닙니다.
이러한 개인 간 차이로 인해
생물학적 연령(출생 이후 경과한 시간으로 정의되는 개인의 나이)과
생물학적 나이(분자 및 세포 손상 축적과 같은 생물학적 변화 수준)가 정의되었습니다.3
chronological age -- biological age
달력나이-- 생물학적 건강나이

수많은 연구에서
분자 및 세포 생체표지자,
전신 표지자,
다중 오믹스 접근법 또는 비분자 생체표지자 등
다양한 수준에서 노화 생체표지자를 발견하려는 시도가 이루어졌습니다.10
노화의 12가지 특징을 세 가지 범주로 분류하여 정의하였다[그림 1]:
주요 특징은 세포의 게놈과 세포소기관 내에서 축적되는 주요 손상,
세포 내에서 축적된 손상의 결과를 반영하는 대립적 특징,
그리고 주요 및 대립적 손상의 축적이 장기 내에서 보상될 수 없을 때 발생하는 통합적 특징이다.12
Fig. 1.
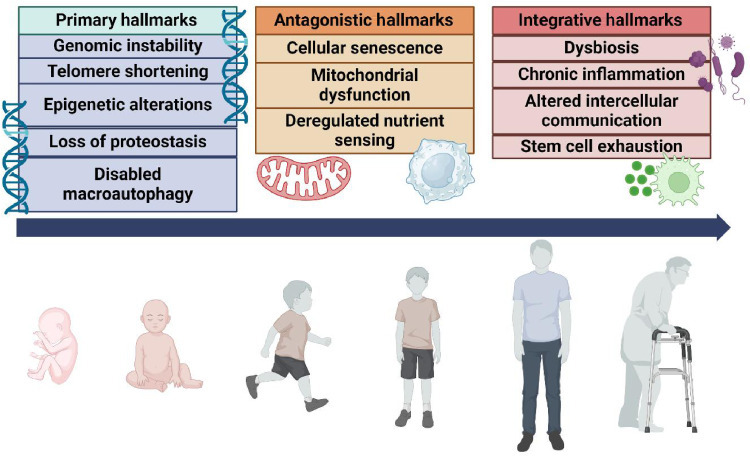
Hallmarks of aging. Twelve hallmarks of aging have been proposed and classified into 3 categories. The primary hallmarks are the primary damages that accumulate within the genome and organelles of the cells. They compile genomic instability, telomere shortening, epigenetic alterations, loss of proteostasis, and disabled macroautophagy. The antagonistic hallmarks reflect the consequences of the damages accumulated within the cells including cellular senescence, mitochondrial dysfunction and deregulated nutrient sensing. Finally, the integrative hallmarks arise when the accumulation of the primary and antagonistic damages cannot be compensated within the organ. They include dysbiosis, chronic inflammation, altered intercellular communication, and stem cell exhaustion.
노화의 특징. 12가지 노화 특징이 제안되었으며 3가지 범주로 분류된다.
1차 특징은
세포의 게놈과 세포소기관 내에 축적되는 1차 손상이다.
여기에는 게놈 불안정성, 텔로미어 단축, 후성유전적 변화, 단백질 항상성 상실, 대식작용 장애가 포함된다.
대립적 특징은
세포 노화, 미토콘드리아 기능 장애, 영양 감지 조절 이상 등 세포 내 축적된 손상의 결과를 반영한다.
마지막으로 통합적 특징은
일차적 및 대립적 손상의 축적이 장기 내에서 보상될 수 없을 때 발생한다.
여기에는 장내 미생물 불균형, 만성 염증, 세포 간 통신 변화, 줄기세포 고갈이 포함된다.
Cellular senescence and its consequences in aging
Aging is characterized by the overall decline of cell functions. The genome is instable with increasing levels of mutations, shortening of telomeres, and epigenetic modification. The proteostasis network has its functions depleted, which leads to the accumulation of misfolded proteins.
Telomeres are portions at the end of linear chromosomes that shorten by 50–200 bp at each somatic division.13 There is a correlation between age and the relative telomere length across tissue: an analysis of telomere length in 25 tissues collected from 952 donors aged from 20 years to 70 years by the genotype-tissue expression (GTEx) showed telomere length was variable depending on the tissue and was inversely correlated with age in 23 tissues out of 25.14 The shortening of telomeres activates the DNA damage response (DDR) which can lead to apoptosis, oncogenic transformation of cells, and to cellular senescence.15
Cellular senescence is a stable and terminal state of growth arrest in which cells are unable to proliferate despite optimal growth and mitogenic conditions. Cellular senescence occurs in response to many different triggers, including DNA damage, telomere shortening, oncogene activation, and organelle stress.16 Cellular senescence can be beneficial especially during the early stages of development, however, the accumulation of senescent cells within tissues and organs promotes the overall aging of the organism.17 Increased number of senescent cells has been reported in older mice and humans. The expression of p21CIP1, p16INK4, and the activity of β-galactosidase are increased in 120-week mice compared to 15-week mice.18 Cellular senescence is initiated and regulated by specific signaling pathways. Telomere attrition or DNA damage activates the DDR and the canonical p53 pathway, whereas cell stress, like oxidative stress, causes cellular senescence via p16INK4–Rb pathway. P53 and p16INK4 may interact and activate the cyclin-dependent kinase inhibitor p21CIP1 which inhibits the formation of the CDK4/6–cyclin D complex, leading to the cell cycle arrest.19 A lysosomal enzyme called senescent-associated (SA)-β-galactosidase is increased in senescent cells which is easily detectable by histochemical staining.17 Senescent cells release a combination of proinflammatory cytokines, chemokines, proteases, growth factors, and bioactive lipids collectively known as senescence-associated secretory phenotype (SASP). The secretion of SASP is mainly controlled by the activation of p38 mitogen-activated protein kinase (MAPK) and Janus kinase (JAK) that leads to the activation of the nuclear factor κB (NF-κB) pathway.20 Other pathways and other upstream factors can also induce cellular senescence and the secretion of SASP. The phosphoinositide-3-kinase (PI3K)-mammalian target of rapamycin (mTOR) pathway can act as a switch where the activation of mTOR causes cellular senescence but inhibition leads to quiescence.21 Moreover, the activation of mTOR induces the production of SASP.22 SASP release leads to low-grade chronic inflammation called inflammaging.23 Immune cells play a crucial role in recognizing and eliminating senescent cells, however, with aging, intercellular communication is altered, and immune cells, in response to SASP, become senescent which leads to a phenomenon called immunosenescence.24 Peripheral blood mononuclear cells (PBMC) from people over 60 years exhibit higher activity of β-galactosidase compared to individuals in their 20s, particularly in CD8+ T cells among which 64% showed high senescence-associated β-galactosidase, associated with increased expression of p16INK4 and telomere damage.25
Cellular senescence is characterized by defective autophagy, defined as the catabolic process that enables the degradation and recycling of cellular components, including damaged organelles, pathogens, and protein aggregates.26 Autophagy is involved in the recognition and the sequestration of these components into the autophagosome. Mature autophagosomes are transported and fused with lysosomes, which leads to the formation of autolysosomes and the ultimate degradation of the cargo.26 Studies suggest that aging is associated with decreased autophagy. The expression of autophagy related 7 (ATG7) and the lipidation of microtubule-associated protein 1A/1B-light chain 3 (LC3) are significantly decreased in the muscle of 26-month-old mice compared to 10-month-old mice. Moreover, this decrease was also found in muscle biopsies of elderly men compared to young adults.27
Stem cells have essential roles in tissue development, renewal, and regeneration. Stem cells are tissue-specific, and they remain in a quiescent state within the tissue, for example, in skin, muscle, blood, bone, or brain. The hallmarks of aging, genome instability, epigenetic modification, increased cellular senescence, and subsequent low-grade inflammation, also affect stem cells. As a result, stem cells undergo a progressive decline in homeostatic and regenerative capacities.28 Transcriptomic analysis of muscle in older mice has shown that the increased number of senescent cells create a microenvironment, a niche that is inflamed and induces the arrest of stem cell proliferation and regeneration. This interaction between senescence and stem cells could be a therapeutic target as the inhibition of SASP production accelerates regeneration by stem cells in old and young mice.29
세포 노화와 노화에서의 그 결과
노화는 전반적인 세포 기능 저하로 특징지어진다.
