beyond reason
https://www.youtube.com/watch?v=kqYWKIM25mM

J Pathol. Author manuscript; available in PMC 2010 Nov 23.
Published in final edited form as:
J Pathol. 2010 May; 221(1): 3–12.
doi: 10.1002/path.2697
PMCID: PMC2990190
NIHMSID: NIHMS251117
PMID: 20225336
Autophagy: cellular and molecular mechanisms
Danielle Glick,1,2 Sandra Barth,1 and Kay F. Macleod1,2,*
Author information Copyright and License information Disclaimer
The publisher's final edited version of this article is available at J Pathol
See other articles in PMC that cite the published article.
Abstract
Autophagy is a self-degradative process that is important for balancing sources of energy at critical times in development and in response to nutrient stress. Autophagy also plays a housekeeping role in removing misfolded or aggregated proteins, clearing damaged organelles, such as mitochondria, endoplasmic reticulum and peroxisomes, as well as eliminating intracellular pathogens.
Thus, autophagy is generally thought of as a survival mechanism, although its deregulation has been linked to non-apoptotic cell death. Autophagy can be either non-selective or selective in the removal of specific organelles, ribosomes and protein aggregates, although the mechanisms regulating aspects of selective autophagy are not fully worked out.
In addition to elimination of intracellular aggregates and damaged organelles, autophagy promotes cellular senescence and cell surface antigen presentation, protects against genome instability and prevents necrosis, giving it a key role in preventing diseases such as cancer, neurodegeneration, cardiomyopathy, diabetes, liver disease, autoimmune diseases and infections. This review summarizes the most up-to-date findings on how autophagy is executed and regulated at the molecular level and how its disruption can lead to disease.
Keywords: autophagy, apoptosis, stress, mechanisms, energy, disease, cancer, neurodegeneration, infection
What is autophagy?
The term ‘autophagy’, derived from the Greek meaning ‘eating of self’, was first coined by Christian de Duve over 40 years ago, and was largely based on the observed degradation of mitochondria and other intra-cellular structures within lysosomes of rat liver perfused with the pancreatic hormone, glucagon [1]. The mechanism of glucagon-induced autophagy in the liver is still not fully understood at the molecular level, other than that it requires cyclic AMP induced activation of protein kinase-A and is highly tissue-specific [2]. In recent years the scientific world has ‘rediscovered’ autophagy, with major contributions to our molecular understanding and appreciation of the physiological significance of this process coming from numerous laboratories [3–6]. Although the importance of autophagy is well recognized in mammalian systems, many of the mechanistic breakthroughs in delineating how autophagy is regulated and executed at the molecular level have been made in yeast (Saccharomyces cerevisiae) [3,7]. Currently, 32 different autophagy-related genes (Atg) have been identified by genetic screening in yeast and, significantly, many of these genes are conserved in slime mould, plants, worms, flies and mammals, emphasizing the importance of the autophagic process in responses to starvation across phylogeny [3].
There are three defined types of autophagy: macro-autophagy, micro-autophagy, and chaperone-mediated autophagy, all of which promote proteolytic degradation of cytosolic components at the lysosome.
Macro-autophagy delivers cytoplasmic cargo to the lysosome through the intermediary of a double membrane-bound vesicle, referred to as an autophagosome, that fuses with the lysosome to form an autolysosome.
In micro-autophagy, by contrast, cytosolic components are directly taken up by the lysosome itself through invagination of the lysosomal membrane. Both macro-and micro-autophagy are able to engulf large structures through both selective and non-selective mechanisms.
In chaperone-mediated autophagy (CMA), targeted proteins are translocated across the lysosomal membrane in a complex with chaperone proteins (such as Hsc-70) that are recognized by the lysosomal membrane receptor lysosomal-associated membrane protein 2A (LAMP-2A), resulting in their unfolding and degradation [8].
Due to recent and increased interest specifically in macroautophagy and its role in disease, this review focuses on molecular and cellular aspects of macro-autophagy (henceforth referred to as ‘autophagy’) and how it is regulated under both healthy and pathological conditions (Table 1).