게놈은 돌연변이 증가, 텔로미어 단축, 후성유전적 변형으로 불안정해진다.
단백질 항상성 네트워크의 기능이 고갈되어 잘못 접힌 단백질이 축적된다.
텔로미어는 선형 염색체 말단에 위치한 부분으로,
체세포 분열 시마다 50~200bp씩 단축됩니다.13
조직 전반에 걸쳐 상대적 텔로미어 길이와 연령 간 상관관계가 존재합니다:
유전자형-조직 발현 (GTEx)를 통해 수집한 20세부터 70세까지의 기증자 952명에서 채취한 25개 조직의 텔로미어 길이를 분석한 결과, 텔로미어 길이는 조직에 따라 변동성이 있었으며 25개 조직 중 23개 조직에서 연령과 역상관 관계를 보였습니다.14 텔로미어의 단축은 DNA 손상 반응(DDR)을 활성화시키며, 이는 세포 사멸, 세포의 발암성 변형 및 세포 노화로 이어질 수 있습니다.15
세포 노화는
최적의 성장 및 세포 분열 조건에도 불구하고
세포가 증식할 수 없는 안정적이고 최종적인 성장 정지 상태입니다.
세포 노화는
DNA 손상, 텔로미어 단축, 발암유전자 활성화, 세포소기관 스트레스 등
다양한 유발 요인에 대한 반응으로 발생한다.16
세포 노화는 특히 발달 초기 단계에서 유익할 수 있으나,
조직 및 장기 내 노화 세포의 축적은 유기체의 전반적인 노화를 촉진한다.17
노화 세포 수는 노화 마우스와 인간에서 증가하는 것으로 보고되었다.
120주령 쥐에서는 15주령 쥐에 비해
p21CIP1, p16INK4 발현과 β-갈락토시다아제 활성이 증가한다.18
세포 노화는
특정 신호 전달 경로에 의해 시작되고 조절된다.
텔로미어 소모 또는 DNA 손상은
DDR 및 표준 p53 경로를 활성화하는 반면,
산화 스트레스와 같은 세포 스트레스는 p16INK4–Rb 경로를 통해 세포 노화를 유발합니다.
p53과 p16INK4는 상호작용하여 사이클린 의존성 키나아제 억제제인 p21CIP1을 활성화할 수 있으며,
이는 CDK4/6-사이클린 D 복합체의 형성을 억제하여 세포 주기 정지를 초래합니다.19
노화 관련 (SA)-β-갈락토시다아제는
노화 세포에서 증가하며, 조직화학 염색을 통해 쉽게 검출할 수 있다.17
노화 세포는
전염증성 사이토카인, 케모카인, 프로테아제, 성장 인자 및 생리활성 지질의 조합을 방출하며,
이를 총칭하여 노화 관련 분비 표현형(SASP)이라 한다.
SASP 분비는
주로 p38 미토겐 활성화 단백질 키나제(MAPK)와 야누스 키나제(JAK)의 활성화에 의해 조절되며,
이는 핵인자 κB(NF-κB) 경로의 활성화를 유도합니다.20
다른 경로와 상류 인자들도 세포 노화와 SASP 분비를 유도할 수 있습니다.
포스포이노시타이드-3-키나제(PI3K)-mTOR 경로는
스위치 역할을 하여,
mTOR의 활성화는 세포 노화를 유발하지만 억제는 휴면 상태로 이끈다. 21
또한
mTOR 활성화는 SASP 생성을 유도한다.22
SASP 방출은 염증성 노화(inflammaging)라 불리는
저등급 만성 염증을 유발한다.23
면역 세포는 노화 세포를 인식하고 제거하는 데 중요한 역할을 하지만,
노화와 함께 세포 간 통신이 변화하고,
면역 세포는 SASP에 반응하여 노화되어 면역 노화(immunosenescence)라는 현상을 초래한다. 24
60세 이상 노인의 말초혈 단핵구(PBMC)는 20대에 비해 β-갈락토시다아제 활성이 높았으며,
특히 CD8+ T 세포에서 64%가 높은 노화 관련 β-갈락토시다아제 활성을 보였는데,
이는 p16INK4 발현 증가 및 텔로미어 손상과 연관되었다.25
세포 노화는
손상된 세포 소기관, 병원체, 단백질 응집체 등
세포 구성 요소의 분해 및 재활용을 가능하게 하는 이화 작용 과정인 오토파지 결핍이 특징이다.26
오토파지는
이러한 구성 요소를 인식하여 오토파고좀으로 격리하는 과정에 관여한다.
성숙한 오토파고좀은 리소좀으로 수송되어 융합되며,
이로 인해 오토리소좀이 형성되고
최종적으로 화물이 분해됩니다.26
연구에 따르면 노화는 오토파지 감소와 관련이 있는 것으로 보입니다.
26개월령 마우스의 근육에서는 10개월령 마우스에 비해 자가포식 관련 단백질 7(ATG7) 발현과 미세소관 관련 단백질 1A/1B-경쇄 3(LC3)의 지질화(lipidation)가 현저히 감소한다. 또한 이 감소는 젊은 성인에 비해 노년 남성의 근육 생검에서도 관찰되었다.27
줄기세포는
조직 발달, 재생 및 회복에 필수적인 역할을 한다.
줄기세포는
조직 특이적이며 피부, 근육, 혈액, 뼈 또는 뇌와 같은 조직 내에서 휴면 상태를 유지한다.
노화의 특징인
게놈 불안정성, 후생유전적 변형, 세포 노화 증가 및 후속적인 저등급 염증은
줄기세포에도 영향을 미친다.