Table 1
Autophagy-deficient mouse models and human diseases linked to defects in specific autophagy genes
Autophagy gene and function (human/mouse)Human disease linked to mutation/inactivationMouse model phenotypeReferences
| ATG16L/Atg16L | 32 – 35 | |
| Atg16L complexes with conjugated Atg5–Atg12 to promote expansion and curvature of the nascent phagophore | T300A mutation in ATG16L linked to Crohn's disease, discovered by GWAS | Loss of Atg16L1 inhibits autophagy in Paneth cells, reducing secretion of granules of antimicrobial peptides that influence intestinal microbiota and causing increased inflammation |
| BECN1/Becn1 | 16,17 | |
| Beclin1 regulates the kinase activity of Vps34 at the ER; complex includes regulatory components UVRAG, Atg14L, Rubicon and Ambra | BECN1 is mono-allelically deleted in breast, ovarian and prostate cancer | Becn1-null mice are embryonic lethal, showing a defect in cavitation of the blastocyst. Becn1 heterozygotes are predisposed to lymphoma, hepatocellular carcinoma and other cancers |
| UVRAG/Uvrag | 18 | |
| UVRAG complexes with Beclin1 and Vps34 at the ER to promote autophagy | UVRAG is mono-allelically deleted in colon cancer | N/A |
| IRGM/Irgm | 36 | |
| Immunity-related GTPase stimulates autophagy and promotes clearance of pathogenic bacteria | Deletion of upstream regulatory sequences segregates with Crohn's disease and is associated with altered IRGM expression | N/A |
| CLN3/Cln3 | 45 | |
| Associated with Golgi, endosomes and lipid rafts and may play a role in transporting ceramide and other sphingolipids to lipid rafts | Accumulation of proteolipids in children with Batten disease leads to neurodegeneration and is due to inactivation of the CLN3 gene, which promotes autophagosome fusion with the lysosome | Immature autophagosomes in tissues from mice with knock-in of mutant forms of Cln3 |
| Parkin | 63,64 | |
| A E3 ubiquitin ligase that localizes to the mitochondria and is required for mitophagy | Parkinson's disease (PD) is associated with cell death of dopaminergic neurons and progressive loss of cognitive and motor function. Mutation of several genes are linked to PD, including Parkin | N/A |
| p62/SQSTM1 | 39,49,50,52,53 | |
| A multifunctional adaptor protein that promotes turnover of polyubiquitinated protein aggregates through interaction with LC3 at the autophagosome | p62 mutations are linked to Paget's disease in which increased bone turnover results in abnormal bone architecture. Associated with deregulated NF-κB signalling and reduced turnover of ubiquitinated proteins | p62-null mice are resistant to Rasdriven lung carcinogenesis. Loss of p62 prevents accumulation of ubiquitin-positive protein aggregates in the liver and neurons of Atg7-deficient mice |
| Lamp2 | 46 | |
| A lysosomal membrane protein required for fusion of the autophagosome with the lysosome | Danon disease is an X-linked disease resulting in hypertrophic cardiomyopathy and accumulation of autophagosomes in the heart muscle | Increased autophagosome numbers in multiple tissues, cardiomyopathy, skeletal myopathy, periodontitis associated with inflammation due to defective clearance of intracellular pathogens |
The basic autophagy machinery
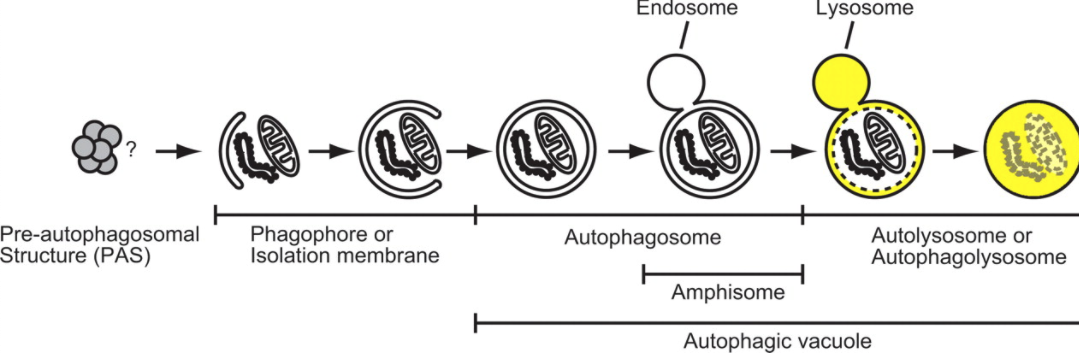
As summarized in Figure 1, autophagy begins with an isolation membrane, also known as a phagophore that is likely derived from lipid bilayer contributed by the endoplasmic reticulum (ER) and/or the trans-Golgi and endosomes [9,10], although the exact origin of the phagophore in mammalian cells is controversial. This phagophore expands to engulf intra-cellular cargo, such as protein aggregates, organelles and ribosomes, thereby sequestering the cargo in a double-membraned autophagosome [5]. The loaded autophagosome matures through fusion with the lysosome, promoting the degradation of autophagosomal contents by lysosomal acid proteases. Lysosomal permeases and transporters export amino acids and other by-products of degradation back out to the cytoplasm, where they can be re-used for building macromolecules and for metabolism [5]. Thus, autophagy may be thought of as a cellular ‘recycling factory’ that also promotes energy efficiency through ATP generation and mediates damage control by removing non-functional proteins and organelles.