결과적으로
줄기세포는
항상성 유지 및 재생 능력이 점진적으로 감소한다.28
노화 마우스 근육의 전사체 분석 결과, 증가된 노화세포 수가 염증이 발생한 미세환경(니치)을 조성하여 줄기세포 증식과 재생을 정지시키는 것으로 나타났다. 노화와 줄기세포 간의 이러한 상호작용은 SASP(노화세포 특이적 분비프로파일) 생산 억제가 노화 및 젊은 마우스에서 줄기세포에 의한 재생을 촉진한다는 점에서 치료 표적이 될 수 있다.29
Normal aging in the lungs
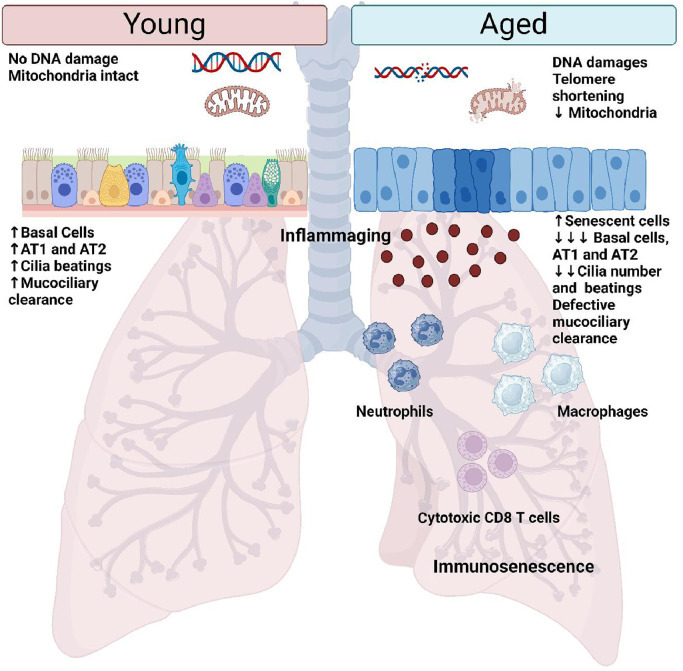
In healthy aging, the respiratory tract undergoes structural, cellular, and immunological changes [Fig. 2]. As a result, the remodeling of the lungs of the elderly leads to the decline of their pulmonary function.30 Lung functions measured with the forced expiratory volume in 1 s (FEV1), forced vital capacity (FVC), and peak expiratory flow rate (PEFR) reach their maximum capacity around 25 years old, and progressively decline with age (rates of FEV1 decline ranged from 17.7 mL/year to 46.4 mL/year).31
폐의 정상 노화
건강한 노화 과정에서 호흡기는
구조적, 세포적, 면역학적 변화를 겪습니다 [그림 2].
이로 인해 노인의 폐 재형성은
폐 기능 저하로 이어집니다. 30
1초 강제호기량(FEV1),
강제활력량(FVC),
최고호기유속(PEFR)으로 측정된 폐 기능은
약 25세에 최대치에 도달한 후 연령이 증가함에 따라 점진적으로 감소합니다(FEV1 감소율은 17.7mL/년에서 46.4mL/년 사이).31
forced expiratory volume in 1 s (FEV1),
forced vital capacity (FVC), and
peak expiratory flow rate (PEFR)
1. FVC (Forced Vital Capacity, 강제 폐활량)
|
지표 의미 주요 관련 질환 정상 범위 예시
| FVC | 폐의 총 용량 | 제한성 폐질환 | 남 4~5 L, 여 3~4 L |
| FEV1 | 첫 1초 동안 나오는 공기량 | 폐쇄성 폐질환 (COPD, 천식) | 남 3~4 L, 여 2~3 L |
| FEV1/FVC | 기도 폐쇄 정도 | <70% → 폐쇄성 | 70~80% 이상 |
| PEFR | 최대 순간 호기 속도 | 천식 모니터링 | 남 400~700 L/min |
Fig. 2.
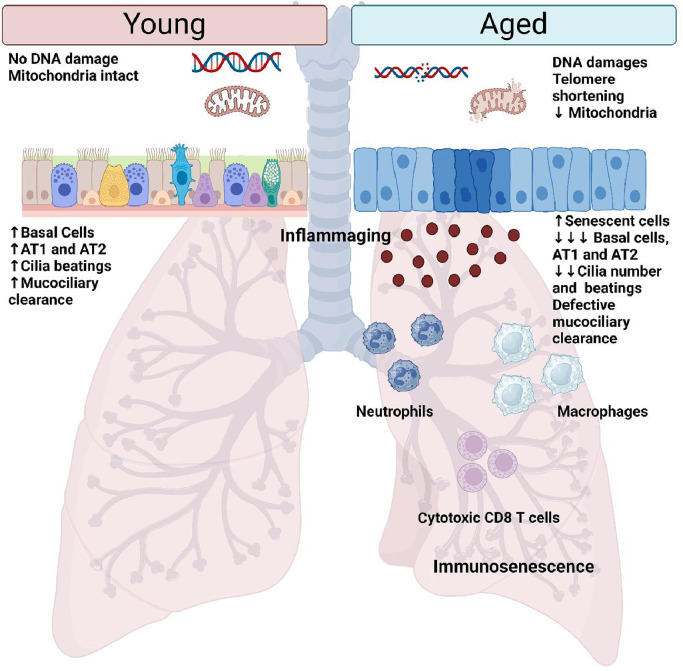
Normal aging of the lungs. Normal aging of the lungs is characterized by the accumulation of DNA damages and oxidative stress that leads to the accumulation of senescent cells, particularly within the small airways. As a result, the epithelium is defective and the number of basal cells, AT1 and AT2 cells diminished. Moreover, the defective mucociliary clearance due to the decreased number of cilia and their defective beating, leads to an increased susceptibility of the elderly to infection. Airway immune response is dysfunctional with aging and is characterized by the accumulation of inflammatory cells such as macrophages, neutrophils, and cytotoxic T cells, within the lungs. These immune cells have decreased functions and may present a senescent phenotype. Aged lungs are also characterized by the production of pro-inflammatory factors, by senescent cells and immune cells, which induce a low-grade inflammation of the lungs called inflammaging. AT1: Alveolar type 1 cell; AT2: Alveolar type 2 cell.