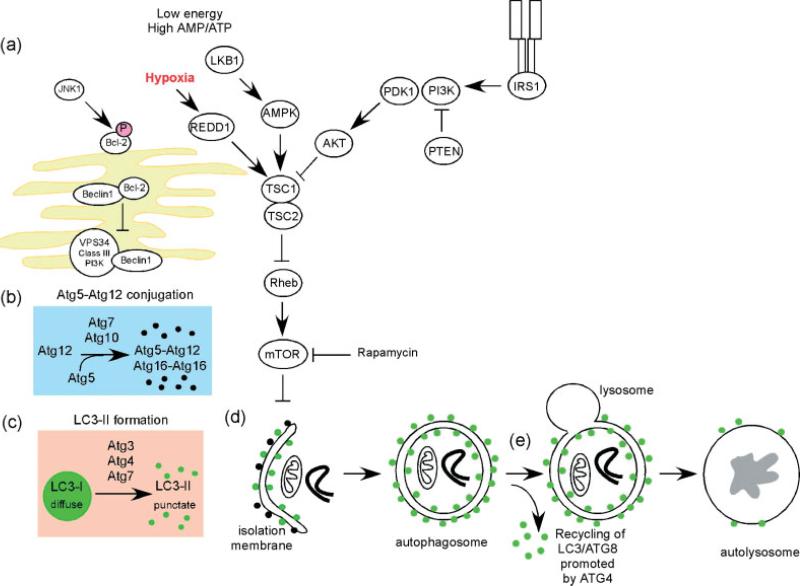
Molecular circuitry and signalling pathways regulating autophagy. Autophagy is a complex self-degradative process that involves the following key steps: (a) control of phagophore formation by Beclin-1/VPS34 at the ER and other membranes in response to stress signalling pathways; (b) Atg5–Atg12 conjugation, interaction with Atg16L and multimerization at the phagophore; (c) LC3 processing and insertion into the extending phagophore membrane; (d) capture of random or selective targets for degradation, completion of the autophagosome accompanied by recycling of some LC3-II/ATG8 by ATG4, followed by; (e) fusion of the autophagosome with the lysosome and proteolytic degradation by lysosomal proteases of engulfed molecules. Autophagy is regulated by important signalling pathways in the cell, including stress-signalling kinases such as JNK-1, which promotes autophagy by phosphorylating Bcl-2, thereby promoting the interaction of Beclin-1 with VPS34 [31]. Perhaps the central signalling molecule in determining the levels of autophagy in cells is the mTOR kinase that likely mediates its effects on autophagy through inhibition of ATG1/Ulk-1/-2 complexes at the earliest stages in phagophore formation from lipid bilayers [6]. mTOR is key to integrating metabolic, growth factor and energy signalling into levels of both autophagy, on the one hand, which is inhibited by mTOR when nutrients are plentiful and, on the other hand, to growth-promoting activities, including protein translation, that are stimulated by mTOR signalling [16]. Autophagy is induced by hypoxia and low cytosolic ATP levels that feed through REDD1 and AMP-kinase to inhibit mTOR activity through reduced Rheb GTPase activity. Conversely, autophagy is inhibited by increased growth factor signalling through the insulin receptor and its adaptor, IRS1, as well as other growth factor receptors that activate the Class I group of PI3-kinases and Akt, to promote mTOR activity through inhibition of TSC1/TSC2 and increased Rheb GTPase activity [16,73].
How is this complex process orchestrated at the molecular level? There are five key stages (Figure 1): (a) phagophore formation or nucleation; (b) Atg5–Atg12 conjugation, interaction with Atg16L and multi-merization at the phagophore; (c) LC3 processing and insertion into the extending phagophore membrane; (d) capture of random or selective targets for degradation; and (e) fusion of the autophagosome with the lysosome, followed by proteolytic degradation by lysosomal proteases of engulfed molecules.
Phagophore formation is under the control of multiple signalling events
Phagophore membrane formation in yeast is formed at, or organized around, a cytosolic structure known as the pre-autophagosomal structure (PAS), but there is no evidence for a PAS in mammals [7]. In mammalian cells, phagophore membranes appear to initiate primarily from the ER [11,12] in dynamic equilibrium with other cytosolic membrane structures, such as the trans-Golgi and late endosomes [5,9,13] and possibly even derive membrane from the nuclear envelope under restricted conditions [14]. However, given the relative lack of transmembrane proteins in autophagosomal membranes, it is not yet possible to completely rule out de novo membrane formation from cytosolic lipids in mammalian cells. The activity of the Atg1 kinase in a complex with Atg13 and Atg17 is required for phagophore formation in yeast, possibly by regulating the recruitment of the transmembrane protein Atg9 that may act by promoting lipid recruitment to the expanding phagophore [7,10,15]. This step is regulated by the energy-sensing TOR kinase that phosphorylates Atg13, preventing it from interacting with Atg1 [16] and rendering initiation of autophagy sensitive to growth factor and nutrient availability. Ulk-1, a mammalian homologue of Atg1 is critical for autophagy in maturing reticulocytes [17] but it remains to be determined whether Ulk-1, or indeed Ulk-2 (a second Atg1 homologue), functions analogously in promoting autophagy in mammalian systems. These early steps in phagophore formation in mammalian systems are an area that requires greater investigation and is likely to lead to many important findings, given that these processes are tightly regulated in yeast and are a nexus for signalling input in higher systems.
The role of class III PI-3 kinases, notably Vps34 (vesicular protein sorting 34) and its binding partner Atg6/Beclin-1, in phagophore formation and autophagy is relatively well understood in mammalian systems. Vps34 is involved in various membrane-sorting processes in the cell but is selectively involved in autophagy when complexed to Beclin-1 and other regulatory proteins [18]. Vps34 is unique amongst PI3-kinases in only using phosphatidylinositol (PI) as substrate to generate phosphatidyl inositol triphosphate (PI3P), which is essential for phagophore elongation and recruitment of other Atg proteins to the phagophore [6]. The interaction of Beclin-1 with Vps34 promotes its catalytic activity and increases levels of PI3P, but how this is regulated in response to starvation signalling is not yet resolved.
Beclin-1 is mono-allelically deleted in human breast, ovarian and prostate cancer, leading various cancer biologists to suggest that autophagy has tumour-suppressor properties [19]. Consistently, while Beclin-1 null mice are embryonic lethal [20], Beclin-1 heterozygous mice are predisposed to lymphoma, hepato-cellular carcinoma and other cancers [21]. Autophagy has been postulated to prevent tumorigenesis by limiting necrosis and inflammation, inducing cell cycle arrest and preventing genome instability [22,23]. Autophagy has also recently been shown to be required for key aspects of the senescent cell phenotype [24], which is known to be anti-tumourigenic. However, as a cell survival mechanism, others have argued that autophagy may promote drug resistance and tumour cell adaptation to stress [25]. Ultimately, the role of autophagy in cancer may be cell type- and/or stage-specific.
Additional regulatory proteins complex with Vps34 and Beclin-1 at the ER and nucleated phagophore to either promote autophagy, such as UVRAG, BIF-1, Atg14L and Ambra [21,26], or to inhibit autophagy, such as Rubicon and Bcl-2 [27–29]. Like Beclin-1, UVRAG has been shown to be mono-allelically deleted in human cancer [21]. The precise subunit composition of complexes at the ER containing Vps34 and Beclin-1 is determined by signalling events in the cell that remain to be fully elucidated but, in many instances, are sensitive to nutrient availability in the microenvironment. One well-characterized regulatory event is the interaction of Beclin-1 with Bcl-2, which disrupts the interaction of Beclin-1 with Vps34 [29,30]. Thus, Beclin-1 activity in autophagy is inhibited by interaction with Bcl-2 (and Bcl-XL) at the ER [30]. This interaction is mediated by the BH3 domain in Beclin-1 and disrupted by Jnk1-mediated phosphorylation of Bcl-2 in response to starvation-induced signalling, thereby allowing autophagy to proceed [31]. Thus, Bcl-2 plays a dual role in determining cell viability that may depend on its subcellular localization: (a) a pro-survival function at mitochondria inhibiting cytochrome c release, thereby blocking apoptosis; and (b) an autophagy-inhibitory activity at the ER, mediated by interaction with Beclin1 that can lead to non-apoptotic cell death [29]. The crosstalk between autophagy and apoptosis extends beyond the regulation of Beclin1 and Bcl-2. For example, calpain-mediated cleavage of Atg5 blocked its activity in autophagy, caused it to translocate to the mitochondria, where its interaction with Bcl-XL resulted in cytochrome c release, caspase activation and apoptosis [32]. How the balance between autophagy and apoptosis is determined in the cellular response to specific stresses is a research area of extreme interest given its relevance for disease progression and treatment, but again is an area that is not resolved [33].
Atg5–Atg12 conjugation
There are two ubiquitin-like systems that are key to autophagy [5,34] acting at the Atg5–Atg12 conjugation step and at the LC3 processing step (see below). In the first of these systems, Atg7 acting like an E1 ubiquitin activating enzyme activates Atg12 in an ATP-dependent manner by binding to its carboxyterminal glycine residue. Atg12 is then transferred to Atg10, an E2-like ubiquitin carrier protein that potentiates covalent linkage of Atg12 to lysine 130 of Atg5. Conjugated Atg5–Atg12 complexes in pairs with Atg16L dimers to form a multimeric Atg5–Atg12–Atg16L complex that associates with the extending phagophore. The association of Atg5–Atg12–Atg16L complexes is thought to induce curvature into the growing phagophore through asymmetric recruitment of processed LC3B-II (see below). Atg5–Atg12 conjugation is not dependent on activation of autophagy and once the autophagosome is formed, Atg5–Atg12–Atg16L dissociates from the membrane, making conjugated Atg5–Atg12 a relatively poor marker of autophagy [35]. Interestingly, genome-wide association studies (GWAS) linked a mutation (T330A) in ATG16L to Crohn's disease, a progressive inflammatory bowel disease in humans [36,37]. Loss of functional Atg16L in mice blocked autophagy in intestinal Paneth cells, resulted in increased inflammasome activation and aberrant inflammatory cytokine production following challenge of Atg16L deficient macrophages with bacterial endotoxin, and reduced secretion of antimicrobial peptides from intestinal Paneth cells in Atg16L hypomorphic mice [38,39]. Similar changes in Paneth cell granule production were observed in Crohn's patients with the ATG16L mutation and this is predicted to alter the diversity of gut microbiota [39]. The IRGM locus was also linked to Crohn's disease by GWAS and, while the specific function of the IRGM GTPase in autophagic turnover of intra-cellular bacteria is not clear, reduced expression of IRGM in Crohn's disease appears to be associated with the identified SNP in its upstream regulatory sequences [40].
LC3 processing
The second ubiquitin-like system involved in auto-phagosome formation is the processing of microtubule-associated protein light chain 3 (LC3B), which is encoded by the mammalian homologue of Atg8. LC3B is expressed in most cell types as a full-length cytosolic protein that, upon induction of autophagy, is proteolytically cleaved by Atg4, a cysteine protease, to generate LC3B-I. The carboxyterminal glycine exposed by Atg4-dependent cleavage is then activated in an ATP-dependent manner by the E1-like Atg7 in a manner similar to that carried out by Atg7 on Atg12 (see above). Activated LC3B-I is then transferred to Atg3, a different E2-like carrier protein before phosphatidylethanolamine (PE) is conjugated to the carboxyl glycine to generate processed LC3B-II. Recruitment and integration of LC3B-II into the growing phagophore is dependent on Atg5–Atg12 and LC3B-II is found on both the internal and external surfaces of the autophagosome, where it plays a role in both hemifusion of membranes and in selecting cargo for degradation. The synthesis and processing of LC3 is increased during autophagy, making it a key readout of levels of autophagy in cells [35]. The related molecule, GABARAP [γ-aminobutyric type A (GABAA)-receptor associated protein] undergoes similar processing during autophagy and GABARAP-II co-localizes with LC3-II at autophagosomes [34]. The significance of LC3-related molecules in autophagy is not clear, although it has been postulated that differences in their protein–protein interactions may determine which cargo is selected for uptake by the autophagosome [41].