Structural changes in the lungs
The respiratory system is composed of the lungs, the thoracic cage, and the diaphragm, and each of these is modified by aging. Lung compliance decreases with age, which is due to a general loss of the elasticity of the respiratory system.32 Indeed, the size of the thoracic cavity and rib spaces can decrease because of bones weakening, and there is an association between the decreased pulmonary function and osteoporosis.33 Respiratory function is also impaired because of muscle atrophy and weakness associated with aging. Intercostal, inspiratory, and expiratory muscles, and the diaphragm lose their contractile capacity, their mass, and their strength with age.34,35 This contributes to reduce cough strength and thus the ability of elderly lungs to clear mucus. Multiple studies have shown that mucociliary transport is slowed in the upper and lower airways of elderly people. In a cohort of 46 healthy subjects aged 19–81 years old that inhaled labeled Teflon particles, the small airway clearance was negatively correlated with age, thus older people present a delayed clearance of particles in the small airways.36 In nasal epithelial cells isolated from people between 11 years and 90 years of age, a negative correlation between the ciliary beat frequency and age has been confirmed.37 Finally, even if the number of alveoli does not seem to be modified by aging, the enlargement and decreased elasticity of alveoli associated with the destruction of lung parenchyma can cause senile emphysema.38
Cellular changes associated with aging in the lungs
Over the course of a lifetime, the respiratory tract is continuously exposed to environmental injuries. Consequently, mechanisms of repair are of utmost importance and their dysfunction can lead to fibrosis and remodeling of the lungs. Aging is associated with decreased epithelial barrier function. In vitro culture of bronchial epithelial cells from subjects aged over 45 showed a decreased transepithelial resistance with increasing age.39 Single-cell analysis has shown that the pulmonary epithelium undergoes dynamic changes during aging. The number of basal cells decreases with age as well as the ratio between epithelial cells and basal cells. Moreover, the function of basal cells, alveolar type (AT)1, and AT2 cells diminishes with aging suggesting the impairment of airway regeneration and repair. In a mouse model, deficiency of the telomerase reverse transcriptase leads to the increased concentration of pro-inflammatory cytokines in the lungs associated with AT2 cell senescence.40 Moreover, AT2 cells isolated from human lungs showed a diminished proliferation rate, elevated cellular senescence, loss of epithelial markers, increased expression of activating protein-1 (AP-1), and chemokine genes in people over 60 years compared to those of 20 years.41 Composition of extracellular matrix (ECM) may also change with aging. ECM from the lungs of old mice (20–24 months) presents a decreased amount of laminin and increased expression of collagen I and fibronectine compared to 4-month-old to 6-month-old mice. Thus, the aged ECM becomes stiffer which leads to biomechanical alterations.42 In aged lungs, endothelial cells function declines due to cellular senescence and a decreased production of endothelial growth factor that impacts angiogenesis.43 The increased cellular senescence of the endothelium has been attributed to an increased vulnerability to oxidative stress, a decline in the number of stem cells leading to defective repair, and impaired nitric oxide signaling.44
Immunological changes associated with aging in the lungsInnate immunity
Pathogen recognition and elimination is impaired with aging, leading to more acute and chronic pathology in the elderly.45 Alveolar macrophages are key effectors in the immune clearance of microbes in the lungs. Alveolar macrophages recognition of pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs) is key to initiate immune response. In a mouse model infected by Streptococcus pneumoniae (S. pneumoniae), alveolar macrophages isolated from old mice (21 months old) present a defective activation of Toll-like receptor 2 (TLR2) signaling leading to a delayed pro-inflammatory cytokines responses associated with enhanced susceptibility to pneumonia compared to young mice (4 months old).46 Moreover, alveolar macrophages change intrinsically with aging, and exhibit a decrease in cell numbers and altered gene expression, which leads to a defective phagocytosis and high production of pro-inflammatory cytokines and chemokines.47,48 In a mouse model of infection with S. pneumoniae, the number of mitochondria decreased in alveolar macrophages isolated from older mice (19 months old) compared to young mice (2 months old). This was associated with decreased adenosine 5′-triphosphate (ATP) production, diminished antioxidant response and increased production of reactive oxygen species (ROS), and was reversed by the administration of pirfenidone, an anti-fibrotic drug with antioxidant properties, suggesting that oxidative stress may drive the aging of macrophages.49 Macrophages work in conjunction with neutrophils to contain and clear infections. Neutrophils are quickly recruited at the sites of infection to carry out their microbicidal activity by phagocytosis, degranulation of antimicrobial proteins, and the release of neutrophil extracellular trap.45 With aging, the recruitment of neutrophils is decreased despite equivalent levels of chemokines chemokine (C-X-C motif) ligand 2 (CXCL2) and C-C motif chemokine ligand 2 (CCL2) at the site of infections, suggesting that neutrophil chemotaxis is dysregulated in aging.50 Thus, recruitment and function of neutrophils are impaired in elderly patients, and this may be due to diminished activity of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase and myeloperoxidase (MPO) that could lead to oxidative stress.51
Adaptive immunity
Similar to the innate immune response, the adaptive response is also considerably modified by aging mechanisms. During aging, hematopoietic stem cells in the bone marrow show reduced self-renewal, leading to the decreased generation of B and T lymphocytes.52 Other studies have reported modification of the phenotype of hematopoietic stem cells with aging with modification of their frequencies, and increased expression of markers of myeloid lineage.53 With advancing age, the naïve B cell pool diminishes while the memory B cell pool expands. Changes in pro-B, pre-B, and immature B cells are associated with a decrease in repertoire diversity leading to low antibody affinity and impaired class switching. B cells are thus more prone to produce auto-antibodies.54 In the same way, the pool of naïve T cells during aging is in favor of the accumulation of differentiated and less proliferative T memory cells and effector memory T cells re-expressing CD45RA (TEMRA).55 The single-cell analysis of T cells isolated from young and old mice showed that CD4+ T cells undergo a gradual reorganization with accumulation of regulatory, exhausted, and cytotoxic phenotype with aging.56 In bronchoalveolar lavage (BAL) fluid, the ratio of CD4+ to CD8+ T lymphocytes increases with age, confirming the expansion of memory T cells.43 A longitudinal study of T-cell receptors (TCR) repertoire of blood CD4+ and CD8+ T cells showed an alteration of TCR repertoire with age. The diminution of TCR repertoire was subset specific with the greatest reduction observed in naïve CD8+ T cells.57 Thus, the adaptive immune system in the elderly has a decreased capacity to recognize microbial agents, which leads to increased susceptibility to infectious stimuli and prolonged and more severe disease.
Lung aging and COPD
COPD is a complex and multifactorial condition that develops following chronic exposure to hazardous compounds. It is the third leading cause of death globally. In 2017, the number of people living with chronic respiratory disease was estimated to be 544.9 million, and approximately 55% of cases can be attributed to COPD.58 The pathophysiology of COPD is driven by cascade of events that include aberrant immune responses and cellular mechanisms that lead to persistent inflammation, fibrosis, and lung damage.59 These molecular and cellular events are similar to those characterized in aging and the acceleration of lung aging is an essential feature of COPD pathophysiology.
Age is a risk factor for developing COPD
The first risk factor identified for COPD is cigarette smoking. According to the Global Initiative for Chronic Obstructive Pulmonary Disease (GOLD), half of smokers will eventually develop COPD.60,61 However, between 25% and 45% of COPD patients have never smoked and therefore other risk factors have been identified: occupational dusts, vapors and fumes, biomass fuels, outdoor pollutants, and age.62 Diagnosis of COPD is made around 60 years old, therefore COPD affects preferentially elderly patients and has dramatic impacts on their health and quality of life.63 Between 40 and 59 years of age, the prevalence of COPD is 9.2% and it increases to 22.6% in people between 60 and 79 years old.64 It was unclear if aging was implicated in COPD pathophysiology. Two biomarkers of biological aging, dehydroepiandrosterone (DHEA) and growth hormone (GH), were measured in the blood of healthy subject and COPD patients. Both hormones were negatively correlated with age, and the levels of DHEA and GH were significantly decreased in the blood of COPD patients compared to aged-matched healthy individuals. The biological age of COPD patients was estimated to be 13–24 years older than healthy donors. Thus, premature aging, particularly of the lungs, is key in COPD pathology.65
Hallmarks of lung aging in COPD
Oxidative stress in COPD lungs
Oxidative stress is the accumulation of free radicals due to the overproduction of ROS that cannot be processed gradually by the cells.66 During normal aging, chronic oxidative stress arises in cells due to the overproduction of ROS, along with a decline in ATP and antioxidants production. This participates in aging through telomere shortening, genetic instabilities, epigenetic modification, alteration of mitochondria functions, and cellular senescence.67 The imbalance between the production of ROS and antioxidants has been characterized in COPD patients. In exhaled breath condensate of COPD patients, increased levels of ethane, H2O2, malondialdehyde (MDA), 4-hydroxynonenal (HNE), or 8-isoprostane have been measured compared to smokers without COPD and healthy non-smokers,68, 69, 70, 71 whereas the levels of antioxidants glutathione (GSH), superoxide dismutase (SOD), and GSH peroxidase (GSH-PX) are reduced.72 In COPD patients, oxidative stress is generated by exogenous products, like cigarette smoke, but also by endogenous production from increased numbers and activation of inflammatory cells, such as leading to a sustained oxidative stress even after smoking cessation.73 In COPD lungs, oxidative species are mainly produced by inflammatory cells. Indeed, the immune response is altered in COPD patients and leads to the recruitment of pro-inflammatory cells within the lungs, leading to the chronic inflammation of the airways. Among these cells, macrophages and neutrophils are activated in COPD and secrete high levels of superoxide (O2−) and hydrogen peroxide (H2O2).74 Moreover, the expression of nuclear factor erythroid 2-related factor (Nrf2), a master transcription factor that has an antioxidant activity, is decreased in alveolar macrophages from COPD patients,75 further suggesting a decline in antioxidant activity of immune cells in COPD.
The chronic imbalance between oxidants and antioxidants leads to cellular damage in the airways and the alveolar spaces of COPD patients. Bronchial epithelial cells, AT2 cells, endothelial cells, macrophages, and fibroblasts exhibit multiple oxidative stress damages, including lipid peroxidation, protein oxidation, DNA damages, telomeres shortenings, inhibition of DNA methylation, and mitochondrial dysfunction, all of which have been identified as aging mechanisms.76 Oxidative stress participates in glucocorticosteroid resistance of COPD patients by driving the phosphorylation of glucocorticoid receptor (GR) as well as the loss of histone deacetylase-2 (HDAC-2).77 Overproduction of ROS participates in chronic bronchitis and the development of mucus plugging by stimulating the production of mucin genes like MUC5b and MUC5ac.78,79 Production of pro-inflammatory factors is promoted by oxidative stress through the NF-κB and the dissociation of the inhibitor of NF-κB (IκB)/NF-κB complex in airway epithelial cells and macrophages of COPD patients.80,81 Oxidative stress influences the proteostasis network, particularly through the endoplasmic reticulum (ER)-stress. ER is the largest organelle in the cell and is a major site of protein synthesis and transport.82 The consequence of ER stress is the misfolding of secretory proteins which induce the unfolded protein response (UPR) controlled by three pathways: ER-resident protein kinase RNA-like ER kinase (PERK), the endoribonuclease inositol requiring enzyme 1 (IRE1α), and the transcription factor activating transcription factor 6 (ATF6).83 In human bronchial epithelial cells from COPD patients, sustained oxidative stress induces the upregulation of UPR markers that can be abrogated when cells are incubated with an antioxidant.84 Moreover, ER stress and autophagy are two important cell processes that are closely related. The activation of PERK promotes the formation of the autophagosome through the Atg5–Atg12–Atg16 complex.85 Lots of studies have shown that autophagy is impaired in COPD, particularly in response to cigarette smoke and oxidative stress.86 However, the interaction between ER stress, autophagy, and COPD is not fully characterized and needs further investigations.
Inflammaging in COPD
COPD is characterized by an abnormal chronic inflammatory response of the lungs due to the repeated and progressive activation of immune cells by cigarette smoke and oxidative stress. This leads to a pulmonary and systemic low-grade inflammation similar to the inflammation characterized in normal aging. As such, aged mice exposed to cigarette smoke exhibit more airway remodeling, emphysema, and decreased lung function than young mice, suggesting that inflammaging increases the susceptibility to cigarette smoke.87
The innate and adaptive immune responses are impaired in COPD and participate in the development of chronic inflammation of the airways. Airway epithelial cells are key players in the development of inflammaging in COPD as they serve as a molecular and physical barrier for particulate matter and microbes. Repeated exposure to oxidative stress and cigarette smoke is associated with decreased epithelial barrier function, abnormalities in cilia structure and function and reduced production of antimicrobial and anti-inflammatory proteins.88 The initial site of these pathological changes in COPD is the small airway epithelial cells where inhaled irritants induce a global reprogramming of small airway epithelial cells toward a phenotype similar to that observed in proximal airways.89 As the disease progresses, this reprogramming leads to the gradual destruction of the small airways.90
Alveolar macrophages are key player in COPD pathophysiology because of their capacity to orchestrate inflammatory response to inhaled irritants. The number of macrophages is increased by approximately 20-fold in COPD lungs,91,92 where they participate in chronic inflammation of COPD airways and emphysema by producing increasing levels of matrix-metalloproteases (MMP) 2 and MMP9.93 The metabolism of alveolar macrophages is modified in COPD: monocytes-derived macrophages isolated from COPD patients showed decreased mitochondrial membrane potential and increased production of ROS that led to defective phagocytosis and may predispose COPD patients to chronic exacerbation particularly in response to bacteria such as Haemophilus influenzae and S. pneumoniae.94,95 Moreover, pulmonary macrophages regulate the inflammatory response by recruiting innate and adaptive immune cells. In response to oxidative stress, macrophages produce transforming growth factor-β (TGF-β) and C-X-C chemokine ligand 8 (CXCL8), inducing the recruitment of neutrophils in COPD airways.96 Efferocytosis is impaired in COPD, leading to an increased number of neutrophils and eosinophils in the airways, correlated with the severity and the frequency of COPD exacerbations.97 Production by macrophages of CXCL9, CXCL10, and CXCL11 induces the recruitment of CD4+ and CD8+ T cells in the lungs of COPD patients. CD4+ and CD8+ T cells produce perforin and granzyme B that drive the apoptosis of alveolar cells contributing to emphysema.98 Altogether, numerous studies have confirmed that immune cell function is impaired in COPD and leads to a low-grade inflammation similar to the inflammatory response described in aging. A study from Maté et al99 has shown that the function of leukocytes was defective in COPD patients and that they appear to age at a faster rate than in healthy age-matched individuals. However, features of aging in immune cells, like immunosenescence, and mechanisms of aging are still unknown and need to be characterized.
Cellular senescence in COPD
Cellular senescence is now considered a major driver of COPD pathophysiology. Higher proportions of senescent AT2 cells, endothelial cells, and smooth muscle cells are detected on lung sections of COPD patients.100 Compared to age-matched healthy individuals, small airway fibroblasts and small airway epithelial cells show increased p21CIP1 and p16INK4 expression along with an increased senescence-associated β-galactosidase staining.101,102
Cellular senescence of the small airway epithelium has been well-characterized and identified as a driving mechanism in COPD pathogenesis.102 Cellular senescence is not restricted to epithelial cells and senescent airway structural cells participate in COPD pathophysiology and lead to lung destruction and emphysema.100 Dysfunction of the endothelium contributes to COPD pathology and severity as well as cardiovascular disease, a common comorbidity that coexists with COPD.103 Endothelial colony-forming cells (ECFC) isolated from healthy smokers and COPD patients express higher levels of senescence markers, p21CIP1, p16INK4, SA-β-galactosidase and increased markers of DDR, gamma histone 2AX (γH2AX), and tumor protein P53 binding protein 1 (53BP1) compared to ECFC from healthy non-smokers.104 Thus, senescent ECFC display impaired angiogenic abilities and participate in the altered pulmonary vasculature observed in COPD. Fibroblasts are key contributors in the development of small airway disease early in COPD pathogenesis. Parenchymal-derived fibroblasts showed altered functions and senescence features in COPD patients with severe emphysema.105 COPD small airway fibroblasts display increased p21CIP1 and p16INK4 expression along with SA-β-galactosidase activity, and mitochondrial dysfunction.101 This phenotype may be associated with fibrotic properties which may participate in small airway disease progression in COPD. Airway smooth muscle cells (ASMC) play a central role in the pathogenesis of COPD as they are crucial components of the airway for their contractile function and contribution to the production of inflammatory mediators, proteases, and growth factors.106 Airway remodeling characterized in COPD involves airway smooth muscle thickening caused by ASMC hypertrophy and/or hyperplasia.107 Impairment of ASMC metabolism has been shown in COPD, particularly the accumulation of lactate, glutamine, fatty acid, and amino acids compared to healthy ASMC.108 These modifications are associated with the alteration of intracellular Ca2+ signaling, also seen in healthy-aged ASMC.109 Moreover, pulmonary-artery smooth muscle cells isolated from COPD patients expressed higher levels of p16INK4, associated with the increased staining of β-galactosidase and decreased proliferation rate.110 Thus, ASMC senescence could participate in COPD and its comorbidities pathophysiology via airways and pulmonary vessel remodeling.