Selection, or not, of cargo for degradation?
In general, autophagy has been viewed as a random process because it appears to engulf cytosol indiscriminantly. Electron micrographs frequently show autophagosomes with varied contents, including mitochondria, ER and Golgi membranes [42]. However, there is accumulating evidence that the growing phagophore membrane can interact selectively with protein aggregates and organelles. It is proposed that LC3B-II, acting as a ‘receptor’ at the phagophore, interacts with ‘adaptor’ molecules on the target (eg protein aggregates, mitochondria) to promote their selective uptake and degradation. The best-characterized molecule in this regard is p62/SQSTM1, a multi-functional adaptor molecule that promotes turnover of poly-ubiquitinated protein aggregates. Mutation of p62/SQSTM1 is linked to Paget's disease, in which abnormal turnover of bone results in bone deformation, arthritis and nerve injury [43]. Osteoclasts in such individuals show deregulated NF-κB signalling and accumulation of ubiquitinated proteins consistent with a key role for autophagy in normal bone development and function. Other molecules, such as NBR1, function similarly to p62/SQSTM1 in promoting turnover of ubiquitinated proteins, while in yeast, Uth1p and Atg32 have been identified as proteins that promote selective uptake of mitochondria, a process known as mitophagy [34,44].
Fusion with the lysosome
When the autophagosome completes fusion of the expanding ends of the phagophore membrane, the next step towards maturation in this self-degradative process is fusion of the autophagosome with the specialized endosomal compartment that is the lysosome to form the ‘autolysosome’ [5]. It has been variously suggested that fusion of the autophagosome with early and late endosomes, prior to fusion with the lysosome, both delivers cargo and also delivers components of the membrane fusion machinery and lowers the pH of the autophagic vesicle before delivery of lysosomal acid proteases [45]. This aspect of the process is relatively understudied but requires the small G protein Rab7 in its GTP-bound state [46,47], and also the Presenilin protein that is implicated in Alzheimer's disease [45]. The cytoskeleton also plays a role in autolysosome formation, since agents such as nocadazole, which are microtubule poisons, block fusion of the autophagosome with the lysosome [48]. Within the lysosome, cathepsin proteases B and D are required for turnover of autophagosomes and, by inference, for the maturation of the autolysosome [49]. Lamp-1 and Lamp-2 at the lysosome are also critical for functional autophagy, as evidenced by the inhibitory effect of targeted deletion of these proteins in mice on autolysosome maturation [50]. Interestingly, inactivation of LAMP-2 is the causative genetic lesion associated with Danon disease in humans, an X-linked condition that causes cardiomyocyte hyper-trophy and accumulation of autophagosomes in heart muscle. Similar cardiac defects are observed in Lamp-2-null mice, as well as skeletal abnormalities and periodontitis associated with inflammation arising from a failure to eliminate intracellular pathogens in the oral mucosa [50].
Atg5/Atg7-independent autophagy
Although Atg5- and Atg7-dependent autophagy has been shown to be critical for survival during the starvation period in the first few days immediately following birth [51,52], recent evidence has identified an alternative Atg5/Atg7-independent pathway of autophagy [53]. This pathway of autophagy was not associated with LC3 processing but appeared to specifically involve autophagosome formation from late endosomes and the trans-Golgi [53]. Atg7-independent autophagy had been implicated in mitochondrial clearance from reticulocytes [54], and it has consistently been shown that Ulk-1 (a mammalian homologue of Atg1) is required for both reticulocyte clearance of mitochondria [17] and, along with Beclin-1, for Atg5/Atg7-independent autophagy [53]. The exact molecular basis of Atg5/Atg7-independent autophagy remains to be elucidated.
Selective autophagy
Here, we focus in more depth on selective autophagy, given its significance for neuropathies, cancer and heart disease. As briefly mentioned above, p62/SQSTM1 associates with polyubiquitinated proteins and aggregates through its ubiquitin-binding domain (UBD) [55], with LC3B-II through its LC3-interacting Region (LIR), but also regulates NF-κB signalling through interaction with Traf-6 [56]. When autophagy is defective, as in mice with targeted deletion of Atg7 [52], p62-associated poly-ubiquitinated aggregates accumulated in cells and the combined knockout of Atg7 and p62 was observed to ‘rescue’ the accumulation of these aberrant cytosolic inclusions [57]. p62 is the major constituent of Mallory bodies in the liver that accumulate in human hepatocellular carcinoma, where recent work indicates that elevated p62 levels play an active role in deregulating NF-κB signalling and inducing inflammation-associated tumorigenesis [58]. Intracellular aggregate accumulation plays a particularly significant role in the aetiology of neurodegenerative diseases, including dementia, Alzheimer's, Huntington's, Parkinson's and Creutzfeldt–Jakob/prion diseases [4,59,60]. For example, polyglutamine-expansion repeats, as seen in mutant huntingtin (Huntington's disease), mutant forms of α-synuclein (familial Parkinson's disease) and different forms of tau (Alzheimer's disease) are dependent on autophagy for their clearance from neurons [59,60]. Consistently, neuronal-specific inactivation of the key autophagy genes Atg5 or Atg7 results in intracellular aggregate accumulation and neurodegeneration in mice [61,62]. This relatively recent link between autophagy and neuropathies has prompted interested in the development of autophagy-inducing drugs to treat these debilitating diseases.
Autophagy-dependent degradation of mitochondria, termed mitophagy, is important for maintaining the integrity of these critical organelles and limiting the production of reactive oxygen species [44]. The first protein identified to be involved in mitophagy was Uth1p, a yeast protein that is required for mitochondrial clearance by autophagy, but it is unknown how Uth1 interacts with the autophagosome and mediates mitophagy, and there are no known mammalian homologues [63]. More recently, Atg32, a mitochondria-anchored protein, was found to be required for mitophagy in yeast, where it functions through interaction with Atg8 and Atg11, suggesting that it functions as a mitochondrial receptor for mitophagy [64,65]. Atg32, like Uth1, has no known homologues in mammals, but contains an amino acid motif, WXXI, that is required for interaction with Atg8 and Atg11 and is conserved in the LIR of p62 [34]. Other molecules that are implicated in mitophagy are BNIP3L, which is involved in mitochondrial clearance in differentiating red blood cells [66,67], Ulk-1, which is the mammalian homologue of Atg1 [17], and Parkin, encoded by a gene that is genetically linked to Parkinson's disease [68]. Parkin is an E3 ubiquitin ligase that is located at the outer mitochondrial membrane, suggesting that key molecules at the mitchondria require to be ubiquitinated in order to promote the uptake of mitochondria by autophagosomes [69].
Both peroxisomes and ribosomes are selectively eliminated via autophagy in yeast [3]. Methylotrophic yeasts use micropexophagy (direct engulfment by the vacuole) and macropexophagy (autophagosome-mediated delivery to the vacuole) to remove peroxisomes during adaptation to an alternative energy source in which Atg30 was essential as an adaptor interacting with peroxisome proteins (Pex3 and Pex14) and with the autophagosome (Atg11 and Atg17) [70]. Ribosomes are also selectively degraded during starvation (ribophagy), a process that is dependent on the catalytic activity of the Ubp3p/Bre5p ubiquitin protease [71]. By comparison with yeast, these specialized forms of autophagy are under-studied in mammalian systems.
Signalling pathways that regulate autophagy
Autophagy is active at basal levels in most cell types where it is postulated to play a housekeeping role in maintaining the integrity of intracellular organelles and proteins [72]. However, autophagy is strongly induced by starvation and is a key component of the adaptive response of cells and organisms to nutrient deprivation that promotes survival until nutrients become available again. How is autophagy induced in response to starvation signals?
A major player in nutrient sensing and in regulating cell growth and autophagy is the target of rapamycin (TOR) kinase, which is a signalling control point downstream of growth factor receptor signalling, hypoxia, ATP levels and insulin signalling. TOR kinase is activated downstream of Akt kinase, PI3-kinase and growth factor receptor, signalling when nutrients are available and acting to promote growth through induction of ribosomal protein expression and increased protein translation [73]. Importantly, TOR acts to inhibit autophagy under such growth-promoting conditions and, while this is mediated through its inhibitory effects on Atg1 kinase activity in yeast and Drosophila, it is not yet clear how this is carried out in mammalian cells.
TOR kinase is repressed by signals that sense nutrient deprivation, including hypoxia. Upstream of TOR, activation of adenosine 5′-monophosphate (AMP)-activated protein kinase (AMPK) in response to low ATP levels promotes the inhibitory activity of the Tsc1/Tsc2 tumour suppressor proteins on Rheb, a small GTase required for mTOR activity [74]. Reduced Akt activity in response to reduced growth factor receptor activity also represses TOR kinase through Tsc1 and Tsc2, while TOR can be artificially inhibited by treatment of cells with rapamycin [73]. Thus, reduced TOR activity induces autophagy, again ensuring that the cell adapts to its changing environment through reduced growth and increased catabolism. Based on these observations and that TOR lies downstream of oncogenes such as Akt, use of rapamycin has been tested in clinical trials for cancer therapy, where it is postulated to be act to inhibit tumour growth by blocking protein translation and by inducing autophagy [75]. However, TOR can function as the catalytic component of two distinct complexes, known as TORC1 and TORC2, and rapamycin appears to have greater inhibitory activity against TORC1, driving the search for so-called ‘rapalogs’ that target both TORC1 and TORC2 [76].
As mentioned, hypoxia also activates autophagy [77] through effects that are both dependent on target genes induced by hypoxia-inducible factor (HIF) [77] and also through HIF-independent effects that are likely mediated through TOR inhibition downstream of AMPK, REDD1 and Tsc1/Tsc2 [74,78]. Given that hypoxia induces ER stress through the unfolded protein response, and that mitochondria have reduced function in oxidative phosphorylation under hypoxia, the induction of autophagy may allow the cell to eliminate portions of compacted ER and to reduce mitochondrial mass at a time when oxygen is not available to accept free electrons from the respiratory chain. This adaptive response to hypoxia would prevent wasteful ATP consumption at the ER and limit production of reactive oxygen species at the mitochondria. Increased autophagy would also allow the cell to generate ATP from catabolism at a time when ATP production by oxidative phosphorylation is limited.