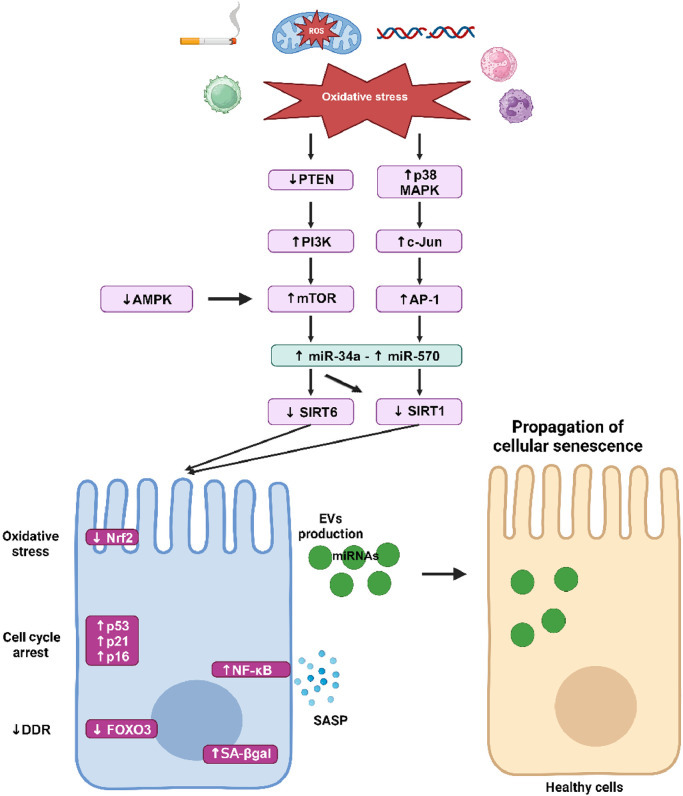
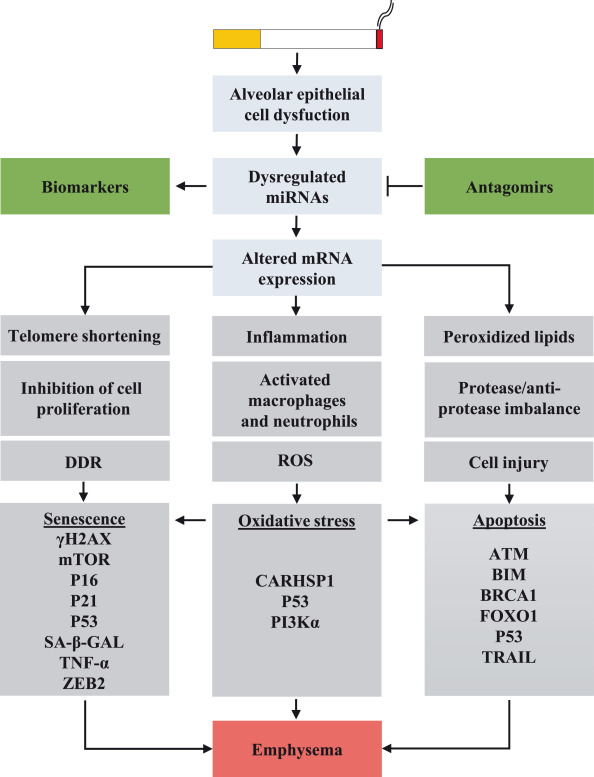
Oxidative stress is an essential inducer of cellular senescence in COPD as it directly activates the p53–p16INK4a pathway as well as damaging the DNA, leading to the activation of p21CIP1 [Fig. 3]. Reduced telomere length has been demonstrated in circulating leukocytes of COPD patients. In lung tissues of COPD patients, an increased number of DNA double-strands breaks, γH2AX were detected in AT1, AT2, and endothelial cells and were associated with the activation of p16INK4 and the NF-κB pathway.111 Failure to repair DNA damage may be related to COPD severity. A DNA repair marker, Ku86, is decreased in parenchymal lung tissue and small airway of COPD patients.112 As such, polymorphisms of DNA repair genes have been described in COPD patients and correlated with DNA damage and disease progression.113 Moreover, oxidative stress leads to the upregulation of peroxisome proliferator-activated receptor-γ coactivator 1-α (PGC1α) and decreased autophagy in COPD airway epithelial cells.102
Fig. 3.
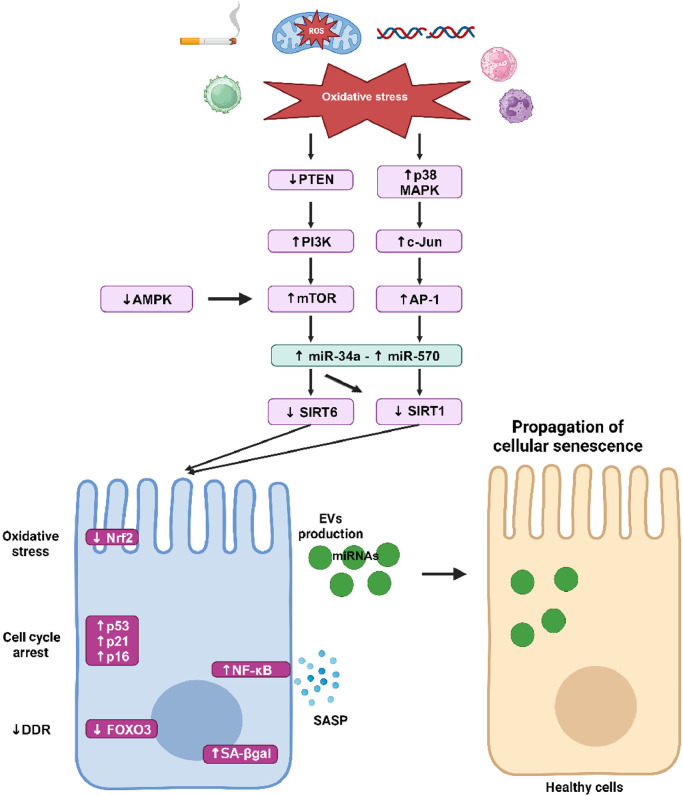
Cellular senescence pathway in COPD. Oxidative stress inhibits PTEN, leading to the activation of PI3K and consequently to the activation of mTOR. Decreased activation of AMPK also increases mTOR activity which leads to the upregulation of microRNA (miR)-34a. Activation of p38 MAPK leads to the upregulation of c-Jun and AP1, which increases miR-570. miR-34a targets SIRT1 and SIRT6 and miR-570 targets SIRT1 but not SIRT6. Decreased SIRT1 and SIRT6 play a key role in the induction of cellular senescence through SASP production via activation of NF-κB and through the increased expression of p53, p21CIP1, and p16INK4. SIRT1 is involved in the diminution of DDR by inhibiting FOXO3 and participates in mitochondria dysfunction by inhibiting autophagy and PGC1α. Downregulation of SIRT6 inhibits the antioxidant Nrf2 which leads to chronic oxidative stress. Senescent cells produce a large number of EVs that contain miRNAs and induce cellular senescence phenotype to healthy cells, thus participating in the propagation of aging within the lungs. AMPK: AMP-activated protein kinase; AP-1: Activating protein-1; COPD: Chronic obstructive pulmonary disease; DDR: DNA damage response; EVs: Extracellular vesicles; FOXO3: Forkhead box O3; miRNAs: MicroRNAs; MAPK: Mitogen-activated protein kinase; mTOR: Mammalian target of rapamycin; NF-κB: Nuclear factor-κB; Nrf2: Nuclear factor erythroid 2-related factor; PTEN: Phosphatase and tensin homolog from chromosome 10; PGC1α: Peroxisome proliferator-activated receptor-γ coactivator 1-α; PI3K: Phosphoinositide 3-kinase; ROS: Reactive oxygen species; SA-βgal: Senescent-associated β-galactosidase; SASP: Senescence-associated secretory phenotype; SIRT1: Sirtuin 1; SIRT6: Sirtuin 6.