Specific HIF targets in autophagy include BNIP3 and BNIP3L that are non-canonical members of the Bcl-2 superfamily of cell death regulators. Although linked to cell death, the normal function of these proteins appears to be in mitophagy [79,80]. As discussed, BNIP3L/NIX plays a physiological role in mitochondrial clearance from maturing reticulocytes [66,67], while BNIP3 has a similar role in cardiac and skeletal muscle in response to oxidative stress [81,82]. The extent to which BNIP3 and BNIP3L are functionally redundant is not resolved and differential regulation of their expression may explain aspects of their non-redundancy in vivo [83]. Various models have been proposed to explain how BNIP3/BNIP3L function in mitophagy [83], including a role for BNIP3 in derepressing Beclin-1 through disruption of its interaction with Bcl-2 [84]. However, a more direct role for BNIP3L in promoting mitochondrial clearance through interaction with the LC3-related molecule GABARAP has also been demonstrated [41], while BNIP3 interacts with Rheb, suggesting an additional indirect role in hypoxia-induced autophagy [85].
Autophagy is known to induce cell cycle arrest and, while it appears that this may be largely driven by nutrient deprivation-induced inhibition of TOR activity and downstream effects on translation of key cell cycle genes, such as cyclin D1 [86], it is not clear whether autophagy can induce cell cycle arrest independent of TOR signalling. This is an area of research that will likely be of increased interest moving forward, given its importance to understanding how and at what stages autophagy acts in tumour progression.
Conclusions
There remain specific challenges to our understanding of autophagy in mammalian cells, including how the phagophore emerges in the first place, how specific cargo is targeted for degradation, and how alternative Atg5/Atg7-independent mechanisms of autophagy are regulated. However, the significance of defects in autophagy for disease and ageing is apparent from growing evidence linking mutation or loss of function of key autophagy genes in cancer, neuropathies, heart disease, auto-immune disease and other conditions. From the perspective of a cancer biologist, it remains controversial whether autophagy is tumour suppressive (through cell cycle arrest, promoting genome and organelle integrity, or through inhibition of necrosis and inflammation) or oncogenic (by promoting cell survival in the face of spontaneous or induced nutrient stress). In other diseases, such as neuropathies (Huntington's, Alzheimer's and Parkinson's diseases) and ischaemic heart disease, autophagy is more widely accepted as beneficial given its role in eliminating ‘toxic assets’ and promoting cell viability. Thus, autophagy has emerged as a new and potent modulator of disease progression that is both scientifically intriguing and clinically relevant.
Acknowledgment
The authors acknowledge financial support from the National Cancer Institute (Grant No. RO1 CA131188; to KFM) and the Swiss National Foundation (Award No. PBZHP3-123296; to SB).
Footnotes
Teaching materials
PowerPoint slides of the Figures from this review are supplied as supporting information in the online version of this article.
References
1. Deter RL, De Duve C. Influence of glucagon, an inducer of cellular autophagy, on some physical properties of rat liver lysosomes. J Cell Biol. 1967;33:437–449. [PMC free article] [PubMed] [Google Scholar]
2. Yin XM, Ding WX, Gao W. Autophagy in the liver. Hepatology. 2008;47:1773–1785. [PubMed] [Google Scholar]
3. Nakatogawa H, Suzuki K, Kamada Y, Ohsumi Y. Dynamics and diversity in autophagy mechanisms: lessons from yeast. Nat Rev Mol Cell Biol. 2009;10:458–467. [PubMed] [Google Scholar]
4. Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27–42. [PMC free article] [PubMed] [Google Scholar]
5. Mizushima N. Autophagy: process and function. Genes Dev. 2007;21:2861–2873. [PubMed] [Google Scholar]
6. Xie Z, Klionsky DJ. Autophagosome formation: core machinery and adaptations. Nat Cell Biol. 2007;9:1102–1109. [PubMed] [Google Scholar]
7. Klionsky DJ. Autophagy: from phenomenology to molecular understanding in less than a decade. Nat Rev Mol Cell Biol. 2007;8:931–937. [PubMed] [Google Scholar]
8. Saftig P, Beertsen W, Eskelinen EL. LAMP-2. Autophagy. 2008;4:510–512. [PubMed] [Google Scholar]
9. Axe EL, Walker SA, Manifava M, Chandra P, Roderick HL, Habermann A, et al. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J Cell Biol. 2008;182:685–701. [PMC free article] [PubMed] [Google Scholar]
10. Simonsen A, Tooze SA. Coordination of membrane events during autophagy by multiple class III PI3–kinase complexes. J Cell Biol. 2009;186:773–782. [PMC free article] [PubMed] [Google Scholar]