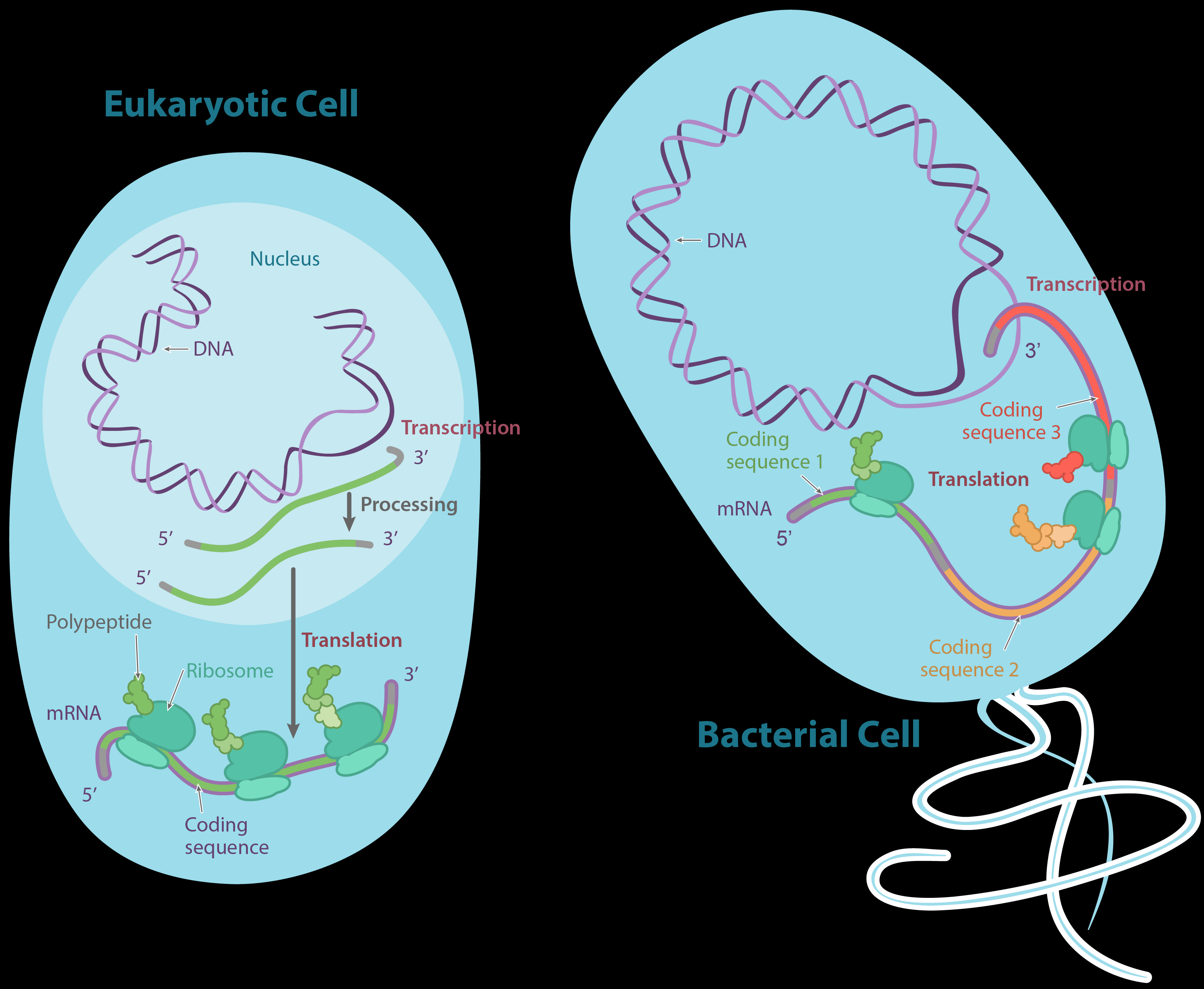
One of the key players in the regulation of cellular senescence in COPD is the anti-aging molecules sirtuins (SIRT). The SIRT family are nicotinamide adenine dinucleotide (NAD)+ dependent histone deacetylases that are highly conserved across species and comprised of 7 members.114 SIRT play a vital role in sustaining genome integrity by maintaining normal chromatin condensation rate, DDR and repair, and modulating oxidative stress and cell metabolism.115 Among SIRT members, SIRT1 and SIRT6 have been linked to cellular senescence and aging because of their capacity to regulate inflammatory responses, oxidative stress, and autophagy.116 SIRT1 and SIRT6 protect cells from senescence by deacetylating the transcription factor forkhead box O3 (FOXO3) and by inhibiting the PI3K–mTOR pathway.117,118 A reduction of SIRT1 expression has been described in serum, peripheral lungs, airway epithelial cells, and circulating PBMC of COPD patients.119, 120, 121 This reduction of SIRT expression is induced by oxidative stress through the inhibition of the phosphatase and tensin homolog from chromosome 10 (PTEN), which leads to the activation of PI3K–mTOR pathway.122 SIRT1 and 6 are downregulated by microRNAs (miRNAs) in COPD.121,123 miRNAs are short, regulatory RNAs that act as post-transcriptional repressor of gene expression.124 Dysregulation of miRNAs expression has been highlighted in COPD, as a response to cigarette smoke injury. Among them, upregulation of miR-15b, miR-22, miR-638, and miR-494 and downregulation of miR-24, miR-126, and miR-149 have been described in COPD lung tissues, fibroblasts, and airway epithelial cells.125 In a mouse model of emphysema induced by cigarette smoke and in human lung tissues from COPD patients, elevated levels of miR-125a have been detected and were negatively correlated with lung function. Moreover, miR-125a induced cellular senescence by inhibiting a transcription factor called Sp1, which leads to the downregulation of SIRT1.126 In small airway epithelial cells from COPD patients, miR-34a and miR-570 are upregulated and their targets SIRT1 and SIRT6 are subsequently downregulated. The activation of the PI3K pathway by oxidative stress leads to the upregulation of miR-34a whereas the activation of p38 MAPK and c-Jun N-terminal kinase signaling leads to the upregulation of miR-570. Most importantly, transfection of COPD small airway epithelial cells with an antagomiR against miR-34a or miR-570 not only restores expression of SIRT1 and SIRT6, but also reduces markers of cellular senescence (p16INK4, p21CIP1, SASP production, and SA-β-galactosidase).121,123 miRNAs are mainly located in the cytoplasm where they exert their biological function. However, recent studies have localized them in different cell compartments, including their secretion outside the cell in extracellular vesicles (EVs).125 EVs are lipid bilayer vesicles secreted by cells and contain a specific cargo composed of proteins, lipids, DNA, RNA, or miRNAs. By carrying and transporting this cargo to other cells, EVs trigger a molecular and/or phenotypic response in recipient cells.127 Modification of EVs cargo has been shown in COPD: EVs isolated from plasma of COPD patients contain higher levels of miR-22, miR-99a, miR-151a, miR-320b, and miR-320d compared to EVs isolated from smokers and non-smokers without COPD. Gene set enrichment analysis revealed that the target of these miRNAs plays crucial roles in cytokine signaling and in tissue remodeling,128 however, their function in COPD pathophysiology needs further study. Senescent cells produce more EVs and their cargo can be enriched with miR-34a, miR-23b, or miR-494, which can induce cellular senescence.129 Thus, EVs can participate in the propagation of cellular senescence within the lungs, which may account for disease severity and comorbidities.
Conclusions
Considerable progress in understanding the role of accelerated lung aging in COPD has been made during the last decade. Aging is an inexorable process. However, for the best or the worst, people aged differently, suggesting that genetic, epigenetic, and environmental factors interact and contribute to healthy aging or to the development of age-related disease, like COPD. The characterization of biomarkers of aging and a more accurate definition of the biological age would help differentiate healthy from unhealthy aging and will allow the early detection of COPD. Moreover, the current studies mainly fail to integrate data from different organs to allow an early detection of age-related disease in an individual. COPD frequently coexists with other diseases and comorbidities that have significant health and economic consequences.130 A recent study by Tian et al131 performed a multimodal analysis to measure the biological age of seven organs and the brain and showed that lung aging strongly influenced the aging of the cardiovascular system. Thus, the future perspective of COPD is to further understand the pathophysiology of COPD and its comorbidities and to understand how organs communicate and influence each other's aging markers.
The identification of aging biomarkers and signaling pathways involved in COPD has led to new therapeutic targets. Among them, the use of senotherapeutics has become an interesting approach. Senomorphics are agents that block senescence pathways and the production of SASP, including telomerase activators, sirtuin activators, mTOR inhibitors, antioxidants, anti-inflammatory agents, autophagy and proteasome activators. Treatment of lung cells from COPD patients with rapamycin, an mTOR inhibitor, prevents cellular senescence and inhibits the production of SASP. However, SASP factors are not only deleterious, they can have a role in tissue homeostasis, thus senomorphics may lead to serious side effects. The elimination of senescent cells may stop disease progression and favor lung regeneration. Clearance of senescent cells in aged mice attenuated the age-related deterioration of several organs and expanded lifespan by 17% to 42%.132,133 Senolytics are small molecules that eliminate senescent cells by inducing apoptosis. Seven classes of senolytics have been described: natural compounds, kinase inhibitors, B-cell lymphoma-2 (Bcl-2) family inhibitors/Bcl-2 homology 3 (BH3) mimetics, inhibitors of mouse double minute 2 homolog (MDM2)/p53 interactions, heat-shock protein 90 (Hsp90) inhibitors, p53 binding inhibitors, and HDAC inhibitors.134 Navitoclax (ABT-263) targets the antiapoptotic proteins Bd-2, B-cell lymphoma-extra large (BCL-XL), and BCL-w and reduces the viability of senescent human umbilical vein endothelial cells (HUVECs), IMR90 human lung fibroblasts, and murine embryonic fibroblasts.135 Studies in mice have shown beneficial effect on aged mice, notably their brain function, however, potential harmful effects have also been described on osteoprogenitors.136,137 These novel therapies may have major effects on COPD and other chronic age-related diseases in the future. However, further studies are needed to understand the effect of this therapy on senescent cells and on the regeneration of cells and tissues.
Declaration of competing interest
None.
Edited by: Peifang Wei
References
- 1.Agustí A, Celli BR, Criner GJ, et al. Global initiative for chronic obstructive lung disease 2023 report: GOLD executive summary. Eur Respir J. 2023;61 doi: 10.1183/13993003.00239-2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jarhyan P, Hutchinson A, Khaw D, Prabhakaran D, Mohan S. Prevalence of chronic obstructive pulmonary disease and chronic bronchitis in eight countries: a systematic review and meta-analysis. Bull World Health Organ. 2022;100:216–230. doi: 10.2471/BLT.21.286870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Moqri M, Herzog C, Poganik JR, et al. Biomarkers of aging for the identification and evaluation of longevity interventions. Cell. 2023;186:3758–3775. doi: 10.1016/j.cell.2023.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Florea M. Aging and immortality in unicellular species. Mech Ageing Dev. 2017;167:5–15. doi: 10.1016/j.mad.2017.08.006. [DOI] [PubMed] [Google Scholar]
- 5.Campisi J, Kapahi P, Lithgow GJ, Melov S, Newman JC, Verdin E. From discoveries in ageing research to therapeutics for healthy ageing. Nature. 2019;571:183–192. doi: 10.1038/s41586-019-1365-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barnett K, Mercer SW, Norbury M, Watt G, Wyke S, Guthrie B. Epidemiology of multimorbidity and implications for health care, research, and medical education: a cross-sectional study. Lancet. 2012;380:37–43. doi: 10.1016/S0140-6736(12)60240-2. [DOI] [PubMed] [Google Scholar]
- 7.Britt HC, Harrison CM, Miller GC, Knox SA. Prevalence and patterns of multimorbidity in Australia. Med J Aust. 2008;189:72–77. doi: 10.5694/j.1326-5377.2008.tb01919.x. [DOI] [PubMed] [Google Scholar]
- 8.Formiga F, Ferrer A, Sanz H, et al. Patterns of comorbidity and multimorbidity in the oldest old: the Octabaix study. Eur J Intern Med. 2013;24:40–44. doi: 10.1016/j.ejim.2012.11.003. [DOI] [PubMed] [Google Scholar]
- 9.Polidori MC. Aging hallmarks, biomarkers, and clocks for personalized medicine: (re)positioning the limelight. Free Radic Biol Med. 2024;215:48–55. doi: 10.1016/j.freeradbiomed.2024.02.012. [DOI] [PubMed] [Google Scholar]
- 10.Kudryashova KS, Burka K, Kulaga AY, Vorobyeva NS, Kennedy BK. Aging biomarkers: from functional tests to multi-omics approaches. Proteomics. 2020;20 doi: 10.1002/pmic.201900408. [DOI] [PubMed] [Google Scholar]
- 11.López-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013;153:1194–1217. doi: 10.1016/j.cell.2013.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.López-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G. Hallmarks of aging: an expanding universe. Cell. 2023;186:243–278. doi: 10.1016/j.cell.2022.11.001. [DOI] [PubMed] [Google Scholar]
- 13.Vaiserman A, Krasnienkov D. Telomere length as a marker of biological age: state-of-the-art, open issues, and future perspectives. Front Genet. 2020;11 doi: 10.3389/fgene.2020.630186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Demanelis K, Jasmine F, Chen LS, et al. Determinants of telomere length across human tissues. Science. 2020;369:eaaz6876. doi: 10.1126/science.aaz6876. [DOI] [PMC free article] [PubMed] [Google Scholar]