11. Hayashi-Nishino M, Fujita N, Noda T, Yamaguchi A, Yoshimori T, Yamamoto A. A subdomain of the endoplasmic reticulum forms a cradle for autophagosome formation. Nat Cell Biol. 2009;11:1433–1437. [PubMed] [Google Scholar]
12. Yla-Anttila P, Vihinen H, Jokitalo E, Eskelinen EL. 3D tomography reveals connections between the phagophore and endoplasmic reticulum. Autophagy. 2009;5:1180–1185. [PubMed] [Google Scholar]
13. Mizushima N, Klionsky DJ. Protein turnover via autophagy: implications for metabolism. Ann Rev Nutr. 2007;27:19–40. [PubMed] [Google Scholar]
14. English L, Chemali M, Duron J, Rondeau C, Laplante A, Gingras D, et al. Autophagy enhances the presentation of endogenous viral antigens on MHC class I molecules during HSV-1 infection. Nat Immunol. 2009;10:480–487. [PMC free article] [PubMed] [Google Scholar]
15. Kundu M, Thompson CB. Macroautophagy versus mitochondrial autophagy: a question of fate? Cell Death Diff. 2005;12:1484–1489. [PubMed] [Google Scholar]
16. Diaz-Troya S, Perez-Perez ME, Florencio FJ, Crespo JL. The role of TOR in autophagy regulation from yeast to plants and mammals. Autophagy. 2008;4:851–865. [PubMed] [Google Scholar]
17. Kundu M, Lindsten T, Yang CY, Wu J, Zhao F, Zhang J, et al. Ulk1 plays a critical role in the autophagic clearance of mitochondria and ribosomes during reticulocyte maturation. Blood. 2008;112:1493–1502. [PMC free article] [PubMed] [Google Scholar]
18. Backer JM. The reguation and function of Class III PI3Ks: novel roles for Vps34. Biochem J. 2008;410:1–17. [PubMed] [Google Scholar]
19. Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H, et al. Induction of autophagy and inhibition of tumorigenesis by beclin1. Nature. 1999;402:672–676. [PubMed] [Google Scholar]
20. Qu X, Zou Z, Sun Q, Luby-Phelps K, Cheng P, Hogan RN, et al. Autophagy gene-dependent clearance of apoptotic cells during embryonic development. Cell. 2007;128:931–946. [PubMed] [Google Scholar]
21. Liang C, Feng P, Ku B, Dotan I, Canaani D, Oh BH, et al. Autophagic and tumour suppressor activity of a novel Beclin1-binding protein UVRAG. Nat Cell Biol. 2006;8:688–699. [PubMed] [Google Scholar]
22. Degenhardt K, Mathew R, Beaudoin B, Bray K, Anderson KL, Chen G, et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation and tumorigenesis. Cancer Cell. 2006;10:51–64. [PMC free article] [PubMed] [Google Scholar]
23. Karantza-Wadsworth V, Patel S, Kravchuk O, Chen G, Mathew R, Jin S, et al. Autophagy mitigates metabolic stress and genome damage in mammary tumorigenesis. Genes Dev. 2007;21:1621–1635. [PMC free article] [PubMed] [Google Scholar]
24. Young ARJ, Narita M, Ferreira M, Kirschner K, Sadaie M, Darot JFJ, et al. Autophagy mediates the mitotic senescence transition. Genes Dev. 2009;23:798–803. [PMC free article] [PubMed] [Google Scholar]
25. Amaravadi RK, Yu DS, Lum JJ, Bui T, Christophorou MA, Evan GI, et al. Autophagy inhibition enhances therapy-induced apoptosis in a Myc-induced model of lymphoma. J Clin Invest. 2007;117:326–336. [PMC free article] [PubMed] [Google Scholar]
26. Fimia GM, Stoykova A, Romagnoli AG, Giunta L, Di Bartolomeo S, Nardacci R, Corazzari M, et al. Ambra1 regulates autophagy and development of the nervous system. Nature. 2007;447:1121–1125. [PubMed] [Google Scholar]
27. Matsunaga K, Saitoh T, Tabatra K, Omori H, Satoh T, Kurotori N, et al. Two Beclin1-binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nat Cell Biol. 2009;11:385–396. [PubMed] [Google Scholar]
28. Zhong Y, Wang QJ, Li X, Yan Y, Backer JM, Chait BT, et al. Distinct regulation of autophagic activity by Atg14L and Rubicon associated with Beclin1–phosphatidylinositol 3-kinase complex. Nat Cell Biol. 2009;11:468–476. [PMC free article] [PubMed] [Google Scholar]
29. Pattingre S, Tassa A, Qu X, Garuti R, Linag XH, Mizushima N, et al. Bcl-2 anti-apoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. 2005;122:927–939. [PubMed] [Google Scholar]
30. Maiuri MC, Le Toumelin G, Criollo A, Rain JC, Gautier F, Juin P, et al. Functional and physical interaction between Bcl-XL and a BH3-like domain in Beclin1. EMBO J. 2007;26:2527–2539. [PMC free article] [PubMed] [Google Scholar]
31. Wei G, Pattingre S, Sinha S, Bassik MC, Levine B. JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol Cell. 2008;30:678–688. [PMC free article] [PubMed] [Google Scholar]
32. Yousefi S, Perozzo R, Schmid I, Zieiecki A, Schaffner T, Scapozza L, et al. Calpain-mediated cleavage of Atg5 switches autophagy to apoptosis. Nat Cell Biol. 2006;8:1124–1132. [PubMed] [Google Scholar]
33. Maiuri MC, Zalckvar E, Kimchi A, Kroemer G. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Biol. 2007;8 [PubMed] [Google Scholar]
34. Kirkin V, McEwan DG, Novak I, Dikic I. A role for ubiquitin in selective autophagy. Mol Cell. 2009;34:259–269. [PubMed] [Google Scholar]
35. Barth S, Glick D, Macleod KF. Autophagy: assays and artifacts. J Pathol. 2010 DOI: 10.1002/path.2694. [PMC free article] [PubMed] [Google Scholar]